

Abstract
The gastrointestinal peptide cholecystokinin (CCK) causes the release of pancreatic digestive enzymes and growth of the normal pancreas. Exogenous CCK administration has been used in animal models to study pancreatitis and also as a promoter of carcinogen-induced or Kras-driven pancreatic cancer. Defining CCK receptors in normal human pancreas has been problematic because of its retroperitoneal location, high concentrations of pancreatic proteases, and endogenous RNase. Most studies indicate that the predominant receptor in human pancreas is the CCK-B type, and CCK-A is the predominant form in rodent pancreas. In pancreatic cancer cells and tumors, the role of CCK is better established because receptors are often overexpressed by these cancer cells and stimulation of such receptors promotes growth. Furthermore, in established cancer, endogenous production of CCK and/or gastrin occurs and their actions stimulate the synthesis of more receptors plus growth by an autocrine mechanism. Initially it was thought that the mechanism by which CCK served to potentiate carcinogenesis was by interplay with inflammation in the pancreatic microenvironment. But with the recent findings of CCK receptors on early PanIN (pancreatic intraepithelial neoplasia) lesions and on stellate cells, the question has been raised that perhaps CCK actions are not the result of cancer but an early driving promoter of cancer. This review will summarize what is known regarding CCK, its receptors, and pancreatic cancer, and also what is unknown and requires further investigation to determine which comes first, the chicken or the egg, “CCK or the cancer.”
Keywords: receptor, G protein-coupled, gastrin, cholecystokinin, pancreas, cancer
traditionally hormones exert their effects on target organs through selective receptors, which initiate an intracellular signal that regulates cell function or tissue proliferation. In many instances, hormone receptors are overexpressed or aberrantly expressed in corresponding cancers that arise from target organs. Likewise, hormones that exert their effects in the gastrointestinal tract also are involved in normal physiological functions including regulation of cellular growth and secretion after signaling through selective receptors. Because of easy accessibility to the stomach, the gastrointestinal peptide gastrin has been well studied both in animals and humans, where it regulates gastric acid secretion (15) and gastrointestinal growth (44). A structurally related peptide, cholecystokinin or CCK, exerts its effects on the pancreas (enzyme secretion and growth) and the gallbladder (contraction), and its physiological actions have been well studied in animal models. However, because of the reasonably inaccessible retroperitoneal location of the pancreas and vulnerable nature of the pancreas, the role of CCK in normal human pancreatic function has been less well described. CCK and its receptors have also been shown to play a significant role in pancreatic carcinogenesis and stimulation of pancreatic cancer growth. This review will summarize published information on the role of CCK and its receptors both in animal models and in humans in relation to the development and perpetuation of pancreatic adenocarcinoma.
Normal Physiological Function of CCK in Animal Models (Secretion and Growth)
CCK acts physiologically on CCK receptors to regulate secretion of digestive enzymes (88) and growth of the pancreas (18, 58). Long-chain fatty acids and aromatic amino acids in the duodenum are responsible for stimulation of CCK release from I cells (64) via GPR40 (51) and calcium-sensing receptors (52), respectively. Recent reviews describing vagal responses and the brain-gut pathway involved in regulation of CCK release have been published (11, 19).
In isolated dispersed rodent pancreatic acinar cells, CCK has a direct effect resulting in increased cytosolic calcium (41) and release of amylase (73) (Fig. 1). Since these acinar cell models are denervated and do not have vagal influence, it was thought that the release of digestive enzymes from pancreatic acinar cells was a direct action of CCK on this target organ. Furthermore, in the dispersed acinar cells, atropine did not block the effects of CCK, suggesting that the mechanism of action of CCK was indeed a direct effect and cholinergically independent (107). However, when researchers studied pancreatic secretion in intact animal models, the actions of CCK on pancreatic enzyme release were blocked by atropine and hexamethonium, cholinergic and ganglionic blockers, respectively (66). These important findings led to the concept that CCK-induced pancreatic enzyme secretion is mediated by a cholinergic pathways in vivo (67).
Fig. 1.
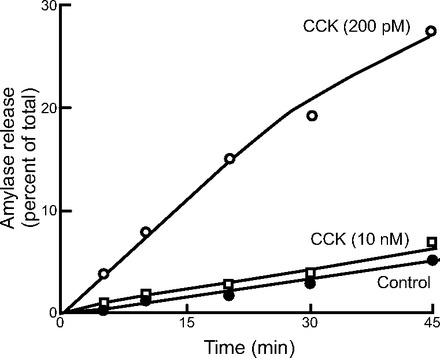
CCK-stimulated amylase release from isolated mouse pancreatic acini. Isolated acini were incubated with CCK at low (200 pM) and high (10 nM) concentrations compared with buffer. At physiological doses, high levels of amylase were released. However, at high concentrations, the amylase release was significantly dampened. From Ref. 73.
Two major physiological forms of CCK exist: CCK8 and CCK58. Whereas both forms exhibit similar actions on calcium signaling and enzyme secretion in the mouse acinar cells (16), only CCK58 induces fluid secretion. It is postulated that, because of its actions on fluid secretion, even at high doses CCK58 does not induce pancreatitis, a known phenomenon of exogenous CCK8 administration. Both peptides are involved in satiety by altering the intermeal interval and animal behavior (29), i.e., CCK8 reduces the time between meals. With careful laboratory techniques to prevent peptide degradation, CCK58 was discovered to be the major form of CCK in canine blood (23) and in human intestine (24).
In addition to their important roles in regulation of digestive enzyme and acid secretion, CCK (58, 90) and gastrin (44) both exhibit trophic effects on the gastrointestinal tract in rodents. CCK has been shown to play an important role in regulating pancreatic growth in both adult (89, 90, 108) and neonatal animals (108). DNA content and 14C-thymidine uptake were increased in the rat pancreas after administration of exogenous CCK (58). Radioactive thymidine uptake localized to pancreatic acinar cells and another “unidentified” cell type (90) after exogenous administration of CCK, indicative of hyperplasia in these cells. In addition to its trophic effect in the adult and neonatal pancreas, CCK has also been shown to play an important role in pancreatic regeneration (22) after pancreatitis or surgical resection (50). The factors involved in pancreatic acinar cell dedifferentiation and proliferation were examined in cultured mouse pancreatic acinar cells, where dedifferentiation was determined to be independent of CCK but proliferation of acinar cells was CCK dependent (33). In this cell culture model, Guo and colleagues (33) found that CCK stimulation increased c-Jun NH2-terminal kinase (JNKs), ERK, and AP-1 activity, which induced cell proliferation. These investigations suggest that cholinergic mechanisms mediate the effects of CCK on pancreatic enzyme secretion in the intact animal; however, the proliferative actions of CCK appear to be a direct effect on pancreatic acinar cells.
Just when we think we have an understanding of the proliferative effects of CCK on the normal pancreas, we are given information from genetically engineered mice lacking CCK. These animals surprisingly still have normal digestion and pancreatic weight (48, 53). Furthermore, in this CCK peptide-deficient transgenic mouse model, the CCK-A receptor density was similar to that of the wild-type mice, suggesting either that circulating CCK concentration does not alter receptor number or that enough gastrin was present to stimulate the receptor. However, the idea that gastrin was responsible for maintaining pancreatic growth in the CCK knockout mouse was disproved by Chen and colleagues (14), who showed that pancreatic weight was normal in a double CCK- and gastrin-knockout mouse. These genetically engineered models, although not physiological, again raise the question of whether CCK has a direct trophic action on the normal pancreas.
Cholecystokinin Receptors
Cholecystokinin receptors belong to the family of G protein-coupled rhodopsin receptors (GPCRs) (101). Two CCK receptor types have been cloned and sequenced: the CCK-A (or CCK-1) receptor (102) and the CCK-B (or CCK-2) receptor (46, 103). The CCK receptors are classic seven-transmembrane-spanning receptors that possess ∼48% homology. Physiologically, the CCK receptors are differentiated by their binding affinities to CCK or gastrin, wherein CCK has 1,000-fold great affinity for the CCK-A receptor compared with gastrin and both CCK and gastrin have equal affinity for the CCK-B receptor (27, 42). Selective CCK receptor antagonists have also been developed with high selectivity for each CCK receptor type; these compounds facilitate functional studies to discern receptor type in tissues or cells (4).
Genetically engineered mice that are deficient in CCK-A receptors have impaired pancreatic enzyme secretion in response to exogenous CCK but still are capable of insulin production from the islets (95). However, in these same CCK-A receptor-deficient mice, there is impaired pancreatic growth and decreased pancreatic DNA content compared with wild-type mice (95). These studies suggest that the CCK peptide-CCK receptor axis must be intact for CCK to exert its direct proliferative effects on pancreatic acinar cells. CCK-A and -B receptors have also been identified on pancreatic stellate cells (68), and stimulation of these receptors promotes collagen production and fibrosis (5, 68), common features in both chronic pancreatitis and pancreatic cancer. Phillips and coworkers (68) demonstrated that pancreatic stellate cells can secrete acetylcholine, which in turn induces secretion from pancreatic acinar cells. Investigators are discovering that there is cross talk among various cell types of the pancreas including stellate, islet, acinar, and duct cells. Intricate control of normal physiological functions of an organ such as growth and secretion are thought to be modulated by the microenvironment created by these interactions.
The study of CCK receptors in normal human pancreas tissue has been problematic and controversial. Despite these obstacles, the general consensus has been that whereas CCK-A receptors are the predominant variety in rodent pancreas (6, 102), CCK-B receptors are the predominant receptor type in human pancreas (35, 80, 105).
Many studies to determine the presence of CCK receptors in the normal human pancreas have examined receptor mRNA. By analyzing RNA, Wank and colleagues (104) demonstrated the presence of the CCK-B but not the CCK-A receptor in normal human tissues by Northern analysis. Using RT-PCR and in situ hybridization, Weinburg and colleagues (105) also demonstrated CCK-B but not CCK-A receptor mRNA in normal human pancreas. By in situ hybridization, Reubi et al. (71) reported that CCK-A receptors could be localized to the nerves in the normal human pancreas whereas CCK-B receptors localized predominantly to the islets and acinar cells. Galindo et al. (26) showed that human pancreatic tissue extracts expressed some, albeit minimal, CCK-A mRNA receptor activity by RT-PCR and much greater expression of the CCK-B receptor type. However, the “normal” human pancreas tissues examined in Galindo's experiments and in some others were obtained from specimens adjacent to pancreatic cancer and therefore these tissues were only macroscopically deemed normal. Although these experiments suggest that the predominant CCK receptor type in normal human pancreas tissue is the CCK-B receptor from the detection of RNA, none of these experiments demonstrated a physiological and functional receptor.
Extending the studies beyond mRNA, Smith et al. (76) showed that the normal human pancreas exhibited CCK-B receptor protein with a binding affinity in the physiological range for the ligand (nM) by radioactive pharmacokinetic receptor binding assays. Although this was the first evidence of receptor protein binding for CCK receptors in the normal human pancreas, the exact cell type bearing the receptors could not be discerned since the binding assays were performed with whole tissue/cell homogenates. To ascertain specific cell types with CCK receptors, Tang et al. (98) examined human pancreas CCK receptor protein by autoradiography with receptor type being specified by competition with selective antagonists. This group compared the binding of CCK in human pancreas specimens to that of human gallbladder smooth muscle and determined the predominant receptor type in the human pancreas to be that of the CCK-B receptor type. Although Ji and coworkers (43) confirmed the presence of CCK-B receptors both in human pancreatic extracts and in isolated human pancreatic acinar preps by RT-PCR techniques, they concluded that these receptors were “nonfunctional” since treatment with CCK failed to stimulate amylase secretion and, thus, that CCK only stimulated pancreatic acinar cells via vagal efferents. These human experiments are further supporting evidence that pancreatic secretory pathways are mediated by cholinergic mechanisms rather than the direct action of CCK on acinar cells. Others have suggested that a possible explanation for Ji's nonfunctional CCK receptors on human acinar cells may be attributed to the tissue transport time from the operating room to the laboratory, resulting in receptor protein degradation. Evidence supporting this speculation of possible receptor degradation includes the fact that supraphysiological doses of carbachol (1 mmol/l) were necessary to elicit a calcium response from the isolated human acinar cells (43). In a study by Murphy and colleagues (65), human pancreatic tissue was suspended immediately in a buffered solution containing protease inhibitors and transported to the laboratory for processing in less than 10 min. With this careful technique, appropriate buffer solution, and rapid tissue handling, these researchers measured calcium fluxes and demonstrated functional CCK receptors in isolated human pancreatic cells after physiological doses of acetylcholine (50 nM) and CCK (2 pM). Atropine successfully blocked the calcium response induced by acetylcholine but not by CCK. The authors also suggest that another possible reason they were able to successfully elicit a response from human acinar cells to CCK was because tissues from any subjects with pancreatic duct obstruction, which may also diminish calcium responses to exogenous stimuli (63), were not used. These investigations suggest that, with appropriate selection and handling of the tissues, functional CCK receptors are present on normal human pancreatic acinar cells.
CCK receptors have also been reported in human pancreatic cancers (80, 81, 87, 105), and the number of receptors in the cancer tissues was markedly increased over normal human pancreas (76). The CCK-B receptor appears to be the predominant receptor type in pancreatic cancer; however, some pancreatic cancers reportedly also have the CCK-A receptor (61, 105). Since CCK-A receptors are not expressed in normal human pancreas tissue, Weinberg et al. (105) proposed that the presence of the CCK-A receptor could be a useful biomarker for the detection of pancreatic cancer. In contrast, despite finding CCK receptor mRNA in human pancreatic cancer (26, 61, 105), others have failed to detect CCK receptors (71), possibly because of the method of tissue handling, experimental conditions, and the sensitivity of the receptors to proteolytic degradation, oxidation, and RNA digestion. With the use of appropriate buffer conditions containing protease inhibitors and controlled temperature, detection of CCK receptors considerably improves (Table 1) (81).
Table 1.
Effects of selective agents and cations on binding of 125I-CCK-8 to PANC-1 human pancreatic cancer cell homogenates
| Tris·HCl (50 mM) | Bacitracin (0.1 mM) | PMSF (1.2 mM) | Leupeptin (10 μg/ml) | BSA (0.2%) | BSA (0.5%) | Leupeptin (5 μg/ml) | EGTA (1 mM) | NaCl (118 mM) | KCl (100 mM) | MgCl2·6H2O (5 mM) | Specific Binding/mg Protein |
|---|---|---|---|---|---|---|---|---|---|---|---|
| + | + | + | + | + | 100 | ||||||
| + | + | + | + | + | 70 ± 10 | ||||||
| + | + | + | + | + | 70 ± 9 | ||||||
| + | + | + | + | + | + | 59 ± 17 | |||||
| + | + | + | + | 48 ± 8 | |||||||
| + | + | + | + | + | + | 23.5 ± 0.3 | |||||
| + | + | + | + | 21 ± 2.5 | |||||||
| + | + | + | + | + | + | 16.5 ± 2.0 | |||||
| + | + | + | + | + | + | 16.0 ± 8.4 |
Values are means ± SE from 3 experiments performed in triplicate. Binding of 25 pM 125I-CCK-8 to PANC-1 cellular homogenates was assayed in buffer containing various additives at 4°C, pH 7.4. Addition of cations or EGTA markedly decreased specific binding and the absence of leupeptin and bovine serum albumin (BSA) markedly decreased binding. Compounds present in buffer are designated by a +. PMSF, phenylmethylsulfonyl fluoride. From Ref. 81.
Further supportive evidence for the presence of CCK receptors on human pancreatic cancer cells comes from growth experiments with ligands and CCK receptor antagonists in vitro and in vivo. Exogenous administration of either gastrin (75) or CCK (79, 83) to cultured human pancreatic cancer cells or xenografted pancreatic tumors in nude mice was found to significantly stimulate cancer growth. One human pancreatic cancer cell line examined in these experiments, PANC-1, has been shown to possess both CCK-A and CCK-B receptors (61, 81). When PANC-1 cells were treated with exogenous CCK (81) or gastrin (75) in the presence of either a selective CCK-A antagonist (L-364,718) (13) or a selective CCK-B receptor antagonist (L-365,260) (12), only the CCK-B receptor antagonist inhibited growth (Fig. 2) (75). These data indicate that the CCK-B receptor is linked to trophic actions on pancreatic cells and the CCK-A receptor may be linked to secretion. Since pancreatic cancer cells do not secrete digestive enzymes, the purpose of the CCK-A receptors on these cells is unknown. In PANC-1 human pancreatic cancer cells, downregulation of the CCK-B receptor with RNA interference while leaving the CCK-A receptor intact inhibits proliferation and induces apoptosis and cell cycle arrest (Fig. 3) (25). These data suggest that the predominant receptor type in human pancreatic cancer is the CCK-B variety and that this receptor, rather than the CCK-A receptor, is the one associated with proliferative effects. Further evidence to support a role of the CCK receptor in growth of human pancreatic cancer comes from the results of a clinical trial in which immunization to gastrin improved survival in subjects that seroconverted (8). CCK-B receptors have also been identified in other gastrointestinal adenocarcinomas (2) including esophageal cancer (62) and stomach cancer (40).
Fig. 2.
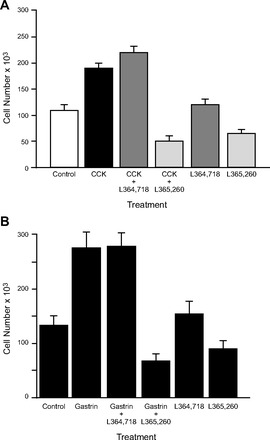
The proliferative actions of CCK and gastrin on PANC-1 pancreatic cancer cell are mediated through the CCK-B receptor, not the CCK-A receptor. A: growth of PANC-1 cells, which have both CCK-A and CCK-B receptors, was tested in the presence of saline (control), CCK (10−9 M) alone, or CCK (10−9 M) in the presence of the CCK-A receptor antagonist L-364,718 (10−9 M) or the CCK-B/gastrin receptor antagonist L-365,260 (10−9 M), and each antagonist alone. CCK stimulated cell growth that was only blocked by the CCK-B receptor antagonist, not the CCK-A antagonist. From Ref. 81. B: gastrin exerts a similar but even greater proliferative effect on growth of PANC-1 cells via the CCK-B receptor. Gastrin (10−9 M) significantly increases cell number compared with control cells. The proliferative effects of gastrin are not altered in the presence of the CCK-A receptor antagonist L-364,718 (10−9 M) but are blocked by the CCK-B/gastrin receptor antagonist L-365,260 (10−9 M), at concentrations of antagonist that do not effect growth alone. Studies were performed in PANC-1 cells in serum-free medium over 6 days (P < 0.005). From Ref. 75.
Fig. 3.
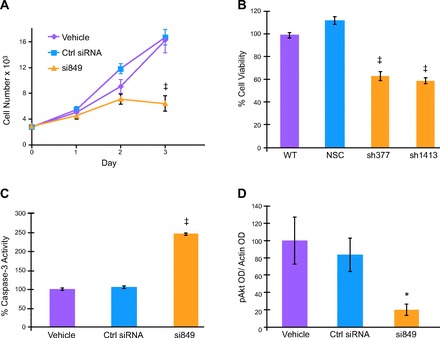
Downregulation of the CCK-B receptor in human PANC-1 pancreatic cancer cells by siRNA (A) or stable shRNA clones (B) significantly decreases cell growth. Apoptosis is also increased as evidenced by increased caspase-3 levels (C). And phosphorylated Akt as quantitated by densitometry from Western analysis of PANC-1 extracts treated with CCK-B receptor siRNA (D) is significantly diminished compared with controls. *P < 0.05, ‡P < 0.001. From Ref. 25.
A third CCK receptor, called the CCK-C receptor for its association only with cancer, has also been identified as a splice variant of the CCK-B receptor (85, 86). This receptor variation is the result of a single nucleotide polymorphism (SNP) in position 32 of the fourth intron causing missplicing of the fourth intron resulting in translation of 69 additional amino acids (77). By transfecting this spliced variant receptor into Balb3T3 cells (37) or into non-CCK-C expressing pancreatic cancer AsPC-1 cells (77), increased growth rate compared with vector transfected cells. Humans subjects with pancreatic cancer that express the A-allele SNP of the CCK-B receptor exhibit a more aggressive phenotype and shortened survival (77).
Intracellular Signaling
After ligand binding to the CCK-A or CCK-B receptors, GTP-coupled responses stimulate activation of either adenylate cyclase or PLC, respectively (20). Depending on the cell type, various intracellular signaling pathways are initiated after CCK binding that result in enzyme/acid secretion, cellular proliferation and antiapoptosis, and cell migration (20, 32). Intracellular signaling pathways activated involve the hydrolysis of phosphatidylinositol bisphosphate by PLC to generate inositol trisphosphate and diacylglycerol, which subsequently induce calcium mobilization and activation of PKC (Fig. 4) (21). Several of these pathways involve activation or cross talk with tyrosine kinase receptors (EGF) and proliferative pathways associated with cell growth [mammalian target of rapamycin (mTOR), Akt, ERK, etc.]. Mitogen-activated protein kinases (MAPKs) including ERK and JNK may be regulated by GPCRs including CCK receptors (21, 91). The epidermal growth factor receptor (EGFR) is also regulated by gastrin through the CCK-B receptor mediated by the Src family kinases (34), and oncogenic Kras in pancreatic cancer cells has also been shown to upregulate EGFR expression (1). In studies on human cancer cells, mitogenic cellular kinases such as phosphorylation of Akt are significantly decreased when the CCK-B receptor is downregulated by RNA interference techniques (Fig. 3D) (25). These studies and others show the importance of intracellular mechanisms involved in cell signaling and activation of mitogenic pathways after activation of CCK receptors. These studies are also supportive of an important role of CCK and gastrin in the proliferative mechanisms of pancreatic cancer.
Fig. 4.
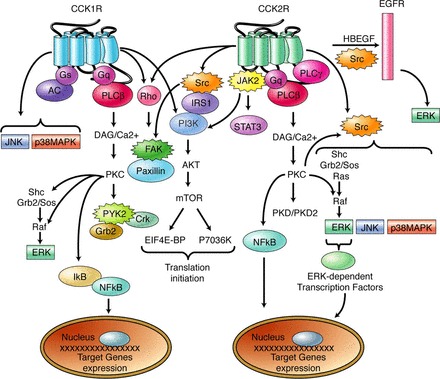
Schematic description of the signaling pathways known to be activated by CCK1R and CCK2R. In addition to signaling pathways classically activated by G protein-coupled receptors such as the PLC-β/diacylglycerol (DAG)/Ca2+/PKC cascade or the adenyl cyclase (AC) pathway, CCK receptors also induce several other signaling pathways known to be activated by tyrosine kinase receptors: 1) the MAPK pathways including ERK, JNK, and p38-MAPK; 2) the phosphatidylinositol 3-kinase (PI3K) pathway; or 3) the PLC-β pathway. Activation of nonreceptor tyrosine kinases including Src, JAK2, FAK, and PYK2 is also an early event in CCK receptor signaling. These tyrosine kinases are involved in particular upstream of the MAP kinase or PI3K pathways and in intracellular events related to cell adhesion (see text for details). Finally, CCK2R are also known to induce epidermal growth factor (EGF) receptor transactivation. Arrows correspond to positive stimulations. From Ref. 21.
Cocarcinogenic Potential of Cholecystokinin in Models of Pancreatic Cancer
Several animal models have been used to study pancreatic cancer (93), including N-nitrosobis(2-oxopropyl)amine (BOP)-induced ductal cancers (69) in Syrian golden hamsters, an azaserine-induced acinar cell carcinoma (54) model in rats, and N-delta-(N-methyl-N-nitrosocarbamoyl)-L-ornithine (MNCO)-induced pancreatic acinar cell carcinomas in rats (56) and ductal carcinomas in hamsters (55). In these models, CCK-B is the receptor type associated with the cancer cells, even in the azaserine-induced cancers like cell line AR4–2J (70). Immunodeficient animal models such as athymic nude or SCID mice have also been used to study human pancreatic cancer growth by employing either subcutaneous xenografting of cancer (75, 83) or orthotopic transplantation (60, 72). Exogenous administration of CCK or a CCK analog caerulein has been used for decades as a classic model for experimental pancreatitis (30, 45). In these experimental models, CCK administration has been shown to accelerate carcinogenesis. Using the same dose of CCK that induced hypertrophy and hyperplasia of the pancreas, Howatson and Carter (38) showed that the latency period to develop pancreatic cancer was shortened and the incidence of pancreatic cancer was significantly increased in the BOP-carcinogen cancer hamster model (Fig. 5). Additionally, exogenous treatment with CCK has been shown to accelerate precursor PanIN (pancreatic intraepithelial neoplasia) lesion progression to cancer in the Kras transgenic mouse model (10). Although the KrasG12D-engineered model of pancreatic cancer develops PanIN lesions and may develop pancreatic cancer (100) slowly over time, it has been suggested that a second “hit” is needed for progression to cancer in these mice (39). In a study by Carriere and coworkers (10), it was suggested that inflammation induced by CCK was the factor that accelerated the production of premalignant and malignant transformation in the Kras mice. Our laboratory has recently identified CCK receptors in early PanIN lesions in mice with Kras mutations and also in humans (unpublished observation). Treatment with CCK receptor antagonists completely halts the progression of PanIN lesions, confirming the important role of CCK in early carcinogenesis and a potential pathway for therapeutic intervention for high-risk populations.
Fig. 5.
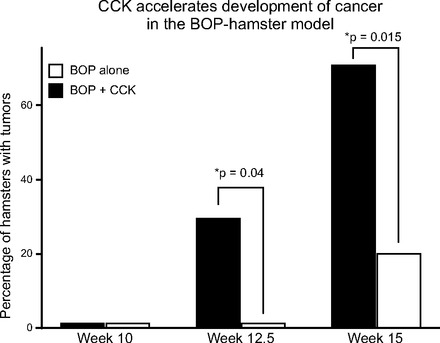
Administration of CCK to hamsters in conjunction with N-nitrosobis (2-oxopropyl)amine (BOP; 5 mg/kg weekly) reduces tumor latency period and increases in induction rate of tumor development compared with BOP alone (*P < 0.05). Data from Ref. 38.
Role of Endogenous CCK in Potentiating Pancreatic Cancer Development
Epidemiological studies have reported that the incidence of pancreatic cancer is increased in countries that consume high-fat diets (28, 36, 57). In a large prospective study, an association was found between dietary intake of fat and pancreatic cancer (99). Many researchers have focused on insulin, leptin, adiposity, and glucose as the most critical factors contributing to an increased risk of pancreatic cancer in obese individuals. Most theories connect obesity and pancreatic cancer risk through one of the following mechanisms: 1) increased insulin and IGF stimulate proliferation via signaling through IGF receptors; or 2) reactive oxygen species and oxidative stress directly damage DNA or indirectly lead to chronic inflammation. Dawson et al. (17) showed that, in the KrasG12D mouse model, PanIN lesion progression was accelerated with a diet high in polyunsaturated fats. Their findings were attributed to many factors such as hyperinsulinemia, hyperglycemia, pancreatic inflammation, elevated IGF1 levels, and elevated leptin levels; however, CCK plasma levels were not measured. Lavine et al. (49) recently showed that in the pancreas of 4- and 14-wk-old obese mice (C57BL/6-Leptinob/ob mice), CCK mRNA is upregulated 500-fold compared with lean mice and expressed in both the α and β cells of the pancreatic islets. Strikingly, CCK was the most upregulated gene in this model. Furthermore, CCK peptide was undetectable in the pancreas of lean mice, whereas obese mice had high levels of the bioactive form of CCK peptide. Endogenous overproduction of CCK in animals receiving a diet high in fat has been shown to accelerate pancreatic cancer growth and the effect is blocked by concomitant treatment with a CCK receptor antagonist (78). A proposed schema regarding how a diet high in fat can induce pancreatic cancer and fibrosis is shown (Fig. 6).
Fig. 6.
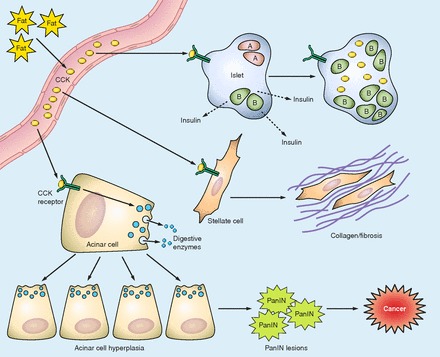
Proposed pathways by which chronic dietary fat increases pancreatic cancer risk through a CCK-mediated mechanism. Chronic consumption of high fat has been shown to increase blood CCK levels. In turn, CCK can act on the CCK receptors on islet, acinar, and stellate cells. On the acinar cells, CCK induces the release of digestive enzymes that may induce a smoldering pancreatitis. CCK also induces pancreatic acinar cell hyperplasia and increases DNA synthesis after cell signaling and induction of mitogenic cell processes involving activation of epidermal growth factor receptor (EGFR), ERK, mammalian target of rapamycin (mTOR), and Akt. The chronic inflammatory state may trigger oncogenic Kras and lead to the transformation of cells and development of PanIN (pancreatic intraepithelial neoplasia) lesions. Over time and with the reactivation of endogenous gastrin, a cancer cell forms. CCK also interacts with receptors on pancreatic stellate cells to release ACh and induce more pancreatic enzyme release with inflammation. Stellate cells also respond with the production of collagen and fibrosis. In the presence of high dietary fat and obesity, CCK interacts with the islet cells and has a role in the release of insulin. Over time CCK synthesis occurs within the islets and beta cell mass increases.
The Chicken or the Egg
In the pancreas of the fetal rat or mouse, gastrin mRNA and peptide are detected but levels rapidly decrease to zero within 20 days after birth, and gastrin expression is then only detected in the gastric antrum (3, 7, 92, 92). A similar pattern is seen in the developing human pancreas where low levels of gastrin RNA may be present in the fetus (92, 96), but there is no gastrin peptide by radioimmunoassay in the adult pancreas (76). Using genetic mouse models, Suissa et al. (92) showed that in the developing pancreas, gastrin-positive cells were derived from Ngn3+ endocrine progenitor cells and expressed transcription factors Nkx2.2 and Nkx6.1 but low levels of Pdx1. Similarly, CCK mRNA has also been detected in the human fetal pancreas where it appears to be localized to the islet cells (97), but this peptide is also not found in the adult pancreas. As described above, Lavine et al. (49) reported that CCK mRNA in the pancreas of obese mice (C57BL/6-Leptinob/ob mice) was upregulated 500-fold compared with lean mice, and CCK was the most upregulated gene in the obese mice pancreas. CCK peptide levels were also increased in the islets of these obese mice and were associated with an expanded beta cell mass in the islets. Takaishi and colleagues (94) used a mouse reporter model with green fluorescent protein (GFP) cloned into a bacterial artificial chromosome containing the entire gastrin gene to study gastrin gene expression in developing tissues. In this model, GFP-positive cells were detected in the fetal pancreas islet cells but again not in the adult pancreas.
In contrast to the absence of CCK and gastrin in the normal human pancreas, our laboratory (75, 76, 82) and others (9) have reported gastrin immunoreactivity in tissues from patients with pancreatic adenocarcinoma and in pancreatic cancer cell lines. The physiological form of gastrin produced and released into the growth medium from pancreatic cancer cells was determined to be gastrin-17 (82). The role of gastrin expression or reexpression in the cancer cells is related to proliferation since growth is impaired when gastrin is downregulated in pancreatic cancer cells in vitro (31, 84). When stable gastrin shRNA knockdown human pancreas cancer clones are grown orthotopically in athymic nude mice, both primary tumor growth and metastases were inhibited, demonstrating the importance of gastrin in regulating growth and spread of pancreas cancer (60). Additionally, the growth rate of human pancreatic tumors in nude mice is directly proportional to the level of gastrin mRNA expression (60). Although CCK peptide has also been associated with some human cancers, the rate of pancreatic tumor growth is unaltered by downregulation of CCK (61), indicating that gastrin peptide reexpression in cancer is the peptide involved in autocrine regulation of growth rather than endogenous CCK. The autocrine mechanism of gastrin fueling its own fire is substantiated by the finding that endogenous gastrin from cancer cells has been shown to induce its own transcription by activating the CCK-B receptor (47). Thus pancreatic cells that produce gastrin and CCK peptides embryologically become “silenced” in the normal adult pancreas until something changes to reactivate expression and transform the cells into cancer. It is also uncertain whether these reactivated cells are stem cells, normal pancreatic acinar cells, or progenitor cells that have been in senescence or suppressed.
There has been a long-time concern about the potential risk of gastric acid proton pump inhibitors raising gastrin levels and inducing cancer; however, numerous studies have demonstrated that subjects with high gastrin levels for other reasons (pernicious anemia, Zollinger-Ellison syndrome) are not at increased risk to develop gastrointestinal cancers. And although exogenous administration of CCK or gastrin promotes pancreatic cancer in animal models, there usually is a concomitant chemical given such as a carcinogen or alcohol, or a coexisting state such as inflammation, pancreatitis, obesity, high dietary fat, or other predisposing genetic risk factors (i.e., mutated Kras). Understanding the underlying triggers that cause reactivation of CCK and gastrin reexpression from their postnatal silenced state to active state in cancer may unlock the door to many questions. Perhaps after an injury like pancreatitis, CCK activation is a normal physiological event in an attempt to restore and repair damaged acini, but with repetitive attacks of relapsing or chronic pancreatitis CCK levels remain elevated and with unopposed stimulation induce transformation. Indeed patients with chronic pancreatitis have been found to have elevated CCK blood levels, and treatment of these patients with a CCK receptor antagonist reduces pain (74). It is unknown, however, whether high-risk patients such as those with hereditary pancreatitis (106) would benefit from treatment with a CCK receptor antagonist or whether this therapy could lower the risk of malignant transformation in this population.
Research demonstrating the presence of CCK receptors on pancreatic stellate cells (5) and the reversal of fibrosis in the Kras engineered mouse by using CCK receptor antagonists (59) suggests that CCK has a role in the formation of the desmoplastic microenvironment surrounding pancreatic cancer. Whether elevated CCK blood levels associated with high dietary fat or chronic pancreatic inflammation act on normal CCK receptors to induce proliferation and activation of a transformed cell leading to cancer is unknown. However, consideration for clinical trials using CCK receptor antagonists as an adjuvant to chemotherapy or a preventative therapy in subjects with risk factors such as chronic or hereditary pancreatitis should be considered. And whether CCK's role in pancreatic cancer is the chicken or the egg, the best way to serve it is “fried.”
DISCLOSURES
J. P. Smith has a pending patent application on a monoclonal antibody to the CCK-C receptor.
AUTHOR CONTRIBUTIONS
J.P.S. conception and design of research; J.P.S. prepared figures; J.P.S. drafted manuscript; J.P.S. and T.E.S. edited and revised manuscript; J.P.S. and T.E.S. approved final version of manuscript.
REFERENCES
- 1.Ardito CM, Gruner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK, Delgiorno KE, Carpenter ES, Halbrook CJ, Hall JC, Pal D, Briel T, Herner A, Trajkovic-Arsic M, Sipos B, Liou GY, Storz P, Murray NR, Threadgill DW, Sibilia M, Washington MK, Wilson CL, Schmid RM, Raines EW, Crawford HC, Siveke JT. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 22: 304–317, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baldwin GS, Shulkes A. CCK receptors and cancer. Curr Top Med Chem 7: 1232–1238, 2007 [DOI] [PubMed] [Google Scholar]
- 3.Bardram L, Hilsted L, Rehfeld JF. Progastrin expression in mammalian pancreas. Proc Natl Acad Sci USA 87: 298–302, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berna MJ, Jensen RT. Role of CCK/gastrin receptors in gastrointestinal/metabolic diseases and results of human studies using gastrin/CCK receptor agonists/antagonists in these diseases. Curr Top Med Chem 7: 1211–1231, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berna MJ, Seiz O, Nast JF, Benten D, Blaker M, Koch J, Lohse AW, Pace A. CCK1 and CCK2 receptors are expressed on pancreatic stellate cells and induce collagen production. J Biol Chem 285: 38905–38914, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bourassa J, Laine J, Kruse ML, Gagnon MC, Calvo E, Morisset J. Ontogeny and species differences in the pancreatic expression and localization of the CCK(A) receptors. Biochem Biophys Res Commun 260: 820–828, 1999 [DOI] [PubMed] [Google Scholar]
- 7.Brand SJ, Fuller PJ. Differential gastrin gene expression in rat gastrointestinal tract and pancreas during neonatal development. J Biol Chem 263: 5341–5347, 1988 [PubMed] [Google Scholar]
- 8.Brett BT, Smith SC, Bouvier CV, Michaeli D, Hochhauser D, Davidson BR, Kurzawinski TR, Watkinson AF, Van Someren N, Pounder RE, Caplin ME. Phase II study of anti-gastrin-17 antibodies, raised to G17DT, in advanced pancreatic cancer. J Clin Oncol 20: 4225–4231, 2002 [DOI] [PubMed] [Google Scholar]
- 9.Caplin M, Savage K, Khan K, Brett B, Rode J, Varro A, Dhillon A. Expression and processing of gastrin in pancreatic adenocarcinoma. Br J Surg 87: 1035–1040, 2000 [DOI] [PubMed] [Google Scholar]
- 10.Carriere C, Young AL, Gunn JR, Longnecker DS, Korc M. Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem Biophys Res Commun 382: 561–565, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chandra R, Liddle RA. Recent advances in pancreatic endocrine and exocrine secretion. Curr Opin Gastroenterol 27: 439–443, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang RS, Chen TB, Bock MG, Freidinger RM, Chen R, Rosegay A, Lotti VJ. Characterization of the binding of [3H]L-365,260: a new potent and selective brain cholecystokinin (CCK-B) and gastrin receptor antagonist radioligand. Mol Pharmacol 35: 803–808, 1989 [PubMed] [Google Scholar]
- 13.Chang RS, Lotti VJ, Chen TB, Kunkel KA. Characterization of the binding of [3H]-(±)-L-364,718: a new potent, nonpeptide cholecystokinin antagonist radioligand selective for peripheral receptors. Mol Pharmacol 30: 212–217, 1986 [PubMed] [Google Scholar]
- 14.Chen D, Zhao CM, Hakanson R, Samuelson LC, Rehfeld JF, Friis-Hansen L. Altered control of gastric acid secretion in gastrin-cholecystokinin double mutant mice. Gastroenterology 126: 476–487, 2004 [DOI] [PubMed] [Google Scholar]
- 15.Chu S, Schubert ML. Gastric secretion. Curr Opin Gastroenterol 28: 587–593, 2012 [DOI] [PubMed] [Google Scholar]
- 16.Criddle DN, Booth DM, Mukherjee R, McLaughlin E, Green GM, Sutton R, Petersen OH, Reeve JR., Jr Cholecystokinin-58 and cholecystokinin-8 exhibit similar actions on calcium signaling, zymogen secretion, and cell fate in murine pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol 297: G1085–G1092, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dawson DW, Hertzer K, Moro A, Donald G, Chang HH, Go VL, Pandol SJ, Lugea A, Gukovskaya AS, Li G, Hines OJ, Rozengurt E, Eibl G. High-fat, high-calorie diet promotes early pancreatic neoplasia in the conditional KrasG12D mouse model. Cancer Prev Res (Phila) 6: 1064–1073, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dembinski AB, Johnson LR. Stimulation of pancreatic growth by secretin, caerulein, and pentagastrin. Endocrinology 106: 323–328, 1980 [DOI] [PubMed] [Google Scholar]
- 19.Dockray GJ. Cholecystokinin. Curr Opin Endocrinol Diabetes Obes 19: 8–12, 2012 [DOI] [PubMed] [Google Scholar]
- 20.Dockray GJ, Moore A, Varro A, Pritchard DM. Gastrin receptor pharmacology. Curr Gastroenterol Rep 14: 453–459, 2012 [DOI] [PubMed] [Google Scholar]
- 21.Dufresne M, Seva C, Fourmy D. Cholecystokinin and gastrin receptors. Physiol Rev 86: 805–847, 2006 [DOI] [PubMed] [Google Scholar]
- 22.Elsasser HP, Adler G, Kern HF. Time course and cellular source of pancreatic regeneration following acute pancreatitis in the rat. Pancreas 1: 421–429, 1986 [DOI] [PubMed] [Google Scholar]
- 23.Eysselein VE, Eberlein GA, Hesse WH, Singer MV, Goebell H, Reeve JR., Jr Cholecystokinin-58 is the major circulating form of cholecystokinin in canine blood. J Biol Chem 262: 214–217, 1987 [PubMed] [Google Scholar]
- 24.Eysselein VE, Eberlein GA, Schaeffer M, Grandt D, Goebell H, Niebel W, Rosenquist GL, Meyer HE, Reeve JR., Jr Characterization of the major form of cholecystokinin in human intestine: CCK-58. Am J Physiol Gastrointest Liver Physiol 258: G253–G260, 1990 [DOI] [PubMed] [Google Scholar]
- 25.Fino KK, Matters GL, McGovern CO, Gilius EL, Smith JP. Downregulation of the CCK-B receptor in pancreatic cancer cells blocks proliferation and promotes apoptosis. Am J Physiol Gastrointest Liver Physiol 302: G1244–G1252, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Galindo J, Jones N, Powell GL, Hollingsworth SJ, Shankley N. Advanced qRT-PCR technology allows detection of the cholecystokinin 1 receptor (CCK1R) expression in human pancreas. Pancreas 31: 325–331, 2005 [DOI] [PubMed] [Google Scholar]
- 27.Gardner JD, Walker MD, Martinez J, Priestly GP, Natarajan S, Bodanszky M. The importance of the amino acid in position 27 of cholecystokinin in determining its biological activity on pancreatic acini. Biochim Biophys Acta 630: 323–329, 1980 [DOI] [PubMed] [Google Scholar]
- 28.Ghadirian P, Lynch HT, Krewski D. Epidemiology of pancreatic cancer: an overview. Cancer Detect Prev 27: 87–93, 2003 [DOI] [PubMed] [Google Scholar]
- 29.Goebel-Stengel M, Stengel A, Wang L, Ohning G, Tache Y, Reeve JR., Jr CCK-8 and CCK-58 differ in their effects on nocturnal solid meal pattern in undisturbed rats. Am J Physiol Regul Integr Comp Physiol 303: R850–R860, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gorelick FS, Thrower E. The acinar cell and early pancreatitis responses. Clin Gastroenterol Hepatol 7: S10–S14, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grabowska AM, Hughes J, Watson SA. Use of interfering RNA to investigate the role of endogenous gastrin in the survival of gastrointestinal cancer cells. Br J Cancer 96: 464–473, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grabowska AM, Watson SA. Role of gastrin peptides in carcinogenesis. Cancer Lett 257: 1–15, 2007 [DOI] [PubMed] [Google Scholar]
- 33.Guo L, Sans MD, Hou Y, Ernst SA, Williams JA. c-Jun/AP-1 is required for CCK-induced pancreatic acinar cell dedifferentiation and DNA synthesis in vitro. Am J Physiol Gastrointest Liver Physiol 302: G1381–G1396, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo YS, Cheng JZ, Jin GF, Gutkind JS, Hellmich MR, Townsend CM., Jr Gastrin stimulates cyclooxygenase-2 expression in intestinal epithelial cells through multiple signaling pathways. Evidence for involvement of ERK5 kinase and transactivation of the epidermal growth factor receptor. J Biol Chem 277: 48755–48763, 2002 [DOI] [PubMed] [Google Scholar]
- 35.Heald EB, Kramer ST, Smith JP. Trophic effects of unsulfated cholecystokinin on mouse pancreas and human pancreatic cancer. Pancreas 7: 530–535, 1992 [DOI] [PubMed] [Google Scholar]
- 36.Heinen MM, Verhage BA, Goldbohm RA, van den Brandt PA. Meat and fat intake and pancreatic cancer risk in the Netherlands Cohort Study. Int J Cancer 125: 1118–1126, 2009 [DOI] [PubMed] [Google Scholar]
- 37.Hellmich MR, Rui XL, Hellmich HL, Fleming RY, Evers BM, Townsend CM., Jr Human colorectal cancers express a constitutively active cholecystokinin-B/gastrin receptor that stimulates cell growth. J Biol Chem 275: 32122–32128, 2000 [DOI] [PubMed] [Google Scholar]
- 38.Howatson AG, Carter DC. Pancreatic carcinogenesis-enhancement by cholecystokinin in the hamster-nitrosamine model. Br J Cancer 51: 107–114, 1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang H, Daniluk J, Liu Y, Chu J, Li Z, Ji B, Logsdon CD. Oncogenic K-Ras requires activation for enhanced activity. Oncogene 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hur K, Kwak MK, Lee HJ, Park DJ, Lee HK, Lee HS, Kim WH, Michaeli D, Yang HK. Expression of gastrin and its receptor in human gastric cancer tissues. J Cancer Res Clin Oncol 132: 85–91, 2006 [DOI] [PubMed] [Google Scholar]
- 41.Jensen RT, Wank SA, Rowley WH, Sato S, Gardner JD. Interaction of CCK with pancreatic acinar cells. Trends Pharmacol Sci 10: 418–423, 1989 [DOI] [PubMed] [Google Scholar]
- 42.Jensen SL, Holst JJ, Nielsen OV, Rehfeld JF. Effect of sulfation of CCK-8 on its stimulation of the endocrine and exocrine secretion from the isolated perfused porcine pancreas. Digestion 22: 305–309, 1981 [DOI] [PubMed] [Google Scholar]
- 43.Ji B, Bi Y, Simeone D, Mortensen RM, Logsdon CD. Human pancreatic acinar cells lack functional responses to cholecystokinin and gastrin. Gastroenterology 121: 1380–1390, 2001 [DOI] [PubMed] [Google Scholar]
- 44.Johnson LR, McCormack SA. Regulation of gastrointestinal mucosal growth. In: Physiology of the Gastrointestinal Tract, edited by Johnson LR. New York: Raven, 1994, p. 611–642 [Google Scholar]
- 45.Kim H. Cerulein pancreatitis: oxidative stress, inflammation, and apoptosis. Gut Liver 2: 74–80, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kopin AS, Lee YM, McBride EW, Miller LJ, Lu M, Lin HY, Kolakowski LF, Jr, Beinborn M. Expression cloning and characterization of the canine parietal cell gastrin receptor. Proc Natl Acad Sci USA 89: 3605–3609, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kovac S, Xiao L, Shulkes A, Patel O, Baldwin GS. Gastrin increases its own synthesis in gastrointestinal cancer cells via the CCK2 receptor. FEBS Lett 584: 4413–4418, 2010 [DOI] [PubMed] [Google Scholar]
- 48.Lacourse KA, Swanberg LJ, Gillespie PJ, Rehfeld JF, Saunders TL, Samuelson LC. Pancreatic function in CCK-deficient mice: adaptation to dietary protein does not require CCK. Am J Physiol Gastrointest Liver Physiol 276: G1302–G1309, 1999 [DOI] [PubMed] [Google Scholar]
- 49.Lavine JA, Raess PW, Stapleton DS, Rabaglia ME, Suhonen JI, Schueler KL, Koltes JE, Dawson JA, Yandell BS, Samuelson LC, Beinfeld MC, Davis DB, Hellerstein MK, Keller MP, Attie AD. Cholecystokinin is up-regulated in obese mouse islets and expands beta-cell mass by increasing beta-cell survival. Endocrinology 151: 3577–3588, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lehv M, Fitzgerald PJ. Pancreatic acinar cell regeneration. IV. Regeneration after resection. Am J Pathol 53: 513–535, 1968 [PMC free article] [PubMed] [Google Scholar]
- 51.Liou AP, Lu X, Sei Y, Zhao X, Pechhold S, Carrero RJ, Raybould HE, Wank S. The G-protein-coupled receptor GPR40 directly mediates long-chain fatty acid-induced secretion of cholecystokinin. Gastroenterology 140: 903–912, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liou AP, Sei Y, Zhao X, Feng J, Lu X, Thomas C, Pechhold S, Raybould HE, Wank SA. The extracellular calcium-sensing receptor is required for cholecystokinin secretion in response to l-phenylalanine in acutely isolated intestinal I cells. Am J Physiol Gastrointest Liver Physiol 300: G538–G546, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lo CM, Samuelson LC, Chambers JB, King A, Heiman J, Jandacek RJ, Sakai RR, Benoit SC, Raybould HE, Woods SC, Tso P. Characterization of mice lacking the gene for cholecystokinin. Am J Physiol Regul Integr Comp Physiol 294: R803–R810, 2008 [DOI] [PubMed] [Google Scholar]
- 54.Longnecker DS, Curphey TJ. Adenocarcinoma of the pancreas in azaserine-treated rats. Cancer Res 35: 2249–2258, 1975 [PubMed] [Google Scholar]
- 55.Longnecker DS, Curphey TJ, Kuhlmann ET, Schaeffer BK. Experimental induction of pancreatic carcinomas in the hamster with N delta-(N-methyl-N-nitrosocarbamoyl)-L-ornithine. J Natl Cancer Inst 71: 1327–1336, 1983 [PubMed] [Google Scholar]
- 56.Longnecker DS, Curphey TJ, Lilja HS, French JI, Daniel DS. Carcinogenicity in rats of the nitrosourea amino acid N delta-(N-methyl-N-nitrosocarbamoyl)-L-ornithine. J Environ Pathol Toxicol 4: 117–129, 1980 [PubMed] [Google Scholar]
- 57.Lowenfels AB, Maisonneuve P. Epidemiology and risk factors for pancreatic cancer. Best Pract Res Clin Gastroenterol 20: 197–209, 2006 [DOI] [PubMed] [Google Scholar]
- 58.Mainz DL, Black O, Webster PD. Hormonal control of pancreatic growth. J Clin Invest 52: 2300–2304, 1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matters GL, Cooper TK, Gilius EL, McGovern C, Liao J, Smith JP. Blockade of the CCK receptor inhibits progression of early PanIN lesions to pancreatic cancer in the Pdx1-Cre, LSL/KrasG12D mouse (Abstract). Pancreas 41: 1384, 2012 [Google Scholar]
- 60.Matters GL, Harms JF, McGovern CO, Jayakumar C, Crepin K, Smith ZP, Nelson MC, Stock H, Fenn CW, Kaiser J, Kester M, Smith JP. Growth of human pancreatic cancer is inhibited by down-regulation of gastrin gene expression. Pancreas 38: e151–e161, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Matters GL, McGovern C, Harms JF, Markovic K, Anson K, Jayakumar C, Martenis M, Awad C, Smith JP. Role of endogenous cholecystokinin on growth of human pancreatic cancer. Int J Oncol 38: 593–601, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moore TC, Jepeal LI, Boylan MO, Singh SK, Boyd N, Beer DG, Chang AJ, Wolfe MM. Gastrin stimulates receptor-mediated proliferation of human esophageal adenocarcinoma cells. Regul Pept 120: 195–203, 2004 [DOI] [PubMed] [Google Scholar]
- 63.Mooren FC, Hlouschek V, Finkes T, Turi S, Weber IA, Singh J, Domschke W, Schnekenburger J, Kruger B, Lerch MM. Early changes in pancreatic acinar cell calcium signaling after pancreatic duct obstruction. J Biol Chem 278: 9361–9369, 2003 [DOI] [PubMed] [Google Scholar]
- 64.Moran TH. Gut peptides in the control of food intake. Int J Obes (Lond) 33, Suppl 1: S7–S10, 2009 [DOI] [PubMed] [Google Scholar]
- 65.Murphy JA, Criddle DN, Sherwood M, Chvanov M, Mukherjee R, McLaughlin E, Booth D, Gerasimenko JV, Raraty MG, Ghaneh P, Neoptolemos JP, Gerasimenko OV, Tepikin AV, Green GM, Reeve JR, Jr, Petersen OH, Sutton R. Direct activation of cytosolic Ca2+ signaling and enzyme secretion by cholecystokinin in human pancreatic acinar cells. Gastroenterology 135: 632–641, 2008 [DOI] [PubMed] [Google Scholar]
- 66.Owyang C. Physiological mechanisms of cholecystokinin action on pancreatic secretion. Am J Physiol Gastrointest Liver Physiol 271: G1–G7, 1996 [DOI] [PubMed] [Google Scholar]
- 67.Owyang C, Logsdon CD. New insights into neurohormonal regulation of pancreatic secretion. Gastroenterology 127: 957–969, 2004 [DOI] [PubMed] [Google Scholar]
- 68.Phillips PA, Yang L, Shulkes A, Vonlaufen A, Poljak A, Bustamante S, Warren A, Xu Z, Guilhaus M, Pirola R, Apte MV, Wilson JS. Pancreatic stellate cells produce acetylcholine and may play a role in pancreatic exocrine secretion. Proc Natl Acad Sci USA 107: 17397–17402, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pour P, Althoff J, Kruger FW, Mohr U. A potent pancreatic carcinogen in Syrian hamsters: N-nitrosobis(2-oxopropyl)amine. J Natl Cancer Inst 58: 1449–1453, 1977 [DOI] [PubMed] [Google Scholar]
- 70.Povoski SP, Zhou W, Longnecker DS, Bell RH., Jr Novel expression of gastrin (CCK-B) receptors in pancreatic carcinomas and dysplastic pancreas from transgenic mice. Am J Surg 167: 120–126, 1994 [DOI] [PubMed] [Google Scholar]
- 71.Reubi JC, Waser B, Gugger M, Friess H, Kleeff J, Kayed H, Buchler MW, Laissue JA. Distribution of CCK1 and CCK2 receptors in normal and diseased human pancreatic tissue. Gastroenterology 125: 98–106, 2003 [DOI] [PubMed] [Google Scholar]
- 72.Saluja AK, Dudeja V. Relevance of animal models of pancreatic cancer and pancreatitis to human disease. Gastroenterology 144: 1194–1198, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sankaran H, Goldfine ID, Bailey A, Licko V, Williams JA. Relationship of cholecystokinin receptor binding to regulation of biological functions in pancreatic acini. Am J Physiol Gastrointest Liver Physiol 242: G250–G257, 1982 [DOI] [PubMed] [Google Scholar]
- 74.Shiratori K, Takeuchi T, Satake K, Matsuno S. Clinical evaluation of oral administration of a cholecystokinin-A receptor antagonist (loxiglumide) to patients with acute, painful attacks of chronic pancreatitis: a multicenter dose-response study in Japan. Pancreas 25: e1–e5, 2002 [DOI] [PubMed] [Google Scholar]
- 75.Smith JP, Fantaskey AP, Liu G, Zagon IS. Identification of gastrin as a growth peptide in human pancreatic cancer. Am J Physiol Regul Integr Comp Physiol 268: R135–R141, 1995 [DOI] [PubMed] [Google Scholar]
- 76.Smith JP, Hamory MW, Verderame MF, Zagon IS. Quantitative analysis of gastrin mRNA and peptide in normal and cancerous human pancreas. Int J Mol Med 2: 309–315, 1998 [DOI] [PubMed] [Google Scholar]
- 77.Smith JP, Harms JF, Matters GL, McGovern CO, Ruggiero FM, Liao J, Fino KK, Ortega EE, Gilius EL, Phillips JA., III A single nucleotide polymorphism of the cholecystokinin-B receptor predicts risk for pancreatic cancer. Cancer Biol Ther 13: 164–174, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Smith JP, Kramer S, Bagheri S. Effects of a high-fat diet and L364,718 on growth of human pancreas cancer. Dig Dis Sci 35: 726–732, 1990 [DOI] [PubMed] [Google Scholar]
- 79.Smith JP, Kramer ST, Solomon TE. CCK stimulates growth of six human pancreatic cancer cell lines in serum-free medium. Regul Pept 32: 341–349, 1991 [DOI] [PubMed] [Google Scholar]
- 80.Smith JP, Liu G, Soundararajan V, McLaughlin PJ, Zagon IS. Identification and characterization of CCK-B/gastrin receptors in human pancreatic cancer cell lines. Am J Physiol Regul Integr Comp Physiol 266: R277–R283, 1994 [DOI] [PubMed] [Google Scholar]
- 81.Smith JP, Rickabaugh CA, McLaughlin PJ, Zagon IS. Cholecystokinin receptors and PANC-1 human pancreatic cancer cells. Am J Physiol Gastrointest Liver Physiol 265: G149–G155, 1993 [DOI] [PubMed] [Google Scholar]
- 82.Smith JP, Shih A, Wu Y, McLaughlin PJ, Zagon IS. Gastrin regulates growth of human pancreatic cancer in a tonic and autocrine fashion. Am J Physiol Regul Integr Comp Physiol 270: R1078–R1084, 1996 [DOI] [PubMed] [Google Scholar]
- 83.Smith JP, Solomon TE, Bagheri S, Kramer S. Cholecystokinin stimulates growth of human pancreatic adenocarcinoma SW-1990. Dig Dis Sci 35: 1377–1384, 1990 [DOI] [PubMed] [Google Scholar]
- 84.Smith JP, Verderame MF, Ballard EN, Zagon IS. Functional significance of gastrin gene expression in human cancer cells. Regul Pept 117: 167–173, 2004 [DOI] [PubMed] [Google Scholar]
- 85.Smith JP, Verderame MF, McLaughlin P, Zagon IS. Characterization of the CCK-C (cancer) receptor in human pancreatic cancer. Digestion 60: 401, 1999 [PubMed] [Google Scholar]
- 86.Smith JP, Verderame MF, McLaughlin P, Martenis M, Ballard E, Zagon IS. Characterization of the CCK-C (cancer) receptor in human pancreatic cancer. Int J Mol Med 10: 689–694, 2002 [PubMed] [Google Scholar]
- 87.Smith JP, Zagon IS. Cholecystokinin receptors and human pancreatic adenocarcinomas. Int J Pancreatol 16: 243–246, 1994 [Google Scholar]
- 88.Solomon TE. Control of exocrine pancreatic secretion. In: Physiology of the Gastrointestinal Tract, edited by Johnson LR. New York: Raven, 1994, p. 1499–1530 [Google Scholar]
- 89.Solomon TE, Petersen H, Elashoff J, Grossman MI. Interaction of caerulein and secretin on pancreatic size and composition in rat. Am J Physiol Endocrinol Metab Gastrointest Physiol 235: E714–E719, 1978 [DOI] [PubMed] [Google Scholar]
- 90.Solomon TE, Vanier M, Morisset J. Cell site and time course of DNA synthesis in pancreas after caerulein and secretin. Am J Physiol Gastrointest Liver Physiol 245: G99–G105, 1983 [DOI] [PubMed] [Google Scholar]
- 91.Stepan VM, Dickinson CJ, del Valle J, Matsushima M, Todisco A. Cell type-specific requirement of the MAPK pathway for the growth factor action of gastrin. Am J Physiol Gastrointest Liver Physiol 276: G1363–G1372, 1999 [DOI] [PubMed] [Google Scholar]
- 92.Suissa Y, Magenheim J, Stolovich-Rain M, Hija A, Collombat P, Mansouri A, Sussel L, Sosa-Pineda B, McCracken K, Wells JM, Heller RS, Dor Y, Glaser B. Gastrin: a distinct fate of neurogenin3 positive progenitor cells in the embryonic pancreas. PLoS One 8: e70397, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Takahashi M, Hori M, Mutoh M, Wakabayashi K, Nakagama H. Experimental animal models of pancreatic carcinogenesis for prevention studies and their relevance to human disease. Cancers (Basel) 3: 582–602, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Takaishi S, Shibata W, Tomita H, Jin G, Yang X, Ericksen R, Dubeykovskaya Z, Asfaha S, Quante M, Betz KS, Shulkes A, Wang TC. In vivo analysis of mouse gastrin gene regulation in enhanced GFP-BAC transgenic mice. Am J Physiol Gastrointest Liver Physiol 300: G334–G344, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Takiguchi S, Suzuki S, Sato Y, Kanai S, Miyasaka K, Jimi A, Shinozaki H, Takata Y, Funakoshi A, Kono A, Minowa O, Kobayashi T, Noda T. Role of CCK-A receptor for pancreatic function in mice: a study in CCK-A receptor knockout mice. Pancreas 24: 276–283, 2002 [DOI] [PubMed] [Google Scholar]
- 96.Tamiolakis D, Venizelos I, Simopoulos C, Kotini A, Jivannakis T, Papadopoulos N. Does neoplastic gastrin expression remodel the embryonal pattern of the protein? A study in human pancreas. Hepatogastroenterology 51: 249–252, 2004 [PubMed] [Google Scholar]
- 97.Tamiolakis D, Venizelos I, Simopoulos C, Lambropoulou M, Kotini A, Jivannakis T, Alexiadis G, Boglou P, Papadopoulos N. Does neoplastic cholecystokinin expression reflect the embryonal pattern of the protein? A study in human pancreas. Acta Medica (Hradec Kralove) 47: 101–105, 2004 [PubMed] [Google Scholar]
- 98.Tang C, Biemond I, Lamers CB. Cholecystokinin receptors in human pancreas and gallbladder muscle: a comparative study. Gastroenterology 111: 1621–1626, 1996 [DOI] [PubMed] [Google Scholar]
- 99.Thiebaut AC, Jiao L, Silverman DT, Cross AJ, Thompson FE, Subar AF, Hollenbeck AR, Schatzkin A, Stolzenberg-Solomon RZ. Dietary fatty acids and pancreatic cancer in the NIH-AARP diet and health study. J Natl Cancer Inst 101: 1001–1011, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, Mercer KL, Grochow R, Hock H, Crowley D, Hingorani SR, Zaks T, King C, Jacobetz MA, Wang L, Bronson RT, Orkin SH, DePinho RA, Jacks T. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell 5: 375–387, 2004 [DOI] [PubMed] [Google Scholar]
- 101.Wank SA. G protein-coupled receptors in gastrointestinal physiology. I. CCK receptors: an exemplary family. Am J Physiol Gastrointest Liver Physiol 274: G607–G613, 1998 [DOI] [PubMed] [Google Scholar]
- 102.Wank SA, Harkins R, Jensen RT, Shapira H, de Weerth A, Slattery T. Purification, molecular cloning, and functional expression of the cholecystokinin receptor from rat pancreas. Proc Natl Acad Sci USA 89: 3125–3129, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wank SA, Pisegna JR, de Weerth A. Brain and gastrointestinal cholecystokinin receptor family: structure and functional expression. Proc Natl Acad Sci USA 89: 8691–8695, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wank SA, Pisegna JR, de Weerth A. Cholecystokinin receptor family. Molecular cloning, structure, and functional expression in rat, guinea pig, and human. Ann NY Acad Sci 713: 49–66, 1994 [DOI] [PubMed] [Google Scholar]
- 105.Weinberg DS, Ruggeri B, Barber MT, Biswas S, Miknyocki S, Waldman SA. Cholecystokinin A and B receptors are differentially expressed in normal pancreas and pancreatic adenocarcinoma. J Clin Invest 100: 597–603, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Whitcomb DC. Genetic risk factors for pancreatic disorders. Gastroenterology 144: 1292–1302, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Williams JA, Korc M, Dormer RL. Action of secretagogues on a new preparation of functionally intact, isolated pancreatic acini. Am J Physiol Endocrinol Metab 235: E517–E524, 1978 [DOI] [PubMed] [Google Scholar]
- 108.Zucker KA, Adrian TE, Bilchik AJ, Modlin IM. Effects of the CCK receptor antagonist L364,718 on pancreatic growth in adult and developing animals. Am J Physiol Gastrointest Liver Physiol 257: G511–G516, 1989 [DOI] [PubMed] [Google Scholar]