

Abstract
Consequences of prenatal exposure to ethanol (E) include morphological, physiological, and cognitive deficits and are collectively classified as fetal alcohol spectrum disorders. Adult prenatal E exposed offspring show insulin resistance, and given that in utero hyperglycemic environment can cause metabolic disorders in subsequent generations; we investigated the effects of grandmaternal E on functional glucose and insulin responses of the second generation. Sprague-Dawley (S) rat dams, mated with S males, received E-containing liquid diet and two different control diets between gestational days 8 and 20. Additionally, because prenatal E-induced behavioral deficits can be reversed by simultaneous thyroxine (T4) treatment, another group of dams received 0.3 mg/l T4 in their E diet. Their first-generation (F1) offspring were mated with control Brown Norway (B) males or females to produce SB and BS F2 progeny. Dams consuming E during pregnancy were hyperglycemic, and their F1 offspring showed insulin resistance in the glucose tolerance test (GTT). However, F2 responses to GTT varied based on the sex of prenatal E-exposed parent. BS F2 females, and both male and female SB F2 progeny, displayed hypoglycemic and hyperinsulinemic GTT response patterns. Although administering T4 to E dams normalized thyroid function of the F1 generation, it did not reverse their prenatal E-induced metabolic dysfunction. In contrast, administration of T4 to the alcohol-consuming grandmother reversed or alleviated the aberrant GTT responses of the F2 progeny. Prenatal E-induced dysregulation of glucose metabolism can affect the next generation, possibly via ethanol effects on the germline of the F1 fetus.
Keywords: glucose challenge test, insulin response, grandmaternal ethanol, thyroid function
since the introduction of the fetal origins of adult disease hypothesis, it has been recognized that adverse intrauterine environment can have long-lasting effects on adult health (18, 35). Prenatal ethanol (E) exposure is one of these adverse events that is known to lead to morphological, physiological and cognitive anomalies that are collectively classified as fetal alcohol spectrum disorders (47).
Among the many harmful effects caused by maternal E consumption during pregnancy, offspring show altered glucose homeostasis. From early postnatal days (14, 44) through adulthood (13, 52) these offspring show higher blood glucose levels than controls, which may or may not be accompanied by higher insulin levels. These findings in animal studies parallel glucose metabolism in humans exposed to E in utero, in one study to date (11). Because adult female offspring who were exposed to E in utero have impaired glucose tolerance, their progeny can be exposed to an intrauterine environment that puts them at risk for developing their own metabolic problems (18). As glucose dysregulation and hyperinsulinemia often lead to lifelong insulin resistance, this intergenerational cycle may increase the risk and/or accelerate the onset of Type 2 diabetes, even in the absence of additional E exposure.
Not only may vulnerability to prenatal E effects be mediated by the amount of E consumed and the time of exposure during pregnancy, but genetic makeup may also influence the vulnerability of the mother and the fetus. We have demonstrated this latter phenomenon by showing that the strain of the E-consuming pregnant rats determines behavioral vulnerabilities of their offspring (38) and that another phenotype, fetal E-induced decreases in birth weight of the offspring, is affected interactively by both paternal and maternal genotypes.
The goals of the present study were to investigate the effects of grandmaternal alcohol consumption, the role of genotype in vulnerability to prenatal E, and the effects of prenatal T4 treatment on metabolic phenotypes. Our selected measure, glucose and insulin response to glucose tolerance test (GTT), is a classic challenge test that provides useful information regarding the state of the metabolic function of the organism, while also being predictive toward its current or future diabetic conditions (22). These measures, together with body weights, could indicate the vulnerability, or resilience, of offspring with different genetic backgrounds to grandmaternal exposure to ethanol. First, to scrutinize the intergenerational hypothesis, we produced two generations, the first of which was exposed to E in utero. We envisioned that vulnerability and resilience to prenatal E effects due to maternal and paternal genotype may occur in metabolic functions as it did in behavior (38); therefore, we elected to cross the F1 generation Sprague-Dawley (S) rats with naïve male or female Brown Norways (B) as shown in Fig. 1. The pregnant dams (P0) consumed E or control diets as described previously (38, 42, 48), and the mating of their offspring led to SB F2 progeny whose mothers were exposed to E or control diets in utero, as well as BS F2 offspring whose fathers were exposed to alcohol in utero. Finally, since we have shown previously that prenatal E-induced behavioral deficits can be attenuated with administration of thyroxin (T4) to the alcohol-consuming dams (48), we examined whether T4 administration to alcohol-consuming grandmothers could reverse the prenatal E-induced alterations in glucose metabolism.
Fig. 1.
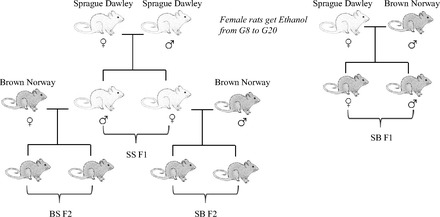
Schematic experimental design. Sprague-Dawley (S) females were mated with either S or Brown Norway (B) males. From gestational (G) day 8 through 20 these dams were exposed to 1 of 4 prenatal treatments (C, PF, E, or E+T4). The resulting SS and SB F1 were used for testing. Naïve male and female SS F1 offspring were paired with B mates. The resulting BS and SB F2 offspring did not experience in utero E exposure and were used for testing. C, control; PF, pair fed; E, ethanol; E+T4, ethanol + thyroxin.
METHODS
Animals.
All procedures were approved by the Institutional Animal Care and Use Committee of Northwestern University. This study employed S (Harlan, Indianapolis, IN) and B (Charles River, Wilmington, MA) rats and their reciprocal crosses. We chose to use the B, the most phylogenetically divergent inbred rat strain, and the S, the most commonly utilized outbred strain (41). The B and S genomes have been sequenced by the Rat Genome Project and Celera, respectively, and differences between these strains in litter size, temperament, appearance, and attributes of sensorimotor gating have been reported (41). In future experiments addressing the molecular differences that create susceptibilities in specific strains, being able to track parent of origin, allele-specific expression of imprinted genes will be necessary. All rats were housed in a temperature- and humidity-controlled environment with a 12 h light cycle (lights on at 6 AM). Throughout the experiments water was available ad libitum. Female S rats were mated with male S or male B. The day when sperm was found in vaginal smears was considered gestational day (GD) 1. Dams were divided into four prenatal treatment groups: control (C), pair-fed (PF), ethanol (E), and ethanol + thyroxin (E+T4). C dams were kept on conventional laboratory chow ad libitum throughout pregnancy. The remaining three prenatal treatment groups received liquid diet (Lieber-DeCarli ′82; Bio-Serv, Frenchtown, NJ) starting at GD4. E diet began at GD8, and from GD8 to 10 the percentage of E in the diet was increased until it reached 5% (wt/vol) and then was kept constant until GD20, as reported previously (42). The E+T4 group received 0.3 mg/l thyroxin (Sigma-Aldrich, St. Louis, MO) in the E-containing liquid diet, which, based on the daily diet consumption, is equivalent to ∼8 μg/100 g body wt/day of T4. Each individual PF rat received a liquid diet that was isocaloric to the amount consumed by an individual E or E+T4 (since there was no difference in diet consumption between these two groups) on the previous GD. On GD21, all rats were provided with regular laboratory chow ad libitum. Offspring were weaned at postnatal day (PD) 24 and testing began at PD70. One or two male and female rats from each litter were used for the GTT to avoid potential litter effects.
From each SS F1 prenatal treatment group, females and males that were experimentally naïve were chosen as breeders for the second generation. These rats were paired with B males or females to produce SB and BS F2 rats. As in the F1, offspring were weaned at PD 24 days and testing began at PD 70. Male and female rats from each prenatal treatment group received GTT as was done in the first generation.
GTT.
Animals were fasted for ∼16 h overnight, and tail blood was collected in the morning into tubes containing EDTA (0 min or fasting time point) to collect the plasma. Immediately following the first blood collection, an intraperitoneal injection of 2 g/kg body wt dextrose (Sigma-Aldrich) was given as described previously (39). Tail blood was collected in the same manner at the 30 and 60 min time points after dextrose injection. Rats were killed at 120 min after injection and trunk blood was collected (nC = 5–13, nPF = 4–19, nE = 5–16, nE+T4 = 5–15 per generation, sex, and maternal or grandmaternal treatments). In another set of animals, trunk blood was collected from pregnant dams at GD21 for glucose and insulin measures of different prenatal diet groups (nC = 7–8, nPF = 3–6, nE = 6–7, nE+T4 = 7).
Assays.
Glucose levels were measured in duplicates by Stanbio glucose liquicolor kit (Stanbio laboratory, Boerne, TX). Insulin levels were measured in duplicate by Ultra sensitive rat insulin ELISA kit (Crystal Chem, Downers Grove, IL). Sensitivity of the insulin assay was 0.1 ng/ml with an inter- and intra-assay coefficient of variation (CV) of ≤ 10%.
Experimentally naïve rats from different prenatal diet groups were killed by decapitation for tissue and blood collection. Plasma was collected from trunk blood was used for thyroid stimulating hormone (TSH) enzyme immunoassay (ELISA) (Immuno-Biological Laboratories, Minneapolis, MN) (nC = 3–12, nPF = 4–11, nE = 4–11, nE+T4 = 3–10). Sensitivity was 0.1 ng/ml with an assay CV of ≤ 12.4%.
Statistical analysis.
Graphs represent means ± SE. Data were compared by two-way or three-way ANOVA with sex, prenatal treatment, and time point as factors. Time points were considered as a repeated measure. Area under the curve (AUC) was calculated by the trapezoidal method. If there was no significant effect of sex, male and female data were combined. If there was no significant effect between C and PF, occasionally data were combined. For body weight data, nested ANOVAs were done with litter as the nested factor. We found that litter had a significant effect on the variability seen in prenatal treatment groups in birth and weaning weights, but this effect usually dissipated by adulthood. When appropriate, Bonferroni post hoc comparisons were used. Hypothesis testing was carried out by Student's t-test in some cases as noted. Significance was considered P < 0.05. Statistical analyses were done by Systat 11 (Chicago, IL).
RESULTS
Body weights of SS F1 males and females and their progeny at different developmental time points are shown in Tables 1 and 2. SS F1 males that were exposed to E and E+T4 in utero weighted less than controls throughout their lives, but these differences reached significance only at birth (F(3,134) = 30.42, P < 0.01; Table 1). The BS F2 offspring of SS F1 E males weighed significantly less than controls at birth (male and female data combined F(3,95) = 4.41, P < 0.01). At weaning, only males showed a decrease in body weight (F(3,159) = 4.4, P < 0.01), but no effects were seen in adulthood. These data suggest that SS F1 males and their BS F2 progeny experience catch up in growth, such that they are comparable with controls at adulthood. The prenatal E-induced decreases in birth weight were not reversed by prenatal T4 treatment, neither in the first nor the second generation.
Table 1.
Effects of maternal E consumption on male progeny and their offspring's body weight
| Generation | Prenatal Treatment | Sex | Body Weight P0, g | Body Weight P21–24, g | Body Weight Adult, g |
|---|---|---|---|---|---|
| SS F1 | C | M | 6.57 ± 0.07 | 61.9 ± 0.72 | 312.8 ± 3.37 |
| PF | 6.34 ± 0.08 | 61.0 ± 1.23 | 306.2 ± 2.39 | ||
| E | 5.82 ± 0.06** | 58.9 ± 1.51 | 300.4 ± 4.04 | ||
| E+T4 | 5.91 ± 0.09^^ | 59.4 ± 1.34 | 308.0 ± 3.37 | ||
| BS F2 | C | M | 7.67 ± 0.14 | 71.7 ± 1.11 | 328.6 ± 6.79 |
| F | 7.24 ± 0.18 | 66.5 ± 1.01 | 206.8 ± 5.17 | ||
| PF | M | 7.02 ± 0.29 | 70.9 ± 1.47 | 319.7 ± 3.86 | |
| F | 7.21 ± 0.23 | 67.9 ± 1.32 | 209.6 ± 3.29 | ||
| E | M | 6.84 ± 0.17* | 66.2 ± 0.77* | 319.3 ± 4.65 | |
| F | 6.51 ± 0.15* | 62.9 ± 0.86 | 196.2 ± 3.38 | ||
| E+T4 | M | 6.59 ± 0.20^^ | 68.1 ± 1.17 | 331.5 ± 6.50 | |
| F | 6.33 ± 0.21^^ | 64.0 ± 1.05 | 202.6 ± 3.69 |
Data are represented as means ± SE; n = 5–51/group. Bonferroni post hoc.
P < 0.05,
P < 0.01 C vs. E,
P < 0.01 C vs. E+T4. BS F2 P0 body weight was analyzed by combining male and female data. P, postnatal day; S, Sprague-Dawley; B, Brown Norway; C, control; PF, pair fed; E, ethanol; T4, thyroxin; M, male; F, female.
Table 2.
Effects of maternal E consumption on female progeny and their offspring's body weights
| Generation | Prenatal Treatment | Sex | Body Weight P0, g | Body Weight P21–24, g | Body Weight Adult, g |
|---|---|---|---|---|---|
| SS F1 | C | F | 6.19 ± 0.07 | 57.1 ± 0.73 | 213.9 ± 3.05 |
| PF | 6.11 ± 0.06 | 56.1 ± 0.89 | 207.3 ± 1.84 | ||
| E | 5.52 ± 0.06** | 54.3 ± 1.52 | 200.6 ± 2.91* | ||
| E+T4 | 5.67 ± 0.08^^ | 55.1 ± 1.30 | 203.4 ± 2.58 | ||
| SB F2 | C | M | 6.33 ± 0.06 | 60.6 ± 0.54 | 421.9 ± 4.58 |
| F | 6.15 ± 0.07 | 57.5 ± 0.47 | 255.8 ± 3.43 | ||
| PF | M | 6.89 ± 0.07 | 57.9 ± 1.45 | 416.3 ± 5.53 | |
| F | 6.49 ± 0.06 | 54.4 ± 1.21 | 252.2 ± 3.04 | ||
| E | M | 6.46 ± 0.11 | 63.4 ± 0.68 | 439.0 ± 3.87*++ | |
| F | 5.98 ± 0.12 | 58.9 ± 0.73 | 264.5 ± 2.73**++ | ||
| E+T4 | M | 6.37 ± 0.10 | 62.6 ± 0.55 | 415.7 ± 6.45 | |
| F | 6.20 ± 0.10 | 59.5 ± 0.43 | 250.0 ± 3.09 |
Data are represented as mean ± SE; n = 11–44/group. Bonferonni post hoc.
P < 0.05,
P < 0.01 C vs. E,
P < 0.01 C vs. E+T4,
P < 0.01 E vs. E+T4. Adult weights were gathered at 60–70 days for F1 and 120 days for SB F2.
Female offspring that were exposed to E and E+T4 in utero weighed less compared with controls throughout their lives, with significant differences at birth for both groups and at adulthood only for the E group (birth F(3,141) = 36.8, P < 0.01; adulthood F(3,67) = 5.76, P < 0.01; Table 2). Surprisingly both male (F(3,80) = 6.47, P < 0.01) and female (F(3,80) = 7.60, P < 0.01) offspring of SS F1 E females weighed significantly more than controls in adulthood (Table 2). This effect was reversed by T4 treatment to the E-consuming grandmother, resulting in the body weight of their SB F2 progeny similar to those of controls (E vs. E+T4, P < 0.05). Unlike the straight transfer of body weight characteristics via the male line, the offspring of prenatal E-exposed females showed a body weight pattern that reversed that of their mother; an underweight mother had heavier adult progeny.
Given this increased body weight of the SB F2 progeny of the prenatally E-exposed females, we tested the functionality of the glucose/insulin system in the P0, F1, and F2 generations. SS females that received E while pregnant had elevated plasma glucose levels at GD21 compared with control dams (F(3,24) = 4.06, P < 0.05; Fig. 2A), and simultaneous treatment with T4 did not reverse this relative hyperglycemia. Interestingly, pregnant PF females showed elevated insulin levels (F(3,19) = 4.61, P = 0.01; Fig. 2B) compared with C dams, while E dams showed comparable insulin levels to C dams. Compared with PF, E dams had significantly lower insulin levels, while T4 administration in the E diet eliminated this difference.
Fig. 2.
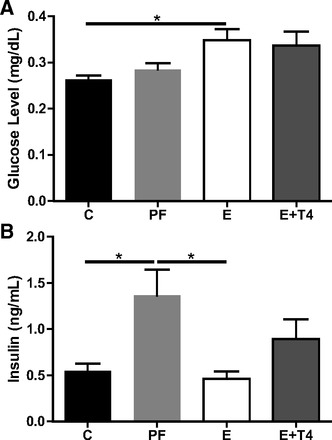
E consumption during pregnancy increases nonfasting maternal plasma glucose without altering insulin levels. Glucose (A) and insulin (B) levels in the plasma of pregnant SS females on gestational day (GD) 21. *P < 0.05 Bonferroni post hoc. Data are represented as means ± SE; n = 3–8/group.
Glucose response to GTT, either the time course of the response or the AUC, did not differ between the SS F1 offspring by sex; therefore, male and female SS F1 data were combined. SS F1 E rats had significantly higher glucose levels than controls 30 min after glucose injection, but their levels returned to control by 60 min (F(9,177) = 2.92, P < 0.01; Figs. 3A and 4A) postinjection. This exaggerated glucose response to GTT was not reversed by T4 treatment of the E dams but was worsened such that the response stayed elevated in the SS F1 E+T4 offspring even at 60 min (F(9,177) = 2.92, P < 0.01). Insulin responses to GTT differed between the male and female SS F1 offspring significantly (F(1,40) = 27.64, P < 0.01). SS F1 E males had significantly increased levels of insulin compared with those of controls at 30 min postglucose that was not reversed by prenatal T4 treatment (F(9,63) = 3.0, P < 0.01; Fig. 3B), similar to the glucose response. Though SS F1 E males showed an insulin-resistant profile, when they were mated with naïve B females their BS F2 offspring experienced a normal in utero glucose environment. Accordingly, the glucose GTT profile of the BS F2 offspring did not differ significantly by grandmaternal diet. However, the insulin responses of BS F2 female offspring of SS F1 E males were significantly higher than controls at 30 min postglucose challenge (F(9,76) = 2.09, P < 0.05; Fig. 3B), which was not attenuated by grandmaternal T4 treatment.
Fig. 3.
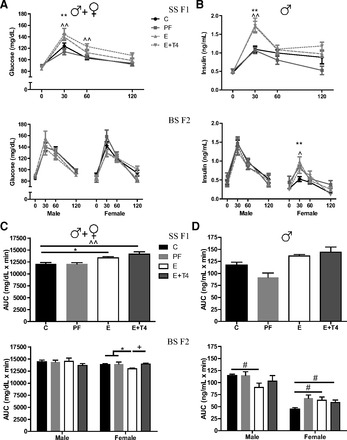
Plasma glucose and insulin levels following glucose challenge test in adult prenatal E and C male rats and their BS F2 offspring. Glucose (A) and insulin (B) time course in response to glucose challenge test in SS F1 offspring who received prenatal treatment, and the BS F2 male and female offspring of the SS males crossed with naïve B females. The area under the curve (AUC) of plasma glucose (C) and insulin (D) levels in response to glucose challenge test in the SS F1 and BS F2 offspring. *C vs E, ^C vs E+T4, +E vs E+T4 by appropriate ANOVA followed by Bonferroni post hoc tests. *, ^, +P < 0.05 or **, ^^, ++P < 0.01. #P < 0.05 by t-test. Data are represented as means ± SE; n = 4–11/group.
Fig. 4.
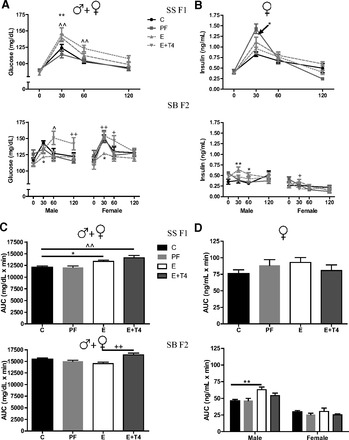
Plasma glucose and insulin levels following glucose challenge test in adult prenatal E and C female rats and their SB F2 offspring. Glucose (A) and insulin (B) time course in response to glucose challenge test in SS F1 offspring who received prenatal treatment, and the SB F2 male and female offspring of the SS females crossed with naïve B males. The AUC of plasma glucose (C) and insulin (D) levels in response to glucose challenge test in the SS F1 and SB F2 offspring. See Fig. 3 for details.
In agreement with the time course of GTT, SS F1 offspring of E and E+T4 dams showed significantly greater overall glucose responses than control, as is seen by the AUC (F(3,60) = 8.58, P < 0.01; Figs. 3C and 4C). Overall insulin response of SS F1 E males did not differ from controls but did from their nutritional control PF group (t(10) = 4.67, P < 0.01; Fig. 3D). The BS F2 male progeny presented normal GTT, while the female offspring of the hyperglycemic SS F1 E males showed significantly lower glucose AUC compared with the combined controls of C and PF, and to the grandoffspring of E+T4 dams (F(2, 18) = 6.65, P < 0.01; Fig. 3C). The physiological relevance of the overall hypoglycemia of the BS F2 E females was suggested by their significantly higher insulin AUC compared with BS F2 C females (t(10) = 2.62, P < 0.05; Fig. 3D). However, their overall insulin responses to GTT did not differ from BS F2 PF females, which were significantly different from controls. In contrast, male BS F2 grandoffspring of E dams showed a decrease in insulin AUC compared with F2 offspring of C dams (t(10) = 2.55, P < 0.05). Thus, male offspring of E and E+T4 dams exhibited hyperinsulinemia with a hyperglycemic profile, which resulted in hyperinsulinemic and hypoglycemic GTT responses in their female offspring. While none of the effects of prenatal E on SS F1 male GTT were reversed by T4 treatment, grandmaternal T4 reversed the effect of E on the GTT responses of the BS F2 progeny (please see female insulin significance in the figure).
As discussed previously, male and female SS F1 combined data had a significant prenatal treatment effect due to higher glucose response to GTT at 30 min in E and E+T4 offspring and 60 min in the E+T4 offspring (Figs. 3A and 4A). Similar to the SS F1 E males, females presented a significantly greater insulin response to GTT at 30 min (F(9,57) = 5.80, P < 0.01; Fig. 4B), but not a significantly increased AUC (Fig. 4D). Both male and female SB F2 offspring of the SS F1 E females displayed significantly lower glucose levels at 30 min postglucose injection, which returned to C levels by 60 min (F(9,128) = 2.59, P < 0.01; F(9,116) = 1.78, P = 0.05; Fig. 4A). However, in contrast to the effects of maternal T4 on SS F1 E females, grandmaternal treatment with T4 reversed the E effect on glucose metabolism in their offspring (E vs. E+T4, P < 0.05). In agreement with the hypoglycemic glucose responses of SB F2 progeny, insulin levels were significantly higher than control at 30 and 60 min postglucose injection in the male offspring (F(9,112) = 2.35, P < 0.05; Fig. 4B), and at 30 min in the female offspring of the SS F1 E females (F(9,104) = 3.19, P < 0.01). The hypoglycemic glucose response to GTT was reflected in the combined SB F2 male and female AUC levels (F(3,62) = 6.80, P < 0.01; Fig. 4C). However, only the SB F2 male offspring of the SS F1 E females showed the hyperinsulinemic AUC (F(3,28) = 5.68, P < 0.01; Fig. 4D), which was reversed by the grandmaternal T4 treatment (E vs E+T4, P < 0.05). Thus, the hyperglycemic and hyperinsulinemic GTT response profile of the SS F1 E and E+T4 females led to hypoglycemic, hyperinsulinemic GTT responses in their SB F2 offspring, specifically in males. While none of the effects of prenatal E on SS F1 female GTT were reversed by T4 treatment, grandmaternal T4 reversed the effect of E on the GTT responses of the SB F2 progeny.
The thyroid hormonal profile of SS F1 males and their BS F2 offspring are shown in Fig. 5A. In the SS F1 males, prenatal E caused elevated plasma TSH levels compared with controls, indicating abnormal thyroid function (F(3,20) = 3.71, P < 0.05; Fig. 5A). This apparent hypothyroid TSH profile was reversed by maternal T4 treatment. In the BS F2 male grandoffspring of dams on the PF, E, and E+T4 diet, there was an apparent decrease in their plasma TSH levels that reached significance in the BS F2 E+T4 males (F(3,13) = 8.6, P < 0.01; Fig. 5A). Thyroid dysfunction in the BS F2 females is unlikely as there were no differences in plasma TSH measures. Although SS F1 E females showed no significant differences in TSH plasma levels (Fig. 5B), their SB F2 male offspring had decreased plasma TSH levels (F(3,16) = 3.25, P < 0.05; Fig. 5B). Thus, in contrast to the normalized E-induced deficit in GTT responses by grandmaternal T4 administration, thyroid function by large was not normalized to control levels.
Fig. 5.
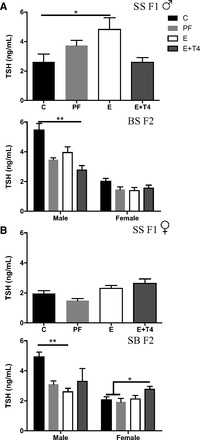
Thyroid function of adult F1 and F2 progeny of dams exposed to different prenatal treatments. A: thyroid stimulating hormone (TSH) levels of SS males who received prenatal treatment and their male and female offspring crossed with naïve B females. B: TSH levels in SS females receiving prenatal treatment and their male and female offspring crossed with naïve B males. *P < 0.05, **P < 0.01 Bonferroni post hoc. Data are represented as means ± SE; n = 3–8/group.
Given our results that grandmaternal T4 treatment restores E-induced changes in body weights, glucose levels, and insulin levels in SB F2 rats, but not in SS F1s (summarized in Table 3), we generated SB F1 offspring of S mothers exposed to the same treatments as the SS F1s. There was no effect of in utero E exposure on SB F1 male offspring's body weights at birth, weaning, or adulthood, but there was a significant effect in the SB F1 female offspring at birth only (F(3,154) = 22.4, P < 0.01) that was exaggerated by T4 treatment (Table 4).
Table 3.
Results summary
| Generation | Sex | Prenatal Treatment | Body Weight | Glucose (30 min) | Insulin (30 min) | TSH |
|---|---|---|---|---|---|---|
| SS F1 | M | E | ↓ | ↑ | ↑ | ↑ |
| E+T4 | ↓ | ↑ | ↑ | ↔ | ||
| BS F2 | M | E | ↓ | ↓ | ||
| E+T4 | ↓ | ↓ | ||||
| F | E | ↑ | ||||
| E+T4 | ↓ | ↑ | ||||
| SS F1 | F | E | ↓ | ↑ | ↑ | |
| E+T4 | ↓ | ↑ | ||||
| SB F2 | M | E | ↑ | ↓ | ↑ | ↓ |
| E+T4 | ↔ | ↔ | ↔ | ↔ | ||
| F | E | ↑ | ↓ | ↑ | ||
| E+T4 | ↔ | ↔ | ↔ | ↑ |
TSH, thyroid stimulating hormone; ↔, T4 no difference from control = reverses.
Table 4.
Effects of maternal E consumption on the SB F1 male and female progeny's body weights
| Sex | Prenatal Treatment | Body Weight P0, g | Body Weight P21–24, g | Body Weight Adult, g |
|---|---|---|---|---|
| M | C | 6.10 ± 0.07 | 59.9 ± 0.65 | 290.3 ± 4.11 |
| PF | 6.00 ± 0.05 | 53.7 ± 0.81 | 278.7 ± 2.78 | |
| E | 5.89 ± 0.07++ | 57.9 ± 0.75 | 292.8 ± 2.83 | |
| E+T4 | 5.23 ± 0.11^^ | 57.7 ± 0.53 | 300.6 ± 2.98 | |
| F | C | 5.92 ± 0.06 | 56.0 ± 0.52 | 190.0 ± 2.35 |
| PF | 5.74 ± 0.07 | 52.0 ± 0.75 | 183.7 ± 2.26 | |
| E | 5.60 ± 0.06*++ | 54.0 ± 0.45 | 185.5 ± 2.25 | |
| E+T4 | 5.1 ± 0.12^^ | 53.1 ± 0.56 | 190.2 ± 1.45 |
Data are represented as means ± SE; n = 22–52/group. Bonferonni post hoc.
P < 0.05 C vs. E,
P < 0.01 C vs. E+T4,
P < 0.01 E vs. E+T4.
SB F1 E males showed a trend toward elevated glucose responses to GTT (F(3,159) = 2.58, P = 0.064; Fig. 6A), and their overall glucose response was significantly higher compared with controls (AUC: F(3,51) = 3.65, P < 0.05; Fig. 6C). The insulin response profile to GTT seemed to be inverse of the hyperglycemic response (F(3,153) = 6.57, P < 0.05; Fig. 6B), although no significant overall difference in AUC was found in males or females (Fig. 6D). The time course of plasma glucose response to GTT differed in SB F1 females by prenatal treatment (F(3,180) = 1.56, P = <.01; Fig. 6C), but the only significant difference in AUC occurred between the E and E + T4 offspring (F(3,57) = 2.73, P < 0.05). As expected, plasma insulin responses to GTT also differed by prenatal treatment (F(9,153) = 2.23, P < 0.05; Fig. 6D) with E+ T4 prenatal treatment resulting in significantly lower insulin response at 30 min postglucose injection compared with C offspring.
Fig. 6.
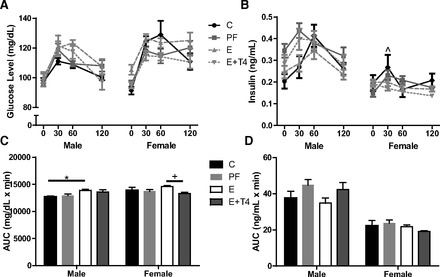
Effects of T4 treatment in SB F1 male and female rats. Glucose (A) and insulin (B) levels in response to a glucose challenge test in SB offspring who received prenatal treatments. The AUC of plasma glucose (C) and insulin (D) following the glucose challenge test in adult SB F1 E males and females. *P < 0.05, ^C vs E+T4, +E vs E+T4, *, ^, +P < 0.05 Bonferonni post hoc. Data are represented as means ± SE; n = 11–19/group.
DISCUSSION
This study confirmed the presence of maternal hyperglycemia after consumption of E during pregnancy. Subsequently, their S offspring showed hyperglycemic, hyperinsulinemic responses to GTT. These aberrant GTT responses of the prenatal E-exposed offspring were not reversed by simultaneous T4 administration to the E-consuming dams. The major novel findings of this study indicate that the second-generation progeny, derived from F1 males and females reciprocally mated with naïve B rats, remained affected by grandmaternal E consumption. In contrast to the first generation, the F2 generation's hypoglycemic and hyperinsulinemic responses to GTT were reversed by grandmaternal T4 treatment, suggesting that T4 prevented, reversed, or interfered with the effects of E on F1 germ cells. This pattern was particularly strong in the SB F2 offspring, where grandmaternal E+T4 treatment led to glucose and insulin responses to GTT that were dramatically different from those with grandmaternal E alone.
Human chronic alcoholics are predominantly hyperglycemic even when abstinent, although some cases present with hypoglycemia (5, 6, 24). These findings extend to rodents, where some, but not all, studies show hyperglycemia with chronic alcohol consumption (23, 27). The pregnant P0 females consumed E from GD8 to GD21 in our study and exhibited chronic alcohol-induced hyperglycemia, consistent with what was found in humans. However, other studies found no hyperglycemia in dams who consumed E (32, 43, 49). The difference in findings may be related to experimental variations in the timing of blood collection relative to their alcohol or food intake. For example, a fed pregnant female has an increase in blood glucose levels when consuming alcohol, whereas a fasted one shows a decrease (45). Since we measured fed glucose levels in the E-consuming dams, the elevated glucose levels, compared with controls, are in agreement with these findings.
Two possible physiological mechanisms could explain the abnormal GTT responses of the F1 generation exposed to E in utero, the hyperglycemic maternal/prenatal environment, or the direct effect of E on the developing fetus. The hyperglycemic in utero environment of the F1 generation could influence fetal development of glucose regulation resulting in hyperglycemic, hyperinsulinemic GTT responses in the adult offspring. Diabetic or prediabetic maternal environment is known to affect metabolic functioning of offspring throughout their lives (8, 26, 31). Similarly, animal studies using exposure to a hyperglycemic in utero environment (15), or transfer to an in utero environment that is hyperglycemic, can lead to hyperglycemia of the progeny (20).
Alternatively, given that E-induced hyperglycemia is not a consistent finding in the pregnant dams, but glucose dysregulation and lower body weights are present in the F1 offspring of these dams, E could affect the developing embryo directly (32, 33, 44, 49). Direct effects of E on the developing embryo or fetus could be conferred from in vitro studies where fetal neuronal cells treated with E show impaired insulin responses (21, 51). Additionally, prenatal E-exposed rats show pancreatic damage, which is specific to E (9, 10). The direct and indirect effects of E on food consumption and metabolism, leading to decreased caloric intake, are difficult to separate (46), particularly since calorie restriction during pregnancy can also cause GTT deficits in the offspring (34). Nevertheless, the hyperglycemic, hyperinsulinemic response to GTT of the F1 offspring of E dams in this study is not related to caloric restriction, since the PF group, which is calorically matched to the E dams, did not produce effects in the F1 offspring similar to those of prenatal E. The current findings cannot conclusively identify whether prenatal programming of adult metabolic function or direct effect of E contribute to the aberrant GTT responses of the F1 offspring. Future experiments need to be designed that can control the nutritional deficits within the E-consuming dams themselves or that can allow for E exposure of the fetus directly.
The potential causes of the intergenerational effect of prenatal E on glucose and insulin regulation could not be due to the intrauterine environment since female offspring of SS F1 E males showed hyperinsulemia and hypoglycemia, even though their B mothers were naïve females that were never exposed to E. In view of this, the phenotype of the SB F2 males and females and the BS F2 females can be explained by the grandmaternal E affecting the germline in the fetal SS F1. Should that be a straightforward explanation, we would expect to see the BS F2 males to be affected as well. Since the matrilinear SB F2 males and females and the patrilinear BS F2 females are all affected, epigenetic effects of grandmaternal E on X chromosomal genes of the F1 germ cells might be involved. Interestingly, insulin-resistant diabetes is found in Turner's (monosomy X) syndrome (36), and Klinefelter's (XXY) syndrome (7, 29). These cases can be found even in the absence of diabetes in the parents of individuals with Turner's or Klinefelter's, suggesting that change in the dosage of some gene or genes on the X chromosome is causative in the increased rate of diabetes in these syndromes (3). A similar pattern of inheritance has been suggested for insulin resistance with polycystic ovarian syndrome (37), where fathers are more likely to have insulin resistance/Type 2 diabetes than mothers. Since several genes related to cognitive function are also mapped to the X chromosome, potential epigenetic alteration of X-linked genes by grandmaternal E consumption could lead to both metabolic and behavioral consequences in the second generation. The mechanisms of these epigenetic changes are likely to include DNA methylation, since prenatal E induces changes in DNA methylation and, subsequently, imprinting (17, 19, 25). Specifically it has been shown to cause changes in the methylation of the X chromosome in the developing neural tube (28). Future work is aimed at determining the epigenetic modifications responsible for the intergenerational effects of E, and the candidate genes on the X chromosome, which may have altered methylation status due to grandmaternal E.
Grandmaternal T4 administration attenuated the E-induced aberrant GTT responses in the F2, but not the F1 generation, thereby further supporting the proposed effect of E and T4 on the germline in the fetal SS F1. Endogenous or exogenous manipulations during the critical period of pregnancy affect not only the mother (P0), but also the developing germ cells (F2) in her offspring (F1). Prior to complete cell differentiation and epigenetic modifications, there is an opportunity for prenatal manipulations to alter the DNA methylation patterning of future generation (16). There are examples of prenatal exposure to endocrine disruptors such as vinclozolin, methoxychlor, and diethylstilbestrol acting as hormonal antagonist or agonist, causing inherited physiological effects through the male or female germline for up to two to four generations (1, 2). The mechanism of transgenerational inheritance include DNA methylation (30) and RNA interference (4). Thyroid hormones might also alter the epigenome, possibly through methylation and histone acetylation (40, 50). In our study T4 could affect the germ cells of F2 generation without direct impact on the F1 through the same mechanism.
One can also argue that the success of grandmaternal T4 treatment is related to the influence of B genetic background on the F2 responses to GTT. The effect of the paternal B genetic background is clear in the differences between the GTT responses of the SS F1 and SB F1 offspring. Both of these F1 offspring had the same SS mothers, consuming the same amount of E diet and having the same blood alcohol levels (38). Nevertheless, their offspring showed differing vulnerability to the effects of maternal E. Furthermore, SB F1 and F2 offspring of control dams have higher fasting glucose levels than any other crosses employed in this study (Figs. 6A and 7A). Inversely, plasma insulin levels are lower in these offspring (Figs. 6B and 7B). These differences suggest that B genetic background, particularly paternal B, is involved in the differences observed in the variations in glucose and insulin responses to GTT across the generations.
Fig. 7.
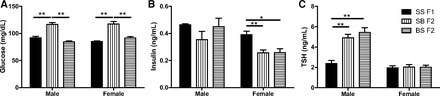
Paternal background determines plasma glucose and thyroid levels. Comparisons of plasma levels of fasting glucose (A), fasting insulin (B), and TSH (C) of C animals from SS F1 and their F2 offspring. *P < 0.05 and **P < 0.01 Bonferroni post hoc. Data are represented as means ± SE; n = 3–12/group.
We assumed that effects of prenatal or grandmaternal E on thyroid hormone regulation would parallel that of glucose and insulin responsiveness to GTT. This was not the case. Interestingly, both the euthyroid TSH profile of the F1 E female and the hypothyroid profile of the F1 E male appeared to lead to a similar pattern of plasma TSH in the SB and BS F2 progeny. Surprisingly, grandmaternal PF, E, and E+T4 diets resulted in a similar hyperthyroid-like plasma TSH profile in male F2 progeny, suggesting an intergenerational effect of these treatments, likely related to calorie restraint on thyroid function.
This is the first demonstration of intergenerational transfer of the prenatal E-induced aberrant glucose and insulin responsiveness. Given that 7.6% of women have been found by the Centers for Disease Control and Prevention to drink while pregnant (12), many children and grandchildren are at risk for developing abnormalities in glucose regulation and vulnerabilities to metabolic disorders. A deeper understanding of this intergenerational transfer is needed with the proposed mechanism of environmental effects on germline in utero.
GRANTS
Funding for this research was provided by National Institute on Alcohol Abuse and Alcoholism Grant AA-017978.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: K.M.H., E.T.-O., and E.N.G. performed experiments; K.M.H. analyzed data; K.M.H., E.T.-O., and E.E.R. interpreted results of experiments; K.M.H. prepared figures; K.M.H. drafted manuscript; K.M.H., E.T.-O., E.N.G., and E.E.R. edited and revised manuscript; K.M.H., E.T.-O., E.N.G., and E.E.R. approved final version of manuscript; E.E.R. conception and design of research.
REFERENCES
- 1.Anway MD, Leathers C, Skinner MK. Endocrine disruptor vinclozolin induced epigenetic transgenerational adult-onset disease. Endocrinology 147: 5515–5523, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anway MD, Memon MA, Uzumcu M, Skinner MK. Transgenerational effect of the endocrine disruptor vinclozolin on male spermatogenesis. J Androl 27: 868–879, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bakalov VK, Cooley MM, Troendle J, Bondy CA. The prevalence of diabetes mellitus in the parents of women with Turner's syndrome. Clin Endocrinol (Oxf) 60: 272, 2004 [DOI] [PubMed] [Google Scholar]
- 4.Buckley BA, Burkhart KB, Gu SG, Spracklin G, Kershner A, Fritz H, Kimble J, Fire A, Kennedy S. A nuclear Argonaute promotes multigenerational epigenetic inheritance and germline immortality. Nature 489: 447–451, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bunout D. Nutritional and metabolic effects of alcoholism: their relationship with alcoholic liver disease. Nutrition 15: 583–589, 1999 [DOI] [PubMed] [Google Scholar]
- 6.Bunout D, Petermann M, Bravo M, Kelly M, Hirsch S, Ugarte G, Iturriaga H. Glucose turnover rate and peripheral insulin sensitivity in alcoholic patients without liver damage. Ann Nutr Metab 33: 31–38, 1989 [DOI] [PubMed] [Google Scholar]
- 7.Burch PR. Klinefelter's syndrome, dizygotic twinning and diabetes mellitus. Nature 221: 175–177, 1969 [DOI] [PubMed] [Google Scholar]
- 8.Bush NC, Chandler-Laney PC, Rouse DJ, Granger WM, Oster RA, Gower BA. Higher maternal gestational glucose concentration is associated with lower offspring insulin sensitivity and altered beta-cell function. J Clin Endocrinol Metab 96: E803–E809, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cano MJ, Ayala A, Murillo ML, Carreras O. Protective effect of folic acid against oxidative stress produced in 21-day postpartum rats by maternal-ethanol chronic consumption during pregnancy and lactation period. Free Radic Res 34: 1–8, 2001 [DOI] [PubMed] [Google Scholar]
- 10.Cano MJ, Garcia-Benitez O, Ojeda ML, Murillo ML, Carreras O. Response of the exocrine pancreas to the CCK on offspring rats of ethanol dams. Effects of folic acid. Alcohol Alcohol 42: 277–284, 2007 [DOI] [PubMed] [Google Scholar]
- 11.Castells S, Mark E, Abaci F, Schwartz E. Growth retardation in fetal alcohol syndrome. Unresponsiveness to growth-promoting hormones. Dev Pharmacol Ther 3: 232–241, 1981 [DOI] [PubMed] [Google Scholar]
- 12.Centers for Disease Control and Prevention. Alcohol use and binge drinking among women of childbearing age–United States, 2006–2010. MMWR Morb Mortal Wkly Rep 61: 534–538, 2012 [PubMed] [Google Scholar]
- 13.Chen L, Nyomba BL. Whole body insulin resistance in rat offspring of mothers consuming alcohol during pregnancy or lactation: comparing prenatal and postnatal exposure. J Appl Physiol 96: 167–172, 2004 [DOI] [PubMed] [Google Scholar]
- 14.Chen L, Zhang T, Nyomba BL. Insulin resistance of gluconeogenic pathways in neonatal rats after prenatal ethanol exposure. Am J Physiol Regul Integr Comp Physiol 286: R554–R559, 2004 [DOI] [PubMed] [Google Scholar]
- 15.Ding GL, Wang FF, Shu J, Tian S, Jiang Y, Zhang D, Wang N, Luo Q, Zhang Y, Jin F, Leung PC, Sheng JZ, Huang HF. Transgenerational glucose intolerance with Igf2/H19 epigenetic alterations in mouse islet induced by intrauterine hyperglycemia. Diabetes 61: 1133–1142, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dolinoy DC, Weidman JR, Waterland RA, Jirtle RL. Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ Health Perspect 114: 567–572, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Downing C, Johnson TE, Larson C, Leakey TI, Siegfried RN, Rafferty TM, Cooney CA. Subtle decreases in DNA methylation and gene expression at the mouse Igf2 locus following prenatal alcohol exposure: effects of a methyl-supplemented diet. Alcohol 45: 65–71, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eberle C, Ament C. Diabetic and metabolic programming: mechanisms altering the intrauterine milieu. ISRN Pediatr 2012: 975685, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garro AJ, McBeth DL, Lima V, Lieber CS. Ethanol consumption inhibits fetal DNA methylation in mice: implications for the fetal alcohol syndrome. Alcohol Clin Exp Res 15: 395–398, 1991 [DOI] [PubMed] [Google Scholar]
- 20.Gill-Randall R, Adams D, Ollerton RL, Lewis M, Alcolado JC. Type 2 diabetes mellitus–genes or intrauterine environment? An embryo transfer paradigm in rats. Diabetologia 47: 1354–1359, 2004 [DOI] [PubMed] [Google Scholar]
- 21.He L, Simmen FA, Mehendale HM, Ronis MJ, Badger TM. Chronic ethanol intake impairs insulin signaling in rats by disrupting Akt association with the cell membrane. Role of TRB3 in inhibition of Akt/protein kinase B activation. J Biol Chem 281: 11126–11134, 2006 [DOI] [PubMed] [Google Scholar]
- 22.Heikkinen S, Argmann CA, Champy MF, Auwerx J. Evaluation of glucose homeostasis. Curr Protoc Mol Biol Chapter 29: Unit 29B 23, 2007 [DOI] [PubMed] [Google Scholar]
- 23.Jung KI, Ju A, Lee HM, Lee SS, Song CH, Won WY, Jeong JS, Hong OK, Kim JH, Kim DJ. Chronic ethanol ingestion, type 2 diabetes mellitus, and brain-derived neurotrophic factor (BDNF) in rats. Neurosci Lett 487: 149–152, 2011 [DOI] [PubMed] [Google Scholar]
- 24.Kallas P, Sellers EM. Blood glucose in intoxicated chronic alcoholics. Can Med Assoc J 112: 590–592, 1975 [PMC free article] [PubMed] [Google Scholar]
- 25.Kaminen-Ahola N, Ahola A, Maga M, Mallitt KA, Fahey P, Cox TC, Whitelaw E, Chong S. Maternal ethanol consumption alters the epigenotype and the phenotype of offspring in a mouse model. PLoS Genet 6: e1000811, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelstrup L, Damm P, Mathiesen ER, Hansen T, Vaag AA, Pedersen O, Clausen TD. Insulin resistance and impaired pancreatic beta-cell function in adult offspring of women with diabetes in pregnancy. J Clin Endocrinol Metab 98: 3793–3801, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim SJ, Ju A, Lim SG, Kim DJ. Chronic alcohol consumption, type 2 diabetes mellitus, insulin-like growth factor-I (IGF-I), and growth hormone (GH) in ethanol-treated diabetic rats. Life Sci 93: 778–782, 2013 [DOI] [PubMed] [Google Scholar]
- 28.Liu Y, Balaraman Y, Wang G, Nephew KP, Zhou FC. Alcohol exposure alters DNA methylation profiles in mouse embryos at early neurulation. Epigenetics 4: 500–511, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Menzinger G, Fallucca F, Andreani D, Wais S, Salvati E, Nielsen J. Klinefelter's syndrome and diabetes mellitus. Lancet 2: 747–748, 1966. 4162069 [Google Scholar]
- 30.Morgan HD, Sutherland HG, Martin DI, Whitelaw E. Epigenetic inheritance at the agouti locus in the mouse. Nat Genet 23: 314–318, 1999 [DOI] [PubMed] [Google Scholar]
- 31.Plagemann A, Harder T, Kohlhoff R, Rohde W, Dorner G. Glucose tolerance and insulin secretion in children of mothers with pregestational IDDM or gestational diabetes. Diabetologia 40: 1094–1100, 1997 [DOI] [PubMed] [Google Scholar]
- 32.Probyn ME, Parsonson KR, Gardebjer EM, Ward LC, Wlodek ME, Anderson ST, Moritz KM. Impact of low dose prenatal ethanol exposure on glucose homeostasis in Sprague-Dawley rats aged up to eight months. PLoS One 8: e59718, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Probyn ME, Zanini S, Ward LC, Bertram JF, Moritz KM. A rodent model of low- to moderate-dose ethanol consumption during pregnancy: patterns of ethanol consumption and effects on fetal and offspring growth. Reprod Fertil Dev 24: 859–870, 2012 [DOI] [PubMed] [Google Scholar]
- 34.Ravelli AC, van der Meulen JH, Michels RP, Osmond C, Barker DJ, Hales CN, Bleker OP. Glucose tolerance in adults after prenatal exposure to famine. Lancet 351: 173–177, 1998 [DOI] [PubMed] [Google Scholar]
- 35.Roseboom TJ, Painter RC, van Abeelen AF, Veenendaal MV, de Rooij SR. Hungry in the womb: what are the consequences? Lessons from the Dutch famine. Maturitas 70: 141–145, 2011 [DOI] [PubMed] [Google Scholar]
- 36.Salgin B, Amin R, Yuen K, Williams RM, Murgatroyd P, Dunger DB. Insulin resistance is an intrinsic defect independent of fat mass in women with Turner's syndrome. Horm Res 65: 69–75, 2006 [DOI] [PubMed] [Google Scholar]
- 37.Sir-Petermann T, Angel B, Maliqueo M, Carvajal F, Santos JL, Perez-Bravo F. Prevalence of Type II diabetes mellitus and insulin resistance in parents of women with polycystic ovary syndrome. Diabetologia 45: 959–964, 2002 [DOI] [PubMed] [Google Scholar]
- 38.Sittig LJ, Shukla PK, Herzing LB, Redei EE. Strain-specific vulnerability to alcohol exposure in utero via hippocampal parent-of-origin expression of deiodinase-III. FASEB J 25: 2313–2324, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Solberg Woods LC, Ahmadiyeh N, Baum A, Shimomura K, Li Q, Steiner DF, Turek FW, Takahashi JS, Churchill GA, Redei EE. Identification of genetic loci involved in diabetes using a rat model of depression. Mamm Genome 20: 486–497, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sui L, Li BM. Effects of perinatal hypothyroidism on regulation of reelin and brain-derived neurotrophic factor gene expression in rat hippocampus: role of DNA methylation and histone acetylation. Steroids 75: 988–997, 2010 [DOI] [PubMed] [Google Scholar]
- 41.Swerdlow NR, Breier M, Mora AB, Ko D, Shoemaker JM. A novel rat strain with enhanced sensitivity to the effects of dopamine agonists on startle gating. Pharmacol Biochem Behav 88: 280–290, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tunc-Ozcan E, Ullmann TM, Shukla PK, Redei EE. Low-dose thyroxine attenuates autism-associated adverse effects of fetal alcohol in male offspring's social behavior and hippocampal gene expression. Alcohol Clin Exp Res 37: 1986–1995, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Villarroya F, Mampel T. Effects of chronic ethanol treatment on glucose tolerance, insulin response and circulating metabolites in the pregnant rat. Gen Pharmacol 16: 591–596, 1985 [DOI] [PubMed] [Google Scholar]
- 44.Villarroya F, Mampel T. Glucose tolerance and insulin response in offspring of ethanol-treated pregnant rats. Gen Pharmacol 16: 415–417, 1985 [DOI] [PubMed] [Google Scholar]
- 45.Villarroya F, Mampel T, Herrera E. Similar metabolic response to acute ethanol intake in pregnant and non-pregnant rats either fed or fasted. Gen Pharmacol 16: 537–540, 1985 [DOI] [PubMed] [Google Scholar]
- 46.Weinberg J. Effects of ethanol and maternal nutritional status on fetal development. Alcohol Clin Exp Res 9: 49–55, 1985 [DOI] [PubMed] [Google Scholar]
- 47.Weinberg NZ. Cognitive and behavioral deficits associated with parental alcohol use. J Am Acad Child Adolesc Psychiatry 36: 1177–1186, 1997 [DOI] [PubMed] [Google Scholar]
- 48.Wilcoxon JS, Kuo AG, Disterhoft JF, Redei EE. Behavioral deficits associated with fetal alcohol exposure are reversed by prenatal thyroid hormone treatment: a role for maternal thyroid hormone deficiency in FAE. Mol Psychiatry 10: 961–971, 2005 [DOI] [PubMed] [Google Scholar]
- 49.Witek-Janusek L. Maternal ethanol ingestion: effect on maternal and neonatal glucose balance. Am J Physiol Endocrinol Metab 251: E178–E184, 1986 [DOI] [PubMed] [Google Scholar]
- 50.Wong NC, Schwartz HL, Strait K, Oppenheimer JH. Thyroid hormone-, carbohydrate, and age-dependent regulation of a methylation site in the hepatic S14 gene. Mol Endocrinol 3: 645–650, 1989 [DOI] [PubMed] [Google Scholar]
- 51.Xu J, Yeon JE, Chang H, Tison G, Chen GJ, Wands J, de la Monte S. Ethanol impairs insulin-stimulated neuronal survival in the developing brain: role of PTEN phosphatase. J Biol Chem 278: 26929–26937, 2003 [DOI] [PubMed] [Google Scholar]
- 52.Yao XH, Nyomba BL. Hepatic insulin resistance induced by prenatal alcohol exposure is associated with reduced PTEN and TRB3 acetylation in adult rat offspring. Am J Physiol Regul Integr Comp Physiol 294: R1797–R1806, 2008 [DOI] [PubMed] [Google Scholar]