

Abstract
Increased medial arterial thickness is a structural change in pulmonary arterial hypertension (PAH). The role of smooth muscle hypertrophy in this process has not been well studied. Bone morphogenetic proteins (BMPs), transforming growth factor (TGF)-β1, serotonin (or 5-hydroxytryptamine; 5-HT), and endothelin (ET)-1 have been implicated in PAH pathogenesis. We examined the effect of these mediators on human pulmonary artery smooth muscle cell size, contractile protein expression, and contractile function, as well on the roles of glycogen synthase kinase (GSK)-3β and p70 ribosomal S6 kinase (p70S6K), two proteins involved in translational control, in this process. Unlike epidermal growth factor, BMP-4, TGF-β1, 5-HT, and ET-1 each increased smooth muscle cell size, contractile protein expression, fractional cell shortening, and GSK-3β phosphorylation. GSK-3β inhibition by lithium or SB-216763 increased cell size, protein synthesis, and contractile protein expression. Expression of a non-phosphorylatable GSK-3β mutant blocked BMP-4-, TGF-β1-, 5-HT-, and ET-1-induced cell size enlargement, suggesting that GSK-3β phosphorylation is required and sufficient for cellular hypertrophy. However, BMP-4, TGF-β1, 5-HT, and ET-1 stimulation was accompanied by an increase in serum response factor transcriptional activation but not eIF2 phosphorylation, suggesting that GSK-3β-mediated hypertrophy occurs via transcriptional, not translational, control. Finally, BMP-4, TGF-β1, 5-HT, and ET-1 treatment induced phosphorylation of p70S6K and ribosomal protein S6, and siRNAs against p70S6K and S6 blocked the hypertrophic response. We conclude that mediators implicated in the pathogenesis of PAH induce pulmonary arterial smooth muscle hypertrophy. Identification of the signaling pathways regulating vascular smooth muscle hypertrophy may define new therapeutic targets for PAH.
Keywords: bone morphogenetic proteins, transforming growth factor-β1, serotonin, endothelin-1
pulmonary vascular remodeling is an important pathological feature of pulmonary arterial hypertension (PAH). Remodeling is characterized by thickening of all three layers of the pulmonary artery wall: the adventitia, the media, and the intima. In addition, there is extension of new smooth muscle into the partially muscular and nonmuscular peripheral arteries (31). Little information exists regarding the basis for the observed increase in vascular smooth muscle mass (hyperplasia vs. hypertrophy). In rats exposed to hypobaric hypoxia, hilar pulmonary artery medial thickness and individual myocyte diameter increase two- to threefold (31), whereas the number of myocytes per unit area nearly halves (32). There is only a small increase in DNA synthesis by medial smooth muscle cells, in contrast to robust increases in fibroblast and endothelial cell labeling. More recently, it has been shown that bovine distal artery media is composed of myocytes that are resistant to proliferation and instead hypertrophy in response to growth-promoting stimuli (42). These data suggest that cellular hypertrophy, as well as hyperplasia, may contribute to medial thickening in PAH.
Bone morphogenetic protein (BMP), transforming growth factor (TGF)-β, serotonin or 5-hydroxytryptamine (5-HT), and endothelin (ET)-1 have each been implicated in the pathogenesis of PAH (4, 5, 7, 12, 13, 23, 26, 30, 45–47, 53). Although the effects of these mediators on pulmonary artery smooth muscle proliferation has been well studied, little is known about the potential effects of BMPs, TGF-β, 5-HT, and ET-1 on vascular smooth muscle cell contractile protein expression or contractility. ET-1 (21, 54) and 5-HT (11, 29) each exert mitogenic activity for pulmonary artery smooth muscle cells. Myocytes from patients with PAH grow faster than controls when stimulated by 5-HT or serum (30). In human pulmonary artery smooth muscle cells, TGF-β initially promotes contractile protein expression, followed by proliferation at a later time point (43). In contrast, BMP-4 inhibits the proliferation of pulmonary artery smooth muscle cells from proximal pulmonary arteries (52). In myocytes from patients with PAH, TGF-β and BMPs fail to inhibit serum-stimulated DNA synthesis as in normal cells (34). Finally, serotonin increases the cell size of bovine pulmonary artery smooth muscle cells in culture (27).
In myocytes from patients with PAH, myofilament area and the proportions of Golgi and rough sarcoplasmic reticulum are increased (6), consistent with increased protein synthesis. Increases in cellular protein synthetic rates, in turn, can result from: 1) augmentation of transcription; 2) increased mRNA stability; and 3) accelerated translation rates. Numerous studies have shown that changes in the rate of transcription can account for qualitative changes in the expression of specific genes during hypertrophic growth. For example, in the heart, a majority of proteins that comprise the “fetal gene program,” i.e., β-myosin heavy chain (MHC), skeletal α-actin, and cardiac α-actin, are regulated at the level of transcription (3, 36, 37). On the other hand, electrical stimulation of adult feline cardiocytes acutely increases β-MHC synthesis without a corresponding change in steady-state mRNA levels, and β-MHC synthesis is accompanied by a shift of mRNA into larger polysomes, indicative of increased translational efficiency (25). Conversely, mechanical inactivity, which depresses protein expression, blocks translation at initiation, increasing the nonpolysomal RNA fraction and decreasing the amount in the polysomal fraction (35). Thus, accelerated translation rate, as well as augmented transcription, contributes to cardiac myocyte hypertrophy. Translational control mechanisms also modulate skeletal muscle gene expression during hypertrophy (16, 39).
The translational control mechanisms regulating protein synthesis in vascular smooth muscle cells are not completely understood. There are three highly regulated steps in mRNA translation, each of which is controlled by a distinct biochemical signaling pathway. The first is binding of initiator methionyl tRNA to the 40S ribosomal subunit to form the 43S preinitiation complex, which requires formation of the eukaryotic initiation factor (eIF)2·GTP·Met-tRNAi ternary complex. eIF2 GTP loading is determined by the activity of eIF2B, a guanine nucleotide exchange factor. eIF2Bε Ser539 phosphorylation by the constitutively active serine-threonine kinase glycogen synthase kinase (GSK)-3β inhibits its GDP/GTP exchange activity, thereby limiting binding of methionyl tRNA to the 40S ribosomal subunit (50). Phosphorylation of GSK-3β by the serine-threonine kinase Akt inactivates it, increasing formation of the ternary and 43S preinitiation complexes. In rat aortic smooth muscle cells, ET-1 stimulates phosphorylation and inactivation of GSK-3β (44). The second step involves mRNA binding to the 43S preinitiation complex, mediated via a 7-methylguanosine cap at the 5′ end of mRNAs. Phosphorylation of eIF-4E binding protein (4E-BP) by mammalian target of rapamycin (mTOR) releases it from eIF-4E, allowing eIF-4E to bind to the mRNA cap (17, 55). Angiotensin II induces phosphorylation of eIF-4E in rat aortic smooth muscle cells (38). Rapamycin, an inhibitor of mTOR, blocks angiotensin II-induced hypertrophy of rat aortic smooth muscle cells (15, 51). Mnk1, an eIF4E kinase, is required for angiotensin II-induced protein synthesis in rat aortic smooth muscle cells (24). Third, protein synthesis may also be upregulated by an increase in translational capacity, i.e., ribosome synthesis. Translation of mRNAs with 5′-terminal oligopyrimidine (5-TOP) tracts, most of which encode ribosomal proteins, is upregulated by successive phosphorylation of mTOR, p70 ribosomal S6 kinase (p70S6K)-1, and S6 ribosomal protein. In rat aortic smooth muscle, chemical inhibitors of p70S6K (tosylphenylalanine chloromethyl ketone and tosyllysine chloromethyl ketone) had no effect on angiotensin II-induced protein synthesis (51), suggesting that p70S6K is not involved in vascular smooth muscle hypertrophy driven by angiotensin II. Any or all of these three pathways may be required (or sufficient) for hypertrophy.
GSK-3β may also regulate smooth muscle cell size by transcriptional mechanisms. GSK-3β negatively regulates transcription factors involved in muscle-specific gene expression, including NFAT (nuclear factors of activated T cells), GATA4, β-catenin, and serum response factor (SRF) (1, 2, 10, 19, 20, 33, 48).
The aim of the study was to evaluate whether BMP-4, TGF-β1, 5-HT, or ET-1 induce hypertrophy in pulmonary artery smooth muscle cells. In addition, we sought to determine the signaling mechanisms regulating mRNA translation in this system, focusing on the GSK-3β and p70S6K pathways.
METHODS
Cell culture.
Human pulmonary artery smooth muscle cells were obtained from Lonza (Conshohocken, PA). All cytokines were obtained from PeproTech (Rocky Hill, NJ). LiCl and SB-216763 were obtained from Sigma-Aldrich (St. Louis, MO). Cells were cultured in DMEM with 10% FBS and penicillin/streptomycin (InVitrogen, Carlsbad, CA). Cells were seeded on uncoated plastic culture plates at ∼50% confluence. Before experiments, cells were serum-deprived for 24 h. Cells were treated with BMP-4 (10 ng/ml), TGF-β1 (10 ng/ml), 5-HT (1 μmol/l), ET-1 (1 μmol/l), LiCl (10 mM), SB-216763 (50 nM), and EGF (50 ng/ml) for 4 days. This relatively long incubation time was required for the observed phenotypic changes. Fresh medium and chemicals were added 48 h after initial treatment. Experiments were performed in the absence of serum. For selected experiments, A7R5 rat aortic smooth muscle cells (American Type Culture Collection, Manassas, VA) were studied.
Cell size analysis.
Cell size was measured by fluorescence-activated cell sorting. Cells were treated with BMP-4, TGF-β, 5-HT, ET-1, LiCl, SB-216763, or EGF. Cells were collected and fixed with 75% ethanol and stored at −20°C before staining. Cells were centrifuged and stained with propidium iodide (50 μg/ml) and RNase (100 μg/ml) solution for 1 h. Cells in G0/G1 phase were gated for forward scatter measurement using a FACSCalibur flow cytometer (BD Biosciences).
Protein and DNA synthesis.
Cells were serum-starved for 24 h before experiments. Cells were plated at 5 × 105 cells/well (or 3 × 105 cells/well for experiments involving transfection) and incubated in [3H]leucine or [3H]thymidine (0.5 μCi, PerkinElmer Life Sciences) for 48 h. Cells were lysed, and proteins were precipitated with 10% trichloroacetic acid. After washing with cold ethanol and solubilization with 1% Triton X-100 in 0.5 mol/l NaOH, radioactivity was measured by a scintillation counter.
Cell contraction.
Individual cell length before and after KCl-induced contraction was measured by computerized image micrometry, as described (17). Cells were seeded in 100-mm dishes and grown to confluence in serum-free medium or medium supplemented with BMP-4, TGF-β1, 5-HT, or ET-1. At confluence, cells were scraped off with a rubber policeman, triturated, and transferred to polypropylene tubes. At this stage, cells tend to maintain a contracted state due to mechanical stimulation. The cells were treated with 8-bromo-cAMP and then allowed to float freely and relax for 24 h with occasional swirling to prevent settling or sticking to the sides of the tube. During this period, cells regain a spindle shape and extend processes. Aliquots of cultured cell suspension (2.5 × 104 cells/0.5 ml) were stimulated with 75 mM KCl. The reaction was allowed to proceed for 4 min and was stopped by the addition of 0.1 ml of glutaraldehyde at a final concentration of 1% (vol/vol). Fixed cells were allowed to settle and were then transferred by wide-mouth pipette to a microscope slide for analysis. The average length of cells before or after the addition of test agents was obtained from 20 cells encountered in successive microscopic fields.
Immunoblotting.
Cell lysates were matched for protein concentration, resolved by SDS-PAGE, and transferred to nitrocellulose or polyvinylidene difluoride membrane. Membranes were blocked in 5% milk for 1 h and probed with either mouse anti-α-smooth muscle actin (Calbiochem), mouse anti-smMHC (Sigma), rabbit anti-phospho-Ser9 GSK-3β, rabbit anti-GSK-3β, rabbit anti-phospho-p70 S6 kinase, rabbit anti-p70 S6 kinase, rabbit anti-phospho-ribosomal protein S6, rabbit anti-ribosomal protein S6 (each from Cell Signaling, Danvers, MA), rabbit anti-phospho-Ser539 eIF2Bϵ (Biosource, Camarillo, CA), or anti-phosphotyrosine mouse monoclonal 4G10 (Millipore, Billerica, MA). Antibody binding was detected with a peroxidase-conjugated anti-rabbit or anti-mouse IgG and chemiluminescence. Pixel densitometry was performed using NIH Image.
Fluorescence microscopy.
Cells were grown on collagen-coated glass slides (BD Biosciences) and fixed in 1% paraformaldehyde. To stain filamentous actin, slides were incubated with Alexa Fluor 488-conjugated phalloidin (Molecular Probes, Eugene, OR). For immunocytochemistry, slides were probed with Cy3-conjugated mouse anti-α-smooth muscle actin-Cy3 (Sigma) followed by Alexa Fluor 594-labeled goat anti-mouse IgG (Molecular Probes) or phospho-GSK-3β antibody followed by Alexa Fluor 488-labeled goat anti-rabbit IgG (Molecular Probes).
Retroviral transduction of A7R5 cells.
DNA encoding a nonphosphorylatable GSK-3β (GSK-3β-A9), with Ser9 replaced by alanine, was provided by Dr. Anne Vojtek (Univ. of Michigan). Expression of GSK-3β-A9 acts as a “dominant-negative,” decreasing the binding of upstream kinases and scaffolding proteins to native GSK-3β. This leads to a relative reduction of phosphorylated, inactive GSK-3β and an increase in GSK-3β activity. GSK-3β-A9 cDNA was subcloned into the pMSCVpuro retroviral vector (BD Biosciences). The Phoenix-GP retrovirus packaging cell line, a 293-cell derivative line that expresses only the gag-pol viral components (provided by G. Nolan, Stanford University), was transiently transfected with pHCMV-G, which contains the vesicular stomatitis virus envelope glycoprotein, and either pMSCVpuro-AA-GSK-3β-A9 or pMSCV alone. Viral supernatant was collected, filtered, and supplemented with Polybrene (8 μg/ml). A7R5 cells were infected with viral supernatant (4 times for 4 h each). Infected cells were selected with puromycin (2 μg/ml). After selection, cells were grown to confluence, split into six-well plates, and incubated in the absence or presence of BMP-4, TGF-β, 5-HT, ET-1, LiCl, or SB-216763.
Reporter assay.
A7R5 cells were used for these experiments because of their superior transfection efficiency. Cells were transiently transfected with 200 ng of SRF-luc (from J. Solway, Univ. of Chicago). Three nanograms of the SV40 Renilla luciferase vector was used as a transfection control. Cells were transfected using Lipofectamine 2000 (Invitrogen). The following day, cells were serum-deprived for 2 h and treated with BMP-4, TGF-β1, 5-HT, or ET-1 for 48 h. Cells were subsequently lysed, and luciferase activity was measured using the Promega luciferase assay system (Madison, WI).
Quantitative PCR of α-actin mRNA.
Human pulmonary artery smooth muscle cells were treated with BMP-4, TGF-β1, 5-HT, ET-1, LiCl, or SB-216763, processed for mRNA, and first-strand cDNA synthesized as described (10). qPCR was conducted using SYBR Green 1 fluorescence (human α-actin forward primer, 5′-GAC CCT GAA GTA CCC GAT AGA AC-3′; reverse primer 5′-GGG CAA CAC GAA GCT CAT TG-3′). GAPDH mRNA was used as an internal control (forward primer, 5′ CTT CAC CAC CAT GGA GAA GGC 3′; reverse primer, 5′ GGC ATG GAC TGT GGT CAT GAG 3′). Samples were run in triplicate, and the cycle threshold (CT) was determined. Relative gene expression was calculated as previously described (10).
Transfection of siRNA against p70S6K and ribosomal protein S6.
21-bp duplexes of either p70S6K or ribosomal protein S6 siRNA (both from Dharmacon, Lafayette, CO) were transfected into subconfluent human pulmonary artery smooth muscle cells using RNAiMAX in OptiMEM (Invitrogen). For p70S6K, a pool of double-stranded siRNAs containing equal parts of the following antisense sequences was used: 1, 1, 5′-CAAGGUCAUGUGAAACUAA-3′; 2, 5′-GAGAGUCAAUGUCAUUACA-3′; 3, 5′-CUCGCGACAUCUUUCUCAA-3′; 4, 5′-PCAAAGAUCAACUCUGGUGCUU-3′. For ribosomal protein S6 siRNA, a pool of double-stranded siRNAs containing equal parts of the following antisense sequences was used: 1, 5′-GAAGCAGCGUACCAAGAAA-3′; 2, 5′-CUGCGAGCUUCUACUUCUA-3′; 3, 5′-GUCUGAAUCCAGUCAGAAA-3′. The corresponding nontargeting siRNA sequence was 5′-CGAACUCACUGGUCUGACCdtdt-3′ (sense), 5′-GGUCAGACCAGUGAGUUCGdtdt-3′ (antisense). Six hours later, DMEM and FBS were added. The next morning, cells were incubated in fresh DMEM containing 10% FBS for 24 h. Finally, cells were treated with the relevant stimulus in serum-free medium for 2 days before harvest.
RESULTS
BMP-4, TGF-β1, 5-HT, ET-1, and GSK-3β inhibitors increase pulmonary artery smooth muscle cell size and protein synthesis.
We first characterized the effects of BMP-4, TGF-β1, 5-HT, and ET-1 on cell size, protein synthesis, and DNA synthesis. We also examined the effects of EGF, a potent mitogen for pulmonary artery smooth muscle cells (40), which we would not expect to cause cellular hypertrophy. We found that cell size was increased by treatment with BMP-4, TGF-β1, 5-HT, and ET-1, as indicated by the rightward shift of the forward scatter compared with the control (Fig. 1A). In contrast, EGF treatment did not alter the size of cells in G0/G1 phase. BMP-4, TGF-β1, 5-HT, and ET-1 also potently stimulated protein synthesis (Fig. 1B). No effect on DNA synthesis except for ET-1 was found in these cells (Fig. 1C), indicating that besides stimulating cell enlargement, ET-1 also promotes cell proliferation. We also examined the effect of GSK-3β inhibition on cell size and protein synthesis using two GSK-3β inhibitors, LiCl and SB-216763. LiCl and SB-216763 each caused an enlargement of cell size relative to control (Fig. 1A) and an increase in protein synthesis (Fig. 1B) but not DNA synthesis (Fig. 1C).
Fig. 1.
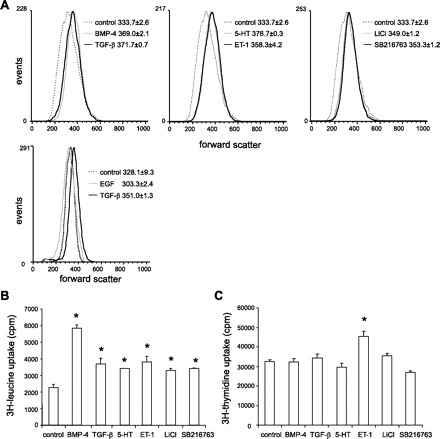
Bone morphogenetic protein (BMP)-4, transforming growth factor (TGF)-β1, serotonin (or 5-hydroxytryptamine; 5-HT), endothelin (ET)-1, and glycogen synthase kinase (GSK)-3β inhibitors increase pulmonary smooth muscle cell size and protein synthesis. A: change in forward scatter in human pulmonary artery smooth muscle cells treated with PBS, BMP-4, TGF-β1, 5-HT, ET-1, LiCl, SB-216763, and EGF. B: overall protein synthesis of cells treated with PBS, BMP-4, TGF-β1, 5-HT, ET-1, LiCl, or SB-216763, as assessed by [3H]leucine incorporation (cpm/well). C: Overall DNA synthesis of cells treated with PBS, BMP-4, TGF-β1, 5-HT, ET-1, LiCl, or SB-216763, as assessed by [3H]thymidine incorporation (cpm/well); n = 3, means ± SE; *P < 0.05, ANOVA.
BMP-4, TGF-β, 5-HT, and ET-1 increase contractile protein expression.
Expression of contractile proteins was measured by immunoblot. BMP-4, TGF-β1, 5-HT, and ET-1 all increased smooth muscle α-actin and MHC protein expression without affecting that of β-actin (Fig. 2A). GSK-3β inhibitors LiCl and SB-216763 also increased α-actin and MHC expression (Fig. 2B). On the other hand, the growth factor EGF decreased the amount of α-actin relative to β-actin while increasing tyrosine phosphorylation of a protein the size of the EGF receptor (Fig. 2C). Immunocytochemical stains showed increased α-smooth muscle actin content in cells treated with agents causing hypertrophy (Fig. 2D).
Fig. 2.
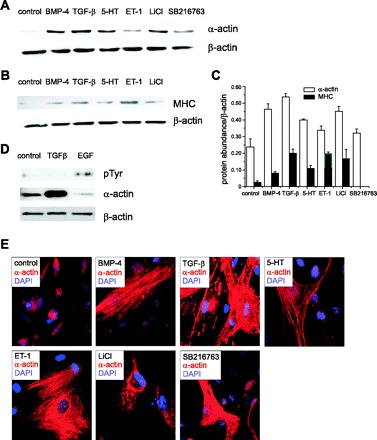
BMP-4, TGF-β1, 5-HT, and ET-1 increase contractile protein expression. Representative immunoblots for α-smooth muscle actin (A) and MHC (B). β-actin expression was employed as a control. C: group mean data (± SE). D: EGF does not increase α-actin expression. EGF treatment induced tyrosine phosphorylation of a 120-kDa protein, likely the EGF receptor. As a positive control, the α-actin response to TGF-β is shown. E: cells were preincubated with BMP-4, TGF-β, 5-HT, ET-1, LiCl, or SB-216763 and stained with Cy3-conjugated anti-α-actin. Nuclei were stained with DAPI.
BMP-4, TGF-β, 5-HT, and ET-1 each increase cell shortening in response to KCl.
To determine whether the hypertrophic effect induced by BMP-4, TGF-β1, 5-HT, and ET-1 was accompanied by an increase in contractility, we compared the shortening response to KCl in cells with or without treatment with BMP-4, TGF-β1, 5-HT, and ET-1. Treatment of cells with BMP-4, TGF-β1, 5-HT, and ET-1 each significantly increased resting length and fractional change in length compared with PBS (Fig. 3).
Fig. 3.
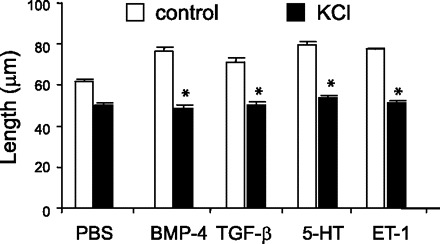
BMP-4, TGF-β1, 5-HT, and ET-1 each increase cell shortening in response to KCl. Compared with control cells, treatment with BMP-4, TGF-β1, 5-HT, and ET-1 each increased resting length and fractional shortening in response to 75 mM KCl. N = 20 for each group; means ± SE; *greater than control cells without KCl treatment, P < 0.05, ANOVA.
Effects of BMP-4, TGF-β1, 5-HT, ET-1, and GSK-3β inhibitors on GSK-3β phosphorylation.
To gain insight into the mechanisms by which BMP-4, TGF-β1, 5-HT, and ET-1 increase cell size and protein synthesis, we examined the ability of these compounds to induce phosphorylation of GSK-3β, a kinase that negatively regulates cell hypertrophy. Immunoblot analysis showed increased phosphorylation of GSK-3β following treatment with BMP-4, TGF-β1, 5-HT, ET-1, and LiCl (Fig. 4A). As expected, SB-21673, a permeable, structurally distinct maleimide that inhibits GSK-3 activity (8), did not induce phosphorylation.
Fig. 4.
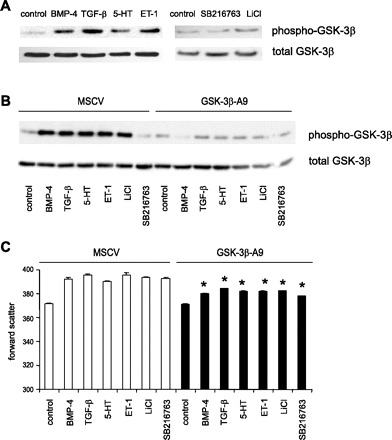
Phosphorylation of GSK-3β is required for BMP-4-, TGF-β1-, 5-HT-, and ET-1-induced hypertrophy. A: representative immunoblots for phospho-GSK-3β and total GSK-3β in human pulmonary artery smooth muscle cells treated with BMP-4, TGF-β1, 5-HT, ET-1, LiCl, and SB-216763. B: GSK-3β-A9 was expressed in A7R5 cells via retroviral gene transfer. Expression of GSK-3β-A9 acts as a “dominant-negative,” decreasing the binding of upstream kinases and scaffolding proteins to native GSK-3β. This leads to a relative reduction of phosphorylated, inactive GSK-3β, and an increase in GSK-3β activity. C: effect of GSK-3β-A9 overexpression on the size of cells treated with BMP-4, TGF-β1, 5-HT, ET-1, LiCl, or SB-216763 (*different from MSCV-transduced cells, P < 0.05, ANOVA).
Phosphorylation of GSK-3β is required for BMP-4, TGF-β1, 5-HT, and ET-1-induced hypertrophy.
To determine the requirement of GSK-3β phosphorylation for BMP-4, TGF-β1, 5-HT, and ET-1-induced cell enlargement, we expressed GSK-3β-A9, a GSK-3β mutant that cannot be phosphorylated at Ser9, in A7R5 cells via retroviral gene transfer, and determined its effect on cell size. In cells infected with empty MSCV vector, BMP-4, TGF-β1, 5-HT, ET-1, and LiCl but not SB-216763 increased GSK-3β phosphorylation (Fig. 4B). As expected, in cells infected with GSK-3β-A9, phosphorylation of GSK-3β was attenuated. BMP-4, TGF-β1, 5-HT, ET-1, and the GSK-3β inhibitors each caused an increase in the forward scatter of cells infected with empty vector, which was blocked by GSK-3β-A9 overexpression (Fig. 4C). These data provide evidence that phosphorylation of GSK-3β is required for BMP-4-, TGF-β1-, 5-HT-, and ET-1-induced cell enlargement.
Mechanism of GSK-3β-mediated cellular hypertrophy.
To explore whether the GSK-3β translational pathway mediates the hypertrophic effect, we measured the expression of phospho-eIF2B, the downstream phosphorylation target of GSK-3β that recruits methionyl tRNA to the 40S ribosomal subunit. We found that BMP-4, TGF-β1, 5-HT, and ET-1 did not affect eIF2B phosphorylation, whereas LiCl and SB-21673 caused a striking reduction in phosphorylation (Fig. 5A). These data indicate that BMP-4, TGF-β1, 5-HT, and ET-1 induce cell hypertrophy by a mechanism other than GSK-3β/eIF2-mediated translational control.
Fig. 5.
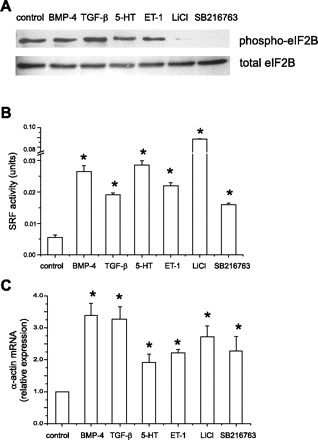
Mechanism of GSK-3β-mediated cell hypertrophy. A: representative immunoblots for phospho- and total eIF2B in pulmonary artery smooth muscle cells treated with BMP-4, TGF-β1, 5-HT, ET-1, and GSK-3β inhibitors. B: effect of BMP-4, TGF-β1, 5-HT, ET-1, LiCl, and SB-216763 on serum response factor (SRF) reporter activity. A7R5 cells were transiently transfected with SV40 Renilla luciferase vector and SRF-luc. Forty-eight hours after treatment, cells were lysed and luciferase activity determined. Each stimulus increased SRF activity (n = 8, means ± SE; *different from control cells, P < 0.05, ANOVA). C: effect of BMP-4, TGF-β1, 5-HT, ET-1, LiCl, and SB-216763 on α-actin mRNA in human pulmonary artery cells. Cells were treated for 4 days and processed for qPCR analysis of α-actin mRNA levels relative to GAPDH mRNA. Each stimulus increased α-actin mRNA (n = 3, means ± SE, *different from control cells, P < 0.05, ANOVA).
To determine whether BMP-4, TGF-β1, 5-HT, and ET-1 regulate contractile protein gene expression in a transcriptional rather than a translational manner, we examined the effects of these soluble mediators on the transcriptional activity of SRF, a regulator of smooth muscle-specific gene expression (28, 41). We have previously shown that inhibition of GSK-3β induces transactivation of SRF in cultured human airway smooth muscle cells (10). In the current study, we found that BMP-4, TGF-β1, 5-HT, ET-1, and the GSK-3β inhibitors each increased the reporter activity of SRF (Fig. 5B), suggesting that the requirement of GSK-3β phosphorylation for cellular hypertrophy relates to its role in the transcription of genes encoding smooth muscle contractile proteins, rather than accelerated formation of the 43S preinitiation complex. This is further supported by significant increases in α-actin mRNA relative to GAPDH (Fig. 5C).
Activation of the p70S6 kinase pathway is required for BMP-4-, TGF-β1-, 5-HT-, and ET-1-induced hypertrophy.
Since GSK-3β-mediated hypertrophy does not involve translational control, we investigated the contribution of p70S6K signaling to BMP-4-, TGF-β1-, 5-HT-, and ET-1-mediated cell hypertrophy. BMP-4, TGF-β1, 5-HT, and ET-1 each increased the phosphorylation of p70S6K and its downstream substrate, ribosomal S6 (Fig. 6A), indicating an increase in p70S6K activity.
Fig. 6.
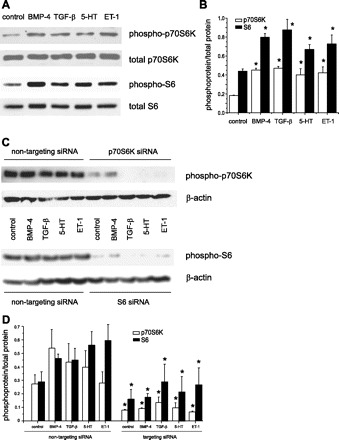
BMP-4, TGF-β1, 5-HT, and ET-1 activate the p70S6K signaling pathway. A: representative immunoblots for phospho-p70S6K, total p70S6K (top), phospho-S6, and total S6 (bottom) in pulmonary artery smooth muscle cells treated with BMP-4, TGF-β1, 5-HT, and ET-1. B: group mean data (n = 3, ± SE, *different from unstimulated cells, P < 0.05, ANOVA). C: specific siRNAs against p70S6K (top) and S6 (bottom) block phosphorylation of these proteins. D: group mean data (n = 3, ± SE, *different from nontargeting siRNA, P < 0.05, ANOVA).
To determine the requirement of p70S6K for cell hypertrophy and contractile protein expression, we used specific siRNAs against p70S6K and S6. As anticipated, the specific siRNAs decreased total p70S6K and S6 protein expression (Fig. 6B). siRNA against p70S6K blocked the increases in cell size caused by BMP-4, TGF-β1, 5-HT, or ET-1 treatment (Fig. 7A). Cell enlargement was also blocked by ribosomal protein S6 siRNA (Fig. 7B). These data suggest that p70S6K signaling is required for mediator-induced cell enlargement.
Fig. 7.
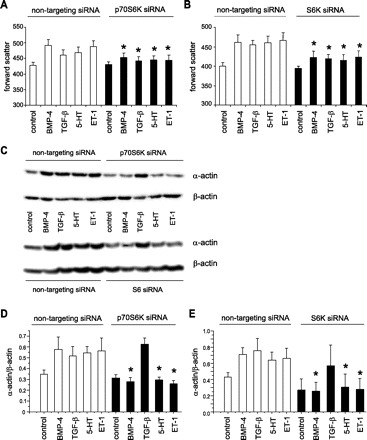
Activation of the p70S6K pathway is required for cell hypertrophy. Pulmonary artery smooth muscle cells were transfected with either nontargeting siRNA, specific siRNA against p70S6K (A), or siRNA against S6 (B), and treated with BMP-4, TGF-β1, 5-HT, or ET-1. Cell size was measured by flow cytometry. C: representative immunoblots for α-actin and β-actin from cells transfected with either nontargeting siRNA, p70S6K siRNA, or S6 siRNA. D: group mean data for p70S6K siRNA experiments (n = 3, ± SE, *different from nontargeting siRNA, P < 0.05, ANOVA). E: group mean data for S6 siRNA experiments (n = 3, ± SE, *different from nontargeting siRNA, P < 0.05, ANOVA).
We also examined the requirements of p70S6K and ribosomal S6 for BMP-4, TGF-β1, 5-HT, and ET-1-induced α-actin expression. siRNAs against p70S6K and S6 blocked the increases in contractile protein expression caused by BMP-4, 5-HT, and ET-1, but not TGF-β1 (Fig. 7C).
DISCUSSION
BMPs, TGF-β1, 5-HT, and ET-1 have each been implicated in the pathogenesis of PAH. Subsets of patients with familial and sporadic PAH may harbor related mutations or polymorphisms, most notably in BMP receptor (BMPR)-2 (26, 46), the TGF-β type 1 receptor, ALK1 (47), and the 5-HT transporter (12, 30). Genetic ablation of the BMPR2 gene in pulmonary endothelium is sufficient to predispose to PAH in mice (23). Patients with primary pulmonary hypertension show increased expression of TGF-β isoforms in the media and neointima of hypertensive muscular arteries (4), and TGF-β-dependent signaling is required for monocrotaline-induced pulmonary hypertension in rats (53). Mice expressing a dominant negative mutation of the TGF-β type II receptor fail to undergo hypoxia-induced pulmonary arterial hypertrophy (7). TGF-β/activin-like kinase 5 mediates abnormal proliferation of vascular smooth muscle cells from patients with familial pulmonary arterial hypertension and is involved in the progression of experimental pulmonary arterial hypertension induced by monocrotaline (45). The expression of 5-HT transporter is elevated in the lung tissues and pulmonary arteries of patients with PAH (12). Treatment with 5-HT potentiates the development of pulmonary hypertension in chronically hypoxic rats (13). ET-1 is increased in the lungs of patients with PAH (5).
Given the potential importance of BMPs, TGF-β, 5-HT, and ET-1 in the pathogenesis of PAH, and the fundamental significance of pulmonary artery thickening in this disease, we examined the effects of these mediators on human pulmonary artery smooth muscle cell size, protein and DNA synthesis, contractile protein expression, and fractional cell shortening. We show for the first time that BMP-4, TGF-β1, and ET-1 induce human pulmonary artery smooth muscle hypertrophy. Each mediator increased cell size, contractile protein expression, and fractional cell shortening. In contrast, only ET-1 increased DNA synthesis. On this basis, we speculate that pulmonary artery smooth muscle hypertrophy, as well as hyperplasia, may contribute to medial thickening in PAH. Increased medial thickening is likely to play an important physiological role in PAH, especially at an early stage before the obliterative arteriopathy characteristic of late state disease.
GSK-3β is a serine/threonine kinase that is constitutively active in unstimulated cells and becomes inactivated upon phosphorylation at Ser9 (9). Phosphorylation of GSK-3β by the serine-threonine kinase Akt inactivates it, leading to activation of eIF-2, which functions to recruit methionyl tRNA and conduct it as a tRNA-eIF2-GTP ternary complex to the 40S ribosomal subunit, leading to a general enhancement of translation initiation (50). GSK-3β also negatively regulates transcription factors involved in muscle-specific gene expression, including NFAT, GATA4, and β-catenin (1, 2, 19, 20, 33, 48). We have shown that inhibition of GSK-3β induces transactivation of SRF in cultured human airway smooth muscle cells (10). In the present study, BMP-4, TGF-β1, 5-HT, and ET-1 each increased the phosphorylation of GSK-3β. Two chemical GSK-3β inhibitors, LiCl and SB-216753, increased cell size, protein synthesis, and contractile protein expression. Overexpression of GSK-3β-A9, which cannot be phosphorylated or inactivated, blocked BMP-4-, TGF-β1-, 5-HT-, and ET-1-induced cell enlargement. Together, these data suggest that inhibition of GSK-3β is required and sufficient for human pulmonary artery smooth muscle cell hypertrophy.
To explore the mechanism by which GSK-3β mediates cellular hypertrophy, we measured the phosphorylation of eIF2B. We found that, while LiCl and SB-216763 decreased the phosphorylation of eIF2B, BMP-4, TGF-β1, 5-HT, and ET-1 had no effect, suggesting that the hypertrophic effect of these factors was translation independent. This result is different from the situation in airway smooth muscle (10). We next investigated whether these mediators activate a transcriptional control pathway. We measured the reporter activity of SRF, a regulator of a large subset of smooth muscle-specific genes (28, 41). We found that inhibition of GSK-3β increased SRF transactivation, supporting the notion that transcription of genes encoding contractile proteins accounts for the hypertrophic effect of BMP-4, TGF-β1, 5-HT, and ET-1.
Since GSK-3β-mediated hypertrophy does not involve translational control, we investigated the contribution of another translational control intermediate, p70S6K, to BMP-4-, TGF-β1-, 5-HT-, and ET-1-mediated cell hypertrophy. p70S6K is a mitogen and amino acid-sensitive serine-threonine kinase that ubiquitously regulates cell size. p70S6K is phosphorylated and activated by mTOR (14). p70S6K, in turn, phosphorylates the 40S ribosomal protein S6. The precise mechanism by which p70S6K controls translation is unclear. In addition to ribosomal protein S6, eukaryotic elongation factor-2 kinase is a phosphorylation target of p70S6K (49). In addition, p70S6K also mediates assembly of eukaryotic initiation factor-3 translation preinitiation complex (22). Rapamycin, an inhibitor of mTOR, blocks angiotensin II-induced aortic smooth muscle hypertrophy (15, 51). However, inhibition by rapamycin does not necessarily implicate p70S6K, since rapamycin also inhibits mTOR-mediated phosphorylation of eIF4E. In aortic smooth muscle, chemical inhibitors of p70S6K (tosylphenylalanine chloromethyl ketone and tosyllysine chloromethyl ketone) had no effect on angiotensin II-induced protein synthesis (51). In the present study, we found that BMP-4, TGF-β, 5-HT, and ET-1 each increased the phosphorylation of p70S6K and ribosomal protein S6 in pulmonary artery smooth muscle cells. We also found that transfection with specific siRNAs against p70S6K and S6 each blocked the cell enlargement induced by BMP-4, TGF-β, 5-HT, and ET-1, indicating that activation of p70S6K is required for the cell size enlargement induced by these factors. Furthermore, these data suggest that ribosomal protein S6 mediates the hypertrophic effect of p70S6K activation in this system. Interestingly, siRNAs against p70S6K and S6 also blocked contractile protein expression induced by BMP-4, 5-HT, and ET-1, but not TGF-β1. Thus, TGF-β1 must activate additional signaling pathways regulating contractile protein expression. For example, we have shown in human airway smooth muscle cells that TGF-β induces cell hypertrophy in part via activation of the 4E-BP/eIF4E pathway (17).
Finally, we should note that the precise ramifications of smooth muscle hypertrophy or hyperplasia on contraction have not been settled. While it stands to reason that increased muscle would result in increased shortening and a reduction in luminal diameter, this assumes that there is no change in smooth muscle function. However, in hypoxia-induced pulmonary hypertension, increased smooth muscle content is accompanied by an increase in connective tissue, leading to increased passive tissue stiffness and reduced active stress (18). In the current study, hypertrophied vascular smooth muscle cells demonstrated a greater fractional shortening, but length at end-contraction was unchanged.
In conclusion, BMP-4, TGF-β1, 5-HT, and ET-1 each induce human pulmonary artery smooth muscle hypertrophy, as evidenced by increases in cell size, protein synthesis, contractile protein expression, and fractional cell shortening. Hypertrophy is dependent on both phosphorylation and inhibition of GSK-3β and activation of p70S6K. Based on the potential contribution of vascular smooth muscle hypertrophy to pulmonary hypertension in the initial stages of the disease, identification of the signaling pathways regulating vascular smooth muscle hypertrophy may define new therapeutic targets for the early treatment of PAH.
GRANTS
This work was supported by the American Heart Association/Cardiovascular Medical Research and Education Fund.
DISCLOSURES
No conflicts of interest are declared by the author(s).
REFERENCES
- 1. Antos CL, McKinsey TA, Frey N, Kutschke W, McAnally J, Shelton JM, Richardson JA, Hill JA, Olson EN. Activated glycogen synthase-3 beta suppresses cardiac hypertrophy in vivo. Proc Natl Acad Sci USA 99: 907–912, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Armstrong DD, Esser KA. Wnt/β-catenin signaling activates growth-control genes during overload-induced skeletal muscle hypertrophy. Am J Physiol Cell Physiol 289: C853–C859, 2005 [DOI] [PubMed] [Google Scholar]
- 3. Boheler K, Chassagne C, Martin X, Wisnewsky C, Schwartz K. Cardiac expressions of alpha- and beta-myosin heavy chains and sarcomeric alpha-actins are regulated through transcriptional mechanisms. Results from nuclear run-on assays in isolated rat cardiac nuclei. J Biol Chem 267: 12979–12985, 1992 [PubMed] [Google Scholar]
- 4. Botney MD, Bahadori L, Gold LI. Vascular remodeling in primary pulmonary hypertension. Potential role for transforming growth factor-beta. Am J Pathol 144: 286–295, 1994 [PMC free article] [PubMed] [Google Scholar]
- 5. Cacoub P, Dorent R, Nataf P, Carayon A, Riquet M, Noe E, Piette JC, Godeau P, Gandjbakhch I. Endothelin-1 in the lungs of patients with pulmonary hypertension. Cardiovasc Res 33: 196–200, 1997 [DOI] [PubMed] [Google Scholar]
- 6. Chazova I, Loyd JE, Zhdanov VS, Newman JH, Belenkov Y, Meyrick B. Pulmonary artery adventitial changes and venous involvement in primary pulmonary hypertension. Am J Pathol 146: 389–397, 1995 [PMC free article] [PubMed] [Google Scholar]
- 7. Chen YF, Feng JA, Li P, Xing D, Zhang Y, Serra R, Ambalavanan N, Majid-Hassan E, Oparil S. Dominant negative mutation of the TGF-β receptor blocks hypoxia-induced pulmonary vascular remodeling. J Appl Physiol 100: 564–571, 2006 [DOI] [PubMed] [Google Scholar]
- 8. Coghlan MP, Culbert AA, Cross DA, Corcoran SL, Yates JW, Pearce NJ, Rausch OL, Murphy GJ, Carter PS, Roxbee Cox L, Mills D, Brown MJ, Haigh D, Ward RW, Smith DG, Murray KJ, Reith AD, Holder JC. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem Biol 7: 793–803, 2000 [DOI] [PubMed] [Google Scholar]
- 9. Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol 2: 769–776, 2001 [DOI] [PubMed] [Google Scholar]
- 10. Deng H, Dokshin GA, Lei J, Goldsmith AM, Bitar KN, Fingar DC, Hershenson MB, Bentley JK. Inhibition of glycogen synthase kinase-3β is sufficient for airway smooth muscle hypertrophy. J Biol Chem 283: 10198–10207, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eddahibi S, Fabre V, Boni C, Martres MP, Raffestin B, Hamon M, Adnot S. Induction of serotonin transporter by hypoxia in pulmonary vascular smooth muscle cells. Relationship with the mitogenic action of serotonin. Circ Res 84: 329–336, 1999 [DOI] [PubMed] [Google Scholar]
- 12. Eddahibi S, Humbert M, Fadel E, Raffestin B, Darmon M, Capron F, Simonneau G, Dartevelle P, Hamon M, Adnot S. Serotonin transporter overexpression is responsible for pulmonary artery smooth muscle hyperplasia in primary pulmonary hypertension. J Clin Invest 108: 1141–1150, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eddahibi S, Raffestin B, Pham I, Launay JM, Aegerter P, Sitbon M, Adnot S. Treatment with 5-HT potentiates development of pulmonary hypertension in chronically hypoxic rats. Am J Physiol Heart Circ Physiol 272: H1173–H1181, 1997 [DOI] [PubMed] [Google Scholar]
- 14. Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev 16: 1472–1487, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Giasson E, Meloche S. Role of p70 S6 protein kinase in angiotensin II-induced protein synthesis in vascular smooth muscle cells. J Biol Chem 270: 5225–5231, 1995 [DOI] [PubMed] [Google Scholar]
- 16. Glass DJ. Molecular mechanisms modulating muscle mass. Trends Mol Med 9: 344–350, 2003 [DOI] [PubMed] [Google Scholar]
- 17. Goldsmith AM, Bentley JK, Zhou L, Jia Y, Bitar KN, Fingar DC, Hershenson MB. Transforming growth factor-beta induces airway smooth muscle hypertrophy. Am J Respir Cell Mol Biol 34: 247–254, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Griffith SL, Rhoades RA, Packer CS. Pulmonary arterial smooth muscle contractility in hypoxia-induced pulmonary hypertension. J Appl Physiol 77: 406–414, 1994 [DOI] [PubMed] [Google Scholar]
- 19. Haq S, Choukroun G, Kang ZB, Ranu H, Matsui T, Rosenzweig A, Molkentin JD, Alessandrini A, Woodgett J, Hajjar R, Michael A, Force T. Glycogen synthase kinase-3beta is a negative regulator of cardiomyocyte hypertrophy. J Cell Biol 151: 117–130, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Haq S, Michael A, Andreucci M, Bhattacharya K, Dotto P, Walters B, Woodgett J, Kilter H, Force T. Stabilization of beta-catenin by a Wnt-independent mechanism regulates cardiomyocyte growth. Proc Natl Acad Sci USA 100: 4610–4615, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hassoun PM, Thappa V, Landman MJ, Fanburg BL. Endothelin 1: mitogenic activity on pulmonary artery smooth muscle cells and release from hypoxic endothelial cells. Proc Soc Exp Biol Med 199: 165–170, 1992 [DOI] [PubMed] [Google Scholar]
- 22. Holz MK, Ballif BA, Gygi SP, Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 123: 569–580, 2005 [DOI] [PubMed] [Google Scholar]
- 23. Hong KH, Lee YJ, Lee E, Park SO, Han C, Beppu H, Li E, Raizada MK, Bloch KD, Oh SP. Genetic ablation of the BMPR2 gene in pulmonary endothelium is sufficient to predispose to pulmonary arterial hypertension. Circulation 118: 722–730, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ishida M, Ishida T, Nakashima H, Miho N, Miyagawa K, Chayama K, Oshima T, Kambe M, Yoshizumi M. Mnk1 is required for angiotensin II-induced protein synthesis in vascular smooth muscle cells. Circ Res 12: 1218–1224, 2003 [DOI] [PubMed] [Google Scholar]
- 25. Ivester CT, Tuxworth WJ, Cooper G, IV, McDermott PJ. Contraction accelerates myosin heavy chain synthesis rates in adult cardiocytes by an increase in the rate of translational initiation. J Biol Chem 270: 21950–21957, 1995 [DOI] [PubMed] [Google Scholar]
- 26. Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, Loyd JE, Nichols WC, Trembath RC. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. The International PPH Consortium. Nat Genet 26: 81–84, 2000 [DOI] [PubMed] [Google Scholar]
- 27. Lee SL, Wang WW, Lanzillo JJ, Fanburg BL. Serotonin produces both hyperplasia and hypertrophy of bovine pulmonary artery smooth muscle cells in culture. Am J Physiol Lung Cell Mol Physiol 266: L46–L52, 1994 [DOI] [PubMed] [Google Scholar]
- 28. Li L, Liu ZC, Mercer B, Overbeek P, Olson EN. Evidence for serum response factor-mediated regulatory networks governing SM22α transcription in smooth, skeletal, and cardiac muscle cells. Dev Biol 187: 311–321, 1997 [DOI] [PubMed] [Google Scholar]
- 29. Liu Y, Fanburg BL. Serotonin-induced growth of pulmonary artery smooth muscle requires activation of phosphatidylinositol 3-kinase/serine-threonine protein kinase B/mammalian target of rapamycin/p70 ribosomal S6 kinase 1. Am J Respir Cell Mol Biol 34: 182–191, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marcos E, Fadel E, Sanchez O, Humbert M, Dartevelle P, Simonneau G, Hamon M, Adnot S, Eddahibi S. Serotonin-induced smooth muscle hyperplasia in various forms of human pulmonary hypertension. Circ Res 94: 1263–1270, 2004 [DOI] [PubMed] [Google Scholar]
- 31. Meyrick B, Reid L. Hypoxia-induced structural changes in the media and adventitia of the rat hilar pulmonary artery and their regression. Am J Pathol 100: 151–178, 1980 [PMC free article] [PubMed] [Google Scholar]
- 32. Meyrick B, Reid L. Hypoxia and incorporation of 3H-thymidine by cells of the rat pulmonary arteries and alveolar wall. Am J Pathol 96: 51–70, 1979 [PMC free article] [PubMed] [Google Scholar]
- 33. Morisco C, Seta K, Hardt SE, Lee Y, Vatner SF, Sadoshima J. Glycogen synthase kinase 3beta regulates GATA4 in cardiac myocytes. J Biol Chem 276: 28586–28597, 2001 [DOI] [PubMed] [Google Scholar]
- 34. Morrell NW, Yang X, Upton PD, Jourdan KB, Morgan N, Sheares KK, Trembath RC. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-β1 and bone morphogenetic proteins. Circulation 104: 790–795, 2001 [DOI] [PubMed] [Google Scholar]
- 35. Nikcevic G, Heidkamp MC, Perhonen M, Russell B. Mechanical activity in heart regulates translation of alpha-myosin heavy chain mRNA but not its localization. Am J Physiol Heart Circ Physiol 276: H2013–H2019, 1999 [DOI] [PubMed] [Google Scholar]
- 36. Ojamaa K, Petrie J, Balkman C, Hong C, Klein I. Posttranscriptional modification of myosin heavy-chain gene expression in the hypertrophied rat myocardium. Proc Natl Acad Sci USA 91: 3468–3472, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Parker TG, Packer SE, Schneider MD. Peptide growth factors can provoke “fetal” contractile protein gene expression in rat cardiac myocytes. J Clin Invest 85: 507–514, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rao GN, Griendling KK, Frederickson RM, Sonenberg N, Alexander RW. Angiotensin II induces phosphorylation of eukaryotic protein synthesis initiation factor 4E in vascular smooth muscle cells. J Biol Chem 269: 7180–7184, 1994 [PubMed] [Google Scholar]
- 39. Sartorelli V, Fulco M. Molecular and cellular determinants of skeletal muscle atrophy and hypertrophy. Sci STKE 244: re11, 2004 [DOI] [PubMed] [Google Scholar]
- 40. Schultz K, Fanburg BL, Beasley D. Hypoxia and hypoxia-inducible factor-1α promote growth factor-induced proliferation of human vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 290: H2528–H2534, 2006 [DOI] [PubMed] [Google Scholar]
- 41. Solway J, Seltzer J, Samaha FF, Kim S, Alger LE, Niu Q, Morrisey EE, Ip HS, Parmacek MS. Structure and expression of a smooth muscle cell-specific gene, SM22. J Biol Chem 270: 13460–13469, 1995 [DOI] [PubMed] [Google Scholar]
- 42. Stiebellehner L, Frid MG, Reeves JT, Low RB, Gnanasekharan M, Stenmark KR. Bovine distal pulmonary arterial media is composed of a uniform population of well-differentiated smooth muscle cells with low proliferative capabilities. Am J Physiol Lung Cell Mol Physiol 285: L819–L828, 2003 [DOI] [PubMed] [Google Scholar]
- 43. Sturrock A, Cahill B, Norman K, Huecksteadt TP, Hill K, Sanders K, Karwande SV, Stringham JC, Bull DA, Gleich M, Kennedy TP, Hoidal JR. Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 290: L661–L673, 2006 [DOI] [PubMed] [Google Scholar]
- 44. Taurin S, Hogarth K, Sandbo N, Yau DM, Dulin NO. Gbetagamma-mediated prostacyclin production and cAMP-dependent protein kinase activation by endothelin-1 promotes vascular smooth muscle cell hypertrophy through inhibition of glycogen synthase kinase-3. J Biol Chem 282: 19518–19525, 2007 [DOI] [PubMed] [Google Scholar]
- 45. Thomas M, Docx C, Holmes AM, Beach S, Duggan N, England K, Leblanc C, Lebret C, Schindler F, Raza F, Walker C, Crosby A, Davies RJ, Morrell NW, Budd DC. Activin-like kinase 5 (ALK5) mediates abnormal proliferation of vascular smooth muscle cells from patients with familial pulmonary arterial hypertension and is involved in the progression of experimental pulmonary arterial hypertension induced by monocrotaline. Am J Pathol 174: 380–389, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Thomson JR, Machado RD, Pauciulo MW, Morgan NV, Humbert M, Elliott GC, Ward K, Yacoub M, Mikhail G, Rogers P, Newman J, Wheeler L, Higenbottam T, Gibbs JSR, Egan J, Crozier A, Peacock A, Allcock R, Corris P, Loyd JE, Trembath RC, Nichols WC. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-beta family. J Med Genet 37: 741–745, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Trembath RC. Mutations in the TGF-beta type 1 receptor, ALK1, in combined primary pulmonary hypertension and hereditary haemorrhagic telangiectasia, implies pathway specificity. J Heart Lung Transplant 20: 175, 2001 [DOI] [PubMed] [Google Scholar]
- 48. Vyas DR, Spangenburg EE, Abraha TW, Childs TE, Booth FW. GSK-3beta negatively regulates skeletal myotube hypertrophy. Am J Physiol Cell Physiol 283: C545–C551, 2002 [DOI] [PubMed] [Google Scholar]
- 49. Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J 20: 4370–4379, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Welsh GI, Miller CM, Loughlin AJ, Price NT, Proud CG. Regulation of eukaryotic initiation factor eIF2B: glycogen synthase kinase-3 phosphorylates a conserved serine which undergoes dephosphorylation in response to insulin. FEBS Lett 421: 125–130, 1998 [DOI] [PubMed] [Google Scholar]
- 51. Yamakawa T, Tanaka Si Kamei J, Kadonosono K, Okuda K. Phosphatidylinositol 3-kinase in angiotensin II-induced hypertrophy of vascular smooth muscle cells. Eur J Pharmacol 478: 39–46, 2003 [DOI] [PubMed] [Google Scholar]
- 52. Yang X, Long L, Southwood M, Rudarakanchana N, Upton PD, Jeffery TK, Atkinson C, Chen H, Trembath RC, Morrell NW. Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ Res 96: 1053–1063, 2005 [DOI] [PubMed] [Google Scholar]
- 53. Zaiman AL, Podowski M, Medicherla S, Gordy K, Xu F, Zhen L, Shimoda LA, Neptune E, Higgins L, Murphy A, Chakravarty S, Protter A, Sehgal PB, Champion HC, Tuder RM. Role of the TGF-beta/Alk5 signaling pathway in monocrotaline-induced pulmonary hypertension. Am J Respir Crit Care Med 177: 896–905, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zamora MA, Dempsey EC, Walchak SJ, Stelzner TJ. BQ123, an ETA receptor antagonist, inhibits endothelin-1-mediated proliferation of human pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol 429–433, 1993 [DOI] [PubMed] [Google Scholar]
- 55. Zhou L, Goldsmith AM, Bentley JK, Jia Y, Rodriguez ML, Abe MK, Fingar DC, Hershenson MB. 4E-binding protein phosphorylation and eukaryotic initiation factor-4E release are required for airway smooth muscle hypertrophy. Am J Respir Cell Mol Biol 33: 195–202, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]