

Abstract
The neuropeptide vasoactive intestinal peptide (VIP) is expressed at high levels in the neurons of the suprachiasmatic nucleus (SCN). While VIP is known to be important to the input and output pathways from the SCN, the physiological effects of VIP on electrical activity of SCN neurons are not well known. Here the impact of VIP on firing rate of SCN neurons was investigated in mouse slice cultures recorded during the night. The application of VIP produced an increase in electrical activity in SCN slices that lasted several hours after treatment. This is a novel mechanism by which this peptide can produce long-term changes in central nervous system physiology. The increase in action potential frequency was blocked by a VIP receptor antagonist and lost in a VIP receptor knockout mouse. In addition, inhibitors of both the Epac family of cAMP binding proteins and cAMP-dependent protein kinase (PKA) blocked the induction by VIP. The persistent increase in spike rate following VIP application was not seen in SCN neurons from mice deficient in Kv3 channel proteins and was dependent on the clock protein PER1. These findings suggest that VIP regulates the long-term firing rate of SCN neurons through a VIPR2-mediated increase in the cAMP pathway and implicate the fast delayed rectifier (FDR) potassium currents as one of the targets of this regulation.
Keywords: circadian system, fast delayed rectifier, potassium, suprachiasmatic nucleus, VIP
daily biological rhythms are intrinsically generated, synchronized, and regulated by networks of circadian oscillators. These oscillations are generated by robust negative feedback mechanisms that occur at the molecular, cytoplasmic, and membrane levels within single cells (Maywood et al. 2007; Takahashi et al. 2008). In mammals, the suprachiasmatic nucleus (SCN) of the hypothalamus contains the master oscillatory neurons necessary for coordinating these rhythms found throughout the body (Dibner et al. 2010; Mohawk and Takahashi 2011; Welsh et al. 2010). These SCN pacemaker neurons receive photic input as well as a number of other timing cues and then integrate this information. One of the hallmark features of SCN neurons is that these cells are spontaneously active and generate neural activity rhythms both in vitro and in vivo (Colwell 2011; Webb et al. 2009; Welsh et al. 2010). Whereas single neurons can generate these rhythms, the SCN cell population normally works together in a circuit to produce a more robust oscillation. While many of the features of this circuit are not yet known, there is compelling evidence that a subset of neurons expressing the neuropeptide vasoactive intestinal peptide (VIP) play a critical role in the SCN circuit (An et al. 2011; Aton et al. 2005; Brown et al. 2007; Ciarleglio et al. 2009; Colwell et al. 2003; Harmar et al. 2002; Maywood et al. 2011).
The mechanism by which VIP regulates the neural activity of SCN neurons is not well understood, and yet this is important information for clarifying the role of this peptide in the SCN circuit. Neural activity is critical for both the inputs and output of the SCN circuit, and VIP regulation of the firing rate of SCN neurons could explain much of its physiological function. While the acute effects (within 5 min) of VIP on SCN firing rates have been examined (Reed et al. 2002), the long-term effects have not been previously examined. In the present study, we used electrophysiology tools to test the hypothesis that VIP regulates long-term (>1 h) electrical activity of neurons in the dorsal SCN (dSCN) region. We focused on this region of the SCN as the rhythms in both electrical activity (Schaap et al. 1999) and clock gene expression (Hamada et al. 2004; Yan and Okamura 2002) are more robust in this region of the SCN. We were interested in clarifying both the receptor as well as the second messenger cascades that mediate the impact of VIP on SCN electrical activity. We examined whether VIP increased PER1 expression in the SCN and the impact of antisense oligodeoxynucleotide (ODN) against Per1 on the VIP regulation of firing rate. Finally, the possibility that the fast delayed rectifier (FDR) potassium (K+) current mediates the VIP regulation of electrical activity was examined.
MATERIALS AND METHODS
Ethical approval.
The experimental protocols used in this study were approved by the University of California, Los Angeles (UCLA) Animal Research Committee, and all recommendations for animal use and welfare, as dictated by the UCLA Division of Laboratory Animals and the guidelines from the National Institutes of Health, were followed.
Animals.
Our studies used 1- to 3-mo-old male C57BL/6 mice. All mice were housed in cages within light-tight chambers with controlled lighting conditions. To obtain mice homozygous for the Kcnc1 and Kcnc2 gene mutations on the ICR background, heterozygous male offspring of Kcnc1−/− mice (Ho et al. 1997) were interbred with homozygote Kcnc2 −/− mice (Lau et al. 2000). Both lines of mice were initially provided by Dr. Bernardo Rudy (New York University, New York, NY). The resulting offspring were genotyped, and Kcnc1−/−, Kcnc2−/− double knockout (dKO) mice were used. The genotype of the mice was determined with genomic tail DNA and PCR. The VIPR2 KO mice (Harmar et al. 2002) used in this study were provided by Dr. James Waschek (UCLA).
Brain slice preparation for electrophysiology.
Methods utilized were similar to those described previously (Gamble et al. 2007, 2011). Animals were killed at zeitgeber time (ZT) 11 in the light-dark (LD) cycle for recording during the night (ZT 17–19). ZT is used to describe the projected time of the circadian clock within the SCN based on the previous light cycle, with lights-on defined as ZT 0. Brain slices were prepared with standard techniques from mice (C57BL/6, VIPR2 KO, and Kv dKO) between 1 and 3 mo of age. Mice were killed by isoflurane (Phoenix Pharmaceutical, Burlingame, CA) anesthesia and rapidly decapitated. To prepare SCN cultures, the brain was excised from the skull and placed in chilled low-Ca2+ ACSF [in mM: 26 NaHCO3, 1.25 NaH2PO4, 10 glucose, 125 NaCl, 3 KCl, 5 MgCl2, and 1 CaCl2, pH 7.2–7.4 (290–310 mOsm)]. After chilling (5 min), the brain was trimmed to a block containing the hypothalamus and optic nerves. The brain was sliced in the coronal plane on a vibratome (Dosaka EM, Kyoto, Japan) at a thickness of 200 μm. The slice was then trimmed to ∼4 × 4-mm squares, transferred directly to culture membranes (Millipore, Billerica, MA) in 35-mm culture dishes with 1.0 ml of MEM (Invitrogen, Carlsbad, CA) containing 30 mM HEPES, 20 mM d-glucose, 5% B27, 5.0 mM l-glutamine, and 25 U/ml streptomycin-penicillin, and maintained at 34°C and 5% CO2 (Han et al. 2006).
Loose-patch electrophysiology.
Slices were placed in a recording chamber attached to the stage of a fixed-stage upright differential interference contrast (DIC) microscope (Olympus, Tokyo, Japan). The slices were superfused continuously (2 ml/min) with ACSF [in mM: 26 NaHCO3, 1.25 NaH2PO4, 10 glucose, 125 NaCl, 3 KCl, 2 MgCl2, 2 CaCl2, pH 7.2–7.4 (290–310 mosmol/kgH2O)] aerated with 95% O2-5% CO2. The loose-patch recordings from the SCN were taken with recording electrodes. These micropipettes (4–6 MΩ) were pulled from glass capillaries (WPI, Sarasota, FL) on a multistage puller (P-97; Sutter Instruments, Novato, CA) and filled with the ACSF solution. Recordings were obtained with an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA) and monitored online with pCLAMP (version 10.0, Molecular Devices). Each of the cells was determined to be within the SCN by direct visualization of the location of the cell with DIC microscopy. In the neurons identified by biocytin labeling (n = 32), 91% (29/33) were confirmed to be located within the Nissl-defined dSCN region. The dorsal region is also confirmed by AVP and VIP immunohistochemistry. Each experimental treatment contained data from 3–5 animals and typically recordings from ∼30 neurons. All recordings were performed at room temperature.
Whole cell patch-clamp electrophysiology.
Recording methods utilized were similar to those described previously (Itri et al. 2005; Kudo et al. 2011; Michel et al. 2006). Slices were placed in a recording chamber (PH-1, Warner Instruments) attached to the stage of a fixed-stage upright DIC microscope (Olympus). The slices were superfused continuously (2 ml/min) with ACSF aerated with 95% O2-5% CO2. The whole cell patch-clamp recordings from the SCN were taken with recording electrodes. These micropipettes (typically 5–7 MΩ) were pulled from glass capillaries (WPI) on a multistage puller (Sutter) and filled with the standard solution. The standard solution contained (in mM) 112.5 K-gluconate, 1 EGTA, 10 HEPES, 5 MgATP, 1 GTP, 0.1 leupeptin, 10 phosphocreatine, 4 NaCl, 17.5 KCl, 0.5 CaCl2, 1 MgCl2, and 1 BAPTA. BAPTA was used to buffer intracellular calcium and inhibit calcium-dependent potassium currents. The pH was adjusted to 7.25–7.30, and the osmolality was adjusted between 290 and 300 mOsm. Recordings were obtained with an Axopatch 200B amplifier (Molecular Devices) and monitored online with pCLAMP (Molecular Devices). Each of the cells was determined to be within the SCN by direct visualization of the cell's location with DIC microscopy. Cells were approached with slight positive pressure (2–3 cmH2O). The pipette was lowered to the vicinity of the membrane while maintaining positive pressure. After a high-resistance seal (2–10 GΩ) was formed by application of negative pressure, a second pulse of negative pressure was used to break the membrane.
The access resistance of these cells ranged from 15 to 40 MΩ in the whole cell voltage-clamp configuration, while the cell capacitance was typically between 6 and 18 pF. Data were not collected if access resistance was >40 MΩ or if the value changed significantly (>20%) during the course of the experiment. In these studies, we used a 70% compensation using positive feedback correction. The junction potentials between the pipette and the extracellular solution were canceled by the voltage offset of the amplifier before a seal was established and were not further corrected. Series and input resistance were monitored repeatedly by checking the response to small pulses in a passive potential range. The standard extracellular solution used for all experiments was ACSF. Drug treatments were performed by dissolving pharmacological agents in the ACSF used to bathe the slices during recording. Solution exchanges within the slice were achieved by a rapid gravity-feed delivery system.
Current traces were recorded with pCLAMP with the whole cell voltage-clamp recording configuration and then analyzed with Clampfit (Molecular Devices). Methods utilized for FDR currents were similar to those described previously (Kudo et al. 2011). The FDR K+ currents were isolated pharmacologically with a voltage-step protocol in the whole cell voltage-clamp configuration with the neurons initially held at −70 mV. The protocol consisted of a 100-ms prepulse at −90 mV (to elicit maximal conductance) followed by 250-ms steps at progressively depolarized potentials (−80 to 50 mV, 10-mV steps). Leak subtraction was performed during acquisition with a p/4 protocol, which utilizes four subpulses with 1/4 of the test pulse amplitude and reversed polarity given from a holding potential of −70 mV. Current traces from treatment were subtracted from control to isolate FDR currents. 4-Aminopyridine (4-AP) was used to isolate FDR currents. Current measurements were performed in control solution and after 5 min of drug treatment in each cell. No current was injected during recording. The ACSF perfusion solution contained bicuculline (25 μM), TTX (0.5 μM), and cadmium (25 μM). After breakthrough of the membrane, data on membrane potential and resistance were obtained within 1 min. The membrane potential was measured in intervals between the action potentials. Each experimental treatment contained data from 3–5 animals and typically recordings from ∼30 neurons. All recordings were performed at room temperature.
Drugs.
VIP (HSDAVFTDNYTRLRKQMAVKKYLNSILN-NH2, final concentration 0.1, 1, or 10 μM) and VIPR2 antagonist (MyrH-SDAVFTDNYTKLRKQMAVKKYLNSI-K-K-G-G-T, final concentration 1 μM) were purchased from GL Biochem (Shanghai, China). Forskolin (final concentration 1 μM), brefeldin A (final concentration 100 μM), N6-benzoyladenosine-3′,5′-cyclic monophosphate sodium salt (6-Bnz-cAMP, final concentration 100 μM), 4-AP (final concentration 0.5 μM), and NMDA were purchased from Sigma-Aldrich (St. Louis, MO). 2,4,6-Trimethyl-N-(m-3-trifluoromethylphenyl)benzenesulfonamide (m-3M3FBS, final concentration 100 μM) was purchased from Merck (Gibbstown, NJ). H89 (final concentration 10 μM) and edelfosine [(7R)-4-hydroxy-7-methoxy-N,N,N-trimethyl-3,5,9-trioxa-4-phosphaheptacosan-1-aminium-4-oxide, final concentration 10 μM] were purchased from Tocris Bioscience (Ellisville, MO). 8-(4-Chlorophenylthio)-2-O-methyl-cAMPS, Sp-isomer (final concentration 100 μM) was purchased from Axxora (San Diego, CA). We used concentrations of drugs considered selective based on prior literature.
For Per1 gene knockdown, either antisense ODN sequence against the 5′ translational start site of Per1 (taggggaccactcatgtct; Sigma-Aldrich) or a random ODN with equivalent GC content (ccgttagtactgagctgac) (Akiyama et al. 1999; Tischkau and Gillette 2005) was applied at a final concentration of 10 μM. Vehicle treatments were used as controls. The ODN was applied to 35-mm dishes at ZT 2 until ZT 4 (day) and ZT 14 until ZT 16 (night).
Immunohistochemistry.
Methods utilized were similar to those described previously (Wang et al. 2009). Immunohistochemistry (IHC) was performed on male wild-type (WT) mice at 1–3 mo of age. Mouse SCN slices were made with the same method described above for the electrophysiology. Antisense ODN for Per1 was added at ZT 14, and the slices were fixed by 4% (wt/vol) paraformaldehyde (Sigma-Aldrich) in PBS (pH 7.4) at 4°C overnight and cryoprotected in 20% sucrose in PBS. IHC using a rabbit anti-PER1 antibody (1:500; Thermo Scientific, Rockford, IL) or rabbit anti-phospho-cAMP response element-binding protein (pCREB) (Ser133) antibody (1:1,000; Millipore, Temecula, CA) was performed on free-floating 20-μm cryostat coronal brain sections from the slice. Sections were washed three times (5 min) with PBS. Sections were dipped in 3% normal goat serum in PBS with 0.1% Triton X-100 for 1 h and then incubated with PER1 antibody with a solution of Triton X-100-PBS with 3% normal goat serum. Sections were incubated with the PER1 antibody for 72 h at 4°C. Other sections were incubated with a rabbit polyclonal antibody raised against VIP (ImmunoStar, no. 20077) diluted 1:1,000 and guinea pig polyclonal antibody raised against AVP (Peninsula, no. T-5048) with a solution of Triton X-100-PBS with 3% normal goat serum. Sections were incubated with the VIP and AVP antibodies for 24 h at 4°C. Sections were rinsed three times (5 min) in PBS and incubated for 2 h with an Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen) diluted 1:200 with Triton X-100-PBS with 3% normal goat serum. After incubation, sections were rinsed in PBS three times (5 min) and immediately mounted on slides. Sections were then dried and coverslipped. Then sections were imaged on the Axio Vision camera systems (Apotome, Carl Zeiss). Four sections from each slice were chosen and images taken. All immune-positive cells within the SCN were counted manually at ×40 with the aid of a grid. All immune-positive cells within the grid were counted equally without regard to the intensity of the staining. Counts were performed by two observers blind to the treatment protocol, and the results were averaged.
Statistical measurements.
The data sets were analyzed by test for equal variance and normal distribution to help select the appropriate test. The data sets were analyzed by one-way, two-way, or two-way repeated-measures ANOVA. If significant group differences were detected by the ANOVA, then a post hoc analysis was applied. Statistical significances between two groups were determined by Student's unpaired t-test. Values were considered significantly different if P < 0.05. All tests were performed with SigmaStat (version 3.5; Systat Software, Chicago, IL). Values are shown as means ± SE. Sample sizes are reported as the number of neurons, with the data from each group coming from between three and five mice.
RESULTS
VIP increased electrical activity of SCN neurons.
Using the voltage-clamp recording technique in the loose-patch configuration, we measured the spontaneous firing rate (SFR) in dSCN neurons. Each of these cells was determined to be within the dorsal region of the SCN by direct visualization of the location of the cell with infrared DIC videomicroscopy. Recordings (1 min) were made during the night (ZT 17–19, 2–4 h after the treatment and 1–3 h after VIP washout), and the resulting data were analyzed by one-way ANOVA. This analysis revealed a significant effect of treatments (H4 = 42.815, P < 0.01, Kruskal-Wallis 1-way ANOVA on ranks). Application of VIP (1, 10 μM) produced a significant increase in firing rate in dSCN neurons (Fig. 1; n = 30 cells, post hoc Dunn's method, P < 0.05). This increase in firing rate returned to control levels within 4–6 h after VIP application (see below). In contrast, a lower concentration of VIP (0.1 μM) did not alter the SFR (Fig. 1). Therefore, we used 1 μM VIP for the remainder of the experiments.
Fig. 1.
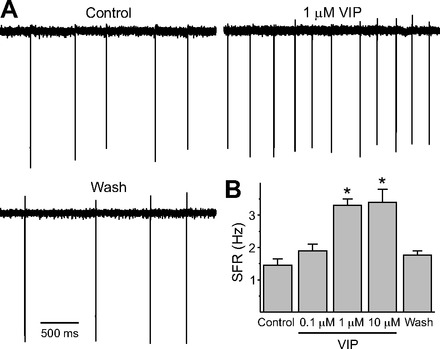
Application of vasoactive intestinal peptide (VIP) increased the firing rate of dorsal suprachiasmatic nucleus (dSCN) neurons during the night. A: representative examples illustrating the VIP-induced increase in firing rate in a dSCN neuron. B: average firing rate [spontaneous firing rate (SFR)] for each group (+SE). *Significant difference (P < 0.05) compared with controls analyzed by 1-way ANOVA followed by Dunn's method for multiple comparisons. The washout data were collected 4–6 h after drug application. For each group, n = 29 or 30 neurons. Throughout this study, sample sizes are reported as the number of neurons, with the data from each group coming from between 3 and 5 mice.
Excitatory effects of VIP were long-lasting.
To examine the duration of the VIP regulation in the dSCN, we sampled SFR at 0.5–2, 2–4, and 4–6 h after the 1-h treatment. The resulting data were analyzed by two-way ANOVA. This analysis revealed a significant effect of both condition [F(1,171) = 36.875, P < 0.01] and time [F(3,171) = 17.689, P < 0.01]. There was also a significant condition × time interaction [F(3,17) = 23.462, P < 0.01]. The impact of VIP was seen 2–4 h after treatment (Fig. 2; post hoc Holm-Sidak method, P < 0.05). The firing rate returned to control levels after 4 h. To check the condition of the neurons after the wash, we applied NMDA (10 μM) to confirm that the cells were still able to generate action potentials (Fig. 2). In a separate experiment, we recorded from a set of VIP-treated neurons (n = 12) for up to 3 h. Of the eight neurons that showed a VIP-induced increase in firing, seven neurons exhibited increased SFR that was maintained over the full 3 h. These findings demonstrate that VIP can produce long-lasting changes to the electrical activity of SCN neurons.
Fig. 2.
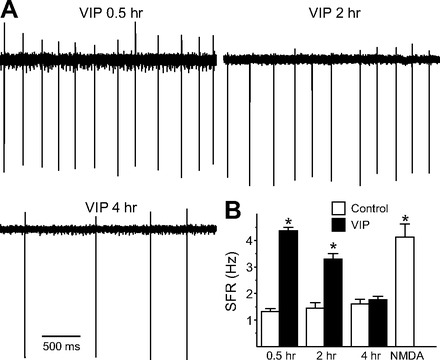
VIP produced long-lasting changes in firing rate in dSCN neurons during the night. A: representative examples illustrating the time course of VIP (1 μM)-evoked increase in firing rate. B: average firing rate for each group (+SE). *Significant difference (P < 0.05) analyzed by 2-way ANOVA followed by Holm-Sidak method for multiple comparisons (vs. control). For each group, n = 21–30 neurons.
VIP stimulation was mediated by VIPR2 pathway.
To determine the receptor that mediates VIP's regulation of SCN electrical activity, we determined the impact of VIP in the presence of a VIPR2 antagonist (MyrH-SDAVFTDNYTKLRKQMAVKKYLNSI-K-K-G-G-T; Moreno et al. 2000) as well as in VIPR2 KO mice. Recordings (1 min) were made during the night (ZT 17–19), and the resulting data were analyzed by one-way ANOVA. This analysis revealed a significant effect of treatments [F(3,116) = 13.026, P < 0.01]. The application of VIPR2 antagonist significantly reduced the effect of VIP on spike frequency (Fig. 3), while the antagonist alone did not produce a significant effect on the firing rate compared with untreated controls. The application of VIP failed to increase SFR in the VIPR2 KO [control (Ctl): 1.4 ± 0.3 Hz, n = 30; VIP: 1.8 ± 0.2 Hz, n = 30]. These results indicate that VIP regulates firing rate in the dSCN through a VIPR2-dependent mechanism.
Fig. 3.
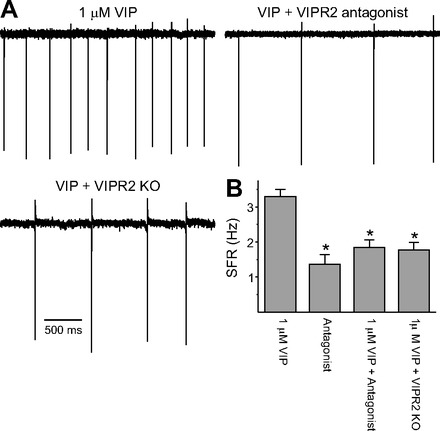
VIP-induced increase in spike frequency is mediated by VIPR2. A: representative examples showing that VIP does not increase firing in the presence of the VIPR2 antagonist (MyrH-SDAVFTDNYTKLRKQMAVKKYLNSI-K-K-G-G-T, 1 μM) as well as in VIPR2 knockout (KO) mice. B: average firing rate for each group (+SE). *Significant difference (P < 0.05) analyzed by 1-way ANOVA followed by Holm-Sidak method for multiple comparisons (vs. 1 μM VIP). For each group, n = 30.
Multiple signal transduction cascades mediate effect of VIP.
The VIPR2 is positively coupled to both the adenylyl cyclase (AC) and phospholipase C (PLC) signaling pathways (An et al. 2011; Harmar et al. 2012), and we sought to use pharmacological tools to differentiate the contribution of each pathway. As a first step, we applied a series of agents that stimulated these pathways to determine which could mimic the impact of VIP on firing rate (Fig. 4A). Recordings were made during the night (ZT 17–19, 2–4 h after the treatment) from neurons in the dSCN region. A comparison of treatments by Kruskal-Wallis one-way ANOVA on ranks revealed a significant effect (H4 = 32.186, P < 0.01). The application of the AC activator forskolin (n = 14; post hoc Dunn's method, P < 0.05) as well as the membrane-permeant and selective PKA activator 6-Bnz-cAMP increased the firing frequency (n = 30; post hoc Dunn's method, P < 0.05). Similarly, the application of 8-(4-chlorophenylthio)-2-O-methyl-cAMPS, Sp-isomer, which is a membrane-permeant and metabolically stable activator of the Epac cAMP receptor (O'Neill et al. 2008), increased the firing rate (n = 29; post hoc Dunn's method, P < 0.05), as did the PLC activator m-3M3FBS (O'Neill et al. 2008; n = 30; post hoc Dunn's method, P < 0.05). Therefore, it appears that activation of either the AC or PLC signaling pathway can increase firing rate in these neurons and mimic the effect of VIP.
Fig. 4.
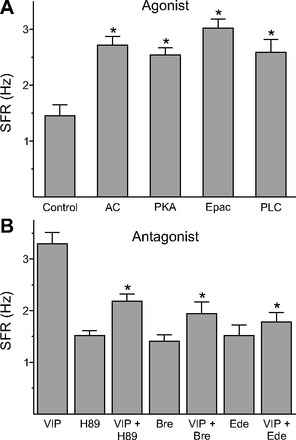
VIP-induced increase in spike frequency was mediated by both adenylyl cyclase (AC) and phospholipase C (PLC) pathways. A: average firing rate for the activators (+SE). *Significant difference (P < 0.05) analyzed by 1-way ANOVA followed by Dunn's method for multiple comparisons (vs. control). We evaluated several agents: AC activator forskolin (n = 14); cAMP-dependent protein kinase (PKA) activator N6-benzoyladenosine-3′,5′-cyclic monophosphate (6-Bnz-cAMP, n = 30); Epac activator cAMP receptor 8-(4-chlorophenylthio)-2′-O-methyl-cAMPS (n = 29); PLC activator m-3M3FBS (n = 30). B: average firing rate (+SE) for groups treated with the inhibitors alone and the inhibitors + VIP (1 μM). *Significant difference (P < 0.05) analyzed by 1-way ANOVA followed by Dunn's method for multiple comparisons (vs. VIP). We evaluated several agents: PKA inhibitor H89 (10 μM, n = 23); Epac inhibitor brefeldin A (Bre, 100 μM, n = 28); PLC inhibitor edelfosine (Ede, 10 μM, n = 24).
We next evaluated a panel of blockers of these same cascades (Fig. 4B). A comparison of treatments by Kruskal-Wallis one-way ANOVA on ranks revealed a significant effect (H6 = 50.523, P < 0.01). In the presence of the PKA inhibitor H89, there was significant reduction in the VIP increase in firing rate (n = 23; post hoc Dunn's method, P < 0.05). Similarly, the Epac antagonist brefeldin A (Huang and Hsu 2006) inhibited the VIP-induced spike frequency (n = 28; post hoc Dunn's method, P < 0.05). Finally, we examined the effects of a specific inhibitor of PLC, edelfosine (Powis et al. 1992), and found that this compound inhibited the effects of VIP (n = 24; post hoc Dunn's method, P < 0.05). By themselves, none of the inhibitors produced a significant change in the firing rates in the dSCN. Therefore, it appears that inhibition of either the AC or PLC signaling pathway can reduce the effects of VIP. The residual activity (H89 vs. H89+VIP, Bre vs. Bre+VIP, and Ede vs. Ede+VIP) was also examined, but there were no significant differences (data not shown). The coapplication of H89 and brefeldin A significantly blocked VIP-induced firing rates (VIP: 3.3 ± 0.2 Hz, n = 29; H89+Bre+VIP: 2.0 ± 0.2 Hz, n = 34; Student's t-test, P < 0.01).
VIP-induced increase in spike frequency was not driven by a change in membrane potential.
In some neurons VIP has been reported to depolarize the membrane, and we sought to determine whether membrane depolarization could underlie the increase in electrical activity measured in our preparation 2–4 h after treatment. We found that bath-applied VIP (1 μM) did not alter the resting potential membrane (Vm) in dSCN neurons recorded during the night (Ctl: −50 ± 3 mV, VIP: −51 ± 3 mV; n = 13; Student's t-test, P > 0.05). In addition, we did not see a significant change in the input resistance in the VIP-treated neurons (Ctl: 1.0 ± 0.2 GΩ, VIP: 1.1 ± 0.2 GΩ; n = 13; Student's t-test, P > 0.05). Thus the long-term VIP-induced increase in firing did not appear to be driven by changes in the Vm of the SCN neurons.
VIP regulation of firing rate is dependent on PER1.
Previous work has shown that VIP application induces Per1 expression in SCN as measured by in situ hybridization (Nielsen et al. 2002) and has raised the possibility that increasing PER1 expression can alter the firing rate of SCN neurons (Gamble et al. 2007, 2011). Using IHC, we found that application of VIP (1 μM, 60 min) at ZT 15 induced a significant increase in the number of pCREB- and PER1-expressing neurons in the SCN [Fig. 5, A and B; pCREB: 2-way ANOVA, time effect: F(5,24) = 1.887, P = 0.134, condition effect: F(1,24) = 23.624, P < 0.01, interaction: F(5,24) = 3.459, P < 0.05, post hoc: Holm-Sidak, P < 0.05 at ZT 15.25, 15.5, and 16 vs. control; PER1: 2-way ANOVA, time effect: F(3,18) = 0.231, P = 0.874, condition effect: F(1,18) = 10.425, P < 0.01, interaction: F(3,18) = 3.214, P < 0.05, post hoc: Holm-Sidak, P < 0.05 at ZT 17 vs. control]. Both ventral and dorsal PER1 were increased by VIP at ZT 17 (ventral: control 27 ± 4, VIP 47 ± 4 counts/area, Student's t-test, P < 0.05; dorsal: control 53 ± 7, VIP 96 ± 9 counts/area, Student's t-test, P < 0.05). Slices in control experiments in which the primary antibody was not added or the primary antibody was preabsorbed with a peptide control did not exhibit any positive staining (data not shown). Antisense ODN against Per1 blocked VIP-induced PER1 levels, whereas a scrambled ODN was without effect (Kruskal-Wallis 1-way ANOVA on ranks, H3 = 9.051, P < 0.05, post hoc: Tukey test, control vs. antisense + VIP: P > 0.05., control vs. scrambled + VIP: P < 0.05; Fig. 5C). Importantly, blocking the PER1 induction with antisense ODN also prevented the VIP induction of firing rate as measured 2–4 h after treatment (Kruskal-Wallis 1-way ANOVA on ranks, H5 = 89.671, P < 0.01, post hoc: Dunn's method, control vs. antisense + VIP: P > 0.05; Fig. 5D). The application of VIP also increased firing rate when measured 0.5–1 h after treatment. The application of the antisense ODN treatment did not alter the ability of VIP to increase firing rate as measured 0.5–1 h after treatment (control: 1.4 ± 0.2 Hz, n = 30; VIP: 4.0 ± 0.4 Hz, n = 30; antisense: 1.4 ± 0.2 Hz, n = 32; antisense + VIP: 4.2 ± 0.3 Hz, n = 31; Kruskal-Wallis 1-way ANOVA on ranks: H3 = 68.298, P < 0.01, post hoc: Dunn's method, VIP vs. antisense + VIP, P > 0.05). These results indicate that while PER1 was not necessary for short-term changes in firing rate, the VIP regulation of neural activity over the long term (>1 h) is dependent on induction of PER1 in the SCN.
Fig. 5.
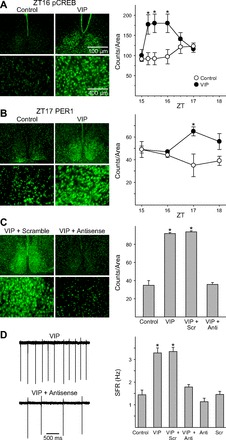
Persistent VIP-induced increases in electrical activity are dependent on PER1. A: photomicrographs illustrate that the application of VIP (1 μM) increases phospho-cAMP response element-binding protein (p-CREB) as measured by immunohistochemistry (IHC). Bar graphs show p-CREB-positive cell counts for control and VIP-treated groups (±SE). *Significant difference (P < 0.05) analyzed by 1-way ANOVA followed by Dunn's method for multiple comparisons (vs. control). ZT, zeitgeber time. B: photomicrographs illustrate that application of VIP (1 μM) increases PER1 protein as measured by IHC. Bar graphs show PER1-positive cell counts for control and VIP-treated groups (±SE). *Significant difference (P < 0.05) analyzed by 1-way ANOVA followed by Dunn's method for multiple comparisons (vs. control). C: photomicrographs illustrate that antisense against Per1 blocked the VIP induction of PER1. Bar graphs show PER1-positive cell counts for control and VIP-treated groups (+SE) exposed to antisense or scrambled message. Anti, group treated with antisense; Scr, group treated with scrambled message. D: representative examples showing that the application of VIP (1 μM) does not cause persistent increase in neural activity in the presence of antisense against Per1. Bar graphs show average firing rate for each group (+SE). *Significant difference (P < 0.05) analyzed by 1-way ANOVA followed by Dunn's method for multiple comparisons.
VIP-induced increase in spike frequency was mediated by FDR current.
The Kv3 (Shaw-related) family of K+ channels is thought to be primarily responsible for generation of the FDR current (Rudy and McBain 2001), and we have shown that this current is absent in the SCN of mice lacking both Kv3.1 and 3.2 channels (Kudo et al. 2011). To determine whether the FDR current mediates the effect of VIP, we used Kv3 dKO mice as well as a pharmacological blocker (4-AP) of these currents (Fig. 6A). Recordings were made during the night (ZT 17–19, 2–4 h after the drug application), and the resulting data were analyzed by Kruskal-Wallis one-way ANOVA on ranks. This analysis revealed a significant effect of treatments (H2 = 30.417, P < 0.01). VIP did not produce a significant increase in firing rate in the Kv3 dKO mice (n = 30; post hoc Dunn's method, P > 0.05) or in WT mice in the presence of 4-AP (n = 30; post hoc Dunn's method, P > 0.05).
Fig. 6.
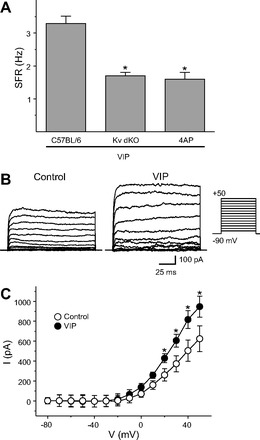
VIP-induced increase spike frequency was mediated by fast delayed rectifier (FDR) current. A: average firing rate for each group (+SE). *Significant difference (P < 0.05) analyzed by 1-way ANOVA followed by Dunn's method for multiple comparisons (vs. VIP). For each, VIP was applied at 1 μM and 4-aminopyridine (4-AP) at 0.5 mM. For each group, n = 30. B: representative examples showing that application of VIP (1 μM) increases the magnitude of FDR currents in dSCN. C: current (I)-voltage (V) relationship of FDR currents in SCN neurons. Data are shown as means ± SE. *Significant difference (P < 0.05) analyzed by 2-way repeated-measures ANOVA followed by Holm-Sidak method for multiple comparisons (vs. control). For each group, n = 11 or 12. FDR currents were isolated by subtraction (baseline, 0.5 mM 4-AP) using a voltage-step protocol with a prepulse potential of −90 mV and test pulse potentials (see materials and methods). Holding potential was −70 mV.
Next, the possibility that VIP can regulate the magnitude of FDR currents in SCN neurons was examined (Fig. 6, B and C). The resulting data were analyzed by two-way repeated-measures ANOVA and indicated a significant effect of both condition [F(1,273) = 6.19, P < 0.05] and voltage [F(13,273) = 31.982, P < 0.01]. There was also a significant condition × voltage interaction [F(13,273) = 4.957, P < 0.01]. VIP enhanced the magnitude of FDR currents in dSCN neurons at 20, 30, 40, and 50 mV (post hoc: Holm-Sidak method, P < 0.05). Together, these results indicated that VIP regulates the FDR current and the regulation of firing rate in the night is dependent on FDR currents.
DISCUSSION
VIP causes long-lasting changes in firing rate in dSCN neurons.
In this study, we found that the application of VIP increases the electrical activity of dSCN neurons during the night (Fig. 1). The dSCN is not a uniform population of cells, although the responses that we observed were consistent. As a percentage of control the increase of firing rate by VIP was robust, but the actual frequency changes were relatively small. These VIP-induced increases in firing rate developed over 30 min and lasted for 2–4 h (Fig. 2). These long-term physiological changes in response to VIP have not been previously reported in the SCN or in other regions of the central nervous system. Prior studies using extracellular recordings in the rat found that brief application (5 min) of VIP acutely alters the firing rate of about half of SCN neurons, with most responding neurons exhibiting a decreased firing rate, although a few cells are activated (Reed et al. 2002). The present study focused on long-term changes, and we did not measure the short-term impact of VIP on SCN firing.
In the present study, we found that micromolar concentrations of VIP were necessary to induce these long-term changes in neural activity in the SCN slice culture. Similar concentrations of VIP were needed to evoke changes in the expression of PER2::LUC in SCN explants (An et al. 2011). Earlier work in acute brain slices found that nanomolar concentrations of this peptide were effective in both acutely suppressing SCN neuronal firing (Reed et al. 2002) and increasing inhibitory postsynaptic currents (IPSCs) (Itri and Colwell 2003), with an EC50 of 42 nM. The most direct explanation for these differences is that higher concentrations of VIP may be required to evoke a response when the peptide is applied in a static culture compared with a flow-through system. In the static culture, the SCN tissue is fixed onto a membrane that can form a permeability barrier. It is also possible that receptor internalization or other biological processes are responsible for the differences between the concentration response observed in static and flow-through culture conditions. In each of these studies, the VIPR2 receptor appears to mediate the response in the SCN (see below).
Given the time course of the VIP action observed in the present study, one explanation for the increase in excitability of SCN neurons during the night is that the peptide causes a large phase shift in the SCN clock. For example, a large VIP-induced phase advance could move the SCN neuron from an electrically inactive night state to an electrically active day state. We cannot rule out this possibility but think that it is unlikely. Prior work examined the impact of VIP treatment on the phase of the SCN molecular clockwork as measured by PER2::LUC rhythms (An et al. 2011). With this assay, VIP treatment can indeed cause phase shifts but only when the peptide is applied during the subjective day time. More likely is that the impact of VIP in exciting SCN neurons during the night will modulate how the cells respond to stimulation (see discussion below).
Both light exposure as well as treatment with gastrin-releasing peptide (GRP) can also cause persistent increases in neural activity within the SCN at night (Gamble et al. 2007, 2011; Kuhlman et al. 2003; LeSauter et al. 2011). Thus peptide transmitters can drive long-lasting changes in the excitability within the SCN network. We speculate that VIP and GRP may work together functionally to regulate the excitability of the SCN circuit. VIP is expressed with a subset of GABAergic interneurons found throughout the central nervous system and has been shown to alter the excitability of several neural populations (Hermes et al. 2009; Jeftinija et al. 1982; Lee and Cox 2006; Pawelzik et al. 1992). It is not known whether VIP-evoked changes lasting hours are a common feature of this peptide's action in the nervous system.
Multiple signaling pathways mediate impact of VIP.
The VIPR2 (VPAC2) receptor is highly expressed within the SCN (An et al. 2012; Cagampang et al. 1998; Sheward et al. 1995; Usdin et al. 1994; Vertongen et al. 1998). Using both a receptor KO and a pharmacological receptor antagonist, we were able to confirm that the VIPR2 receptor mediates the regulation of electrical activity in the dSCN (Fig. 3). In the SCN, and other brain regions, VIP binding is followed by AC activity leading to increases in cAMP and PKA (Meyer-Spasche and Piggins 2004; Rea 1990; Vanecek and Watanabe 1998). Imaging techniques have shown that VIP rapidly increases intracellular cAMP in most SCN neurons (An et al. 2011). VIP and the VIPR2 are also known to be positively coupled to PLC (Harmar et al. 2012). In the present study, we examined a panel of activators and inhibitors of these second messenger pathways. We found evidence that activators of both AC and PLC could mimic the effects of VIP while antagonists of both cascades inhibited the effects of VIP (Fig. 4). One caveat of this type of analysis is uncertainty about the specificity of the various pharmacological agents. Still, taken at face value, our results suggest that VIP activation of both AC and PLC signaling cascades mediate the regulation of excitability. A similar conclusion was reached in a recent study examining the mechanisms underlying VIP's impact on the phase of circadian rhythms in gene expression (An et al. 2011), which reported that blockade of both AC and PLC activities is required to inhibit VIP-induced phase shifts.
VIP induction of PER1 supports long-term increases in firing rate.
VIP causes the rapid induction of pCREB (within 15 min) and a slower induction (within 2 h) of PER1 (Fig. 5, A and B). Together with the prior observation that VIP application induces Per1 message in the SCN (Nielsen et al. 2002), this study shows that VIP can directly alter the molecular clockwork in SCN neurons. Previous work has shown that Per1 levels and SCN neuronal activity are tightly correlated (Kuhlman et al. 2003). Prior work has also shown that blocking Per1 with antisense ODN inhibits GRP regulation of spike frequency (Gamble et al. 2007). These studies raise the possibility of a direct mechanistic link between PER1 and electrical activity in the SCN (Colwell 2011). We now find that blocking Per1 with antisense ODN blocks the persistent increase in firing rate evoked by VIP application (Fig. 5D). These results suggest that these peptides (GRP, VIP) can act through the regulation of clock gene expression to produce long-term changes in the firing rate in SCN neurons. In contrast, the shorter-term VIP-induced increase in firing occurred in the presence of the antisense. Application of VIP increased levels of PER1, but this increase could not be detected until 2 h after treatment. Therefore, we believe that VIP can regulate excitability of SCN neurons through at least two mechanisms, with the longer-term changes dependent on PER1. Recent work focusing on ventral SCN neurons also found evidence that photic regulation of excitability occurs through two distinct mechanisms (LeSauter et al. 2011).
Ionic mechanisms.
A number of mechanisms could underlie these long-lasting changes in excitability. In neurons where VIP acutely alters membrane excitability, the peptide causes a membrane depolarization by either increasing an inward current or decreasing an outward current. For example, in cultured SCN neurons application of VIP induces an inward current mediated by the apparent decrease in K+ current (Pakhotin et al. 2006). In neurons in the subparaventricular zone (SCN targets), bath-applied VIP results in a membrane depolarization caused by a nonselective cation conductance (Hermes et al. 2009). Both mechanisms have also been proposed to underlie a long-term increase in excitability in SCN neurons in response to light exposure (Kuhlman et al. 2003; LeSauter et al. 2011). In our preparation, the long-term increase in firing was not associated with a change in Vm or conductance at resting Vm measured 2–4 h after treatment. We did see evidence that VIP increased the magnitude of the FDR current in the dSCN and that VIP does not increase firing rate in the absence of the FDR currents (Fig. 6).
The FDR current is known to exhibit a set of unique physiological properties including high thresholds of activation, rapid activation, and large conductance in the SCN (Itri et al. 2005; Kudo et al. 2011) as well as in many other brain regions (reviewed by Baranauskas 2007; Joho and Hurlock 2009; Rudy and McBain 2001). These properties enable neurons with this current to rapidly repolarize after the generation of action potentials. On the basis of the present data, we are proposing that the VIP-induced enhancement of the FDR current is at least part of the mechanism that allows dSCN neurons to fire more frequently. Previous work has shown that application of GRP also increases the magnitude of the FDR current in the SCN (Gamble et al. 2011). Kcnc genes are widely expressed in the nervous system (Joho and Hurlock 2009; Rudy and McBain 2001), and it may be that FDR currents can serve to regulate the excitability and response to sensory stimulation in other regions.
Functional significance.
A schematic illustrating our view of the impact of VIP on dSCN neurons is shown in Fig. 7. The findings that VIP can produce long-term changes in the firing rate of dSCN neurons during the night raise questions about the function of this regulation. Within the SCN circuit, VIP synchronizes individual cell-autonomous oscillators (Maywood et al. 2011; Webb et al. 2009; Welsh et al. 2010), and it is possible that the VIP-induced changes in firing rate are part of the mechanism by which this coupling occurs. Aside from coupling SCN oscillators, prior data suggest that VIP has a critical role in regulating how the circadian system responds to light (Dragich et al. 2010; Hughes et al. 2004; Vosko et al. 2007). During the night, SCN neurons are normally silent but do respond to photic stimulation transduced by mRGCs that generate action potentials up to 20 Hz (Berson et al. 2002; Irwin and Allen 2007; Meijer et al. 1998; Tu et al. 2005; Warren et al. 2003). This light-induced increase in neural activity drives synaptic communication with the rest of the cells in the circuit.
Fig. 7.
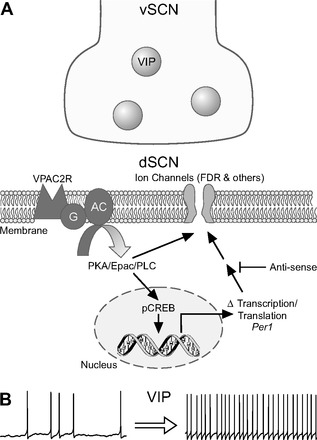
An illustration of our model of VIP regulation of firing rate in the dSCN. A: VIP is held in dense core synaptic vesicles in a subset of neurons in the ventral SCN (vSCN) region. Upon release, VIP activates the VPAC2 (VIPR2) receptors and activates a network of signaling pathways. These pathways produce short-term changes in firing rate presumably through posttranslational modification of intrinsic ion channels. In addition, these signaling pathways phosphorylate CREB and increase the transcription and translation of the clock gene Period1 (Per1). The increase in PER1 regulates the FDR current among other intrinsic ion channels to increase the firing rate of dSCN neurons. B: the net result of VIP application is a long-lasting (2–4 h) increase in the firing rate of dSCN neurons. It remains to be seen whether the ability of VIP to modulate ongoing electrical activity over the course of hours is restricted to the SCN or whether this is a common mechanism by which this peptide regulates nervous system function.
The long-term increases in excitability in SCN neurons driven by GRP (Gamble et al. 2011) and VIP (present study) are likely to play a critical role in determining the response of SCN neurons to light. Prior work in both mollusks and mammals suggests that the electrical activity of circadian pacemaker neurons determines how these cells respond to photic stimulation (Colwell 2011; Irwin and Allen 2007; McMahon and Block 1987). This regulation can explain why the loss of VIP or its receptor has such a dramatic effect on photic entrainment of the circadian system (Colwell et al. 2003; Harmar et al. 2002). VIP is expressed in a subset of GABAergic interneurons throughout the central nervous system. It remains to be seen whether the ability of VIP to modulate ongoing electrical activity over the course of hours is restricted to the SCN or whether this is a common mechanism by which this peptide regulates nervous system function.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: T.K., K.L.G., D.G.M., G.D.B., and C.S.C. conception and design of research; T.K., Y.T., and K.L.G. performed experiments; T.K., Y.T., and K.L.G. analyzed data; T.K. and C.S.C. interpreted results of experiments; T.K. and C.S.C. prepared figures; T.K. drafted manuscript; T.K. and C.S.C. edited and revised manuscript; T.K., Y.T., K.L.G., D.G.M., G.D.B., and C.S.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Danny Truong and Yingfei Wu for technical assistance.
REFERENCES
- Akiyama M, Kouzu Y, Takahashi S, Wakamatsu H, Moriya T, Maetani M, Watanabe S, Tei H, Sakaki Y, Shibata S. Inhibition of light- or glutamate-induced mPer1 expression represses the phase shifts into the mouse circadian locomotor and suprachiasmatic firing rhythms. J Neurosci 19: 1115–1121, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- An S, Irwin RP, Allen CN, Tsai C, Herzog ED. Vasoactive intestinal polypeptide requires parallel changes in adenylate cyclase and phospholipase C to entrain circadian rhythms to a predictable phase. J Neurophysiol 105: 2289–2296, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- An S, Tsai C, Ronecker J, Bayly A, Herzog ED. Spatiotemporal distribution of vasoactive intestinal polypeptide receptor 2 in mouse suprachiasmatic nucleus. J Comp Neurol 520: 2730–2741, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aton SJ, Colwell CS, Harmar AJ, Waschek J, Herzog ED. Vasoactive intestinal polypeptide mediates circadian rhythmicity and synchrony in mammalian clock neurons. Nat Neurosci 8: 476–483, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranauskas G. Ionic channel function in action potential generation: current perspective. Mol Neurobiol 35: 129–150, 2007 [DOI] [PubMed] [Google Scholar]
- Berson DM, Dunn FA, Takao M. Phototransduction by retinal ganglion cells that set the circadian clock. Science 295: 1070–1073, 2002 [DOI] [PubMed] [Google Scholar]
- Brown TM, Colwell CS, Waschek JA, Piggins HD. Disrupted neuronal activity rhythms in the suprachiasmatic nuclei of vasoactive intestinal polypeptide-deficient mice. J Neurophysiol 97: 2553–2558, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagampang FR, Sheward WJ, Harmar AJ, Piggins HD, Coen CW. Circadian changes in the expression of vasoactive intestinal peptide 2 receptor mRNA in the rat suprachiasmatic nuclei. Brain Res Mol Brain Res 54: 108–112, 1998 [DOI] [PubMed] [Google Scholar]
- Ciarleglio CM, Gamble KL, Axley JC, Strauss BR, Cohen JY, Colwell CS, McMahon DG. Population encoding by circadian clock neurons organizes circadian behavior. J Neurosci 29: 1670–1676, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwell CS. Linking neural activity and molecular oscillations in the SCN. Nat Rev Neurosci 12: 553–569, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwell CS, Michel S, Itri J, Rodriguez W, Tam J, Lelievre V, Hu Z, Liu X, Waschek JA. Disrupted circadian rhythms in VIP- and PHI-deficient mice. Am J Physiol Regul Integr Comp Physiol 285: R939–R949, 2003 [DOI] [PubMed] [Google Scholar]
- Dibner C, Schibler U, Albrecht U. The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annu Rev Physiol 72: 517–549, 2010 [DOI] [PubMed] [Google Scholar]
- Dragich JM, Loh DH, Wang LM, Vosko AM, Kudo T, Nakamura TJ, Odom IH, Tateyama S, Hagopian A, Waschek JA, Colwell CS. The role of the neuropeptides PACAP and VIP in the photic regulation of gene expression in the suprachiasmatic nucleus. Eur J Neurosci 31: 864–875, 2010 [DOI] [PubMed] [Google Scholar]
- Gamble KL, Allen GC, Zhou T, McMahon DG. Gastrin-releasing peptide mediates light-like resetting of the suprachiasmatic nucleus circadian pacemaker through cAMP response element-binding protein and Per1 activation. J Neurosci 27: 12078–12087, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble KL, Kudo T, Colwell CS, McMahon DG. Gastrin-releasing peptide modulates fast delayed rectifier potassium current in Per1-expressing SCN neurons. J Biol Rhythms 26: 99–106, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada T, Antle MC, Silver R. Temporal and spatial expression patterns of canonical clock genes and clock-controlled genes in the suprachiasmatic nucleus. Eur J Neurosci 19: 1741–1748, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MH, Bolanos CA, Green TA, Olson VG, Neve RL, Liu RJ, Aghajanian GK, Nestler EJ. Role of cAMP response element-binding protein in the rat locus ceruleus: regulation of neuronal activity and opiate withdrawal behaviors. J Neurosci 26: 4624–4629, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmar AJ, Fahrenkrug J, Gozes I, Laburthe M, May V, Pisegna JR, Vaudry D, Vaudry H, Waschek JA, Said SI. Pharmacology and functions of receptors for vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide: IUPHAR review 1. Br J Pharmacol 166: 4–17, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmar AJ, Marston HM, Shen S, Spratt C, West KM, Sheward WJ, Morrison CF, Dorin JR, Piggins HD, Reubi JC, Kelly JS, Maywood ES, Hastings MH. The VPAC2 receptor is essential for circadian function in the mouse suprachiasmatic nuclei. Cell 109: 497–508, 2002 [DOI] [PubMed] [Google Scholar]
- Hermes ML, Kolaj M, Doroshenko P, Coderre E, Renaud LP. Effects of VPAC2 receptor activation on membrane excitability and GABAergic transmission in subparaventricular zone neurons targeted by suprachiasmatic nucleus. J Neurophysiol 102: 1834–1842, 2009 [DOI] [PubMed] [Google Scholar]
- Ho CS, Grange RW, Joho RH. Pleiotropic effects of a disrupted K+ channel gene: reduced body weight, impaired motor skill and muscle contraction, but no seizures. Proc Natl Acad Sci USA 94: 1533–1538, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Hsu KS. Presynaptic mechanism underlying cAMP-induced synaptic potentiation in medial prefrontal cortex pyramidal neurons. Mol Pharmacol 69: 846–856, 2006 [DOI] [PubMed] [Google Scholar]
- Hughes AT, Fahey B, Cutler DJ, Coogan AN, Piggins HD. Aberrant gating of photic input to the suprachiasmatic circadian pacemaker of mice lacking the VPAC2 receptor. J Neurosci 24: 3522–3526, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin RP, Allen CN. Calcium response to retinohypothalamic tract synaptic transmission in suprachiasmatic nucleus neurons. J Neurosci 27: 11748–11757, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itri J, Colwell CS. Regulation of inhibitory synaptic transmission by vasoactive intestinal peptide (VIP) in the mouse suprachiasmatic nucleus. J Neurophysiol 90: 1589–1597, 2003 [DOI] [PubMed] [Google Scholar]
- Itri J, Michel S, Waschek JA, Colwell CS. Circadian rhythm in inhibitory synaptic transmission in the mouse suprachiasmatic nucleus. J Neurophysiol 92: 311–319, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itri JN, Michel S, Vansteensel MJ, Meijer JH, Colwell CS. Fast delayed rectifier potassium current is required for circadian neural activity. Nat Neurosci 8: 650–656, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeftinija S, Murase K, Nedeljkov V, Randic M. Vasoactive intestinal polypeptide excites mammalian dorsal horn neurons both in vivo and in vitro. Brain Res 243: 158–164, 1982 [DOI] [PubMed] [Google Scholar]
- Joho RH, Hurlock EC. The role of Kv3-type potassium channels in cerebellar physiology and behavior. Cerebellum 8: 323–333, 2009 [DOI] [PubMed] [Google Scholar]
- Kudo T, Loh DH, Kuljis D, Constance C, Colwell CS. Fast delayed rectifier potassium current: critical for input and output of the circadian system. J Neurosci 31: 2746–2755, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman SJ, Silver R, Le Sauter J, Bult-Ito A, McMahon DG. Phase resetting light pulses induce Per1 and persistent spike activity in a subpopulation of biological clock neurons. J Neurosci 23: 1441–1450, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau D, Vega-Saenz de Miera EC, Contreras D, Ozaita A, Harvey M, Chow A, Noebels JL, Paylor R, Morgan JI, Leonard CS, Rudy B. Impaired fast-spiking, suppressed cortical inhibition, and increased susceptibility to seizures in mice lacking Kv3.2 K+ channel proteins. J Neurosci 20: 9071–9085, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Cox CL. Excitatory actions of vasoactive intestinal peptide on mouse thalamocortical neurons are mediated by VPAC2 receptors. J Neurophysiol 96: 858–871, 2006 [DOI] [PubMed] [Google Scholar]
- LeSauter J, Silver R, Cloues R, Witkovsky P. Light exposure induces short- and long-term changes in the excitability of retinorecipient neurons in suprachiasmatic nucleus. J Neurophysiol 106: 576–588, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maywood ES, Chesham JE, O'Brien JA, Hastings MH. A diversity of paracrine signals sustains molecular circadian cycling in suprachiasmatic nucleus circuits. Proc Natl Acad Sci USA 108: 14306–14311, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maywood ES, O'Neill JS, Reddy AB, Chesham JE, Prosser HM, Kyriacou CP, Godinho SI, Nolan PM, Hastings MH. Genetic and molecular analysis of the central and peripheral circadian clockwork of mice. Cold Spring Harb Symp Quant Biol 72: 85–94, 2007 [DOI] [PubMed] [Google Scholar]
- McMahon DG, Block GD. The Bulla ocular circadian pacemaker. I. Pacemaker neuron membrane potential controls phase through a calcium-dependent mechanism. J Comp Physiol A 161: 335–346, 1987 [DOI] [PubMed] [Google Scholar]
- Meijer JH, Watanabe K, Schaap J, Albus H, Detari L. Light responsiveness of the suprachiasmatic nucleus: long-term multiunit and single-unit recordings in freely moving rats. J Neurosci 18: 9078–9087, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Spasche A, Piggins HD. Vasoactive intestinal polypeptide phase-advances the rat suprachiasmatic nuclei circadian pacemaker in vitro via protein kinase A and mitogen-activated protein kinase. Neurosci Lett 358: 91–94, 2004 [DOI] [PubMed] [Google Scholar]
- Michel S, Itri J, Han JH, Gniotczynski K, Colwell CS. Regulation of glutamatergic signalling by PACAP in the mammalian suprachiasmatic nucleus. BMC Neurosci 7: 15, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohawk JA, Takahashi JS. Cell autonomy and synchrony of suprachiasmatic nucleus circadian oscillators. Trends Neurosci 34: 349–358, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno D, Gourlet P, De Neef P, Cnudde J, Waelbroeck M, Robberecht P. Development of selective agonists and antagonists for the human vasoactive intestinal polypeptide VPAC2 receptor. Peptides 21: 1543–1549, 2000 [DOI] [PubMed] [Google Scholar]
- Nielsen HS, Hannibal J, Fahrenkrug J. Vasoactive intestinal polypeptide induces per1 and per2 gene expression in the rat suprachiasmatic nucleus late at night. Eur J Neurosci 15: 570–574, 2002 [DOI] [PubMed] [Google Scholar]
- O'Neill JS, Maywood ES, Chesham JE, Takahashi JS, Hastings MH. cAMP-dependent signaling as a core component of the mammalian circadian pacemaker. Science 320: 949–953, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakhotin P, Harmar AJ, Verkhratsky A, Piggins H. VIP receptors control excitability of suprachiasmatic nuclei neurones. Pflügers Arch 452: 7–15, 2006 [DOI] [PubMed] [Google Scholar]
- Pawelzik H, Dodt HU, Zieglgansberger W. Actions of vasoactive intestinal polypeptide (VIP) on neocortical neurons of the rat in vitro. Neurosci Lett 147: 167–170, 1992 [DOI] [PubMed] [Google Scholar]
- Powis G, Seewald MJ, Gratas C, Melder D, Riebow J, Modest EJ. Selective inhibition of phosphatidylinositol phospholipase C by cytotoxic ether lipid analogues. Cancer Res 52: 2835–2840, 1992 [PubMed] [Google Scholar]
- Rea MA. VIP-stimulated cyclic AMP accumulation in the suprachiasmatic hypothalamus. Brain Res Bull 25: 843–847, 1990 [DOI] [PubMed] [Google Scholar]
- Reed HE, Cutler DJ, Brown TM, Brown J, Coen CW, Piggins HD. Effects of vasoactive intestinal polypeptide on neurones of the rat suprachiasmatic nuclei in vitro. J Neuroendocrinol 14: 639–646, 2002 [DOI] [PubMed] [Google Scholar]
- Rudy B, McBain CJ. Kv3 channels: voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci 24: 517–526, 2001 [DOI] [PubMed] [Google Scholar]
- Schaap J, Bos NP, de Jeu MT, Geurtsen AM, Meijer JH, Pennartz CM. Neurons of the rat suprachiasmatic nucleus show a circadian rhythm in membrane properties that is lost during prolonged whole-cell recording. Brain Res 815: 154–166, 1999 [DOI] [PubMed] [Google Scholar]
- Sheward WJ, Lutz EM, Harmar AJ. The distribution of vasoactive intestinal peptide2 receptor messenger RNA in the rat brain and pituitary gland as assessed by in situ hybridization. Neuroscience 67: 409–418, 1995 [DOI] [PubMed] [Google Scholar]
- Takahashi JS, Hong HK, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet 9: 764–775, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischkau SA, Gillette MU. Oligodeoxynucleotide methods for analyzing the circadian clock in the suprachiasmatic nucleus. Methods Enzymol 393: 593–610, 2005 [DOI] [PubMed] [Google Scholar]
- Tu DC, Zhang D, Demas J, Slutsky EB, Provencio I, Holy TE, Van Gelder RN. Physiologic diversity and development of intrinsically photosensitive retinal ganglion cells. Neuron 48: 987–999, 2005 [DOI] [PubMed] [Google Scholar]
- Usdin TB, Bonner TI, Mezey E. Two receptors for vasoactive intestinal polypeptide with similar specificity and complementary distributions. Endocrinology 135: 2662–2680, 1994 [DOI] [PubMed] [Google Scholar]
- Vanecek J, Watanabe K. Melatonin inhibits the increase of cyclic AMP in rat suprachiasmatic neurons induced by vasoactive intestinal peptide. Neurosci Lett 252: 21–24, 1998 [DOI] [PubMed] [Google Scholar]
- Vertongen P, Schiffmann SN, Gourlet P, Robberecht P. Autoradiographic visualization of the receptor subclasses for vasoactive intestinal polypeptide (VIP) in rat brain. Ann NY Acad Sci 865: 412–415, 1998 [DOI] [PubMed] [Google Scholar]
- Vosko AM, Schroeder A, Loh DH, Colwell CS. Vasoactive intestinal peptide and the mammalian circadian system. Gen Comp Endocrinol 152: 165–175, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LM, Dragich JM, Kudo T, Odom IH, Welsh DK, O'Dell TJ, Colwell CS. Expression of the circadian clock gene Period2 in the hippocampus: possible implications for synaptic plasticity and learned behaviour. ASN Neuro 1: e00012, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren EJ, Allen CN, Brown RL, Robinson DW. Intrinsic light responses of retinal ganglion cells projecting to the circadian system. Eur J Neurosci 17: 1727–1735, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb AB, Angelo N, Huettner JE, Herzog ED. Intrinsic, nondeterministic circadian rhythm generation in identified mammalian neurons. Proc Natl Acad Sci USA 106: 16493–16498, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh DK, Takahashi JS, Kay SA. Suprachiasmatic nucleus: cell autonomy and network properties. Annu Rev Physiol 72: 551–577, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Okamura H. Gradients in the circadian expression of Per1 and Per2 genes in the rat suprachiasmatic nucleus. Eur J Neurosci 15: 1153–1162, 2002 [DOI] [PubMed] [Google Scholar]