

Abstract
We and others have shown that epithelial Na+ channels (ENaC) in alveolar type 2 (AT2) cells are activated by β2 agonists, steroid hormones, elevated oxygen tension, and by dopamine. Although acetylcholine receptors (AChRs) have been previously described in the lung, there are few reports of whether cholinergic agonists alter sodium transport in the alveolar epithelium. Therefore, we investigated how cholinergic receptors regulate ENaC activity in primary cultures of rat AT2 cells using cell-attached patch-clamp recordings to assess ENaC activity. We found that the muscarinic agonists, carbachol (CCh) and oxotremorine, activated ENaC in a dose-dependent manner but that nicotine did not. CCh-induced activation of ENaC was blocked by atropine. Western blotting and immunohistochemistry suggested that muscarinic M2 and M3 receptors (mAChRs) but not nicotinic receptors were present in AT2 cells. Endogenous RhoA and GTP-RhoA increased in response to CCh and the increase was reduced by pretreatment with atropine. We showed that Y-27632, an inhibitor of Rho-associated protein kinase (ROCK), abolished endogenous ENaC activity and inhibited the activation of ENaC by CCh. We also showed that ROCK signaling was necessary for ENaC stability in 2F3 cells, a model for AT2 cells. Our results showed that muscarinic agonists activated ENaC in rat AT2 cells through M2 and/or M3 mAChRs probably via a RhoA/ROCK signaling pathway.
Keywords: cholinergic receptor, epithelial sodium channels, single channel recording, alveolar type 2 epithelial cell
cholinergic receptors (AChR) are present in lung tissue. Muscarinic M1 and M3 receptors are present in bronchial epithelial cells (23), and M2 and M3 receptors in bronchial smooth muscle are therapeutic targets in certain respiratory diseases like chronic obstructive pulmonary disease (COPD) and asthma (10) because their activation can lead to bronchoconstriction (9, 24). Nicotinic receptors are present throughout the airways (23), and recently nicotinic receptors in macrophages have been implicated in anti-inflammatory responses that result in better survival in some experimental models of sepsis (1, 27).
Other neurotransmitters including dopamine and β2 agonists activate epithelial Na+ channels (ENaC) in alveolar type 2 (AT2) cells (7, 11, 12). Although there have been some reports that acetylcholine alters sodium transport in some epithelial cells, description of the effects in alveolar epithelial cells is limited (32). However, we felt that our single channel and biochemical approaches could provide evidence for or against cholinergic regulation of ENaC in alveolar epithelial cells.
We report here that muscarinic agonists activated ENaC through mAChR M2 and/or M3 and that it was likely mediated by Rho-GTPase and Rho-associated protein kinase (ROCK) signaling downstream of the muscarinic receptors.
MATERIALS AND METHODS
Chemicals.
Carbachol or carbamylcholine chloride, oxotremorine, atropine, and Y-27632 were purchased from Sigma-Aldrich (St. Louis, MO). Mouse anti-RhoA and anti-AChRα(D6), rabbit anti-mAChR M2, and anti-mAChR M3, were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The lamellar body marker, lyso-tracker red, was purchased from Invitrogen (Grand Island, NY).
Cell culture.
AT2 cells were isolated by enzymatic digestion of lung tissue from adult Sprague-Dawley rats (200–250 g) and then placed in primary cultures using published techniques (14). All procedures involving animals were reviewed and approved by our Institutional Animal Care and Use Committee. Briefly, the rats were anesthetized with pentobarbital and heparinized (100 mg/kg). Resected rat lung was digested by tracheal instillation of elastase (0.4 mg/ml). Lung cell purification was based on differential adherence of cells to dishes coated with rat IgG. Nonadherent AT2 cells in the supernatant were collected, centrifuged, and seeded onto permeable supports in a highly enriched medium (3 parts Coon's modification of Ham's F-12 and 7 parts Liebovitz's L-15 with 1 μM dexamethasone). Cells were grown on a specialized culture support (14), which is optimized for patch-clamp recording and allows the cells to grow on a permeable support (Millipore CM) while they are submerged in medium. After 18 h the cells had attached to the culture surface and medium was drained from the apical surface. Cells were allowed to grow with medium on the basolateral surface and air on the apical side in 95% air and 5% CO2.
With this method, we have been able to obtain an AT2 cell population with 95% viability, 95% purity, and less than 5% of contamination with macrophages and fibroblasts (4, 14). Cells were used for patch-clamp experiments during the first 24–72 h in culture while they maintain an AT2 cell phenotype as we have previously reported (14).
Tracheal instillation of saline and radiographic assessment of lung fluid clearance.
Tracheal instillations were performed as previously described (5). Briefly, mice were anesthetized with intraperitoneal (IP) injection of 15 mg/kg xylazine (Anased 100 mg/ml; Lloyd Laboratories, Shenandoah, IA) and 125 mg/kg ketamine (ketamine hydrochloride injection USP 50 mg/ml; Bioniche Pharma Lake Forest, IL) prepared in PBS (pH 7.4; Invitrogen, Grand Island, NY) for a final IP injection volume of 250 μl/20 g mouse. The instillate vehicle for all experiments consisted of 140 mM NaCl, 5 mM KCl, 1 mM CaCl2, and 10 mM HEPES with pH = 7.4, hereafter referred to as control saline. Tracheal instillation volumes of 5 μl/g body wt (100 μl; final volume in 20 g C57Bl/6J mice) were delivered using a 26-gauge, 5/8-inch needle, and 1-ml syringe. IP injections of 20 μM β-agonists occurred immediately before tracheal instillation of saline, and all animals were immediately assayed for lung fluid clearance using X-ray imaging.
X-ray imaging.
Immediately after a mouse received a tracheal instillation, lung fluid clearance was assessed using radiographic imaging as previously described (5). Briefly, animals were placed in a Kodak In-Vivo Multispectral Imaging System (Carestream Health, Rochester, NY) connected to a rodent nose cone with continuous anesthesia (1.5 l/min isoflurane) and supplemental oxygen (100%). Animals were X-ray imaged at 5-min intervals up to 240 min with an acquisition period of 120 s. X-ray settings were as follows: 2X2 binding, 180-mm field of view, 149-μA X-ray current, 35 kVp, and 0.4-mm aluminum filter. X-ray density was quantified using Carestream Health MI software with a 5-mm2 region of interest (ROI). In all whole animal studies, the ROI was the left upper lobe of the lung as described in Ref. 5.
A model for in vivo fluid clearance.
To describe the change in lung fluid balance in response to a saline challenge after administration of agents which might alter fluid reabsorption from or fluid secretion into the airways we developed a simple model in which the rate of change in lung surface fluid consisted of three components. Fluid absorption depended upon the amount of fluid on the surface of the airways and the rate of absorption; fluid secretion is defined by a single rate constant; and the overall change in fluid absorption also depends upon the rate of clearance of drug with a single time constant. The rate of change of surface fluid is given in Eq. 1
| (1) |
where ks is the rate of secretion, ka is the rate of reabsorption, and F is the volume of airway lung fluid. The steady-state value of fluid balance, K, is the ratio of the rate of absorption to the rate of secretion. Integration of this differential equation with the addition of the rate of elimination of drug gives the final model in Eq. 2
| (2) |
where F(t) is the amount of surface fluid in the lung at any time, t; K is the steady-state or peak amount of lung fluid; ka is the rate of absorption of fluid; and ke is the rate of drug elimination. The form of this function is consistent with fluid rapidly rising at rate ka to a peak value, K, before gradually decreasing toward initial levels as drug is eliminated at rate ke. Values for the rate of secretion, ks, can be obtained as the ratio of ka to K. The additional advantage of this fit is that it provides values that are a measure of the effect of carbachol. In particular, the value, K, provides a measure of the maximal effect of any of the treatments and can be statistically compared.
Single-channel recordings.
The cell-attached configuration was used in all patch-clamp studies (Fig. 1). Micropipettes were pulled from filamented borosilicate glass capillaries (TW-150, World Precision Instruments) with a two-stage vertical puller (Narishige, Tokyo, Japan). The resistances of the pipettes were between 14 and 19 MΩ for AT2 cell and between 7 and 10 MΩ for 2F3 cells when filled with and immersed in patch solution containing (in mM) 140 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, and 10 HEPES for AT2 cells, and 96 NaCl, 3.4 KCl, 0.81 CaCl2, 0.8 MgCl2, and 10 HEPES for 2F3 cells with pH adjusted to 7.4 with NaOH. Pipettes were back-filled with patch solution. After formation of a high-resistance seal of about 5–10 GΩ, the channel currents were recorded at 1 kHz with an Axopatch 1-D amplifier (Molecular Devices) with a low-pass 100-Hz Bessel filter. The channel activity NPo was calculated from pClampfit 10.1 data analysis software (Molecular Devices).
Fig. 1.
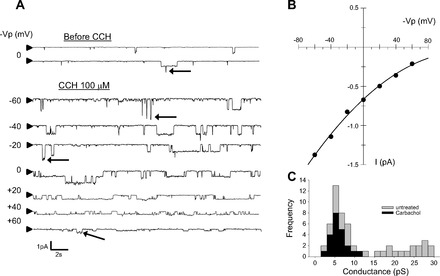
Carbachol (CCh) activates epithelial sodium channel (ENaC) activity in primary cultured rat AT2 cells. A: a representative recording of cell-attached patch clamp from AT2 cells. Downward deflections indicate inward current; arrowhead at left = closed level. Carbachol (100 μM) activated ENaC within 1 min. As shown in C, there are 2 categories of observable conductance events, low conductance and high conductance; but high-conductance events are much less frequent. Several of the high-conductance events are marked with arrows in the records in A. B: current-voltage (I-V) relation for AT2 cell after carbachol. Values were obtained from current events from patch-clamp recording in A. Conductance with no potential applied to the pipette was 5.5 pS consistent with a highly Na+-sensitive cation (HSC) channel similar to ENaC measured in many other cells. C: distribution of single-channel conductances before and after carbachol. AT2 cells are known to have a distribution of conductances with one peak value near 6 pS and a second at higher conductances (∼22 pS). However, after carbachol all of the conductances are clustered near the lower values.
Western blot analysis.
Rat lung tissue was quickly homogenized on ice in RIPA lysis buffer (150 mM NaCl, 10 mM NaPO4, 0.1% SDS, 1% NP-40, 0.25% deoxycholate) containing protease inhibitors (antipain, leupeptin, TLCK/TPCK, and PMSF) and centrifuged at 12,000 rpm for 15 min at 4°C. Protein amounts in the supernatant were determined (Bio-Rad) after which sample buffer (glycerol, SDS, and DTT) was added to the supernatant and the lysate boiled at 95°C for 10 min. Proteins were separated by SDS-PAGE (10% gels) and subsequently transferred to nitrocellulose membrane. Western blot analysis was performed by blocking the nitrocellulose in 5% milk and then probing with each primary antibody [anti-mAChR M2, M3, and anti-AChRα(D6)] at 1:1,000 dilution overnight. The secondary antibody goat (anti-rabbit IgG, anti-mouse IgG and anti-mouse-IgM) coupled to alkaline phosphatase (Kirkegaard and Perry Laboratories) was used at 1:5,000 dilution. Blots were developed using CDP-Star (Applied Biosystems, Foster City, CA).
Immunohistochemistry.
For immunohistochemistry, AT2 cells were purified as described above, fixed with 4% paraformaldehyde, and permeabilized with 0.1% Triton X-100. Primary antibodies [anti-mAChR M2, M3, anti-AChα(D6), anti-surfactant protein A (SP-A), and anti-RhoA antibodies] were used at 1:50 dilutions. The secondary antibody conjugated with Alexa Fluor 488 for rabbit IgG or 548 for murine IgG from Santa Cruz Biotechnology (Santa Cruz, CA) was applied and incubated at dilution of 1:1,000. The nuclei of AT2 cells were stained using Vectashield with DAPI (Vector Laboratories). Cells were mounted onto glass and imaged using a confocal laser scanning microscope (FLUOVIEWFV1000, Olympus).
RhoA activation assay.
RhoA activation was assayed by measuring GTP-association with RhoA. Activation was determined with the G-LISA RhoA Activation Assay Biochem Kit, luminescence Based (Cytoskeleton). After purification, AT2 cells were aliquoted into groups and treated with protocols consistent with the patch-clamp experiments. Samples were then lysed with protease inhibitors and a protein assay was performed. GTP-loaded RhoA was measured as fluorescence according to the manufacturer's instruction with an ELISA-based method. Specifically, the relative fluorescence was given as
where R is the relative fluorescence, F is the measured fluorescence of treated cells, F0 is the minimum or background fluorescence, and Fmax is the fluorescence of maximally activated RhoA.
Data analysis.
Data presented in this paper are usually means ± SE except when explicitly stated as otherwise. Differences between groups were evaluated with one-way ANOVA, and differences of properties of the same cell at different membrane potentials were tested by paired t-test. Parameter estimation from curve fitting was done with Sigmaplot and Sigmastat (San Rafael, CA).
RESULTS
Muscarinic agonists increase ENaC NPo in rat AT2 cells.
AChRs are categorized into two subgroups: muscarinic acetylcholine receptors (mAChRs) and nicotinic acetylcholine receptors. There are generic agonists and specific agonists for each class of receptors. Carbachol (CCh) activates both classes while oxotremorine (OXO)and nicotine (NICO) activate muscarinic and nicotinic receptors, respectively. Atropine (ATR) is a muscarinic antagonist. We performed cell-attached patch clamp and found that ENaC was activated within 1 min after we applied CCh (Fig. 1A). The current-voltage relationship showed that the channel was a 5-pS channel that reversed at positive potentials (Fig. 1B). These characteristics were consistent with the characteristics of epithelial sodium channels (ENaC) described by us and others in a variety of tissues including lung (3, 5, 12–14, 19, 22). However, in previous work, we showed that AT2 cells contained two types of amiloride-sensitive cation channels. Therefore, we examined the different types of channels in untreated and carbachol-treated cells. As expected based on our previous observations (11–14), we observed two categories of channels; however, after carbachol treatment all of the channels we observed fell in the lower range of conductances reenforcing the idea that carbachol induces 5- to 6-pS ENaC (Fig. 1C). Carbachol activates both nicotinic and muscarinic cholinergic receptors, but a muscarinic agonist, oxotremorine, also activates ENaC (Fig. 2). Both carbachol and oxotremorine activate ENaC in a dose-dependent manner: carbachol with a K0.5 of 18 μM (Fig. 3A) and oxotremorine with a K0.5 of 11 μM (Fig. 3B). On the other hand, even 100 μM NICO did not increase ENaC NPo (Fig. 4), implying that AT2 cells contained mAChRs but not nicotinic receptors and that the mAChRs could activate ENaC in rat AT2 cells.
Fig. 2.
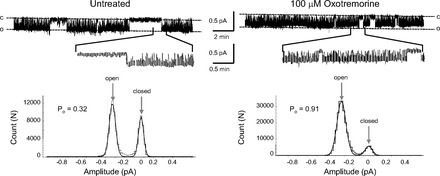
Representative recordings of cell-attached patches and amplitude histograms from AT2 cells before and after the muscarinic agonist, oxotremorine (100 μM). Downward deflections are inward current; “c” indicates the closed level; “o” indicates open events. Expanded segments of each trace are shown below the full-length record. Oxotremorine activated ENaC within 1 min. The amplitude histograms show that oxotremorine significantly increases the open probability of ENaC.
Fig. 3.
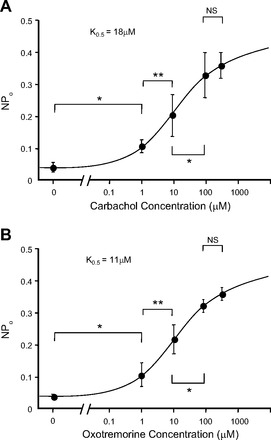
Dose-response of ENaC NPo in response to cholinergic agents. A and B: carbachol and oxotremorine activated ENaC in a dose-dependent manner. K0.5 for carbachol was 18 μM and for oxotremorine was 11 μM. Values are means ± SE. *P < 0.05, **P < 0.01. NSP > 0.05.
Fig. 4.
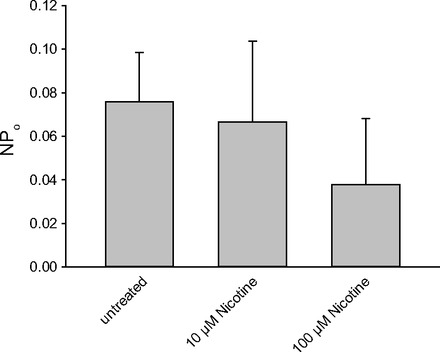
Nicotine did not activate ENaC. NPo was analyzed from recordings over 3 min before application and from the point 3 min after applications with nicotine at 10 and 100 μM. The activity of the channel was measured as NPo. There is no significant effect of nicotine.
Atropine inhibited carbachol-induced activation of ENaC.
To make sure that the muscarinic agonists activated ENaC through muscarinic cholinergic receptors in AT2 cells, we incubated AT2 cells with ATR (100 μM) for 5 min before forming a patch and applying CCh (100 μM). Pretreatment with ATR prevented any significant CCh-induced increase in ENaC NPo [after pretreatment with ATR, CCh only increased NPo to 0.099 ± 0.010, n = 6 (P < 0.01), significantly less than in the absence of ATR (NPo 0.372 ± 0.037, n = 10)] (Fig. 5). In other words, ATR inhibited more than 85% of the CCh-induced increase in ENaC NPo (Fig. 6, A and B).
Fig. 5.
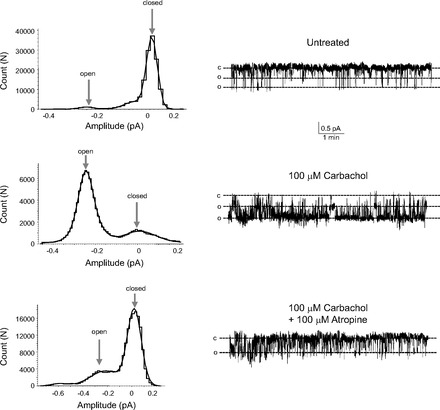
Representative recordings of cell-attached patches and amplitude histograms from AT2 cells before and after carbachol (100 μM) and then after addition of carbachol and the cholinergic antagonist, atropine (100 μM). Downward deflections are inward current; “c” indicates the closed level; “o” indicates open events. Carbachol activated ENaC within 1 min, and atropine inhibited the carbachol activation within 3–5 min. The amplitude histograms show that atropine significantly reduces the carbachol-induced increase in the open probability of ENaC.
Fig. 6.
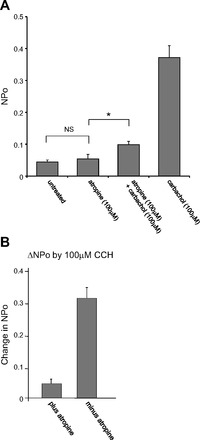
Atropine (ATR) inhibits carbachol-induced ENaC activation. A: treatment with ATR for 5 min did not affect basal activity of ENaC (Ctr: 0.044 ± 0.007, n = 17. ATR: 0.054 ± 0.013, n = 6), but pretreatment with ATR (100 μM) diminished carbachol (100 μM)-induced increase in ENaC NPo (ATR + CCh: 0.099 ± 0.010, n = 6; CCh: 0.371 ± 0.037, n = 10). B: without ATR, carbachol-induced ΔNPo (100 μM) was 0.318 but with ATR ΔNPo was 0.045, 85.8% of ΔNPo caused by carbachol. Values are means ± SE. *P < 0.05, NSP > 0.05.
IP injection of carbachol promotes lung fluid clearance.
We assessed the effect of stimulation with the cholinergic agonist, carbachol, on lung fluid clearance using X-ray fluoroscopy of anesthetized mice, which enabled quantification of airspace fluid content in C57Bl/6J mouse lung following a tracheal instillation of saline (5). In Fig. 7 we injected intraperitoneally several different concentrations of carbachol (or carbachol plus atropine) immediately before tracheally instilling 5 μl/g body wt (∼100 μl) instillate. Freely breathing, anesthetized animals were X-ray imaged every 5 min for up to 4 h (5-min exposure times) to compare the rate of alveolar fluid clearance against uninjected (control) groups of mice that also received a saline challenge. Figure 7 shows that IP injections of carbachol increased the rate of alveolar fluid clearance compared with uninjected mice all receiving a 100-μl tracheal instillation of saline. In Fig. 7, we fit the data to a simple model described in materials and methods to give us the peak value for lung fluid and the rate of fluid absorption for different concentrations of carbachol or carbachol plus atropine. The peak value for the change in relative fluid amount and the rate of absorption, secretion, and drug elimination derived from the model are given in Table 1. The peak value is a measure of the maximal effect of carbachol. Comparison of the peak values and their standard deviations show that the maximal effect of each dose of carbachol is statistically different from control and from every other dose (P < 0.05). The effect of 500 nM atropine reduces the peak level to a value of lung fluid that is statistically less than the level of 500 nM carbachol alone (P < 0.001).
Fig. 7.
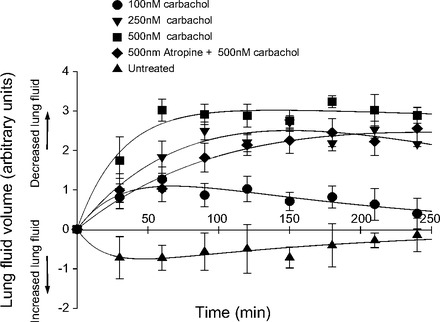
Carbachol increases lung fluid reabsorption as determined by a fluorometric determination of lung fluid (5). This figure shows lung fluid clearance following saline challenge in mice receiving an intraperitoneal (IP) injection of 100, 250, or 500 nM carbachol or 500 nM atropine plus 500 nM carbachol. Control group represents mice receiving only a tracheal instillation of saline (values are means ± SD, n = 4 per group).
Table 1.
Peak fluid volume and rates of fluid reabsorption and secretion and rate of carbachol elimination based on the model described in materials and methods
| Drug Concentration (nM)* | Peak Fluid Volume (K) | Rate of Fluid Reabsorption (ka) | Rate of Fluid Secretion (ks) | Rate of Elimination of Drug (ke) |
|---|---|---|---|---|
| 0 | −1.12 ± 0.399† | 0.0462 ± 0.0375 | 0.0060 ± 0.0024 | |
| 100 Carbachol | 2.05 ± 0.103† | 0.0239 ± 0.0178 | 85.6 ± 76.9 | 0.0060 ± 0.0027 |
| 250 Carbachol | 2.65 ± 0.104† | 0.0231 ± 0.00560 | 106. ± 26.4 | 0.0020 ± 0.0017 |
| 500 Carbachol | 3.22 ± 0.323† | 0.0316 ± 0.0108 | 102. ± 38.6 | 0.00040 ± 0.00090 |
| 500 Carbachol +500 Atropine | 2.66 ± 0.196‡ | 0.0119 ± 0.00230 | 223 ± 46.2 | 0.00040 ± 0.00090 |
Values are means ± SE. The model provides values for the peak amount of lung fluid, the initial rate of fluid absorption, and the rate at which fluid levels return to normal (i.e., the rate of drug elimination). The maximal effect of any of the treatments (K) can be statistically compared to measure the effect of different doses of carbachol.
Concentration in 0.1 ml of saline injected intraperitoneally,
All of these values are significantly different from one another (P < 0.05).
Peak fluid volume for 500 nM atropine +500 nM carbachol is significantly less than for 500 nM carbachol alone.
The effect of atropine on single ENaC (Figs. 5 and 6) appears to be greater than the effect of atropine on carbachol-induced fluid accumulation. Of course, the experiments are fundamentally different and therefore it is difficult to compare the doses of carbachol and atropine. However, we suggest that the dose of carbachol we attempted to reverse with atropine in the in vivo experiments was very high, but the amount in the patch-clamp experiments was lower relative to complete block. Since atropine is a competitive inhibitor, we would not expect it to be as effective against a high concentration of carbachol as against a relatively lower concentration. The competitive nature of atropine can actually be seen in Figs. 5 and 6.
In previous work, we have shown that instillation of 1 mM amiloride (reconstituted in saline) immediately following IP injection of agents that increase fluid reabsorption blocks the action of these agents. Therefore, we concluded that the parenteral route (IP injection of carbachol) was more successful than a local delivery in promoting lung fluid clearance in mice following a saline challenge.
AT2 cells have muscarinic M2 and M3 receptors.
Western blotting of lung lysate detected mAChRs M2 and M3 but not nicotinic receptor, AChRα(D6) (Fig. 8). Results with 2F3 cells were the same (data not shown). Anti-AChRα(D6) is an antibody against the α subunit present in most nicotinic receptors. Nicotinic receptors are known to be present in macrophages and airway but most macrophage and airway cells would have been removed during the process of preparing the AT2 cells. Thus it appears that AT2 cells have muscarinic receptors but few, if any, nicotinic receptors.
Fig. 8.
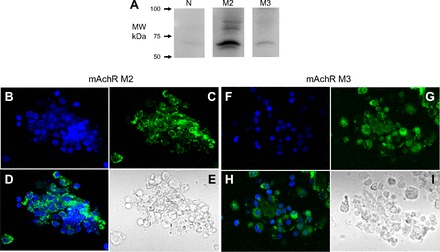
Muscarinic M2 and M3 receptors (mAChR) but not α subunit of nicotinic receptors are present in lung lysate. A: results of Western blotting were positive for mAChR M2 and M3 but not AChRα (anti-α-subunit of nicotinic receptors) in lung lysate. B–E and F–I: M2 an M3 receptors also can be detected in AT2 cells using fluorescently labeled antibodies and confocal microscopy in AT2 cells [labeled with primary rabbit anti-mAChR M2 or M3 antibody and secondary antibody conjugated with FITC (green)]. B and F: DAPI staining of cell nuclei. C and G: M2 or M3 antibodies. D and H: merge of DAPI and M2/M3 antibodies. E and I: differential interference contrast images.
Immunohistochemistry detects M2 and M3 cholinergic receptors and the small G protein, RhoA, in AT2 cells.
Results of Western blotting indicated mAChRs M2 and M3 are present in AT2 cells while nicotinic receptors are not (Fig. 8A). When we used immunohistochemistry to detect the receptors in the AT2 cells, we found positive immunofluorescence for mAChR M2 and M3 cells compared with negative results when only secondary antibody was used (Fig. 8, bottom left and right). We found no detectable AChRα (data not shown). Since muscarinic receptor signaling is often linked through the small G protein, RhoA, we also used immunohistochemistry to localize RhoA. Immunofluorescence for RhoA was positive in AT2 cells where Lysotracker red, a biological marker of AT2 cells, was also positive (Fig. 9), indicating there was endogenous RhoA in AT2 cells.
Fig. 9.
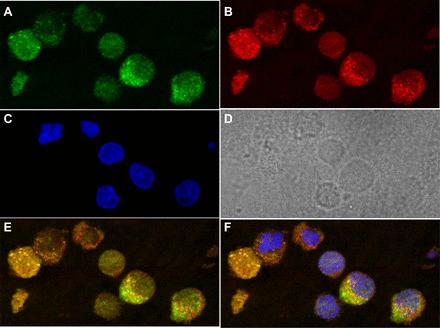
Immunohistochemistry shows RhoA is present in AT2 cells. Endogenous RhoA (green), in the cytosolic space of nonstimulated AT2 cells (A) identified by costaining with Lysotracker red (red) (B), a biological marker for AT2 cells. DAPI staining of cell nuclei is shown in C and the DIC image is shown in D. A merged Lysotracker and RhoA image is at bottom right in E, and an image merging RhoA, Lysotracker red, and DAPI is shown in F.
Cholinergic agonists activate RhoA.
Others have shown that muscarinic agonists can activate the small G protein, RhoA (15, 25), and that RhoA activation can increase ENaC activity (20, 21, 26). To determine whether muscarinic cholinergic agonists activated RhoA in AT2 cells, we used a commercially available ELISA assay for RhoA activation. Groups of AT2 cells were either treated with CCh (100 μM), pretreated with ATR (100 μM) for 5 min before applying CCh, or left untreated. After 5 min all the groups of cells were lysed with the lysis buffer of the assay kit. Figure 10 shows that carbachol significantly increases RhoA activity above that of untreated cells. Atropine completely blocked the effect of carbachol and, in fact, reduced RhoA activity below that of untreated cells implying that there is a low level of baseline activity in the absence of a muscarinic antagonist.
Fig. 10.
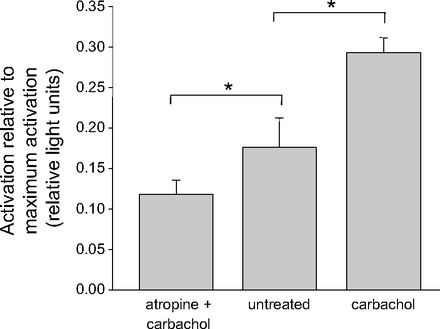
RhoA was activated by carbachol application and inhibited by atropine. GTP-loaded RhoA was measured with RhoA Activation Assay Biochem kit and shown as RLU (relative luminescence unit) in Y-axis. Five-minute exposure to carbachol (100 μM) increased RLU compared with AT2 cells in basal condition. Five-minute pretreatment with ATR (100 μM) inhibited the increase caused by carbachol (100 μM). Values are means ± SE for 4 experiments; *P < 0.05.
The RhoA kinase (ROCK) inhibitor, Y-27632, inhibited carbachol-induced activation of ENaC.
Incubation of AT2 cells with Y-27632 (1 μM) for 30 min abolished not only the activation by carbachol (200 μM) but also intrinsic ENaC activity (Fig. 11, A and B). Although we could not find any openings characteristic of ENaC after the preincubation of AT2 cells with Y-27632, there still remained 20- to 25-pS channels whose properties were unlike ENaC and appeared to be a nonselective cation channel. Since we observed no ENaC openings after the preincubation with Y-27632 in AT2 cells, we also applied Y-27632 acutely to the AT2 cell model, 2F3 cells, to determine if the inhibitor would also decrease ENaC activity. Y-27632 gradually reduced ENaC activity over a 25-min period after application. Addition of forskolin (10 μM), which normally strongly activates ENaC (3, 5, 6, 17, 18), failed to increase channel activity. Thus RhoA appeared to be an essential factor for ENaC activity.
Fig. 11.
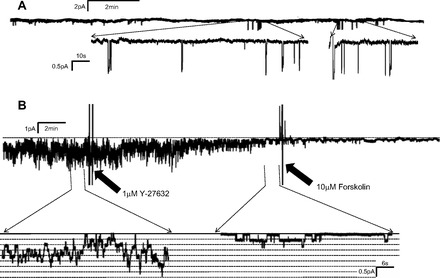
RhoA-associated protein kinase inhibitor, Y-27632, abolished endogenous ENaC in AT2 cells (A) and reduced ENaC activity and inhibited ENaC activation by forskolin in 2F3 cells (B). Application of Y-27632 (1 μM) abolished endogenous ENaC activity in AT2 cells except for some occasional high-conductance channels (not ENaC) and gradually reduced ENaC activity when the inhibitor was applied acutely to an AT2 cell model, 2F3 cells. After inhibition, ENaC could not be activated by forskolin (10 μM). In the absence of Y-27632, we and others have shown that ENaC is always activated by forskolin. These results imply that RhoA kinase plays a role in maintaining ENaC activity.
DISCUSSION
Regulation of ion transport and mucus secretion in the airway mediated by autonomic activity has been previously reported; excitation of parasympathetic nerves increases mucus secretion from goblet cells (30) and also induces fluid secretion through epithelial cells in the central airway apparently by generating a transepithelial potential difference(29). Cholinergic antagonists like atropine are, therefore, commonly prescribed to relieve symptoms of excess secretion/sputum production, prior to endoscopy, or as palliative care for terminally ill patients with lungs flooded with sputum. However, there are few reports of the effects of the direct action of cholinergic agonists on sodium transport in the alveoli. However, Woods et al. (32) have reported that instillation of acetylcholine produced lung liquid reabsorption in fetal pig lungs that was blocked by atropine, suggesting neural control of lung liquid reabsorption in alveoli, a result consistent with our results in vitro.
In our work, we chose three agonists, carbachol, nicotine, or oxotremorine, to electrophysiologically characterize the cholinergic receptors in AT2 cells. We found that muscarinic but not nicotinic agonists activate ENaC. We also observed muscarinic but not nicotinic receptors by Western blot and immunohistochemistry.
Nicotinic receptors have been previously reported in the airway (23), in human and murine macrophages (27, 31), and in human AT2 cells (27), but there has been no report of nicotinic receptors in rat alveolar epithelial cells. In the present study, we were unable to detect nicotinic receptors by Western blots of rat lung lysate. This would be a relatively dramatic species difference, but we have no reason to conclude that nicotinic receptors were present in rat AT2 cells since Western blotting, immunohistochemistry, and patch-clamp electrophysiology were consistently negative.
Based on the effect of oxotremorine, it appears that muscarinic receptors can activate ENaC in AT2 cells. Based on immunohistochemistry, both M2 and M3 receptors seem to be present in the cells. Western blotting, on the other hand, suggests that the predominant receptor is M2. Nonetheless, we cannot rule out a contribution of both receptors leading to ENaC activation.
RhoA and subsequent components of its signaling cascade (RhoA kinase, 4-PIP-5-kinase, and PI-3-kinase) have been reported to increase ENaC activity in CHO cells through effects on the cytoskeleton and promoting ENaC trafficking to the plasma membrane (20, 21, 26). In the present study, we were able to measure a significant increase in channel NPo but were unable to determine whether the change was due primarily to a change in N or Po. This is consistent with the observation that increases in inositol phospholipids increase both Po and N. However, it is also true that if the initial Po is relatively low, counting the number of channels per patch will be difficult because there would be a lower chance of all channels opening simultaneously. After an increase in Po, it may appear that there is an increase in N (since the chance of simultaneous opening increases). Nonetheless, the increase in N appeared larger than could be accounted for based on an increase in Po alone (17).
Endogenous RhoA has been reported to increase basal activity of ENaC in CHO cells expressing exogenous ENaC (20). Such a contribution also appears to be true for AT2 cells based on the results of our RhoA activation assay, immunohistochemistry, and patch-clamp studies of AT2 and 2F3 cells. We were unable to identify any difference in ENaC activity (NPo) between untreated AT2 cells and AT2 cells treated with atropine, implying little endogenous muscarinic activity, but again low basal levels of NPo could mask such an effect. That there might be some endogenous muscarinic receptor activation is suggested by the RhoA activation assay. Atropine by itself reduced endogenous RhoA activity below the activity of untreated cells while also completely inhibiting the increase by carbachol. This idea that there is basal RhoA (ROCK) activity that is necessary for basal ENaC activity seems also to be implied by the fact that the ROCK inhibitor, Y-27632, eliminated basal ENaC activity. Further activation of RhoA/ROCK by muscarinic agonists then further increases ENaC activity above basal levels. This mechanism is illustrated in Fig. 12.
Fig. 12.
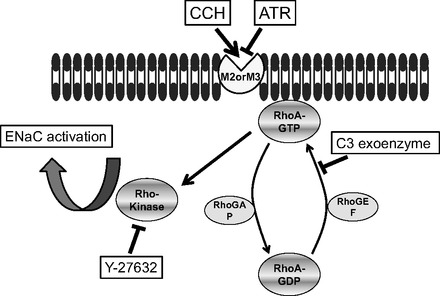
Proposed mechanism for the activation of ENaC by muscarinic agonists. Muscarinic agonists act through G-coupled mAChR M2 and/or M3 in the membrane of AT2 cells and then activate the signal transduction of RhoA/Rho kinase signaling, which will effect/promote activation of ENaC in the plasma membrane.
Anticholinergic bronchodilators are often used to treat COPD and asthma. They are reported to reduce sputum production presumably through the inhibition of mucus secretion or water transport (28). On the other hand, our study suggests that an alternative and possibly adverse effect is to increase fluid secretion into the alveolar space. But other factors may be important in these patients. Under a pathological condition like COPD where cholinergic regulation is dominant, there will be hypersecretion in the airway, but our results would suggest that additional reabsorption in the alveoli may partially compensate. Moreover, while some fraction of inhaled anticholinergic bronchodilator may reach the alveoli and affect alveolar sodium reabsorption, most of the agent is likely to stay in the airway and inhibit secretion. Thus it will be necessary to determine net fluid balance over the whole airway with the implication that final homeostasis may be determined by a coordinated response of secretion and absorption at different locations in the lung. This idea is reminiscent of the β2 agonists which activate ENaC in alveolar epithelial cells but promote secretion via CFTR in the airway (2, 8, 16).
DISCLOSURES
No conflicts of interest, financial or otherwise are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: Y.T., M.N.H., A.F.E., J.S., S.R., and H.-F.B. performed experiments; Y.T., M.N.H., A.F.E., L.J., H.-F.B., and D.C.E. analyzed data; Y.T., M.N.H., and D.C.E. drafted manuscript; Y.T., M.N.H., A.F.E., J.S., S.R., L.J., H.-F.B., and D.C.E. approved final version of manuscript; M.N.H., L.J., H.-F.B., and D.C.E. conception and design of research; M.N.H., L.J., H.-F.B., and D.C.E. interpreted results of experiments; M.N.H., A.F.E., H.-F.B., and D.C.E. prepared figures; M.N.H., A.F.E., L.J., H.-F.B., and D.C.E. edited and revised manuscript.
REFERENCES
- 1. Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 405: 458–462, 2000 [DOI] [PubMed] [Google Scholar]
- 2. Boucher RC. Molecular insights into the physiology of the “thin film” of airway surface liquid. J Physiol 516: 631–638, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen XJ, Eaton DC, Jain L. β-Adrenergic regulation of amiloride-sensitive lung sodium channels. Am J Physiol Lung Cell Mol Physiol 282: L609–L6220, 2002 [DOI] [PubMed] [Google Scholar]
- 4. Chen XJ, Seth S, Yue G, Kamat P, Compans RW, Guidot D, Brown LA, Eaton DC, Jain L. Influenza virus inhibits ENaC and lung fluid clearance. Am J Physiol Lung Cell Mol Physiol 287: L366–L373, 2004 [DOI] [PubMed] [Google Scholar]
- 5. Downs CA, Kriener LH, Yu L, Eaton DC, Jain L, Helms MN. β-Adrenergic agonists differentially regulate highly selective and nonselective epithelial sodium channels to promote alveolar fluid clearance in vivo. Am J Physiol Lung Cell Mol Physiol 302: L1167–L1178, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Eaton DC, Chen XJ, Ramosevac S, Matalon S, Jain L. Regulation of Na+ channels in lung alveolar type II epithelial cells. Proc Am Thorac Soc 1: 10–16, 2004 [DOI] [PubMed] [Google Scholar]
- 7. Eaton DC, Helms MN, Koval M, Bao HF, Jain L. The contribution of epithelial sodium channels to alveolar function in health and disease. Annu Rev Physiol 71: 403–423, 2009 [DOI] [PubMed] [Google Scholar]
- 8. Engelhardt JF, Yankaskas JR, Ernst SA, Yang Y, Marino CR, Boucher RC, Cohn JA, Wilson JM. Submucosal glands are the predominant site of CFTR expression in the human bronchus. Nat Genet 2: 240–248, 1992 [DOI] [PubMed] [Google Scholar]
- 9. Gosens R, Zaagsma J, Meurs H, Halayko AJ. Muscarinic receptor signaling in the pathophysiology of asthma and COPD. Respir Res 7: 73, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gross NJ, Skorodin MS. Role of the parasympathetic system in airway obstruction due to emphysema. N Engl J Med 311: 421–425, 1984 [DOI] [PubMed] [Google Scholar]
- 11. Helms MN, Chen XJ, Ramosevac S, Eaton DC, Jain L. Dopamine regulation of amiloride-sensitive sodium channels in lung cells. Am J Physiol Lung Cell Mol Physiol 290: L710–L722, 2006 [DOI] [PubMed] [Google Scholar]
- 12. Helms MN, Self J, Bao HF, Job LC, Jain L, Eaton DC. Dopamine activates amiloride-sensitive sodium channels in alveolar type I cells in lung slice preparations. Am J Physiol Lung Cell Mol Physiol 291: L610–L618, 2006 [DOI] [PubMed] [Google Scholar]
- 13. Jain L, Chen XJ, Malik B, Al-Khalili OK, Eaton DC. Antisense oligonucleotides against the α-subunit of ENaC decrease lung epithelial cation-channel activity. Am J Physiol Lung Cell Mol Physiol 276: L1046–L1051, 1999 [DOI] [PubMed] [Google Scholar]
- 14. Jain L, Chen XJ, Ramosevac S, Brown LA, Eaton DC. Expression of highly selective sodium channels in alveolar type II cells is determined by culture conditions. Am J Physiol Lung Cell Mol Physiol 280: L646–L658, 2001 [DOI] [PubMed] [Google Scholar]
- 15. Keller J, Schmidt M, Hussein B, Rumenapp U, Jakobs KH. Muscarinic receptor-stimulated cytosol-membrane translocation of RhoA. FEBS Lett 403: 299–302, 1997 [DOI] [PubMed] [Google Scholar]
- 16. Knowles M, Murray G, Shallal J, Askin F, Ranga V, Gatzy J, Boucher R. Bioelectric properties and ion flow across excised human bronchi. J Appl Physiol 56: 868–877, 1984 [DOI] [PubMed] [Google Scholar]
- 17. Marunaka Y, Eaton DC. Effects of vasopressin and cAMP on single amiloride-blockable Na channels. Am J Physiol Cell Physiol 260: C1071–C1084, 1991 [DOI] [PubMed] [Google Scholar]
- 18. Matalon S, Lazrak A, Jain L, Eaton DC. Biophysical properties of sodium channels in lung alveolar epithelial cells. J Appl Physiol 93: 1852–1859, 2002 [DOI] [PubMed] [Google Scholar]
- 19. Pochynyuk O, Bugaj V, Stockand JD. Physiologic regulation of the epithelial sodium channel by phosphatidylinositides. Curr Opin Nephrol Hypertens 17: 533–540, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pochynyuk O, Medina J, Gamper N, Genth H, Stockand JD, Staruschenko A. Rapid translocation and insertion of the epithelial Na+ channel in response to RhoA signaling. J Biol Chem 281: 26520–26527, 2006 [DOI] [PubMed] [Google Scholar]
- 21. Pochynyuk O, Staruschenko A, Bugaj V, Lagrange L, Stockand JD. Quantifying RhoA facilitated trafficking of the epithelial Na+ channel toward the plasma membrane with total internal reflection fluorescence-fluorescence recovery after photobleaching. J Biol Chem 282: 14576–14585, 2007 [DOI] [PubMed] [Google Scholar]
- 22. Pochynyuk O, Tong Q, Staruschenko A, Stockand JD. Binding and direct activation of the epithelial Na+ channel (ENaC) by phosphatidylinositides. J Physiol 580: 365–372, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Racke K, Juergens UR, Matthiesen S. Control by cholinergic mechanisms. Eur J Pharmacol 533: 57–68, 2006 [DOI] [PubMed] [Google Scholar]
- 24. Roffel AF, Elzinga CR, Van Amsterdam RG, De Zeeuw RA, Zaagsma J. Muscarinic M2 receptors in bovine tracheal smooth muscle: discrepancies between binding and function. Eur J Pharmacol 153: 73–82, 1988 [DOI] [PubMed] [Google Scholar]
- 25. Seasholtz TM, Majumdar M, Brown JH. Rho as a mediator of G protein-coupled receptor signaling. Mol Pharmacol 55: 949–956, 1999 [DOI] [PubMed] [Google Scholar]
- 26. Staruschenko A, Nichols A, Medina JL, Camacho P, Zheleznova NN, Stockand JD. Rho small GTPases activate the epithelial Na+ channel. J Biol Chem 279: 49989–49994, 2004 [DOI] [PubMed] [Google Scholar]
- 27. Su X, Lee JW, Matthay ZA, Mednick G, Uchida T, Fang X, Gupta N, Matthay MA. Activation of the alpha7 nAChR reduces acid-induced acute lung injury in mice and rats. Am J Respir Cell Mol Biol 37: 186–192, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Takeyama K, Tamaoki J, Nakata J, Konno K. Effect of oxitropium bromide on histamine-induced airway goblet cell secretion. Am J Respir Crit Care Med 154: 231–236, 1996 [DOI] [PubMed] [Google Scholar]
- 29. Tamaoki J, Chiyotani A, Tagaya E, Takemura H, Konno K. Cholinergic control of rabbit tracheal transepithelial potential difference in vivo. Eur Respir J 9: 1632–1636, 1996 [DOI] [PubMed] [Google Scholar]
- 30. Tokuyama K, Kuo HP, Rohde JA, Barnes PJ, Rogers DF. Neural control of goblet cell secretion in guinea pig airways. Am J Physiol Lung Cell Mol Physiol 259: L108–L115, 1990 [DOI] [PubMed] [Google Scholar]
- 31. Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421: 384–388, 2003 [DOI] [PubMed] [Google Scholar]
- 32. Woods BA, Ng W, Thakorlal D, Liu AL, Perks AM. Effects of acetylcholine on lung liquid production by in vitro lungs from fetal guinea pigs. Can J Physiol Pharmacol 74: 918–927, 1996 [DOI] [PubMed] [Google Scholar]