

Abstract
The genetic mechanisms underlying the susceptibility to acute respiratory distress syndrome (ARDS) are poorly understood. We previously demonstrated that sphingosine 1-phosphate (S1P) and the S1P receptor S1PR3 are intimately involved in lung inflammatory responses and vascular barrier regulation. Furthermore, plasma S1PR3 protein levels were shown to serve as a biomarker of severity in critically ill ARDS patients. This study explores the contribution of single nucleotide polymorphisms (SNPs) of the S1PR3 gene to sepsis-associated ARDS. S1PR3 SNPs were identified by sequencing the entire gene and tagging SNPs selected for case-control association analysis in African- and ED samples from Chicago, with independent replication in a European case-control study of Spanish individuals. Electrophoretic mobility shift assays, luciferase activity assays, and protein immunoassays were utilized to assess the functionality of associated SNPs. A total of 80 variants, including 29 novel SNPs, were identified. Because of limited sample size, conclusive findings could not be drawn in African-descent ARDS subjects; however, significant associations were found for two promoter SNPs (rs7022797 −1899T/G; rs11137480 −1785G/C), across two ED samples supporting the association of alleles −1899G and −1785C with decreased risk for sepsis-associated ARDS. In addition, these alleles significantly reduced transcription factor binding to the S1PR3 promoter; reduced S1PR3 promoter activity, a response particularly striking after TNF-α challenge; and were associated with lower plasma S1PR3 protein levels in ARDS patients. These highly functional studies support S1PR3 as a novel ARDS candidate gene and a potential target for individualized therapy.
Keywords: acute respiratory distress syndrome, sphingosine 1-phosphate, single nucleotide polymorphism, association study, disease predisposition
acute respiratory distress syndrome (ARDS) is a life-threatening condition characterized by profound inflammation, increased vascular permeability, and alveolar flooding, leading to severe hypoxemia, respiratory failure, and >30% mortality (11, 23, 44). Despite advances in care of the critically ill, there is an urgent need to improve our understanding of the biochemical/genetic basis of ARDS to broaden the limited therapeutic options currently available. ARDS occurs in response to diverse inciting stimuli including sepsis, aspiration, trauma, etc. with only a small fraction of patients with these risk factors developing ARDS. Clinical and epidemiological studies suggest that individual genetic variation may contribute to the observed variability in ARDS susceptibility (6, 10, 16). It is now appreciated that ARDS pathogenesis is influenced by the host genotype, the environment, and the nature of the process that produces ARDS (45). To increase insights into ARDS susceptibility, genes contributing to ARDS development and novel ARDS biomarkers need to be identified (17). In addition, large interethnic differences also significantly contribute to population health disparity in ARDS susceptibility and outcomes.
We employed complementary approaches to identify genetic factors (9, 14, 15, 55) involved in ARDS susceptibility and severity, including strategies designed to leverage the role of increased tissue permeability in ARDS (16). In prior work we identified S1PR3 as a gene whose protein product is critically involved in the regulation of lung vascular permeability and in the development and progression of ARDS (16). S1PR3 elicits multiple aspects of the inflammatory response, including apoptosis, vascular barrier regulation, permeability, and leukocyte diapedesis, by binding to its natural ligand, sphingosine-1-phosphate (S1P) (25, 32–34). Studies in murine models of ARDS showed that S1PR3 plays an important role in promoting pulmonary inflammation, disrupting cell-cell junctions, and increasing lung permeability and alveolar flooding during inflammation (24, 25, 40). More recently, we found that plasma S1PR3 levels were significantly increased in sepsis-associated ARDS patients and in ARDS mice, and that levels of S1PR3 were identified to highly correlate with disease severity among septic patients (39). Therefore, we considered S1PR3 as a novel ARDS biomarker and significant contributor to ARDS susceptibility and severity. In the present study, we explored the association of gene variants of S1PR3 with ARDS susceptibility and investigated the association of S1PR3 variants with ARDS risk in two independent case-control studies from the US and Spain.
MATERIALS AND METHODS
Human S1PR3 Gene Resequencing
DNA samples from 27 unrelated individuals, 14 of African descent (AD) and 13 of European descent (ED), were subjected to S1PR3 sequencing to search for common variants. Sequencing protocols and polymorphism identification were performed by Polymorphic DNA Technologies (Alameda, CA). PCR primers were designed to amplify the entire gene and 2 kb of the predicted promoter (Genomatix Software, Ann Arbor, MI) and 2-kb downstream regions of the gene. A list of primer pairs utilized is provided in Table 1. Details of S1PR3 variants identified are reported in Supplemental Table S1 following established guidelines (2) (supplemental material for this article is available online at the Journal website).
Table 1.
Primers used for polymorphism discovery in S1PR3 gene
| Amplicon | Nest Forward Primer (5′→3′)* | Nest Reverse Primer (5′→3′) | Size† | Boost Forward Primer (5′→3′) | Boost Reverse Primer (5′→3′) | Size† |
|---|---|---|---|---|---|---|
| 1 | GAGGAGACTTATTCAACAGAATTGT | AATTAGTTGCTTCCCGTGATG | 494 | GTGAAACAGGATGCAGAGGA | TAGGTTCATCACACCTCTGTT | 534 |
| 2 | ATAAAGCATGAGAGCCACATTAC | TGGGGTACTTCCCAGCGTT | 506 | TCTCTTAGGGAAACCTGCCA | CTAGAGGCTCTGGCCC | 549 |
| 3 | AGCATGTTTCCTTCTTTTCTTGACT | GATTGGGCCGCAGGG | 496 | TGACCAATGCCAAAGGAGC | CGACCGTGCATTCCTGG | 537 |
| 4 | GCGTTTCTCTGCAGGGAAC | CTCACGCGGATTTGCCC | 492 | CCTTCAGCTCAATTGCTGAG | TCCTAGACCGGCAGCCA | 568 |
| 5 | AACAGGCCTCTCCTTGCTTT | TCTTTTGTAACTGGCCGAGC | 538 | CAGGAATGCACGGTCGG | GTTGTCCAGATCCTGACCA | 578 |
| 6 | TCTGGTGCACCAGCAGAGG | CCTAGCCTGGACCCCTTC | 483 | TAGGAGGGGCGCTGCTCT | AAGCCGCCTGGTCTCCGT | 547 |
| 7 | TGTCAGGGCGACAGGCG | AGCGCTCGCGACGTCGTT | 512 | GTCAGGATCTGGACAACGATAATGA | ATGGGCTGGGAGGCCC | 607 |
| 8 | AGGAGTGCCGGGTTCGC | CCGGGATCGGTGGGGATT | 462 | CGGCTTGAGCGGGAGGT | GCGCCCAAGTCCCCGT | 510 |
| 9 | CTCCCAGCCCATCTGGC | AGGCTAACGACCACAACCCT | 464 | GCGAGCGCTGAGACTGC | GGCCCTAATTGCCTTGTTTGAGAAA | 565 |
| 10 | TCCCCCAGAGCCGCT | CAAGAAGTTCCTCTCTTATATCCAA | 542 | CGGAGCGCTCCACATCA | AGTGTGTTCCCTTAAAAATCCAACA | 589 |
| 11 | TCCCCACAGTGCAATAATGA | CTTCCCTTGTACTTTCTAGGAATT | 527 | AATCGGTGCTGGAAGACGA | ATGTATCTGCAGAGCATAATCCT | 569 |
| 12 | TAGAAGGTTACTTTTAAATGGAGGC | TTTGTATCTCTGGTGTTAGGTGAT | 530 | AGGAACAGAGAGCTCTTTAG | TCTTAAGGGCTAGCTTCAGT | 566 |
| 13 | ATGGTGCGGGGCATGTGT | CATGCTCAAATTCTCCTTTTTTTGG | 591 | CAGGGCTGGTGTTGGTTGA | CTCAGATTTTTCAAGATTGTGGCAT | 642 |
| 14 | AATGCATGAGTCTCTGATTTTCTAC | GATTCCACTCAGTCTATTCTTTTTC | 587 | AAAGTGCCATTTCTCCCTTTATC | GTAGCTATAACAGTAACATCAACTG | 680 |
| 15 | TGTTTCCCAAGTGTTACCAG | ACTGCTTGAGGAGCGGAC | 525 | CTTTAACTGCCAGGACTTTG | TCTGCAAGGCCGAAGAGCA | 573 |
| 16 | GCATAAAGTCTCCTTCCCTTT | AGGGGGGGTGTCACTTGT | 495 | GTTGATGTTACTGTTATAGCTACTG | TGTACACAGGAGGAGACCG | 550 |
| 17 | TGCTGGCATGGGAGCATGA | CTGCTGCTTCATGACAGCAT | 481 | CCCAGAATGACATGCTAACA | GAGTCTTTTTAAACCTTCTCCATTC | 531 |
| 18 | CACGCATGCCGAGTCAC | ATGCAAGTCAGTGCACTTCC | 489 | CTGCCATGAGGCCATGTGT | AGGGTTTTGTATCTTTTCTCCCTT | 569 |
| 19 | GAAGCTCAGGATGGTGTTCA | CAACAAGCAAAATTTCTTGGAAAGC | 509 | GCCTTCCCACACACAAGTC | GAATCAAAACACCAAACATATGCCA | 553 |
| 20 | GCTGGGTGTGTGAAGCATG | ACATTGCTGGGAGGAGGGT | 537 | CATTAATGGGCCAAGCCTGT | TTCCCCCAACTTCCAAGGA | 582 |
| 21 | TTATTGTGCCTATCTTTTATGCGGA | GAGACTTAAGTATTTGATTGTCTGC | 516 | TGACCAAGATGCTATCTTACTATCT | CCACCCAGCTCTTCAAGAG | 556 |
| 22 | GTCTTATTCCTCCTCGACAAA | AAATAAAGCCAGATGCTGAGAC | 497 | CATACTCATCTTCCAGAAGG | CTTAAATTCCACCTGAACACTTAG | 542 |
| 23 | AAGGATGTGCCGTGGAGG | GATTGATTGAGGATGTTCTCTTG | 502 | TTGGAAGAGAGTGGAACGAC | TTGATTATCATCAAGTAGCACTGCT | 554 |
| 24 | TGCATCAGTCTTGCCCTCT | TGCCCCTCCTAGTCACCA | 505 | GGAATAAAGCAAAGAGTGAACACTA | GGACCCTGCAGCCTTC | 554 |
| 25 | ATGAGAGCAAGAATATTCAGCATG | CTGAGGGGTACCAGCC | 476 | TAGTGTATCTACCCACACCAT | TCATGGACAAGAGTTCCCCT | 521 |
| 26 | TGGGGCAGGTCAGCCT | GAGGGCAAACATATTTTCCCATTAA | 513 | TGCTGGTCTCACTGATCTCT | ACCCATCAAAATCATGAATCTTGAG | 561 |
| 27 | TTAACTCTTGGGATGAGGCC | GTTATGATTCAGACTGTTCTTGTAC | 539 | AATCTGGCCTTTAGCCCAG | TTTTAGGTAATACTGGCATGTCGTT | 583 |
| 28 | TATAAGCCGTGGGTCTATTCA | CCTGTGCCAGCATCCTC | 534 | GCTTGTTTTCTGCTCTGATCAATTT | GAGCAAAGGCAGTCTGTGA | 588 |
| 29 | GGAACCAAGGTTTGTCTGCA | ATTTCTGATTTTCACTTTGCTGGGT | 485 | GCCTCAGGAATCACTGACA | AACAGGAACTGGAGAATCACTATT | 527 |
| 30 | TGGCTGTTCGTGTGGGAAA | AGGCTGGAGTGCAGCG | 517 | CATGCCTTTTCCATGAGCAT | GGTGCTAAAAAGATACTGTCCTTT | 632 |
| 31 | TAGGCCTGGTGTGGTAGTT | CAGCCCCTGGTGACTTTTC | 534 | CAGTTCCTGTTCACAGATAAAGAA | TGCTCTGTAACATTTTCTGGAATCA | 608 |
| 32 | ACAGTATCTTTTTAGCACCCATC | CCAGGCTGATGATGCAGGA | 526 | ACTAGGCCTGGTGTGGTAG | AAAGAAAAAGCAGCCAAGTTCCA | 885 |
| 33 | TGGGGGGAAGGCTGCC | GATTTTTTTTCCAGTGTCCTGGAGC | 484 | AGGCTTGAAGGATGGGCAAGAT | GCCCCTTGCTTCTTTGCAAAGAT | 562 |
| 34 | CACATAAGCCTTTCTGCCAG | CTAAATTTGCTTCAGAGACACATGA | 498 | CAGTTAGCAGAGGGCCTTG | GATGTTTGAACCACAGCCCT | 540 |
| 35 | TCCAGAGGGTCACGAGG | CCTCCTGGGTTCAAGTGC | 534 | GAGGGCTTTGACAGCATTC | GCTTTCAGTGGACTCACTTATT | 689 |
| 36 | GGCTCTTTTAAATTAGCCTCATTTG | AAAGTGGTGAGTGAAGGCC | 558 | ACTGGATACACACAGTAGGC | ATTTGGGAACACAGCGAGAG | 599 |
| 37 | AGCAACACTGTGTCTCAAAAGA | TGGCAGAGAGCCCTGAG | 525 | GAGATCGCACCACTGCATT | GTCTGTATGGGCTGTAGGT | 584 |
| 38 | GTCAGTGTGATTGTCACTGC | TGAATCCTGTAACAAAGATACACAG | 504 | GCTCTGAATTTCAAGCCAGA | GGGAAATTCAGTGCATGAATTATTG | 551 |
| 39 | TAAAACTCACCAGGGAGGTTT | ATTCAGCAAGGCGTGCAGG | 504 | GATCTTCCCACGATGTCACT | AAGCGGGGCTGAGGTTC | 549 |
| 40 | GTGTCCCCCAATCACTTAG | AATCAAAACCATCAGGTTCTCCA | 510 | CAGACACCAAATCGCTTCC | GTACATGCGGTTGTGAAATTTATTG | 572 |
| 41 | TCTGTGAATGCCAAGTGATG | ATCAGGAGGAAGACGCGGT | 490 | TCTCGTGGATTTTGGAGCTAAT | GGCAATGAGCCAGCACATC | 536 |
| 42 | ACCTGCTGGCCGGCA | GAACACGCTCACCACAATCA | 503 | TTTTTCATTGGCAACCTGGCT | TTGAAGAGGATGGGGCACG | 611 |
| 43 | CCCTGACTGCTCTACCATC | CTATTGTTGCTGCTGCTTGA | 489 | GCTGGAACTGCCTGCACAA | TTGACCTTCGGAGAGTGGC | 532 |
| 44 | GGCTCAGTGGTTCATCGTG | TGTTTACACTGAAGCGGGGT | 481 | AGCGTGTTCATCGCCTGCT | CTGGAGCCCAAAGTGGACA | 597 |
| 45 | AATGGGATCTTCTGCAACTGAT | CTGTGTGTTGTATGGAGATTCTA | 503 | ACAAGAACGCAGCACTTCAG | AATCCACTTGTATAACCTGTTACC | 546 |
| 46 | CTGTTGTTCACAGAGAGGGA | CTGGCTCTTCTGTAATTTGCA | 507 | GCTGCATCCTAGACACCCT | AAGCTCCTGGGTGATCTTG | 548 |
| 47 | TGACATGATTCTCATGTTAACCAAG | TCACGATCAGTGCCAGCTT | 504 | CCTGGTCTCCTTAGACTGA | CCAGATCACATTTTCTAACCTGA | 567 |
| 48 | AGTGGGGCAGGGGTCT | TCCTTTGAACGCTGGTTGGT | 516 | AAAGAGCCTATGAAGGAGAAG | TAGGGGACTCTGATGGGAG | 596 |
| 49 | TGAACCCGTAGCAGTCTGTT | CCTGTGTTTTCTCATGCTATTTCA | 530 | AAAATGTGATCTGGCAGTTTATGCA | TTTTACTGTCCTAGAAATGCCC | 635 |
| 50 | TGTCATCTGATGCAATTTGCTCTTA | TTTTGGCTCTCTGATGCATTTTG | 603 | GCCTGAATAAAACCTAAAAGAGTAG | CACCTATGTGAAAATGCAATACTGA | 702 |
| 51 | GACAGTAAAACGTTAAAGTACTGGA | CAAATATTGATACCTCCTAGTGGAA | 604 | ACACAGGGCATTTCTAGGAC | GAGCCTTATGTCATACCACAA | 639 |
| 52 | GTATTGCATTTTCACATAGGTGATG | GTTTCCACAAAGGCTTTTGGTTTTA | 547 | ATCAGAGAGCCAAAACATTCAGTA | TGGACTACGAAGAGTGGGTT | 585 |
| 53 | CTTTGATACCAATGTGCACCAAT | CACTTTTATTACCATGGGTCTTAGA | 526 | ATAGCGCTCATTTGCTAAATGCAAA | AAGAGGTCCCATGTCTTTGA | 585 |
| 54 | AGGCTTGCTACCCACCTG | TGCATCCGGGCAGATGTAC | 515 | AGGGGTGCGCACTGTTG | TCATCAAGGCTGCGGAATGA | 559 |
| 55 | TCCCACACAAACGGTCCATT | CACCTTGTTACATTTCTGTGAAAG | 507 | CAACCTTTGCACACTAGCTT | CAACTTAAAAGCATGAAACTGGTGA | 580 |
| 56 | CACTGAAAATACCTACTTCTTAGAG | TTCATGATCTGTCTGGTGCA | 557 | AGTCCCCCAGGGAATCAC | TGTGGTTTCCAAGCATTAGC | 594 |
| 57 | CATAATCAGAAATGCGCACAC | GATTAGGAGACAACTCCATGTAAAT | 639 | CTAAGTCAGATATCCAAAAGTTAGC | ACATTTGGTGTTAAAATCCCAGGAT | 688 |
| 58 | TGCTGTATTCTCAAATCTCCAAAAC | CCACTCATGTGATCATTTTTACCAT | 597 | ATTTAAGCTAATTCTCTTCACCTGC | AGCTTTGAGGTAATGAATTCCCA | 639 |
| 59 | CTAATCCTGGGATTTTAACACCAAA | AAAAACACACAGGCTCCTTC | 438 | CCAAGACATAGACTGTTACAGTAAT | TCTGAATTGATGAATCCTTCAGG | 630 |
Polymorphism discovery was performed by Polymorphic DNA Technologies (Alameda, CA). The company performs a “boost” amplification reaction, and this product serves as a template for the “nest” amplification and sequencing using the nest primers.
Amplification product in base pairs.
Study Populations and Demographics
Chicago case-control samples.
A total of 71 ED and 34 AD unrelated severe sepsis-associated ARDS patients and 186 ED and 185 AD healthy unrelated controls were collected in Chicago as previously described (22). Sepsis-associated ARDS patient samples used in this study were defined per American-European Consensus Criteria (3) and the Society of Critical Care Medicine Consensus statements (5). Admission to the intensive care units was a requirement for enrollment, and all ARDS patients enrolled into the study experienced severe sepsis or septic shock. All ARDS cases also met the criteria of PaO2/FiO2 ratio ≤ 300 mmHg (31) (corresponding to previous consensus criteria for acute lung injury) (3). Exclusion criteria were allogeneic bone marrow transplant and severe leukopenia (white blood cells < 1,000/μl). Detailed demographic and disease severity characteristics of Chicago samples are depicted in Table 2. Healthy control subjects were defined as individuals without any recent acute illness or any chronic illness requiring a physician's care.
Table 2.
Demographic and clinical characteristics of the Chicago samples
| Sepsis-Associated ARDS |
Controls |
|||
|---|---|---|---|---|
| AD | ED | AD | ED | |
| Number | 34 | 71 | 185 | 186 |
| Sex, male/female | 15/19 | 40/31 | 51/134 | 83/103 |
| Age | 44.6 ± 15.4 | 58.6 ± 16.9 | 54.8 ± 17.4 | 55.6 ± 12.4 |
| APACHE II | 28.0 ± 7.7 | 29.7 ± 7.4 | NA | NA |
| PaO2/FiO2, mmHg | 121 ± 51.9 | 128 ± 62.4 | NA | NA |
| Survival, % | 52.4 | 62.0 | NA | NA |
| Smoking history, % | 34.2 | 40.3 | Unavailable | Unavailable |
| Comorbidity | ||||
| Cancer, % | 11.9 | 21.8 | 0.0 | 0.0 |
| CLD, % | 4.9 | 13.9 | 0.0 | 0.0 |
| Diabetes, % | 16.7 | 19.0 | 0.0 | 0.0 |
| COPD, % | 2.4 | 14.3 | 0.0 | 0.0 |
| AIDS, % | 19.0 | 2.5 | Unavailable | Unavailable |
| Organ failure | ||||
| Renal failure, % | 21.4 | 19.0 | 0.0 | 0.0 |
| Anemia, % | 9.5 | 7.7 | 0.0 | 0.0 |
| Site of infection* | ||||
| Lung, % | 71.4 | 73.4 | NA | NA |
| Abdomen/GI, % | 11.9 | 19.0 | NA | NA |
| UTI, % | 9.5 | 10.1 | NA | NA |
| Other, % | 21.4 | 21.5 | NA | NA |
AD, African descent; ED, European descent; ARDS, acute respiratory distress syndrome; APACHE II, Acute Physiology and Chronic Health Evaluation; PaO2/FiO2, ratio of partial pressure of arterial O2 to the fraction of inspired O2; CLD, chronic liver disease; COPD, chronic obstructive pulmonary disease; AIDS, acquired immunodeficiency syndrome; NA, not applicable; GI, gastrointestinal; UTI, urinary tract infection.
More than 1 site of infection is listed for some patients; sum of percentages exceeds 100%.
Spanish case-control samples.
A total of 96 population controls and 80 severe sepsis-associated ARDS cases were collected from postsurgical and intensive care units (ICUs) (9) and evaluated for replication purpose. The Spanish study enrolled patients within 24 h of the diagnosis of ARDS derived from severe sepsis (20, 46) who were admitted to a Spanish network of postsurgical units and ICUs. ARDS was defined on the basis of 1) acute and sudden onset of severe respiratory distress; 2) bilateral infiltrates on frontal chest radiograph; 3) absence of left atrial hypertension, a pulmonary capillary wedge pressure ≤18 mmHg, or no clinical signs of left heart failure; and 4) severe hypoxemia: a PaO2/FiO2 ≤ 300 mmHg, regardless of FiO2 or positive end-expiratory pressure levels. Chest radiographs were reviewed by radiologists and physicians involved in the study. Detailed demographic and severity characteristics of Spanish samples are listed in Table 3. Unrelated Spanish individuals, independent of sex, between 18 and 75 years old without any known common acute and chronic conditions were randomly selected and served as random population controls. The Institutional Review Boards approved both protocols and written, informed consent was obtained from all participants.
Table 3.
Demographic and clinical features of the Spanish samples
| Variable | Cases (n = 80) | Controls (n = 96) | P Value |
|---|---|---|---|
| Sex | |||
| Male, % | 50.7 | 52.6 | |
| Female, % | 49.3 | 47.4 | 0.806* |
| Age, yr (mean ± SD) | 63.9 ± 15.3 | 44.2 ± 13.9 | 0.001† |
| Predisposition to infection | |||
| Previous surgery, % | 74.5 | 46.3 | 0.001* |
| Hospitalized >24 h, % | 78.2 | 52.6 | 0.002* |
| Past medical history of | |||
| Diabetes, % | 19.1 | 9.5 | 0.076* |
| Hypertension, % | 42.6 | 23.2 | 0.008* |
| Ischemic cardiopathy, % | 9.1 | 2.1 | 0.045* |
| Smoker, % | 29.4 | 30.5 | 0.878* |
| Clinical features | |||
| APACHE II | 20 (15–25)‡ | ||
| Source of infection, % | 73.2 Abdominal | ||
| 18.3 Pulmonary | |||
| 8.5 Other | |||
| Pathogen, % | 19.7 Gram negative | ||
| 4.2 Gram positive | |||
| 4.2 Fungi | |||
| 5.6 More than one | |||
| 66.2 Unknown | |||
| ICU mortality, % | 39.4 |
χ2 test;
Student's t-test;
median (interquartile range).
ICU, intensive care unit.
Genotyping
Genotyping of S1PR3 SNPs in Chicago samples was conducted with the iPLEXGold Platform (Sequenom, San Diego, CA) and TaqMan allelic discrimination assays (Applied Biosystems, Foster City, CA). Briefly, iPLEX assays were scanned by MALDI-TOF mass spectrometry and individual SNP genotype calls were automatically generated by use of Sequenom TYPER 3.4 software. TaqMan genotyping was performed by using a 7900HT Fast Real-Time PCR System (Applied Biosystems) with automated calls generated using the SDS software based on discriminating plots (95% confidence). Approximately 10% of the samples were genotyped by duplicate to monitor genotyping quality. Genotyping was blind to case-control status and the ethnic background of the samples.
To reduce the risk for false positives, 93 European ancestry informative markers (i.e., EuroAIMs) were also genotyped in the Chicago ED samples by use of the iPLEX Gold assay, allowing correction for major population stratification effects among European and Spanish populations (26, 27). Out of 93 EuroAIMs, 90 markers were successfully genotyped and considered for the final analysis. Previous studies have demonstrated that a subset of as few as 65 of these EuroAIMs were effective in controlling false positives in case-control association studies in European populations, allowing adjustment for the major axis of variation (26, 27). Based on these markers, principal component analysis (PCA) was utilized to derive the scores for the first principal component (PC1) as ancestry estimates in cases and controls. This analysis was performed by means of EIGENSOFT 4.2 (28) as explained elsewhere (26).
A follow-up study of the top associated SNP as well as the nearest SNP in strong linkage disequilibrium (LD) with it predicting disruption of a transcription factor binding site (TFBS) (see methods below) was performed in the Spanish sample. These two SNPs were genotyped by means of TaqMan allelic discrimination assays (Applied Biosystems) using the same protocols and equipment described above.
Electrophoretic Mobility Shift Assays
EMSA for DNA transcription factor binding was detected as previously described (19). Briefly, nuclear extract from HeLa cells (Promega, Madison, WI) were used as the source of transcription factors. Prediction of TFBS and effects of SNPs were performed by positional weight matrices search using TRANSFAC Match (http://www.biobase-international.com/) and Genomatix MatInspector 8.0 (http://www.genomatix.de/) analysis. Biotinylated oligonucleotide probes (Table 4), containing transcription factor binding motifs of the S1PR3 promoter regions with rs7022797 alleles (−1899T and G) or the rs11137480 alleles (−1785G and C), were utilized for the detection of nuclear protein complexes bound to the oligonucleotides by using a Light Shift Chemiluminescent EMSA kit (Thermo Scientific, Rockford, IL). Nuclear protein (5 μg) was incubated with 20 fmol of the biotin-labeled oligonucleotide, with or without a 50-fold molar excess (1 pmol) of unlabeled oligonucleotide. After electrophoresis, the DNA-protein complex on Hybond-N+ membrane was detected by horseradish peroxidase and electrochemiluminescence.
Table 4.
Biotinylated/nonbiotinylated oligonucleotide probes for EMSA assays
| Probe Names | Sequences (5′ to 3′) |
|---|---|
| −1899T-biotin | \5Biosg\TGAGGACAGGGATCTTTAGGGAAACGGAGC |
| −1899G-biotin | \5Biosg\TGAGGACAGGGATCGTTAGGGAAACGGAGC |
| −1899del-biotin | \5Biosg\CCCACTGAGGACAGGGAAACGGAGCCTCAC |
| Consensus-Cdx1 | TGAGGACAGGGACCTTTATGGAAACGGAGC |
| −1785G-biotin | \5Biosg\AGAGCCACATTACTCCTCAGGGCTTCTGGGCT |
| −1785C-biotin | \5Biosg\AGAGCCACATTACTCCTCAGGCCTTCTGGGCT |
| −1785del-biotin | \5Biosg\GCATGAGAGCCACATCTTCTGGGCTCCGATTC |
| Consensus-Ebf1 | GCATGAGAGCCACACTACTCCTCAGGGAGGTTGT |
Subcloning, Transfection, and Luciferase Activity Assays
Luciferase promoter activities were detected as previously described (42, 43). Briefly, a 2,261-bp DNA fragment of the S1PR3 promoter region (position −1962 to +287 from transcription start site of the gene as annotated in the NCBI build 36/hg18) containing alleles (−1899T and/or −1785G) were amplified by PCR using a forward primer (5′-MluI-ACGCGTGTGTCTGAGCTTTAAGGCCCAG-3′) and a reverse primer (5′-XhoI-CTCGAGCTCCGTCGGCTGAGGGCGCTGG-3′) and the human genomic DNA as template following a standard protocol. Two additional sepsis-associated ARDS SNPs in EDs also resided within this fragment (rs7022664 and rs4877039). We engineered ED major alleles of these two SNPs at their corresponding positions, but no further studies were performed for them, because their alleles did not predict the interruption of TFBS. Amplicons were subsequently modified by site-directed mutagenesis to generate fragments containing alleles −1899G and/or −1785C at these loci. All allele-containing fragments were fused to a pGL3-basic reporter vector (Promega, Madison, WI) at MluI and XhoI sites and transfected into human pulmonary artery endothelial cells (Cambrex, Walkersville, MD) (18, 33). A plasmid with Renilla luciferase gene (phRL-TK) was cotransfected as a control. Transfected cells were cultured in growth medium with 10% FBS for 48 h, then exposed to medium with or without TNF-α 100 ng/ml for 2 h and lysed in passive lysis buffer. Luciferase activity was measured by Dual-Luciferase Assay Kits and GloMax-Multi Detection System (Promega). The relative activities were expressed as the ratio of firefly luciferase in pGL3 to Renilla luciferase in phRL-TK (relative light units). Five independent transfections and duplicate luciferase assays were performed for each condition.
Enzyme-Linked Immunosorbent Assays
Samples from ICU patients were collected from the Chicago Consortium for Investigating ICU Genetics (CIICUG) study, approved by the institutional review board. Thirty-seven ICU patient samples (13 septic, 11 ARDS, and 13 healthy controls) were randomly chosen and measured for plasma S1PR3 levels by ELISA and genotyped for two S1PR3 promoter variants by TaqMan assays. Patients' characteristics are listed in Table 5. Briefly, 5 ml of blood were collected in EDTA-containing tubes, stored on ice, and transported on the same day to the University of Chicago General Clinical Research Center Laboratory. Cold blood was centrifuged for 15 min at 4°C at 1,000 g within 30 min of collection. The plasma supernatant was transferred to a fresh tube and centrifuged for an additional 10 min at 1,000 g at 4°C to minimize variability of future immunosorbent or biochemical assays introduced by platelet contamination, proteolysis, and other aspects of sample handling. Plasma was stored at −80°C in 125-μl aliquots. The measurement details of S1PR3 by ELISA were described previously (39). Briefly, the plates were coated with murine anti-S1PR3 antibody (Exalpha Biologicals, Watertown, MA); incubated with plasma samples from patients, then rabbit anti-S1PR3 antibody (Exalpha Biologicals) and Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen); detected by Cytofluor 4,000 (Applied Biosystems); and normalized by serial dilutions of S1PR3 recombinant protein (Novus, Littleton, CO). The genotypes of S1PR3 variants and the plasma S1PR3 levels for the same individual were used for paired analysis.
Table 5.
Clinical characteristics of the study population for paired measures in plasma and genetic variants
| ICU Controls | Patients with Sepsis | Patients with ARDS | |
|---|---|---|---|
| N | 13 | 13 | 11 |
| Sex, male/female | 6/7 | 7/6 | 6/5 |
| Race, AD/ED | 7/6 | 7/6 | 6/5 |
| Age | 57 ± 19 | 63 ± 17 | 54 ± 18 |
| APACHE II | 18 ± 6.3 | 27 ± 5.8* | 27 ± 7.6* |
| Mortality, % | 0 | 23 | 45 |
| TOC, day | 3 (1–5) | 5 (4–7) | 4 (3–6) |
| Mechanic ventilation, % | 38 | 54 | 100 |
| Cancer, % | 23 | 46 | 27 |
| CLD, % | 8 | 8 | 9 |
| ESRF, % | 23 | 23 | 36 |
| COPD, % | 15 | 23 | 9 |
| Diabetes, % | 62 | 31 | 18 |
| CHF, % | 38 | 23 | 9 |
| AIDS, % | 8 | 8 | 18 |
| Site of infection | |||
| Lung, % | NA | 54 | 55 |
| Abdomen, % | NA | 15 | 9 |
| UTI, % | NA | 8 | 9 |
| Blood, % | NA | 8 | 9 |
| Other, % | NA | 15 | 18 |
TOC, Time of collection; ESRF, end stage renal failure; CHF, congestive heart failure.
P < 0.05 for significant difference vs. ICU controls.
Statistical Analysis
For the Chicago samples, the multiple-marker selection algorithm based on haplotype r2 included in TagIT 3.03 software (51) was used to select a common set of tagging SNPs (tSNPs) for ADs and EDs as described before (8). Hardy-Weinberg equilibrium (HWE) assessments for cases and controls were done with an exact test using SNPInfostats software (26, 38). MaCH 1.0 (21) was used for SNP imputation on the Phase 1 data (May 2011) deposited in the 1000 Genomes Project Consortium (1). Association testing was conducted by means of logistic regression for allele dosages for SNPs showing minor allele frequency (MAF) ≥ 10% and a squared correlation between imputed and true genotypes (Rsq) > 0.3, as provided by MaCH. For that, we used Mach2dat (21), adjusting the associations for the PC1 scores as ancestry estimates in cases and controls (27). Representation of association results was then performed by using LocusZoom 1.1 (29) based on LD data from hg18 deposited by 1000 Genomes Project.
For the Spanish samples, quality control checks, HWE assessments for cases and controls, and Cochran-Armitage trend tests for association assuming additive models were executed with SNPing. Logistic regression models were used to estimate the allele effects in terms of odds ratios (OR) with their 95% confidence intervals (CI) using SPSS (SPSS, Chicago, IL).
Meta-analysis of association results from EDs from Chicago and Spain was conducted by assuming a fixed-effects model using a Mantel-Haenszel stratified analysis with Epidat 3.0. Association was declared only after this joint analysis was conducted. By this approach, false positives were limited by applying a stringent threshold at this stage, established at P < 3 × 10−3 according to a Bonferroni-like correction.
The t-test was used for the comparison of luciferase activities among different constructs. The one-way ANOVA analysis was used for the correlation of S1PR3 genotypes with plasma S1PR3 protein levels. Statistical significance was defined at P < 0.05 in both tests.
RESULTS
Survey of S1PR3 Gene Variants
Resequencing of the entire S1PR3 gene and flanking regions (constituting a total of 8,774 bp per sample) identified 80 variant sites [67 single base changes and 13 insertions/deletions (indels)], with 29 SNPs indels described for the first time (Supplemental Table S1). A total of 16 tSNPs were sufficient to provide adequate coverage (haplotype r2 > 0.9) of common S1PR3 gene variation in both EDs and ADs (Supplemental Table S1) and were utilized for genotyping the Chicago case and control samples.
S1PR3 Variants Associate with ARDS Susceptibility in European-Descent Subjects from Chicago and Spain
After quality control checks, HWE assessment, and filtering of individuals with low genotyping completion rate (<80%), 14 of 16 tSNPs were retained in the study of Chicago samples (11 for AD, and 8 for ED) (Table 6). After imputation and filtering of SNPs with MAF < 10% and Rsq < 0.3, association analyses were then conducted for 17 SNPs in ED and 25 SNPs in AD. Although no associations were detected for AD (a predictable finding given the limited sample size), 5 SNPs showed nominal significance in EDs (rs7022797, rs11137480, rs7022664, rs4877039, rs10867149). The S1PR3 SNP showing the strongest significance was rs7022797 (ancestry-adjusted risk per allele G was OR = 0.67, 95%CI = 0.46–0.97, P = 0.017), which showed moderate to strong LD with the other 4 S1PR3 SNPs, thereby supporting their nonindependent association (Fig. 1, Table 6).
Table 6.
Summary of S1PR3 SNPs studied for association with ARDS in Chicago samples
| HWE |
Genotype Count |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Position* | SNP rs no. | Controls | Cases | Allele 1/Allele 2 | Allele 1 frequency | Ca | Co | Rsq† | OR (allele 1) | SE (OR) | P Value |
| African descent | |||||||||||
| 90794283 | rs7022797 | 0.763 | 0.218 | G/T | 0.42 | 10/16/14 | 30/93/64 | 0.91 | 0.30 | 0.676 | |
| 90794397 | rs11137480 | 0.749 | 0.114 | C/G | 0.37 | 10/14/14 | 25/83/78 | 1.07 | 0.29 | 0.369 | |
| 90794814 | rs7022664 | G/A | 0.83 | 0.71 | 0.97 | 0.47 | 0.954 | ||||
| 90796023 | rs4877039 | G/C | 0.53 | 0.85 | 1.09 | 0.31 | 0.776 | ||||
| 90797066 | rs73654718 | 1 | 1 | A/G | 0.86 | 29/10/1 | 138/46/3 | 0.76 | 0.39 | 0.947 | |
| 90797534 | rs7863572 | T/C | 0.39 | 0.85 | 1.00 | 0.32 | 0.993 | ||||
| 90797689 | rs6559331 | C/T | 0.43 | 0.79 | 0.96 | 0.33 | 0.909 | ||||
| 90797872 | rs11398943 | 0.372 | 1 | —/A | 0.44 | 6/17/13 | 33/99/54 | 0.77 | 0.33 | 0.415 | |
| 90798448 | rs10867149 | T/C | 0.31 | 0.70 | 1.13 | 0.37 | 0.742 | ||||
| 90800116 | rs73496046 | C/T | 0.87 | 0.55 | 3.75 | 0.82 | 0.108 | ||||
| 90800363 | rs3934594 | G/A | 0.65 | 0.52 | 1.14 | 0.42 | 0.756 | ||||
| 90800415 | rs11137481 | 1 | 0.463 | T/G | 0.92 | 32/8/1 | 162/24/0 | ||||
| 90800749 | rs7045576 | 0.613 | 0.33 | A/G | 0.17 | 3/11/27 | 7/52/128 | 1.55 | 0.37 | 0.240 | |
| 90801401 | rs6559333 | G/C | 0.85 | 0.32 | 0.98 | 0.66 | 0.979 | ||||
| 90802459 | rs7858626 | A/G | 0.24 | 0.76 | 1.91 | 0.35 | 0.062 | ||||
| 90802703 | rs7870888 | G/A | 0.86 | 0.71 | 1.22 | 0.50 | 0.687 | ||||
| 90802886 | rs7038457 | 1 | 0.003 | T/C | 0.70 | 28/6/5 | 91/78/16 | 1.36 | 0.33 | 0.130 | |
| 90803463 | rs58235552 | 1 | 0.336 | C/G | 0.85 | 31/6/1 | 129/50/5 | 1.32 | 0.43 | 0.214 | |
| 90804199 | rs73496049 | 1 | 0.183 | A/C | 0.94 | 35/4/1 | 164/23/0 | ||||
| 90807194 | rs73496050 | 0.201 | 1 | G/A | 0.77 | 25/13/1 | 117/58/12 | 1.23 | 0.34 | 0.780 | |
| 90807402 | rs41287349 | G/A | 0.67 | 0.80 | 1.50 | 0.35 | 0.243 | ||||
| 90807922 | rs7853537 | A/G | 0.77 | 0.81 | 1.47 | 0.42 | 0.359 | ||||
| 90807926 | rs7865415 | G/A | 0.86 | 0.67 | 2.93 | 0.71 | 0.128 | ||||
| 90809094 | rs1129925 | G/A | 0.23 | 0.87 | 1.56 | 0.34 | 0.186 | ||||
| 90809604 | rs1867 | C/T | 0.89 | 0.58 | 1.11 | 0.60 | 0.867 | ||||
| 90809944 | rs9314668 | G/A | 0.87 | 0.73 | 1.03 | 0.48 | 0.949 | ||||
| 90811017 | rs7865979 | 1 | 0.437 | A/G | 0.76 | 19/13/4 | 104/70/12 | 0.77 | 0.32 | 0.399 | |
| European descent | |||||||||||
| 90794283 | rs7022797 | 0.625 | 0.764 | G/T | 0.36 | 5/26/38 | 25/75/61 | 0.67 | 0.22 | 0.017 | |
| 90794397 | rs11137480 | 0.875 | 0.764 | C/G | 0.36 | 5/26/38 | 26/85/74 | 0.67 | 0.22 | 0.042 | |
| 90794814 | rs7022664 | G/A | 0.57 | 0.70 | 0.81 | 0.27 | 0.021 | ||||
| 90796023 | rs4877039 | C/G | 0.63 | 0.90 | 0.63 | 0.28 | 0.045 | ||||
| 90797534 | rs7863572 | C/T | 0.46 | 0.80 | 0.71 | 0.21 | 0.126 | ||||
| 90797689 | rs6559331 | T/C | 0.4 | 0.80 | 0.10 | 1.10 | 0.092 | ||||
| 90798448 | rs10867149 | C/T | 0.52 | 0.70 | 0.61 | 0.34 | 0.048 | ||||
| 90800363 | rs3934594 | A/G | 0.42 | 0.70 | 0.93 | 0.27 | 0.058 | ||||
| 90800749 | rs7045576 | 0.009 | 0.32 | G/A | 0.51 | 9/38/22 | 57/67/46 | 0.57 | 0.25 | 0.063 | |
| 90802459 | rs7858626 | A/G | 0.51 | 0.60 | 1.66 | 0.28 | 0.441 | ||||
| 90802703 | rs7870888 | 0.656 | 1 | G/A | 0.82 | 49/17/1 | 117/59/9 | 0.63 | 0.23 | 0.136 | |
| 90802886 | rs7038457 | 1 | 1 | T/C | 0.91 | 52/13/1 | 153/30/1 | 0.70 | 0.24 | 0.267 | |
| 90807922 | rs7853537 | A/G | 0.91 | 1.00 | 1.76 | 0.28 | 0.137 | ||||
| 90809094 | rs1129925 | G/A | 0.49 | 0.50 | 1.13 | 0.20 | 0.799 | ||||
| 90809604 | rs1867 | 1 | 0.641 | C/T | 0.79 | 51/17/2 | 110/66/9 | 0.67 | 0.24 | 0.096 | |
| 90809944 | rs9314668 | 0.837 | 0.637 | G/A | 0.79 | 52/17/2 | 108/69/9 | 0.61 | 0.25 | 0.069 | |
| 90811017 | rs7865979 | 0.658 | 0.802 | A/G | 0.57 | 23/31/12 | 58/89/39 | 0.61 | 0.26 | 0.581 | |
Statistically significant P values are in bold. SNP, single nucleotide polymorphism; HWE, Hardy-Weinberg equilibrium; Rsq, squared correlation between imputed and true genotypes as an estimate of the imputation quality as provided by MaCH (see Ref. 21); OR, odds ratio; SE, standard error. Tagging SNPs (tSNPs) are denoted in bold.
Chromosome position according to NCBI build 36. SNPs rs11137481 and rs73496049 with minor allele frequency (MAF) <10% were excluded from downstream association analysis based on the filtering criteria (MAF <10% or Rsq <0.3). Analyses were conducted for 17 SNPs in the EDs and 25 in ADs adjusting for ancestry estimated.
Fig. 1.
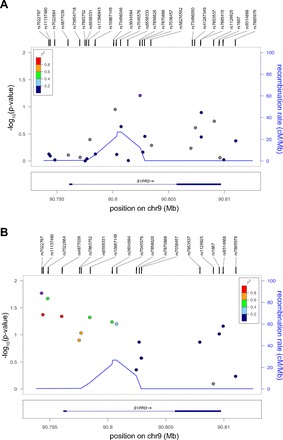
Regional plots of S1PR3 gene association with acute respiratory distress syndrome (ARDS) by chromosome position in African-descent individuals from Chicago (A) and European descent from Chicago (B). P values are expressed in −log10 scale. Associations are color coded to reflect the underlying linkage disequilibrium of the single nucleotide polymorphism (SNP) tested with respect to the top associated SNP on each case (depicted in purple) based on pairwise r2 values from data deposited in the 1000 Genomes Project for European populations. Estimated recombination rates are also plotted on the right axis to reflect the local linkage disequilibrium structure. cM/Mb, centimorgans per megabase.
To independently replicate these findings, we next followed the top SNP, rs7022797, as well as the nearest SNP, rs11137480 in tight LD (r2 = 1), in independent Spanish cases and controls. The rs7022797 genotype counts for GG, GT, and TT were 9/35/34 and 20/48/28 for cases and controls, respectively. The rs11137480 genotype counts for CC, CG, and GG were 9/34/33 and 20/48/26 for cases and controls, respectively. None of the SNPs deviate from HWE expectations (P = 1.0) for each group in these samples. Analysis of the Spanish samples demonstrated SNP-level replication for rs7022797G (OR = 0.61, 95%CI = 0.39–0.94, P = 0.026) and rs11137480C (OR = 0.59, 95%CI = 0.37–0.92, P = 0.019).
Finally, the consistency of allele effects across the two ED study samples was evidenced by a stratified meta-analysis, suggesting per allele effects of OR = 0.61 (95%CI = 0.45–0.83, P = 5.0 × 10−4) for rs7022797G and OR = 0.65 (95%CI = 0.48–0.88, P = 1.1 × 10−3) for rs11137480C. These two associations were considered significant in the context of the multiple tests performed in the study.
ARDS-Associated S1PR3 Promoter SNPs Modify Nuclear Transcription Factor Binding Affinity
In silico analysis using TRANSFAC and Genomatix revealed that Homo sapiens caudal type homeobox transcription factor 1 (Cdx1) was the only transcription factor binding to S1PR3 promoter and potentially interrupted by −1899G (rs7022797), resulting in loss of Cdx1 binding efficiency compared with the −1899T allele. Similarly, the −1785C allele (rs11137480) was predicted to interrupt binding of the only transcription factor, early B-cell factor 1 (Ebf1), to the S1PR3 promoter compared with the −1785G allele. Our analyses did not predict any change in TFBS between the alleles at rs7022664 or rs4877039. To confirm the predicted regulatory roles of the two ARDS-associated SNPs on transcription factor affinity, protein-to-DNA binding and gel retardation assays were performed. As predicted, a DNA fragment containing the −1899 position caused significant shifts with less protein bound to −1899G compared with −1899T, and the binding was specifically blocked by oligonucleotides containing the Cdx1 consensus sequence (Fig. 2A). Similarly, a DNA fragment containing the −1785 position caused significant shifts of DNA-protein complex, with −1785C allele binding less protein of the extract than the −1785G allele (Fig. 2B). Since short oligonucleotides (see Table 4) were used for this study, the presence of other variants in close proximity to SNPs tested did not interfere with results observed.
Fig. 2.
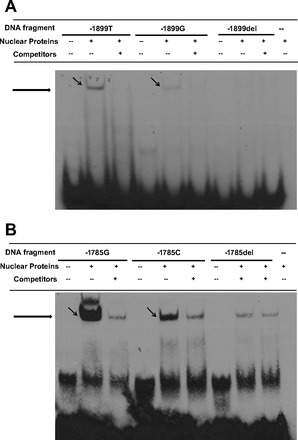
Promoter SNPs −1899T/G and −1785G/C modified nuclear protein binding affinity by electrophoretic mobility shift assays (EMSA). A: examining allele-specific effects of S1PR3 −1899T/G. Biotin-labeled oligonucleotide probes contains either −1899T, −1899G, or −1899del (deletion of −1899T/G), with or without competitor containing sequence for caudal type homeobox 1 (Cdx1) specific binding element. B: examining allele-specific effects of S1PR3 −1785G/C. One milligram of HL60 nuclear extract proteins and 5 pg/ml of biotin-labeled oligonucleotide probes contain either −1785G, −1785C, or −1785del (deletion of −1785G/C), with or without competitor containing sequence for early B-cell factor 1 (Ebf1) specific binding element.
ARDS-Associated S1PR3 SNPs Potentially Alter Inflammatory Factor-Induced S1PR3 Promoter Activity
We utilized a luciferase activity assay to assess the allelic effects of SNPs −1899T/G and −1785G/C on S1PR3 promoter activity. Human lung endothelial cells exhibited significantly less luciferase promoter activity with the −1899G allele (∼40% reduction) compared with the activity with the major allele −1899T (P < 0.05) (Fig. 3). Similarly, the −1785C allele significantly decreased luciferase activity of the S1PR3 promoter (∼50%) compared with −1785G (P < 0.05) (Fig. 3). We next tested whether harboring the alleles −1899G and −1785C simultaneously further affected S1PR3 promoter activity. Coexistence of −1899G and −1785C alleles further decreased luciferase activity by ∼80% compared with the construct containing −1899T and −1785G (P < 0.05) (Fig. 3). Similar differences were observed when endothelial cells were exposed to TNF-α (Fig. 3). These data support that genetic variants of −1899G and −1785C affect S1PR3 gene expression.
Fig. 3.
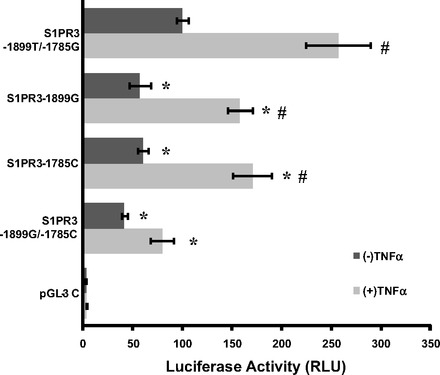
Promoter SNPs −1899T/G and −1785G/C significantly altered S1PR3 promoter activity and responses to inflammatory factors in vitro. The S1PR3 promoter fragments with specific allele as well as the coexistence of 2 alleles at these loci were subcloned into pGL3-basic vector and transfected into human pulmonary artery endothelial cells. Promoter activity was detected by luciferase activity assay upon exposure to growth medium with 10% serum, with or without 100 ng/ml TNF-α challenges. Promoter activity with −1899G or/and −1785C was significantly decreased, especially upon response to TNF-α, compared with the promoter with major alleles −1899T and −1785G (*P < 0.05 vs. S1PR3 −1899T/−1785G with the same concentration of TNF-α; #P < 0.05 vs. S1PR3 −1899T/−1785G without TNF-α).
ARDS-Associated S1PR3 Promoter SNPs Correlate with Plasma S1PR3 Protein Levels in ICU Patients
Plasma S1PR3 protein concentrations were investigated in a limited sample of human ICU patients. Thirty-seven human ICU samples were randomly chosen from all ICU patients, including 13 septic, 11 ARDS, and 13 healthy controls. All samples were genotyped and tested for S1PR3 protein levels. The characteristics of patients including the illness, APACHE score, and mortality as well as the timing of plasma collection are listed in Table 5. Congruent with in vitro results, plasma from subjects carrying −1899G or −1785C alleles exhibited significantly decreased S1PR3 protein levels compared with subjects carrying −1899T or −1785G (*P < 0.05) (Fig. 4). Within patient groups, −1899T and −1785G were associated with higher S1PR3 levels (a trend was observed in controls, and statistical significance was attained in septic and ARDS) (Fig. 5).
Fig. 4.
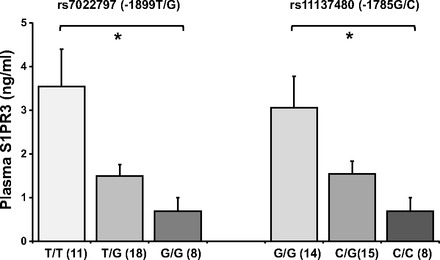
Promoter SNPs −1899T/G and −1785G/C were significantly associated with plasma S1PR3 protein levels of intensive care unit patients. Lower S1PR3 protein plasma levels were reduced with the addition of each of −1899G or −1785C alleles. Levels are indicated as ng/ml. The significance obtained by ANOVA test is indicated (*P < 0.05).
Fig. 5.
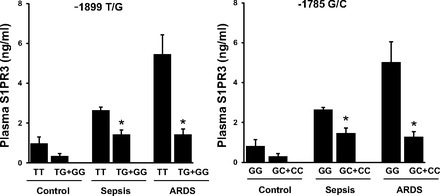
Breakdown of S1PR3 plasma levels by S1PR3 genotypes in each of 3 sample groups (control, sepsis, and ARDS). Alleles −1899T and −1785G are associated with trends of higher S1PR3 levels in controls and significant higher S1PR3 levels in sepsis and ARDS groups (*P < 0.05), compared with the alleles −1899G and −1785C, respectively.
DISCUSSION
S1PRs proteins participate in regulating key pathophysiological features of ARDS, including lung microvascular permeability and host immune responses (18, 30, 34, 36, 48). Our studies, to our knowledge, are the first to investigate the association between S1PR3 genetic variants and risk of sepsis-associated ARDS and were conducted utilizing a combination of S1PR3 sequencing for SNP discovery, two independent case-control association studies, nuclear protein-binding assays, promoter activity assays, and the correlation of genotypes with plasma levels of the S1PR3 protein in ICU patients. We identified two common S1PR3 promoter variants in complete LD in Europeans, evidencing study-wise significant association with ARDS across two independent case-control samples. Alleles −1899G and −1785C associated with protection from sepsis-associated ARDS, reduced nuclear protein binding to S1PR3 promoter, attenuated promoter activity (especially in response to the inflammatory cytokine, TNF-α), and lower plasma levels of the S1PR3 protein in ICU patients.
The two S1PR3 promoter SNPs were predicted to disrupt the binding of Cdx1 and Ebf1 transcription factor to the promoter in silico which was confirmed by protein-DNA binding assay. Cdx1 is an essential transcriptional factor for intestinal differentiation and may be involved in progression of cancer and inflammation (37). Cdx1 plays critical roles in regulating chemokine CCL25 promoter activity (7). Its expression is dramatically enhanced by treatment with 5-azacytidine and TNF-α (53). Cdx2, which recognizes the same DNA binding sequence as Cdx1, was identified as a potentially novel regulator of the response to systemic LPS in the lung, liver, and spleen (54). Ebf1 is critically important for early B lymphocyte development, reprogramming mb-1 (Ig-alpha) promoters by increasing chromatin accessibility and initiating the loss of DNA methylation (13). Recent studies showed that Ebf1 is an early response transcription factor that increases more than threefold during early stages of sepsis (4). Our findings further indicated that enhanced promoter binding to Cdx1 and Ebf1 could contribute to roles of S1PR3 in sepsis and ARDS. Furthermore, the two-SNP haplotype was associated with chromatin accessibility as indicated in HaploReg based on ENCODE data (49). Since Cdx1 and Ebf1 are well recognized to be directly associated with inflammation, the increased S1PR3 transcription could potentially enhance inflammatory effects on vascular barrier dysfunction and severity of ARDS. These findings are consistent with our prior observation that S1PR3 protein expression is significantly increased in plasma microparticles from sepsis-associated ARDS patients (39).
Our additional earlier work revealed that S1P, the natural ligand of S1PR3, is a multifunctional lipid mediator and an angiogenic factor (24, 50). S1PR3 is a dominant S1P receptor in many tissues and an important regulator of lymphocyte trafficking, as well as vascular permeability (24). S1PR3 activates RhoA (34, 48) and serine/threonine kinase, ROCK (12), contributing to vascular barrier disruption (33, 41). Studies in preclinical models of ARDS showed that S1PR3 promotes pulmonary inflammation in the late phase of sepsis via PAR1-S1PR3 cross talk, propagating the dissemination of IL-1β and tissue factor to the lungs (25). Reduced S1PR3 expression in vitro reversed barrier disruption induced by thrombin and low-molecular-weight hyaluronan (34). Similarly, S1PR3 silencing provided barrier protection from LPS compared with controls in vivo (32). Given the multiple mechanisms of S1PR3 regulation in the pulmonary vasculature, functional S1PR3 genetic variants associated with the magnitude of S1PR3 expression could influence ARDS development.
Given the strong LD between the two associated SNPs and their additive effect observed in vitro (both decreased luciferase activity by ∼80%), it is likely that the haplotype rather than individual separate alleles accounts for the observed association. We cannot exclude, however, the potential regulatory roles of SNPs rs7022664 and rs4877039 in gene expression (either alone or as haplotypes), since they also reside in this region. However, we have clearly demonstrated that the sole change at any of the two functionally tested sites was sufficient to obtain an effect in vitro, at least when the ED major alleles of the other two SNPs were present. Moreover, the fact that the two functionally tested SNPs were the only ones with predicted functionality in the 5′ flanking region and that these two SNPs also related with differential plasma protein levels from patients further bolsters the idea that the two associated SNPs are important for susceptibility to development of sepsis-associated ARDS.
A major debate of our study relates to the design itself. The choice of using non-ARDS ICU patients as controls is often seen in genetic association studies (10) because this at risk group allows adjusting for common risk factors. Despite this widespread use, this approach is not entirely free of bias (47). We utilized population controls in both Chicago and Spanish studies as this approach allow reducing the possibility of incurring in sample selection bias (52). Keep in mind that these controls are not appropriate for assessing the environmental risk factors affecting the condition. Therefore, the possibility of detected associations might exist with the underlying critical illness of these patients, rather than with ARDS, remains inconclusive.
Furthermore, we are aware that study power is an additional limiting factor. Excluding AD samples because of insufficient sample size, we calculated (with Power Calculator for Two Stage Association Studies) (35) that a joint analysis provided 60% power to detect an allelic effect (OR) ≥ 1.6 (≤0.63 if protective) for an allele frequency of 0.38 at a significance threshold of P = 3 × 10−3 and disease prevalence of 7 × 10−5. However, the power of the study would be far from appropriate in light of more realistic effects expected for complex traits (ORs ≈ 1.2) and the multiple statistical tests performed. As a result, on the basis of these issues, the associations that we detected should not be considered definitive until further studies in independent and larger samples assess whether common variants from this gene are truly important for the disease. In ELISA studies, we combined ED and AD samples because of limited sample availability for both genotyping and plasma protein measurements. Although S1PR3 plasma level differences between ancestries were not appreciated (not shown), we will further expand our sample sizes to increase the confidence in the association in our next follow-up study. Nevertheless, despite these important caveats, we observed consistent and reproducible SNP-level associations of two S1PR3 functional variants of the predicted promoter across ED samples. Although it is unclear whether other S1PR3 variants may explain such results, the functional evidence provided for these two associated SNPs, the relationship of associated SNPs with S1PR3 protein levels in plasma of ICU patients, as well as the fact that S1PR3 expression correlated with severity of sepsis and ARDS in mice and humans (39), support the likelihood that these two promoter SNPs may be directly involved in ARDS development.
In summary, although the genetic factors underlying inflammatory lung syndromes such as sepsis-associated ARDS remain incompletely understood, our studies demonstrated that common S1PR3 genetic variants constitute risk factors for ARDS or any of the underlying critical illness in the patients we utilized. Thus, in agreement with the evidence supporting S1PR3 as a potential target for amelioration of ARDS pathobiology, the identification of S1PR3 variants that confer altered promoter activity may drive development of a genetic diagnosis for prognosis and individualized therapies for ARDS. We expect our studies to result in the development of precise, genetic-based, individualized ARDS pharmacogenetic therapies for the critically ill.
GRANTS
This work was supported by grants from the National Heart Lung Blood Institute NIH grant P01 HL058064 (J. G. N. Garcia), R01 HL091889 (J. G. N. Garcia), P01 HL098050 (V. Natarajan, J. G. N. Garcia), and GM07019 (X. Sun).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
X.S., S.-F.M., and J.G.G. conception and design of research; X.S., S.-F.M., M.S.W., J.M., and Y.-J.H. performed experiments; X.S., S.-F.M., M.S.W., M.A.-H., J.V., M.P.-Y., T.Z., B.L., and C.F. analyzed data; X.S., S.-F.M., M.S.W., M.A.-H., M.P.-Y., T.Z., B.L., and C.F. interpreted results of experiments; X.S., S.-F.M., M.S.W., M.A.-H., J.V., M.P.-Y., T.Z., B.L., and C.F. prepared figures; X.S., B.L., and C.F. drafted manuscript; X.S., S.-F.M., B.L., P.B., C.F., and J.G.G. edited and revised manuscript; X.S., S.-F.M., R.M., I.N., V.N., S.M.D., J.R.J., and J.G.G. approved final version of manuscript.
Supplementary Material
ACKNOWLEDGMENTS
The authors gratefully acknowledge Lakshmi Natarajan and Steven M. Broderick for expert technical assistance. C. Flores was supported by a specific agreement between Instituto de Salud Carlos III and Gobierno de Canarias (EMER07/001). M. Acosta-Herrera was supported by a fellowship from the Instituto de Salud Carlos III (FI11/00074). M. Pino-Yanes was supported by Fundación Ramón Areces. S.-F. Ma was supported by Core Subsidy Mini-Awards of ITM/CTSA (UL1TR000430).
REFERENCES
- 1.Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature 491: 56–65, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antonarakis SE. Recommendations for a nomenclature system for human gene mutations. Nomenclature Working Group. Hum Mutat 11: 1–3, 1998. [DOI] [PubMed] [Google Scholar]
- 3.Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, Lamy M, Legall JR, Morris A, Spragg R. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med 149: 818–824, 1994. [DOI] [PubMed] [Google Scholar]
- 4.Bhatty M, Fan R, Muir WM, Pruett SB, Nanduri B. Transcriptomic analysis of peritoneal cells in a mouse model of sepsis: confirmatory and novel results in early and late sepsis. BMC Genomics 13: 509, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee American College of Chest Physicians/Society of Critical Care Medicine. Chest 101: 1644–1655, 1992. [DOI] [PubMed] [Google Scholar]
- 6.Cobb JP, O'Keefe GE. Injury research in the genomic era. Lancet 363: 2076–2083, 2004. [DOI] [PubMed] [Google Scholar]
- 7.Ericsson A, Kotarsky K, Svensson M, Sigvardsson M, Agace W. Functional characterization of the CCL25 promoter in small intestinal epithelial cells suggests a regulatory role for caudal-related homeobox (Cdx) transcription factors. J Immunol 176: 3642–3651, 2006. [DOI] [PubMed] [Google Scholar]
- 8.Flores C, Ma SF, Maresso K, Ober C, Garcia JG. A variant of the myosin light chain kinase gene is associated with severe asthma in African Americans. Genet Epidemiol 31: 296–305, 2007. [DOI] [PubMed] [Google Scholar]
- 9.Flores C, Ma SF, Maresso K, Wade MS, Villar J, Garcia JG. IL6 gene-wide haplotype is associated with susceptibility to acute lung injury. Transl Res 152: 11–17, 2008. [DOI] [PubMed] [Google Scholar]
- 10.Flores C, Pino-Yanes Mdel M, Villar J. A quality assessment of genetic association studies supporting susceptibility and outcome in acute lung injury. Crit Care 12: R130, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frutos-Vivar F, Nin N, Esteban A. Epidemiology of acute lung injury and acute respiratory distress syndrome. Curr Opin Crit Care 10: 1–6, 2004. [DOI] [PubMed] [Google Scholar]
- 12.Fujisawa K, Madaule P, Ishizaki T, Watanabe G, Bito H, Saito Y, Hall A, Narumiya S. Different regions of Rho determine Rho-selective binding of different classes of Rho target molecules. J Biol Chem 273: 18943–18949, 1998. [DOI] [PubMed] [Google Scholar]
- 13.Gao H, Lukin K, Ramirez J, Fields S, Lopez D, Hagman J. Opposing effects of SWI/SNF and Mi-2/NuRD chromatin remodeling complexes on epigenetic reprogramming by EBF and Pax5. Proc Natl Acad Sci USA 106: 11258–11263, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao L, Flores C, Fan-Ma S, Miller EJ, Moitra J, Moreno L, Wadgaonkar R, Simon B, Brower R, Sevransky J, Tuder RM, Maloney JP, Moss M, Shanholtz C, Yates CR, Meduri GU, Ye SQ, Barnes KC, Garcia JG. Macrophage migration inhibitory factor in acute lung injury: expression, biomarker, and associations. Transl Res 150: 18–29, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao L, Grant A, Halder I, Brower R, Sevransky J, Maloney JP, Moss M, Shanholtz C, Yates CR, Meduri GU, Shriver MD, Ingersoll R, Scott AF, Beaty TH, Moitra J, Ma SF, Ye SQ, Barnes KC, Garcia JG. Novel polymorphisms in the myosin light chain kinase gene confer risk for acute lung injury. Am J Respir Cell Mol Biol 34: 487–495, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia JG. Focusing on the flood: targeting functional polymorphisms in ALI permeability pathways. Am J Respir Crit Care Med 183: 1287–1289, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garcia JG. Genomic investigations into acute inflammatory lung injury. Proc Am Thorac Soc 8: 167–172, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, Bamberg JR, English D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest 108: 689–701, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han YJ, Ma SF, Wade MS, Flores C, Garcia JG. An intronic MYLK variant associated with inflammatory lung disease regulates promoter activity of the smooth muscle myosin light chain kinase isoform. J Mol Med (Berl) 90: 299–308, 2012. [DOI] [PubMed] [Google Scholar]
- 20.Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, Cohen J, Opal SM, Vincent JL, Ramsay G. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med 31: 1250–1256, 2003. [DOI] [PubMed] [Google Scholar]
- 21.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol 34: 816–834, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma SF, Xie L, Pino-Yanes M, Sammani S, Wade MS, Letsiou E, Siegler J, Wang T, Infusino G, Kittles RA, Flores C, Zhou T, Prabhakar BS, Moreno-Vinasco L, Villar J, Jacobson JR, Dudek SM, Garcia JG. Type 2 deiodinase and host responses of sepsis and acute lung injury. Am J Respir Cell Mol Biol 45: 1203–1211, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacCallum NS, Evans TW. Epidemiology of acute lung injury. Curr Opin Crit Care 11: 43–49, 2005. [DOI] [PubMed] [Google Scholar]
- 24.Marsolais D, Rosen H. Chemical modulators of sphingosine-1-phosphate receptors as barrier-oriented therapeutic molecules. Nat Rev Drug Discov 8: 297–307, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niessen F, Schaffner F, Furlan-Freguia C, Pawlinski R, Bhattacharjee G, Chun J, Derian CK, Andrade-Gordon P, Rosen H, Ruf W. Dendritic cell PAR1–S1P3 signalling couples coagulation and inflammation. Nature 452: 654–658, 2008. [DOI] [PubMed] [Google Scholar]
- 26.Pino-Yanes M, Corrales A, Basaldua S, Hernandez A, Guerra L, Villar J, Flores C. North African influences and potential bias in case-control association studies in the Spanish population. PLoS One 6: e18389, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Price AL, Butler J, Patterson N, Capelli C, Pascali VL, Scarnicci F, Ruiz-Linares A, Groop L, Saetta AA, Korkolopoulou P, Seligsohn U, Waliszewska A, Schirmer C, Ardlie K, Ramos A, Nemesh J, Arbeitman L, Goldstein DB, Reich D, Hirschhorn JN. Discerning the ancestry of European Americans in genetic association studies. PLoS Genet 4: e236, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38: 904–909, 2006. [DOI] [PubMed] [Google Scholar]
- 29.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, Willer CJ. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26: 2336–2337, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pyne S, Pyne N. Sphingosine 1-phosphate signalling via the endothelial differentiation gene family of G-protein-coupled receptors. Pharmacol Ther 88: 115–131, 2000. [DOI] [PubMed] [Google Scholar]
- 31.Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, Camporota L, Slutsky AS. Acute respiratory distress syndrome: the Berlin Definition. JAMA 307: 2526–2533, 2012. [DOI] [PubMed] [Google Scholar]
- 32.Sammani S, Moreno-Vinasco L, Mirzapoiazova T, Singleton PA, Chiang ET, Evenoski CL, Wang T, Mathew B, Husain A, Moitra J, Sun X, Nunez L, Jacobson JR, Dudek SM, Natarajan V, Garcia JG. Differential effects of sphingosine 1-phosphate receptors on airway and vascular barrier function in the murine lung. Am J Respir Cell Mol Biol 43: 394–402, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singleton PA, Dudek SM, Ma SF, Garcia JG. Transactivation of sphingosine 1-phosphate receptors is essential for vascular barrier regulation. Novel role for hyaluronan and CD44 receptor family. J Biol Chem 281: 34381–34393, 2006. [DOI] [PubMed] [Google Scholar]
- 34.Singleton PA, Moreno-Vinasco L, Sammani S, Wanderling SL, Moss J, Garcia JG. Attenuation of vascular permeability by methylnaltrexone: role of mOP-R and S1P3 transactivation. Am J Respir Cell Mol Biol 37: 222–231, 2007. [DOI] [PubMed] [Google Scholar]
- 35.Skol AD, Scott LJ, Abecasis GR, Boehnke M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet 38: 209–213, 2006. [DOI] [PubMed] [Google Scholar]
- 36.Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol 4: 397–407, 2003. [DOI] [PubMed] [Google Scholar]
- 37.Stairs DB, Kong J, Lynch JP. Cdx genes, inflammation, and the pathogenesis of intestinal metaplasia. Prog Mol Biol Transl Sci 96: 231–270, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun X, Ma SF, Wade MS, Flores C, Pino-Yanes M, Moitra J, Ober C, Kittles R, Husain AN, Ford JG, Garcia JG. Functional variants of the sphingosine-1-phosphate receptor 1 gene associate with asthma susceptibility. J Allergy Clin Immunol 126: 241–249, 249.e1–3M, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun X, Singleton PA, Letsiou E, Zhao J, Belvitch P, Sammani S, Chiang ET, Moreno-Vinasco L, Wade MS, Zhou T, Liu B, Parastatidis I, Thomson L, Ischiropoulos H, Natarajan V, Jacobson JR, Machado RF, Dudek SM, Garcia JG. Sphingosine-1-phosphate receptor-3 is a novel biomarker in acute lung injury. Am J Respir Cell Mol Biol 47: 628–636, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swan DJ, Kirby JA, Ali S. Vascular biology: the role of sphingosine 1-phosphate in both the resting state and inflammation. J Cell Mol Med 14: 2211–2222, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tasaka S, Koh H, Yamada W, Shimizu M, Ogawa Y, Hasegawa N, Yamaguchi K, Ishii Y, Richer SE, Doerschuk CM, Ishizaka A. Attenuation of endotoxin-induced acute lung injury by the Rho-associated kinase inhibitor, Y-27632. Am J Respir Cell Mol Biol 32: 504–510, 2005. [DOI] [PubMed] [Google Scholar]
- 42.Tian C, Hinds DA, Shigeta R, Adler SG, Lee A, Pahl MV, Silva G, Belmont JW, Hanson RL, Knowler WC, Gregersen PK, Ballinger DG, Seldin MF. A genomewide single-nucleotide-polymorphism panel for Mexican American admixture mapping. Am J Hum Genet 80: 1014–1023, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tian C, Hinds DA, Shigeta R, Kittles R, Ballinger DG, Seldin MF. A genomewide single-nucleotide-polymorphism panel with high ancestry information for African American admixture mapping. Am J Hum Genet 79: 640–649, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Villar J, Blanco J, Zhang H, Slutsky AS. Ventilator-induced lung injury and sepsis: two sides of the same coin? Minerva Anestesiol 77: 647–653, 2011. [PubMed] [Google Scholar]
- 45.Villar J, Flores C, Perez-Mendez L, Blanco J, Muros M. Genetic determinants of survival in sepsis and acute lung injury. Minerva Anestesiol 74: 341–345, 2008. [PubMed] [Google Scholar]
- 46.Villar J, Manzano JJ, Blazquez MA, Quintana J, Lubillo S. Multiple system organ failure in acute respiratory failure. J Crit Care 6: 75–80, 1991. [Google Scholar]
- 47.Vineis P, McMichael AJ. Bias and confounding in molecular epidemiological studies: special considerations. Carcinogenesis 19: 2063–2067, 1998. [DOI] [PubMed] [Google Scholar]
- 48.Waeber C, Blondeau N, Salomone S. Vascular sphingosine-1-phosphate S1P1 and S1P3 receptors. Drug News Perspect 17: 365–382, 2004. [DOI] [PubMed] [Google Scholar]
- 49.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res 40: D930–D934, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ware LB, Fremont RD, Bastarache JA, Calfee CS, Matthay MA. Determining the aetiology of pulmonary oedema by the oedema fluid-to-plasma protein ratio. Eur Respir J 35: 331–337, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weale ME, Depondt C, Macdonald SJ, Smith A, Lai PS, Shorvon SD, Wood NW, Goldstein DB. Selection and evaluation of tagging SNPs in the neuronal-sodium-channel gene SCN1A: implications for linkage-disequilibrium gene mapping. Am J Hum Genet 73: 551–565, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weiss ST. Association studies in asthma genetics. Am J Respir Crit Care Med 164: 2014–2015, 2001. [DOI] [PubMed] [Google Scholar]
- 53.Wong NA, Wilding J, Bartlett S, Liu Y, Warren BF, Piris J, Maynard N, Marshall R, Bodmer WF. CDX1 is an important molecular mediator of Barrett's metaplasia. Proc Natl Acad Sci USA 102: 7565–7570, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang IV, Alper S, Lackford B, Rutledge H, Warg LA, Burch LH, Schwartz DA. Novel regulators of the systemic response to lipopolysaccharide. Am J Respir Cell Mol Biol 45: 393–402, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ye SQ, Simon BA, Maloney JP, Zambelli-Weiner A, Gao L, Grant A, Easley RB, McVerry BJ, Tuder RM, Standiford T, Brower RG, Barnes KC, Garcia JG. Pre-B-cell colony-enhancing factor as a potential novel biomarker in acute lung injury. Am J Respir Crit Care Med 171: 361–370, 2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.