

Abstract
It is well accepted that TGF-β signaling has critical functional roles in lung development, injury, and repair. We showed previously that null mutation of Smad3, a critical node in the TGF-β pathway, protects mice against fibrosis induced by bleomycin. However, more recently we noticed that abnormal alveolarization also occurs in Smad3-deficient mice and that this is followed by progressive emphysema-like alveolar wall destruction mediated by MMP9. We now know that Smad3 cooperates with c-Jun to synergistically regulate a protein deacetylase SIRT1, by binding to an AP-1 site in the SIRT1 promoter. Consistently, Smad3 knockout lung at postnatal day 28 had reduced SIRT1 expression, which in turn resulted in increased histone acetylation at the binding sites of the transcription factors AP-1, NF-κB, and Pea3 on the MMP9 promoter, as well as increased acetylation of NF-κB. Thus, upon TGF-β activation, phosphorylated Smad3 can be translocated into the nucleus with Smad4, whereat Smad3 in turn collaborates with c-Jun to activate SIRT1 transcription. SIRT1 can deacetylate NF-κB at lysine 30, as well as histones adjacent to the transcription factor AP-1, NF-κB, and Pea3 binding sites of the MMP9 promoter, thereby suppressing MMP9 transcription, hence fixing MMP9 in the OFF mode. Conversely, when Smad3 is missing, this regulatory pathway is inactivated so that MMP9 is epigenetically turned ON. We postulate that these developmental epigenetic mechanisms by which Smad3 regulates MMP9 transcription cell autonomously may be important in modulating both emphysema and pulmonary fibrosis and that this could explain why both pathologies can appear within the same lung specimen.
Keywords: emphysema, epigenetics, fibrosis, Smad3, TGF-β
pulmonary fibrosis, particularly in its idiopathic form (IPF) in adult humans may be an aggressive, fatal disease with as yet no known effective treatment. It has been widely postulated that excessive signaling by TGF-β1 ligand activation of the TGF-β downstream signaling pathway may be the final common pathway causing fibroproliferative disease and fibrosis of many tissues, including the lung, skin, peritoneum, joints, etc. (11, 16), suggesting that blockade or modulation of TGF-β signaling might be a therapeutically tractable target. Importantly, Smad3 functions as a critical node in this canonical signaling pathway downstream of TGF-β ligand-receptor interaction in the lung (reviewed previously in more detail by us in Ref. 17). Here follows a personal view, with an update of recent developments in the field, placed in the context of and expanding on our previous work (19).
We showed previously that null mutation of Smad3 protects mice against fibrosis induced by bleomycin, which appeared to support the concept that TGF-β signaling is the downstream common mediator of fibrosis (19). However, as discussed below, TGF-β signaling is exquisitely finely regulated at both the extracellular and intracellular level. Moreover, the function of TGF-β signaling is not only compartment specific but also temporally specific within the epithelial, mesenchymal, and immune components of the lung. Thus therapeutic manipulation of TGF-β signaling to ameliorate or reverse lung fibrosis remains enticing but extremely challenging.
The lungs of Smad3-deficient mice are by no means developmentally normal, although the phenotype is not neonatal lethal. Rather there are significant but relatively subtle deficiencies in postnatal alveolarization in Smad3−/− mice, which notably include disorganization of elastin fibers and thus failure to correctly complete alveolar septation over the first 28 days of life.
Abnormal alveolarization in Smad3-deficient mice is followed by progressive emphysema-like alveolar wall destruction initiated by MMP9. By 28 days of life, imperfect alveolarization is associated with activation of protease activity in the lung, particularly MMP9. Double crossing of Smad3−/− with MMP9−/− mice resulted in substantial rescue of the alveolar destructive phenotype but not of the developmental hypoalveolarization phenotype (18). Thus we speculated that Smad3 may serve normally to suppress MMP9.
Smad3 participates in epigenetic regulation of MMP9 (Fig. 1). Smad3 functions as a transcriptional cofactor that positively regulates many genes, including, importantly, a protein deacetylase SIRT1, by binding to an AP-1 site in the SIRT1 promoter (18). Furthermore, we found a synergistic regulatory effect on SIRT1 transcriptional expression by Smad3 and c-Jun. Consistent with these findings, Smad3 knockout lung at postnatal day 28 (P28) had reduced SIRT1 expression, which in turn resulted in increased histone acetylation at the transcription factor AP-1, NF-κB, as well as Pea3 binding sites of the MMP9 promoter with increased acetylation of NF-κB. In addition, increased Pea3 expression and nuclear accumulation were also detected in Smad3-null lungs at P28. Consistent with this, binding of acetylated NF-κB and Pea3 to the MMP9 promoter was elevated in Smad3-null lung. Thus deficiency of Smad3 causes downregulation of SIRT1 and hence increased Pea3 expression and nuclear accumulation, respectively. Decreased SIRT1 activity likewise resulted in increased histone and NF-κB acetylation. Hence, increased binding of the transcriptional factors NF-κB and Pea3 to the MMP9 promoter significantly upregulates MMP9 transcription, which mediates increased MMP9 activity and hence emphysema pathogenesis. Moreover the above findings on histone acetylation of MM9 could explain the appearance of proteolytic degradation and hence alveolar destructive function by 28 days of life in Smad3 null lungs. In addition, we have found that exposure to tobacco smoke markedly exacerbates the alveolar wall destruction in Smad3-deficient mice (6).
Fig. 1.
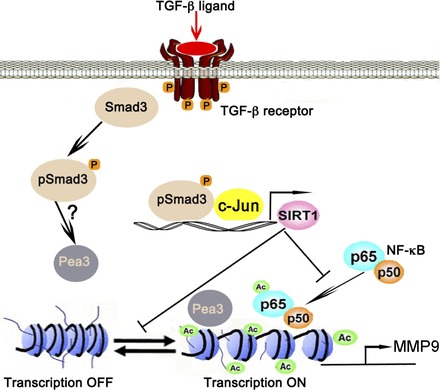
Epigenetic regulation of MMP9 by TGF-β-Smad3 signaling: TGF-β ligand activates TGF-β receptor-Smad3 signaling, which in turn collaboratively activates SIRT1 transcription with c-Jun. SIRT1 in turn deacetylates NF-κB at lysine 30 and histones at the transcription factor AP-1, NF-κB, and Pea3 binding sites of MMP9 promoter to suppresses its transcription, thereby fixing MMP9 in the OFF mode; conversely when Smad3 is missing MMP9 stays in the ON mode. Ac, acetylations.
Timing, dosage, and spatial direction of TGF-β signaling are particularly critical in the lung. Optimal levels of TGF-β-Smad3 signaling appear to be essential for formation of secondary alveolar septae. Compartment-specific abrogation of TGF-β type II receptor signaling in epithelial cells reduces alveolar septation but allows emergence of differentiated type I alveolar epithelial cells (4). However, conversely, TGF-β1 ligand overexpression in early embryonic epithelium strongly inhibits branching morphogenesis (21), whereas misexpression of Sp-C promoter controlled TGF-β1 in embryonic lung epithelium arrests lung growth and epithelial differentiation, while also inhibiting. However, mesenchymal-specific TGF-β type II receptor blockade driven by Dermo1 retards branching after midgestation, whereas, in contrast, epithelial TGF-β signaling abrogation lacked prenatal impact and only disrupted postnatal lung alveolarization (3, 4). Moreover, excess TGF-β ligand induces aggressive pulmonary fibrosis, whereas TGF-β3 ligand excess only induces transient fibrosis (1).
Increased active TGF-β1 ligand bioactivity has been reported in tracheal aspirates of premature infants who develop severe bronchopulmonary dysplasia (BPD) (9, 15). Furthermore, adenoviral vector-mediated misexpression of TGF-β1 ligand in neonatal rat lung causes neonatal alveolar hypoplasia and interstitial fibrosis, pathologically indistinguishable from human BPD (7). Thus in immature lung excessive TGF-β signaling appears to arrest branching and/or alveolar septal development.
Overexpression of active TGF-β1 with use of an adenoviral vector approach in adult rats results in chronic progressive interstitial pulmonary fibrosis (12, 19). In addition, TGF-β1 may be central to lung fibrotic responses to bleomycin, endotoxin, or infection (2). Interestingly, misexpression of TGF-β1 ligand using an adenoviral vector approach causes striking interstitial pulmonary fibrosis and pleural thickening in fetal monkeys at a time when alveolarization is already ongoing (14), thus leading us to speculate that the effects of TGF-β1 ligand excess may depend on developmental timing of exposure: early on it results in developmental arrest, whereas once alveolarization is established it results in fibrosis.
Dominant human mutations in the TGF-β pathway predispose to sudden alveolar rupture, leading to tension pneumothorax and/or aortic dissection in Marfan's syndrome and Loeys-Dietz syndrome (8, 10). Mutations in TGF-β type I receptor (Alk5) or in TGF-β binding proteins such as fibrillin are postulated to underlie the dysplastic matrix and elastin defects that lead to mechanical failure in both the classic Marfan's localized aortic dissection as well as formation of alveolar blebs. In the case of fibrillin mutants, a loss of function of fibrillin leads to a gain of dysfunction of TGF-β signaling. Thus fibrillin mutation inhibits the ability of fibrillin to sequester active TGF-β1 ligand within the tissue matrix. Null mutation of latent TGF-β binding proteins (LTBP-3 or LTBP-4) also cause profound defects in elastin fiber structure and lung alveolarization, similar to those found in the Smad3 knockout mouse (3, 5, 13).
We conclude that TGF-β-Smad3 signaling is very finely regulated during early lung development as well as during alveolarization (20, 21, 22). We have previously termed this the “Goldilocks hypothesis” for TGF-β-Smad signal transduction because everything has to be “just right” for lung development to proceed normally. This concept most likely also applies to other important growth factor signaling pathways.
Canonical TGF-β ligand/receptor/Smad signaling not only is required for the completion of normal alveolarization but also protects against subsequent alveolar proteolytic destruction.
Before alveolarization is well established, excessive TGF-β signaling inhibits this process; this phenotype, together with mild scarring, is similar to that seen in human premature infants with BPD (6, 8). But once alveolarization is well established, excessive TGF-β1 ligand-induced receptor Smad3 signaling leads to aggressive and progressive fibrosis similar to that seen in adult human IPF patients (11, 13).
Thus correct amounts of canonical TGF-β ligand/receptor/Smad3 signaling are absolutely required for the completion of normal alveolarization and maintenance of alveolar homeostasis (2, 3, 16).
We are presently working to further determine the compartment and time-specific functions of TGF-β signaling within the lung during early development, during alveolarization as well as following injury in adults, such as injuries caused by tobacco smoke or bleomycin exposure, as well as to further elucidate the epigenetic mechanisms perturbing the TGF-β-Smad3 signaling pathway in injury and failed repair as now appears likely to be the case in both emphysema and fibrosis (3, 5, 16).
REFERENCES
- 1. Ask K, Bonniaud P, Maass K, Eickelberg O, Margetts PJ, Warburton D, Groffen J, Gauldie J, Kolb M. Progressive pulmonary fibrosis is mediated by TGF-beta isoform 1 but not TGF-beta3. Int J Biochem Cell Biol 40: 484–495, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bonniaud P, Margetts PJ, Ask K, Flanders K, Gauldie J, Kolb M. TGF-beta and Smad3 signaling link inflammation to chronic fibrogenesis. J Immunol 175: 5390–5395, 2005 [DOI] [PubMed] [Google Scholar]
- 3. Chen H, Sun J, Buckley S, Chen C, Warburton D, Wang XF, Shi W. Abnormal mouse lung alveolarization caused by Smad3 deficiency is a developmental antecedent of centrilobular emphysema. Am J Physiol Lung Cell Mol Physiol 288: L683–L691, 2005 [DOI] [PubMed] [Google Scholar]
- 4. Chen H, Zhuang F, Liu YH, Xu B, Del Moral P, Deng W, Chai Y, Kolb M, Gauldie J, Warburton D, Moses HL, Shi W. TGF-beta receptor II in epithelia versus mesenchyme plays distinct roles in the developing lung. Eur Respir J 32: 285–295, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Colarossi C, Chen Y, Obata H, Jurukovski V, Fontana L, Dabovic B, Rifkin DB. Lung alveolar septation defects in Ltbp-3-null mice. Am J Pathol 167: 419–428, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Farkas L, Farkas D, Warburton D, Gauldie J, Shi W, Stampfli MR, Voelkel NF, Kolb M. Cigarette smoke exposure aggravates air space enlargement and alveolar cell apoptosis in Smad3 knockout mice. Am J Physiol Lung Cell Mol Physiol 301: L391–L401, 2011 [DOI] [PubMed] [Google Scholar]
- 7. Gauldie J, Galt T, Bonniaud P, Robbins C, Kelly M, Warburton D. Transfer of the active form of transforming growth factor-beta 1 gene to newborn rat lung induces changes consistent with bronchopulmonary dysplasia. Am J Pathol 163: 2575–2584, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kaartinen V, Warburton D. Fibrillin controls TGF-beta activation. Nat Genet 33: 331–332, 2003 [DOI] [PubMed] [Google Scholar]
- 9. Lecart C, Cayabyab R, Buckley S, Morrison J, Kwong KY, Warburton D, Ramanathan R, Jones CA, Minoo P. Bioactive transforming growth factor-beta in the lungs of extremely low birthweight neonates predicts the need for home oxygen supplementation. Biol Neonate 77: 217–223, 2000 [DOI] [PubMed] [Google Scholar]
- 10. Lindsay ME, Dietz HC. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature 473: 308–316, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Petrides PE, Brenner D. From biochemical analysis to targeted therapies. Fibrogen Tissue Repair 5, Suppl 1: S1, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J Clin Invest 100: 768–776, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sterner-Kock A, Thorey IS, Koli K, Wempe F, Otte J, Bangsow T, Kuhlmeier K, Kirchner T, Jin S, Keski-Oja J, von Melchner H. Disruption of the gene encoding the latent transforming growth factor-beta binding protein 4 (LTBP-4) causes abnormal lung development, cardiomyopathy and colorectal cancer. Genes Dev 16: 2264–2273, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tarantal AF, Chen H, Shi TT, Lu CH, Fang AB, Buckley S, Kolb M, Gauldie J, Warburton D, Shi W. Overexpression of transforming growth factor-beta1 in fetal monkey lung results in prenatal pulmonary fibrosis. Eur Respir J 36: 907–914, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Toti P, Buonocore G, Tanganelli P, Catella AM, Palmeri ML, Vatti R, Seemayer TA. Bronchopulmonary dysplasia of the premature baby: an immunohistochemical study. Pediatr Pulmonol 24: 22–28, 1997 [DOI] [PubMed] [Google Scholar]
- 16. Warburton D. Developmental responses to lung injury: repair or fibrosis. Fibrogen Tissue Repair 5, Suppl 1: S2, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Warburton D, El-Hashash A, Carraro G, Tiozzo C, Sala F, Rogers O, De Langhe S, Kemp PJ, Riccardi D, Torday J, Bellusci S, Shi W, Lubkin SR, Jesudason E. Lung organogenesis. Curr Top Dev Biol 90: 73–158, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xu B, Chen H, Xu W, Zhang W, Buckley S, Zheng SG, Warburton D, Kolb M, Gauldie J, Shi W. Molecular mechanisms of MMP9 overexpression and its role in emphysema pathogenesis of Smad3-deficient mice. Am J Physiol Lung Cell Mol Physiol 303: L89–L96, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhao J, Shi W, Wang YL, Chen H, Bringas P, Jr, Datto MB, Frederick JP, Wang XF, Warburton D. Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am J Physiol Lung Cell Mol Physiol 282: L585–L593, 2002 [DOI] [PubMed] [Google Scholar]
- 20. Zhao J, Sime PJ, Bringas P, Jr, Tefft JD, Buckley S, Bu D, Gauldie J, Warburton D. Spatial-specific TGF-beta1 adenoviral expression determines morphogenetic phenotypes in embryonic mouse lung. Eur J Cell Biol 78: 715–725, 1999 [DOI] [PubMed] [Google Scholar]
- 21. Zhao J, Sime PJ, Bringas P, Jr, Gauldie J, Warburton D. Adenovirus-mediated decorin gene transfer prevents TGF-beta-induced inhibition of lung morphogenesis. Am J Physiol Lung Cell Mol Physiol 277: L412–L422, 1999 [DOI] [PubMed] [Google Scholar]
- 22. Zhao J, Sime PJ, Bringas P, Jr, Gauldie J, Warburton D. Epithelium-specific adenoviral transfer of a dominant-negative mutant TGF-beta type II receptor stimulates embryonic lung branching morphogenesis in culture and potentiates EGF and PDGF-AA. Mech Dev 72: 89–100, 1998 [DOI] [PubMed] [Google Scholar]