

Abstract
IL-18 is an important mediator of obstruction-induced renal fibrosis and renal tubular epithelial cell (TEC) injury. IL-18's proinflammatory properties have been attributed, in part, to NF-κB activation and the stimulation of cytokine gene expression; however, STAT3 has increasingly been shown to mediate renal fibrotic injury. We therefore hypothesized that IL-18 mediates profibrotic TEC injury via STAT3 activation. Male C57BL6 wild-type mice and transgenic mice for human IL-18-binding protein were subjected to unilateral ureteral obstruction or sham operation. The kidneys were harvested 1 or 2 wk afterward and analyzed for active STAT3 (p-STAT3) expression (Western blotting, immunohistochemistry) and suppressor of cytokine signaling 3 (SOCS3) expression. In a separate arm, renal tubular cells (HK-2) were directly stimulated with IL-18 for 2 days with or without the STAT3 inhibitor S3I-201 (50 μM). Cell lysates were then analyzed for p-STAT3 and SOCS3 expression, profibrotic cellular changes (collagen and α-SMA expression), and tubular cell apoptosis. p-STAT3 and SOCS3 expression increased significantly in response to obstruction; however, a significant reduction in p-STAT3 and SOCS3 expression occurred following 1 wk, but not 2 wk, of obstruction in the presence of IL-18 neutralization. In vitro results similarly demonstrate increased p-STAT3, SOCS3, α-SMA, and collagen III expression, and increased collagen production and TEC apoptosis in response to IL-18 stimulation, but the response was significantly diminished in the presence of STAT3 inhibition. These results demonstrate that IL-18-induces profibrotic cellular changes and collagen production in TECs via STAT3 activation.
Keywords: interleukin-18; STAT; kidney; fibrosis; fibrosis, ureteral obstruction
obstructive nephropathy is a major cause of end-stage renal disease (ESRD) in both adults and children. The histological changes associated with chronic renal obstruction include a prominent inflammatory cell infiltrate, followed by progressive tubulointerstitial fibrosis and tubular epithelial cell apoptosis (10). In the kidney, interstitial fibrosis is characterized by de novo activation of α-smooth muscle actin (SMA)-positive myofibroblasts, the principal effector cells responsible for excess extracellular matrix deposition.
IL-18 is a proinflammatory cytokine that has been implicated in the pathophysiology of obstruction-induced renal injury (4) and has been demonstrated to induce both profibrotic changes and apoptosis in tubular epithelial cells in vitro (4, 40). IL-18 is structurally and functionally related to the IL-1 family and is synthesized as an inactive precursor, similar to IL-1β, that requires cleavage into an active molecule by caspase 1 (23). IL-18 is produced by a wide range of cells, including tubular epithelial cells, and binds to its specific receptor IL-18R, resulting in the recruitment of MyD88 to the cytosolic Toll/IL-1R (TIR) domain of the IL-18R itself. This, in turn, activates a signaling cascade leading to the activation of NF-κB, p38 mitogen-activated protein kinases, and AP-1 (6, 28). Through this mechanism, IL-18 can induce the production of other inflammatory cytokines such as IL-1β, TNF-α, and IL-6 (18, 35).
The STAT signaling pathway is an important cascade for signal transduction for a wide variety of growth factors and cytokines (12). This pathway regulates gene expression, as well as cellular activation, proliferation, and differentiation (8, 9). The suppressors of cytokine signaling (SOCS) are a family of proteins that negatively regulate the JAK/STAT signaling pathway by inhibiting STAT phosphorylation (21). STAT signaling has increasingly been implicated in the pathophysiology of fibrotic renal disease, including obstructive nephropathy, ischemia-reperfusion injury, diabetic nephropathy, and glomerulonephritis (1, 2, 3, 5, 19, 22, 25, 27, 29–31, 34, 36, 38, 39, 41). Kuratsune et al. (22) have demonstrated that STAT3 is activated in renal tubular epithelial cells and myofibrobasts in response to 3 or 7 days of ureteral obstruction, and Pang et al. (31) have demonstrated that the STAT3 inhibitor S3I-201 reduces the expression of obstruction-induced profibrotic markers as well as several different proinflammatory mediators. The relationship between IL-18 and STAT3 signaling during obstructive renal injury, however, remains unknown. We therefore hypothesized that IL-18 mediates profibrotic renal tubular cell injury via STAT3 activation. To study this, we examined renal cortical STAT3 activation and SOCS3 expression in male C57BL6 wild-type (WT) mice and mice transgenic for IL-18-binding protein (IL-18BP Tg) using a well-established model of unilateral ureteral obstruction (UUO). In a separate arm, human proximal tubular cells (HK-2) were directly stimulated with IL-18 in the presence or absence of a STAT3 inhibitor and subsequently examined for STAT3 activation, SOCS3 expression, α-SMA expression, collagen production, and apoptosis.
MATERIALS AND METHODS
Animals, experimental groups, and operative techniques.
The animal protocol was reviewed and accepted by the Animal Care and Research Committee of the Indiana University School of Medicine. Male C57BL6 mice transgenic for IL-18 BP Tg were generously donated by Dr. Charles Dinarello (University of Colorado Health Science Center, Denver, CO). These mice overexpresses human IL-18-binding protein isoform a and reliably inhibit IL-18 activity, but do not have any notable phenotype (15). The genotype of the mice was confirmed with a PCR analysis of expected DNA samples from tail snips (5′ primer: 5′-ACA CCT GTC TCG CAG ACC AC-3′ and 3′ primer: 5′-TCA GCT GCT CCA GCA CCA A-3′) as described by Fentuzzi et al. (15), and overexpression of serum levels of human IL-18BP was confirmed using an ELISA (Human IL-18BP Duo SET ELISA; R&D Systems, Minneapolis, MN) before utilization of the animals (3).
Male WT or IL-18BP Tg mice weighing 25–30 g (6 animals/group) were housed in a nonstressful environment for 1 wk before initiation of experiments. The animals were then anesthetized with isoflurane, and the left ureter was isolated and completely ligated with 5-0 silk suture. Sham-operated mice underwent an identical surgical procedure without ureteral ligation. One or two weeks postoperatively, mice were reanesthetized, the left kidneys were removed and snap frozen in liquid nitrogen, and the animals were subsequently euthanized.
Tissue homogenization.
A portion of each renal cortex was homogenized after the samples had been diluted in 10 vol of homogenate buffer/g of tissue [10 mM HEPES (pH 7.9), 10 mM KCl, 0.1 mM EGTA, 0.1 mM DTT, and Complete Protease Inhibitor tabs (Boehringer Manheim, Indianapolis, IN)] using a vertishear tissue homogenizer. Renal homogenates were then centrifuged at 3,000 g for 15 min at 4°C, and the supernatants were stored at −80°C until the ELISAs or Western blots could be performed.
Cell culture, IL-18 stimulation, and S3I-201 treatment.
The human proximal tubular cell line HK-2 was cultured in keratinocyte serum-free medium+5 ng/ml epidermal growth factor and 50 μg/ml bovine extract+100 U/ml penicillin and 100 μg/ml streptomycin. The cells were passaged weekly by trypsinization (0.25% tripsin, 0.02% EDTA) following formation of a confluent monolayer and placed in serum-free media 24 h before IL-18 stimulation. Recombinant mature IL-18 (100 ng/ml; R&D Systems) was added to the cells, with untreated cells serving as controls. The cells were treated with vehicle (0.05% DMSO) or S3I-201 (50 μM; EMD Chemicals, San Diego, CA) 2 h before IL-18 stimulation. Pilot studies indicated that pSTAT3 levels peaked in HK-2 cells after 48 h of IL-18 stimulation; therefore, the cells were exposed to IL-18+vehicle/S3I-201 for 2 days, and both supernatants and cell lysates were harvested (3 plates/treatment group).
Real-time PCR.
Total RNA was extracted from renal cortical tissue or cell lysates by homogenization in TRIzol (GIBCO BRL, Gaithersburg, MD) and then isolated by precipitation with chloroform and isopropanol. Total RNA (0.5 μg) was subjected to cDNA synthesis using iScript (Bio-Rad, Hercules, CA). cDNA from each samples was analyzed for collagen III a1 (Mm00802331_m1) and SOCS3 (Mm00545913_s1) using a TaqMan gene expression assay (RT-PCR; Applied Biosystems, Foster City, CA). FAM/Dye MGB-labeled probes for mouse β-actin (Applied Biosystems) served as endogenous controls.
Cell supernatant total collagen concentration.
The total soluble collagen concentration within cell supernatants (3/group) were measured with a Sircol collagen assay kit (Accurate Chemical and Scientific, Westbury, NY) according to the manufacturer's protocol. The total collagen concentration was measured at 540 nm in all collected supernatants.
STAT3 tissue staining.
Tissue sections (4 μm) were deparaffinized and dehydrated with xylene and alcohol. Antigen was retrieved by incubating the cells with proteinase K for 20 min in an oven. The tissues were then blocked with 1% bovine serum albumin. Slides were incubated with anti-p-STAT3(Tyr705) antibody (1:25; Cell Signaling, Danvers, MA) for 30 min. The slides were washed in TBS and incubated with the secondary antibody (goat anti-rabbit; Dako EnVision, Carpinteria, CA) for 30 min. Peroxidase-stained sections were then developed with 3,3“-diaminobenzidine and counterstained with hemalum. Sections incubated without primary antibody exhibited no staining.
TdT-mediated dUTP nick end labeling assay.
Fluorometric DNA strand breaks representative of apoptosis were detected using terminal deoxynucleotidyl transferase incorporation of fluorescein-dUTP (ApopTag Red in situ Apoptosis Detection kit, Temecula, CA). Adherent HK-2 cells were prepared from each group and fixed in 1% paraformaldehyde for 10 min at room temperature (RT). After cellular permealization with 20 μg/ml proteinase K for 15 min, the slides were exposed to terminal deoxynucleotidyl transferase fluorescein labeling for 1 h at 37°C. The slides were then washed in PBS and exposed to anti-digoxigenin conjugate for 30 min at RT. The slides were washed again and counterstained (10 μg/ml bis-benzimide) for 10 min at RT. The specimens were mounted in an antiquenching medium (ProLong Antifade Kit; Molecular Probes, Eugene, OR) and maintained at −4°C until the microscopic examination could be performed. The number of fluorescent nuclei were quantified per high-power field in each treatment group and compared. Samples were analyzed in triplicate. The characteristic morphological features of apoptosis (i.e., nuclear condensation) were also correlated with nuclear fluorescence. After washing, the slides were mounted using cover glass.
Western blot analysis.
Protein extracts from homogenized samples (30 μg/lane) or cell lysates (20 μg/lane) were subjected to SDS-PAGE on a Tris-glycine gel and transferred to a polyvinylidene fluoride membrane. Immunoblotting was performed by incubating each membrane in 5% dry milk for 1 h, followed by incubation with an anti-α-SMA monoclonal antibody (clone 1A4, 1:500 overnight at 4°C; R&D Systems), an anti-SOCS3 antibody (1:200 overnight at 4°C, Cell Signaling), or an anti-p-STAT3 (1:200 overnight at 4°C; Cell Signaling). After washing three times in TBST, each membrane was incubated for 1 h at RT with a peroxidase-conjugated secondary antibody (1:2,000 for α-SMA, SOCS3, and p-STAT3). Equivalent protein loading in each lane was confirmed by stripping and reblotting each membrane for GAPDH (1:10,000 for 30 min at RT, secondary 1:10,000 for 30 min at RT; Biodesign International, Saco, ME). The membranes were developed using enhanced chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ), and the density of each band was determined using National Institutes of Health image-analysis software and expressed as a percentage of GAPDH density. The location of the pSTAT3 band was confirmed using a positive control obtained from the manufacturer (no. 9133; Cell Signaling).
Statistical analysis.
Data are presented as means ± SE. Differences at the 95% confidence level were considered significant. The experiment groups were compared using one-way ANOVA with a post hoc Bonferroni-Dunn test (JMP 5.0.1).
RESULTS
STAT3 activation during UUO.
To evaluate STAT3 activation during UUO, active STAT3 (pSTAT3) expression and immunohistochemical localization of pSTAT3 were evaluated in renal cortical tissue samples. pSTAT3 expression increased significantly in response to 1 or 2 wk of obstruction (Fig. 1A); however, a marked reduction in pSTAT3 expression was only detected in IL-18BP Tg mice exposed to 1 wk of obstruction. Similarly, a dramatic increase in the number of nuclei staining positive for pSTAT3 was observed in response to 1 or 2 wk of obstruction, with pSTAT3 staining localizing primarily to renal tubular epithelial cells (Fig. 1B). pSTAT3 staining was only significantly reduced in IL-18BP Tg mice exposed to 1 wk of obstruction. These findings suggest that IL-18 is an early mediator of STAT3 activation during obstructive renal injury.
Fig. 1.
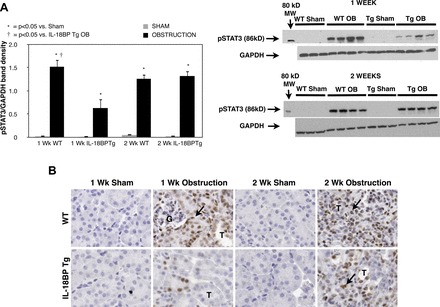
Renal cortical p-STAT3 protein expression and immunolocalization following unilateral ureteral obstruction (UUO). A: gel photographs and densitometric analysis of p-STAT3 expression represented as a percentage of GAPDH in wild-type (WT) and IL-18BP transgenic (IL-18BP Tg) animals exposed to sham operation (Sham) or 1 or 2 wk of UUO (OB). B: photograph depicting renal cortical STAT3 (brown stain; arrows) in WT and IL-18BP Tg animals exposed to sham operation or 1 or 2 wk of UUO. G = glomerulus; T= tubule. Magnification ×400.
SOCS3 expression during UUO.
To evaluate the negative regulation of STAT3 signaling during UUO, SOCS 3 expression was evaluated in response to obstruction. SOCS3 protein and gene expression increased significantly in response to 1 or 2 wk of obstruction, but levels were significantly reduced in IL-18BP Tg mice exposed to 1 wk of obstruction (Fig. 2, A and B). SOCS 3 protein and gene expression were not significantly reduced in IL-18BP Tg mice exposed to 2 wk of obstruction.
Fig. 2.
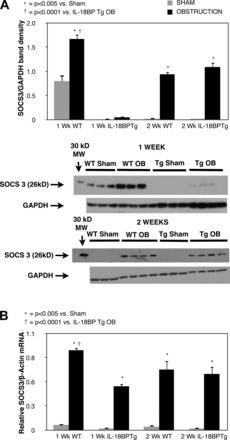
Renal cortical SOCS3 protein expression and quantitative SOCS3 mRNA expression following UUO. A: gel photograph and densitometric analysis of SOCS3 expression represented as a percentage of GAPDH in WT and IL-18BP Tg animals exposed to sham operation (Sham) or 1 or 2 wk of UUO (OB). B: quantitative SOCS3 mRNA expression represented as a percentage of β-actin in WT and IL-18BP Tg animals exposed to sham operation or 1 or 2 wk of UUO.
HK-2 cell STAT3 activation in response to IL-18 stimulation.
To evaluate the direct relationship between IL-18 stimulation and STAT3 activation in renal tubular cells, HK-2 cells were stimulated with IL-18 in the presence or absence of S3I-201. Direct cell stimulation with IL-18 significantly increased pSTAT3 expression, but pSTAT3 expression was reduced to control levels in the presence of S3I-201 (Fig. 3). This supports our observations in vivo and provides further evidence that IL-18 directly stimulates STAT3 activation in TECs.
Fig. 3.
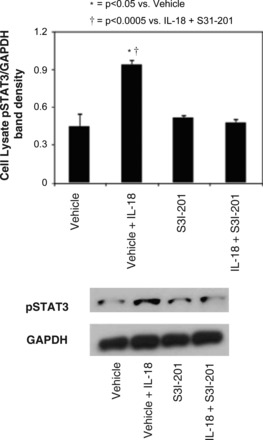
HK-2 cell expression of pSTAT3 following IL-18 stimulation in vitro. Gel photograph (bottom) and densitometric analysis (top) of p-STAT3 expression represented as a percentage of GAPDH in HK-2 cells following 2 days of cell stimulation with recombinant human IL-18 (100 ng/ml) in the presence or absence of STAT3 inhibition (50 μM S3I-201).
Role of STAT3 in IL-18 induced-HK-2 cell expression of SOCS3.
The negative regulation of STAT3 signaling in HK-2 cells following IL-18 stimulation was then evaluated by analyzing SOCS3 expression in the presence or absence of S3I-201. Cell lysate SOCS3 protein and mRNA expression were significantly increased in HK-2 cells exposed to IL-18 stimulation. STAT3 inhibition with S3I-201, however, dramatically reduced SOCS3 expression to control levels in the presence of IL-18 stimulation (Fig. 4, A and B). These findings suggest that IL-18 stimulates STAT3 activation and SOCS3 expression in renal tubular cells in vitro, and that SOCS3 expression is dependent on STAT3 activity.
Fig. 4.
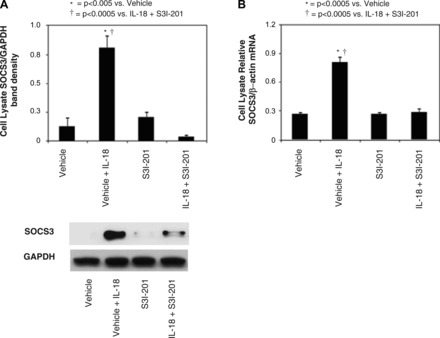
HK-2 cell SOCS3 protein expression and quantitative SOCS3 mRNA expression following IL-18 stimulation in vitro. A: gel photograph and densitometric analysis of SOCS3 protein expression represented as a percentage of GAPDH in HK-2 cells following 2 days of cell stimulation with recombinant human IL-18 (100 ng/ml) in in the presence or absence of STAT3 inhibition (50 μM S3I-201). B: quantitative SOCS3 mRNA expression represented as a percentage of β-actin in HK-2 cells following 2 days of cell stimulation with recombinant human IL-18 (100 ng/ml) in the presence or absence of STAT3 inhibition (50 μM S3I-201).
Role of STAT3 in IL-18 induced-HK-2 cell expression of profibrotic markers.
To evaluate the role of STAT3 inhibition in IL-18-induced renal tubular cell injury, HK-2 cells were stimulated with IL-18 in the presence or absence of S3I-201. The expression of the myofibroblast marker α-SMA was then evaluated in addition to collagen III expression and total collagen production. HK-2 cells subjected to IL-18 stimulation demonstrated a significant increase in α-SMA and collagen III expression as well as total collagen production. Treatment of the cells with S3I-201 during IL-18 stimulation, however, significantly reduced α-SMA and collagen expression (Fig. 5, A–C). This suggests that STAT3 is an important mediator of IL-18-induced profibrotic injury in TECs.
Fig. 5.
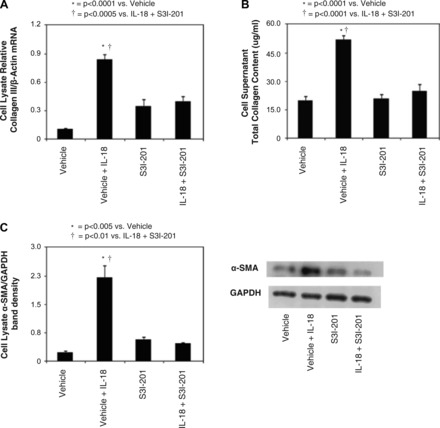
HK-2 cell profibrotic changes following IL-18 stimulation in vitro. A: quantitative collagen III mRNA expression represented as a percentage of β-actin in HK-2 cells following 2 days of cell stimulation with recombinant human IL-18 (100 ng/ml) in the presence or absence of STAT3 inhibition (50 μM S3I-201). B: cell supernatant total collagen concentration in HK-2 cells following 2 days of cell stimulation with recombinant human IL-18 (100 ng/ml) in the presence or absence of STAT3 inhibition (50 μM S3I-201). C: gel photograph and densitometric analysis of α-smooth muscle actin (SMA) expression represented as a percentage of GAPDH in HK-2 cells following 2 days of cell stimulation with recombinant human IL-18 (100 ng/ml) in the presence or absence of STAT3 inhibition (50 μM S3I-201).
Role of STAT3 in IL-18-induced-HK-2 cell apoptosis.
To evaluate the role of STAT3 inhibition in IL-18-induced apoptosis, HK-2 cells were stimulated with IL-18 in the presence or absence of S3I-201. As expected, the number of apoptotic nuclei increased significantly in response to IL-18 stimulation. IL-18-induced apoptosis, however, was dramatically reduced in the presence of STAT3 inhibition (Fig. 6, A and B). This again demonstrates that STAT3 is an important mediator of IL-18-induced renal tubular cell injury.
Fig. 6.
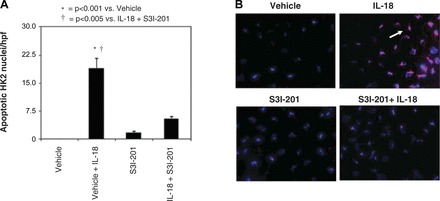
Renal tubular cell apoptosis following IL-18 stimulation in vitro. A: graph depicting the number of apoptotic nuclei (TdT-mediated dUTP nick end labeling) per high-power field (hpf) in HK-2 cells following 2 days of cell stimulation with recombinant human IL-18 (100 ng/ml) in the presence or absence of STAT3 inhibition (50 μM S3I-201). B: photographs (magnification ×400) demonstrating renal tubular cell apoptosis in HK-2 cells following 2 days of cell stimulation with recombinant human IL-18 (100 ng/ml) in the presence or absence of STAT3 inhibition (50 μM S3I-201). Nuclei are stained blue with apoptotic nuclei counterstained red (arrows).
DISCUSSION
IL-18 is a proinflammatory cytokine implicated in the pathogenesis of many inflammatory diseases of the kidney, including urinary tract infections, renal ischemia-reperfusion injury, autoimmune conditions, allograft rejection, and most recently, obstructive nephropathy (4, 11, 14, 16, 33). IL-18 stimulates tubulointerstitial fibrosis and tubular epithelial cell apoptosis during renal obstruction independently of TGF-β or TNF-α. In vitro studies also demonstrate that IL-18 is capable of directly stimulating profibrotic changes and apoptosis in tubular epithelial cells in a dose-dependent fashion (4, 40). While IL-18's proinflammatory effect via downstream NF-κB activation on subsequent cytokine gene upregulation is widely recognized (7, 13), this is the first study to demonstrate that IL-18 stimulates STAT3 activation during renal obstruction, and mediates profibrotic changes and apoptosis in TECs via STAT3 activation in vitro.
The STAT signaling pathway constitutes one of the primary regulatory pathways for cytokine expression (32), and STAT signaling has increasingly been implicated in the pathophysiology of renal disease (27). Among the STAT signaling pathways, STAT3 appears to be central and is best correlated with renal disease (27). Kuratsune et al. (22) have demonstrated STAT3 activation in renal tubular epithelial cells and myofibroblasts in response to obstruction, with peak p-STAT3 expression occurring 7 days after the onset of obstruction. Despite IL-18's significant role in obstruction-induced renal injury, the impact of obstruction-induced IL-18 production on STAT3 activation has not previously been evaluated. Our data reveal that STAT3 expression is significantly increased in response to obstruction, and that active STAT3 expression primarily localizes to renal tubular epithelial cells. Obstruction-induced active STAT3 expression, however, is significantly reduced in the presence of IL-18 neutralization after 1 wk but not 2 wk of renal obstruction. Our observations corroborate the findings of Kuratsune et al. and implicate IL-18 as an important early mediator of STAT3 activation during renal obstruction.
The SOCS family of proteins are an important mechanism for the negative regulation of the JAK/STAT signaling pathway. SOCS proteins negatively regulate JAK/STAT signaling by either binding to and directly inhibiting JAK tyrosine kinase activity or competing with STATs for phosphotyrosine binding sites on cytokine receptors (21). While increased SOCS3 expression has been demonstrated in renal tubular cells in vitro in response to IL-1β stimulation (24), SOCS3 expression during renal obstruction and the effect of obstruction-induced IL-18 production on SOCS3 expression have not previously been evaluated. Our results demonstrate that renal cortical SOCS3 protein and mRNA expression peak in response to 1 wk of renal obstruction and remain significantly elevated in response to 2 wk of obstruction. This response was significantly reduced in the presence of IL-18 neutralization after 1 wk of obstruction, but not 2 wk of obstruction, suggesting that IL-18 is an early mediator of SOCS3 expression during obstructive renal injury. It is unclear; however, whether IL-18 induces SOCS3 expression directly in response to renal obstruction, or if SOCS3 expression is indirectly stimulated as a consequence of IL-18-induced STAT3 activation.
To further investigate the effect of IL-18 on STAT3 activation and signaling in renal tubular cells, HK-2 cells were stimulated with IL-18 in vitro in the presence or absence of the STAT3 inhibitor S3I-201. Direct cell stimulation with IL-18 induced a significant increase in active STAT3 expression as well as SOCS3 expression; however, both pSTAT3 and SOCS3 expression were reduced to control levels in the presence of S3I-201. These findings corroborate our in vivo observations and provide further evidence that IL-18 is an important mediator of STAT3 activation as well as the negative regulation of STAT signaling in renal tubular cells.
IL-18 has previously been demonstrated to induce renal tubular epithelial cell transdifferentiation into myofibroblasts (EMT) in vitro and has been identified as a significant mediator of EMT and obstruction-induced tubulointerstitial fibrosis in vivo (4). The accumulation of matrix-producing myofibroblasts is a prominent feature of tubulointerstitial fibrosis, and a large proportion of these cells appear to derive from tubular epithelial cells (20, 37). Myofibroblasts are characterized by the expression of the mesenchymal marker α-SMA as well as the ability to produce extracellular matrix proteins. We therefore investigated STAT3's impact on IL-18-induced profibrotic renal tubular cell injury in vitro by evaluating α-SMA expression, collagen III expression, and total collagen production in the presence or absence of STAT3 inhibition. Consistent with our previous observations, IL-18 stimulation increased TEC expression of α-SMA and collagen III, and increased total collagen production in cell supernatants. STAT3 inhibition with S3I-201, however, dramatically reduced IL-18-induced α-SMA expression and collagen production in TECs, suggesting that STAT3 mediates IL-18-induced profibrotic renal tubular cell injury. These findings are corroborated by Liu et al. (24), who demonstrated that IL-1β induces EMT in proximal tubular cells via the JAK/STAT signaling pathway.
In addition to fibrotic changes, IL-18 has been demonstrated to induce apoptosis and proapoptotic signaling in renal tubular epithelial cells in vitro in a dose-dependent fashion (40). Previous studies have shown that IL-18 stimulates apoptotic cell death in a variety of cells through both TNF-α and Fas-dependent mechanisms (17, 26, 40), but the impact of STAT3 on IL-18-induced apoptosis in TECs had not previously been evaluated. Our results demonstrate that STAT3 inhibition prevents IL-18-induced renal tubular apoptosis, suggesting that IL-18 stimulates renal tubular cell apoptosis via STAT3 activation.
Urinary tract obstruction stimulates the release of a number of cytokines and growth factors that contribute to tubulointerstitial fibrosis and progressive renal injury. This study demonstrates that IL-18 is an early mediator of STAT3 activation and SOCS3 expression in renal tubular cells and further implicates STAT3 in IL-18-induced profibrotic and apoptotic renal tubular cell injury in vitro. A greater understanding of STAT3's role in acute and chronic renal injury and the interaction between IL-18 and STAT signaling warrants further study.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: F.M., A.R., K.L.H., and H.Z. performed experiments; F.M. and K.K.M. prepared figures; F.M. drafted manuscript; F.M. and K.K.M. edited and revised manuscript; F.M., A.R., K.L.H., H.Z., and K.K.M. approved final version of manuscript; K.K.M. provided conception and design of research; K.K.M. analyzed data; K.K.M. interpreted results of experiments.
REFERENCES
- 1.Arakawa T, Masaki T, Hirai T, Doi S, Kuratsune M, Arihiro K, Kohno N, Yorioka N. Activation of signal transducer and activator of transcription 3 correlates with cell proliferation and renal injury in human glomerulonephritis. Nephrol Dial Transplant 23: 3418–3426, 2008 [DOI] [PubMed] [Google Scholar]
- 2.Arany I, Megyesi JK, Nelkin BD, Safirstein RL. STAT3 attenuates EGFR-mediated ERK activation and cell survival during oxidant stress in mouse proximal tubular cells. Kidney Int 70: 669–674, 2006 [DOI] [PubMed] [Google Scholar]
- 3.Banes AK, Shaw S, Jenkins J, Redd H, Amiri F, Pollock DM, Marrero MB. Angiotensin II blockade prevents hyperglycemia-induced activation of JAK and STAT proteins in diabetic rat kidney glomeruli. Am J Physiol Renal Physiol 286: F653–F659, 2004 [DOI] [PubMed] [Google Scholar]
- 4.Bani-Hani AH, Leslie JA, Asanuma H, Dinarello CA, Campbell MT, Meldrum DR, Zhang H, Hile K, Meldrum KK. IL-18 neutralization ameliorates obstruction-induced epithelial-mesenchymal transition and renal fibrosis. Kidney Int 76: 500–511, 2009 [DOI] [PubMed] [Google Scholar]
- 5.Berthier CC, Zhang H, Schin M, Henger A, Nelson RG, Yee B, Boucherot A, Neusser MA, Cohen CD, Carter-Su C, Argetsinger LS, Rastaldi MP, Brosius FC, Kretzler M. Enhanced expression of Janus kinase-signal transducer and activator of transcription pathway members in human diabetic nephropathy. Diabetes 58: 469–477, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bombardieri M, McInnes IB, Pitzalis C. Interleukin-18 as a potential therapeutic target in chronic autoimmune/inflammatory conditions. Expert Opin Biol Ther 7: 31–40, 2007 [DOI] [PubMed] [Google Scholar]
- 7.Boraschi D, Dinarello CA. IL18 in autoimmunity: review. Eur Cytokine Netw 17: 224–252, 2006 [PubMed] [Google Scholar]
- 8.Bromberg J, Darnell JE., Jr The role of STATs in transcriptional control and their impact on cellular function. Oncogene 19: 2468–2473, 2000 [DOI] [PubMed] [Google Scholar]
- 9.Bromberg JF, Wrzeszczynska MH, Devgan GZ, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr Stat3 as an oncogene. Cell 98: 295–303, 1999 [DOI] [PubMed] [Google Scholar]
- 10.Chevalier RL, Thornhill BA, Forbes MS, Kiley SC. Mechanisms of renal injury and progression of renal disease in congenital obstructive nephropathy. Pediatr Nephrol 25: 687–697, 2010 [DOI] [PubMed] [Google Scholar]
- 11.Daemen MA, van't Veer C, Wolfs TG, Buurman WA. Ischemia/reperfusion-induced IFN-gamma up-regulation: involvement of IL-12 and IL-18. J Immunol 162: 5506–5510, 1999 [PubMed] [Google Scholar]
- 12.Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264: 1415–1421, 1994 [DOI] [PubMed] [Google Scholar]
- 13.Dinarello CA, Novick D, Puren AJ, Fantuzzi G, Shapiro L, Mühl H, Yoon DY, Reznikov LL, Kim SH, Rubinstein M. Overview of interleukin-18: more than an interferon-gamma inducing factor. J Leukoc Biol 63: 658–664, 1998 [PubMed] [Google Scholar]
- 14.Dinarello CA. Interleukin-18 and the pathogenesis of inflammatory diseases. Semin Nephrol 27: 98–114, 2007 [DOI] [PubMed] [Google Scholar]
- 15.Fantuzzi G, Banda NK, Guthridge C, Vondracek A, Kim SH, Siegmund B, Azam T, Sennello JA, Dinarello CA, Arend WP. Generation and characterization of mice transgenic for human IL-18-binding protein isoform a. J Leukoc Biol 74: 889–896, 2003 [DOI] [PubMed] [Google Scholar]
- 16.Faust J, Menke J, Kriegsmann J, Kelley VR, Mayet WJ, Galle PR, Schwarting A. Correlation of renal tubular epithelial cell-derived interleukin-18 up-regulation with disease activity in MRL-Faslpr mice with autoimmune lupus nephritis. Arthritis Rheum 46: 3083–3095, 2002 [DOI] [PubMed] [Google Scholar]
- 17.Finotto S, Siebler J, Hausding M, Chipp M, Wirtz S, Klein S, Protschka M, Doganci A, Lehr HA, Trautwein C, Khosravi-Far R, Strand D, Lohse A, Galle PR, Blessing M, Neurath MF. Severe hepatic injury in interleukin 18 (IL-18) transgenic mice: a key role for IL-18 in regulating hepatocyte apoptosis in vivo. Gut 53: 392–400, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gracie JA, Forsey RJ, Chan WL, Gilmour A, Leung BP, Greer MR, Kennedy K, Carter R, Wei XQ, Xu D, Field M, Foulis A, Liew FY, McInnes IB. A proinflammatory role for IL-18 in rheumatoid arthritis. J Clin Invest 104: 1393–1401, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirai T, Masaki T, Kuratsune M, Yorioka N, Kohno N. PDGF receptor tyrosine kinase inhibitor suppresses mesangial cell proliferation involving STAT3 activation. Clin Exp Immunol 144: 353–361, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 110: 341–350, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krebs DL, Hilton DJ. SOCS proteins: negative regulators of cytokine signaling. Stem Cells 19: 378–387, 2001 [DOI] [PubMed] [Google Scholar]
- 22.Kuratsune M, Masaki T, Hirai T, Kiribayashi K, Yokoyama Y, Arakawa T, Yorioka N, Kohno N. Signal transducer and activator of transcription 3 involvement in the development of renal interstitial fibrosis after unilateral ureteral obstruction. Nephrology 12: 565–571, 2007 [DOI] [PubMed] [Google Scholar]
- 23.Leslie JA, Meldrum KK. The role of interleukin-18 in renal injury. J Surg Res 145: 170–175, 2008 [DOI] [PubMed] [Google Scholar]
- 24.Liu Q, Liu S, Shi Y, Li H, Hao J, Xing L, Cao Y, Duan H. Suppressors of cytokine signaling inhibit tubular epithelial cell-myofibroblast transdifferentiation. Am J Nephrol 34: 142–151, 2011 [DOI] [PubMed] [Google Scholar]
- 25.Lu TC, Wang ZH, Feng X, Chuang PY, Fang W, Shen Y, Levy DE, Xiong H, Chen N, He JC. Knockdown of Stat3 activity in vivo prevents diabetic glomerulopathy. Kidney Int 76: 63–71, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marino E, Cardier JE. Differential effect of IL-18 on endothelial cell apoptosis mediated by TNF-alpha and Fas (CD95). Cytokine 22: 142–148, 2003 [DOI] [PubMed] [Google Scholar]
- 27.Matsui F, Meldrum KK. The role of the Janus kinase family/signal transducer and activator of transcription signaling pathway in fibrotic renal disease. J Surg Res 178: 339–335, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakahira M, Ahn HJ, Park WR, Gao P, Tomura M, Park CS, Hamaoka T, Ohta T, Kurimoto M, Fujiwara H. Synergy of IL-12 and IL-18 for IFN-gamma gene expression: IL-12-induced STAT4 contributes to IFN-gamma promoter activation by up-regulating the binding activity of IL-18-induced activator protein 1. J Immunol 168: 1146–1153, 2002 [DOI] [PubMed] [Google Scholar]
- 29.Neria F, Castilla MA, Sanchez RF, Gonzalez Pacheco FR, Deudero JJ, Calabia O, Tejedor A, Manzarbeitia F, Ortiz A, Caramelo C. Inhibition of JAK2 protects renal endothelial and epithelial cells from oxidative stress and cyclosporin A toxicity. Kidney Int 75: 227–234, 2009 [DOI] [PubMed] [Google Scholar]
- 30.Ortiz-Muñoz G, Lopez-Parra V, Lopez-Franco O, Fernandez-Vizarra P, Mallavia B, Flores C, Sanz A, Blanco J, Mezzano S, Ortiz A, Egido J, Gomez-Guerrero C. Suppressors of cytokine signaling abrogate diabetic nephropathy. J Am Soc Nephrol 21: 763–772, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pang M, Ma L, Gong R, Tolbert E, Mao H, Ponnusamy M, Chin YE, Yan H, Dworkin LD, Zhuang S. A novel STAT3 inhibitor, S3I-201, attenuates renal interstitial fibroblast activation and interstitial fibrosis in obstructive nephropathy. Kidney Int 78: 257–268, 2010 [DOI] [PubMed] [Google Scholar]
- 32.Schindler CW. Series introduction. JAK-STAT signaling in human disease. J Clin Invest 109: 1133–1137, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Striz I, Krasna E, Honsova E, Lacha J, Petrickova K, Jaresova M, Lodererova A, Bohmova R, Valhova S, Slavcev A, Vitko S. Interleukin 18 (IL-18) upregulation in acute rejection of kidney allograft. Immunol Lett 99: 30–35, 2005 [DOI] [PubMed] [Google Scholar]
- 34.Wang S, Yang N, Zhang L, Huang B, Tan H, Liang Y, Li Y, Yu X. Jak/STAT signaling is involved in the inflammatory infiltration of the kidneys in MRL/lpr mice. Lupus 19: 1171–1180, 2010 [DOI] [PubMed] [Google Scholar]
- 35.Yamamura M, Kawashima M, Taniai M, Yamauchi H, Tanimoto T, Kurimoto M, Morita Y, Ohmoto Y, Makino H. Interferon-gamma-inducing activity of interleukin-18 in the joint with rheumatoid arthritis. Arthritis Rheum 44: 275–285, 2001 [DOI] [PubMed] [Google Scholar]
- 36.Yanagita M, Arai H, Nakano T, Ohashi K, Mizuno K, Fukatsu A, Doi T, Kita T. Gas6 induces mesangial cell proliferation via latent transcription factor STAT3. J Biol Chem 276: 42364–42369, 2001 [DOI] [PubMed] [Google Scholar]
- 37.Yang J, Liu Y. Dissection of key events in tubular epithelial to myofibroblast transition and its implications in renal interstitial fibrosis. Am J Pathol 159: 1465–1475, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang N, Luo M, Li R, Huang Y, Zhang R, Wu Q, Wang F, Li Y, Yu X. Blockage of JAK/STAT signalling attenuates renal ischaemia-reperfusion injury in rat. Nephrol Dial Transplant 23: 91–100, 2008 [DOI] [PubMed] [Google Scholar]
- 39.Yokota N, Burne-Taney M, Racusen L, Rabb H. Contrasting roles for STAT4 and STAT6 signal transduction pathways in murine renal ischemia-reperfusion injury. Am J Physiol Renal Physiol 285: F319–F325, 2003 [DOI] [PubMed] [Google Scholar]
- 40.Zhang H, Hile KL, Asanuma H, Vanderbrink B, Franke EI, Campbell MT, Meldrum KK. IL-18 mediates proapoptotic signaling in renal tubular cells through a Fas ligand-dependent mechanism. Am J Physiol Renal Physiol 301: F171–F178, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang W, Chen X, Shi S, Wei R, Wang J, Yamanaka N, Hong Q. Expression and activation of STAT3 in chronic proliferative immune complex glomerulonephritis and the effect of fosinopril. Nephrol Dial Transplant 20: 892–901, 2005 [DOI] [PubMed] [Google Scholar]