

Abstract
Diabetes risk increases significantly with age and correlates with lower oxidative capacity in muscle. Decreased expression of peroxisome proliferator-activated receptor-γ coactivator-1α (Pgc-1α) and target gene pathways involved in mitochondrial oxidative phosphorylation are associated with muscle insulin resistance, but a causative role has not been established. We sought to determine whether a decline in Pgc-1α and oxidative gene expression occurs during aging and potentiates the development of age-associated insulin resistance. Muscle-specific Pgc-1α knockout (MKO) mice and wild-type littermate controls were aged for 2 yr. Genetic signatures of skeletal muscle (microarray and mRNA expression) and metabolic profiles (glucose homeostasis, mitochondrial metabolism, body composition, lipids, and indirect calorimetry) of mice were compared at 3, 12, and 24 mo of age. Microarray and gene set enrichment analysis highlighted decreased function of the electron transport chain as characteristic of both aging muscle and loss of Pgc-1α expression. Despite significant reductions in oxidative gene expression and succinate dehydrogenase activity, young mice lacking Pgc-1α in muscle had lower fasting glucose and insulin. Consistent with loss of oxidative capacity during aging, Pgc-1α and Pgc-1β expression were reduced in aged wild-type mouse muscle. Interestingly, the combination of age and loss of muscle Pgc-1α expression impaired glucose tolerance and led to increased fat mass, insulin resistance, and inflammatory markers in white adipose and liver tissues. Therefore, loss of Pgc-1α expression and decreased mitochondrial oxidative capacity contribute to worsening glucose tolerance and chronic systemic inflammation associated with aging.
Keywords: aging, mitochondria, peroxisome proliferator-activated receptor-γ coactivator-1, muscle, diabetes, inflammation
age is a significant risk factor for sarcopenia, insulin resistance, and the development of type 2 diabetes (37, 40). Mitochondria are central to maintaining muscle health, yet controversy still surrounds whether mitochondrial dysfunction contributes to declining metabolic function and insulin sensitivity associated with advanced age (reviewed in Refs. 23, 41, and 42). Thus, it remains unclear whether deficiencies in mitochondrial health play a significant causative role or whether they are simply a consequence of disease progression.
Altered mitochondrial function can lower ATP synthesis, decrease lipid oxidation, and increase reactive oxygen species, causing energy imbalances and oxidative damage. Many genes encoding mitochondrial electron transport chain (ETC) components are decreased in aged muscle (56), consistent with observed decreases in oxidative capacity (47). Aged muscle has lower mitochondrial density (7), and mitochondria are often depolarized or nonfunctional (41). Similarly, patients with diabetes and/or insulin resistance have decreased expression of gene pathways involved in oxidative phosphorylation (34, 38). Thus, diabetic and aging muscle share similar deficiencies in mitochondrial function, linking ETC dysfunction to glucose intolerance.
Peroxisome proliferator-activated receptor-γ coactivator-1α (Pgc-1α) expression is decreased in aging human muscle (13, 44), and dysregulation of this transcriptional coactivator may underlie lower mitochondrial function and muscle pathologies associated with age (8). PGC-1α is considered a master regulator of mitochondrial biogenesis and function, acting as an upstream regulatory switch for gene pathways controlling oxidative phosphorylation, fatty acid oxidation, reactive oxygen species detoxification, and mitochondrial density. Overexpression of PGC-1α in muscle protects aging mice from age-related muscle wasting and glucose intolerance (50, 52), and yet tight regulation of PGC-1α expression is required to maintain efficient glucose and lipid handling in obese mice (6, 19, 51). Expression of Pgc-1α in muscle is low in type 2 diabetics and related family members (2, 21, 33, 38) but can be increased with exercise to possibly prevent or reverse metabolic abnormalities (3, 25, 44, 49). Thus, a decline in Pgc-1α expression in aging muscle (8, 13), brought on by factors such as diet, obesity, or decreased physical activity, may play a significant role in the age-associated development of glucose intolerance.
In the current study, we address whether decreasing muscle Pgc-1α and oxidative gene expression can serve as a foundation for age-related glucose intolerance. We hypothesized that long-term reductions in muscle Pgc-1α expression, causing persistent yet moderate loss of mitochondrial content and function, would exacerbate glucose intolerance later in life.
METHODS
Generation of a new Pgc-1α floxed mouse line.
Previously generated male Pgc-1α floxed mice are sterile (29), prohibiting generation of homozygous floxed mice (19). As reduced Pgc-1α in multiple tissues can have potentially confounding effects on whole body glucose metabolism (27, 29), a new line of floxed Pgc-1α mice lacking the neomycin selection cassette was generated. Briefly, floxed Pgc-1α mice (29) were bred to mice expressing low-efficiency Cre recombinase (EIIa-cre, no. 003724; Jackson Laboratories). Selected progeny with intact Pgc-1α floxed loci but lacking the neomycin cassette were bred to C57B/6N mice to remove the EIIa-cre transgene. Excision of exons 3–5 in these floxed mice by Cre recombinase prevents protein expression of all currently identified PGC-1α isoforms (B6.129-Ppargc1atm2Brsp/J, no. 009666; Jackson Laboratories).
Muscle-specific Pgc-1α knockout mice.
The regenerated line of Pgc-1αfl/fl mice was bred to a myogenin-Cre line (28) to generate skeletal muscle-specific Pgc-1α knockout (MKO) mice (Pgc-1αfl/fl + myo-Cre). Test groups consisted of male mice homozygous for the floxed Pgc-1α alleles expressing myogenin-Cre (MKOs; 50%) or no transgene (50%, age-matched, littermate controls). Aging groups started with n = 13–17. Mice with the myogenin-Cre transgene alone responded similarly to wild types (WT; not shown). Mice were on a mixed C57B/6J/6N/129 background and maintained on a rodent chow (5008I; PharmaServ) for 2 yr. All experiments were performed in accordance with the BIDMC animal facility Institutional Animal Care and Use Committee regulations.
Expression array.
Genome-wide gene expression profiles were generated using Affymetrix Mouse Genome 430 2.0 Arrays. Gene expression profiles of gastrocnemius muscle mRNA from young (10 wk old) and old (24 mo old) WT or MKO mice were compared (n = 5 mice, 3 arrays/genotype analyzed; arrays 1 and 2 contained RNA pooled from 2 mice, and array 3 was a single mouse). Raw and normalized gene expression data are available on GEO (GEO accession no. GSE52550). A multivariate linear model was fit for each gene. Raw CEL files from Genome 430 2.0 Array chips were normalized using RMA (PMID: 12925520) and annotated using biomaRt (PMID: 16082012). When multiple probe sets matched the same Entrez Gene ID, the probe set exhibiting the highest variance was used for further analysis, reducing the set to 16,617 unique genes:
where Yi is the variable representing expression of gene i, A and G are variables representing age and genotype respectively, A × G is the interaction term for age and genotype, and the β symbols are the regression coefficients. Significance for the contribution of age and genotype are estimated by t-test on the corresponding β. To correct for multiple testing, we computed false discovery rate (FDR) for age and genotype from the nominal P values, using a FDR of 25% as cutoff for significance. Full results are provided in Supplemental Data Set S1 (Supplemental Material for this article can be found on the AJP-Endocrinology and Metabolism web site).
Gene set enrichment analysis.
To identify pathways significantly associated with age and genotype, genes were ranked based on corresponding t-statistics (see statistical comparison of phenotypes), and preranked gene set enrichment analyses were performed [gene set enrichment analysis (GSEA) version 2.0.10 (PMID: 16199517)]. Pathways were defined by gene ontology terms (PMID: 10802651) curated in the mouse genome database (PMID: 22075990). Only pathways whose corresponding gene sets contained at least 15 genes and less than or equal to 500 genes were considered for further analysis (Supplemental Data Set S2). Only pathways with a FDR ≤25% were considered. The full results of the gene set enrichment analyses are provided in Supplemental Data Set S3.
Indirect calorimetry.
Oxygen consumption, carbon dioxide production, movement, respiratory exchange ratios (RER), and cumulative food intake were measured in live mice (ad libitum chow diet with a 12:12-h light-dark cycle) over 3 days using the Comprehensive Laboratory Animal Monitoring System (Columbus Instruments). Air sampling was performed at 32-min intervals. Number of laser breaks (movement along the x- and y-axes) were totaled and plotted for each interval. Food intake is shown as cumulative grams of chow missing from a preweighed chamber at each interval.
Tissue dissection and isolation.
Mice were euthanized by CO2, and the following tissues were isolated by dissection: skeletal muscle (gastrocnemius, quadriceps femoris, tibialis anterior, and soleus) from both legs, epididymal white fat pad, small intestine (jejunum), colon, left brain hemisphere, anterior right lobe of the liver, heart, interscapular brown fat, and pancreas. In general, whole muscle from left leg was used for mRNA and right leg for histology. For gastrocnemius, the lateral head was used.
RNA isolation and quantitative RT-PCR.
Snap-frozen tissue was homogenized in TRIzol (Invitrogen). One microgram of DNAse-treated RNA was reverse transcribed, and cDNA was amplified and quantified using SYBR green PCR master mix (Applied Biosystems). Gene expression levels were normalized to Hprt, and relative expression was calculated by the ΔΔCT threshold cycle method. PCR efficiency was identical for endogenous control and target genes, and Hprt was unaffected by experimental conditions. Primer sequences are listed in Supplemental Table S1.
Succinate dehydrogenase staining.
Succinate dehydrogenase activity was visualized in cross-sections of gastrocnemius muscle frozen in OCT following incubation in a phosphate buffer containing sodium succinate and tetrazoliumand (Sigma). Color development is proportional to succinate dehydrogenase (SDH) activity.
Serum glucose, lipids, body composition, and glucose/insulin tolerance tests.
Blood glucose was measured in tail blood using a standard glucometer at the times indicated. Serum insulin, IL-6, and TNFα were determined by ELISA and triglycerides and nonesterified fatty acids by colorimetric assay (Specialized Assay Core; Joslin Diabetes Center). Mice were fasted for 16 or 6 h prior to intraperitoneal (ip) injection of 1 g/kg d-glucose or 0.8 U/kg insulin (Humulin), respectively. Fat and lean mass was determined by nuclear magnetic resonance in live mice (EchoMRI).
Islet histology and insulitis score.
Formalin-fixed pancreata were mounted and sectioned to capture maximal surface area. Following hematoxylin and eosin staining, lymphocyte infiltration for all islets was scored and an insulitis score calculated as described previously (17). Briefly, scoring was assigned as follows: 0 = lymphocytes being present; 1 = peri-islet infiltration (lymphocytes on the periphery, not fully surrounding the islet); 2 = immune infiltration surrounding the entire islet, but with the islet appearing mostly intact; 3 = one-half of the islet being infiltrated (engulfed) by lymphocytes; and 4 = the entire islet being engulfed (few β-cells evident). Insulitus per islet was calculated as
representing the average inflammation state of each islet per mouse. Two independent reviewers scored blinded sections from each mouse.
Statistical analysis.
Statistical significance (P < 0.05) for tolerance tests and mRNA expression was assessed by two-way ANOVA (with repeated measures for glucose tolerance tests), followed by Fisher's least significant difference post hoc test. Area under the curve (AUC) was calculated with baseline set at fasting glucose. Comparisons between two independent groups were performed using either a two-tailed unpaired Student t-test or a Mann-Whitney U-test if the distribution was not Gaussian (by Shapiro-Wilk normality test). Two-way ANOVA was used when two factors' (age and genotype) effect on gene expression was assessed, followed by a Holm-Sidak's multiple comparison test for significance of each gene. To determine effect of one factor (genotype or age) on multiple genes, we performed an unpaired Student t-test for each gene and corrected our analysis for multiple comparisons (FDR = 10%). Analysis was completed using GraphPad Prism. All data points represent means ± SE.
RESULTS
Aging abolishes compensatory increases in skeletal muscle Pgc-1β expression.
As expected, young male MKO mice exhibited a >95% knockdown of Pgc-1α mRNA in skeletal muscle compared with WT littermates (Fig. 1A). Interestingly, there was a compensatory increase in Pgc-1β mRNA in all skeletal muscles that were tested (Fig. 1B). PGC-1β is a closely related family member that may share significant overlap in function to PGC-1α (45, 57). Expression of Pgc-1α and Pgc-1β was unchanged in other tissues examined, including cardiac muscle, confirming specificity of the knockdown to skeletal muscle only. We next compared muscle RNA from young (6 mo old) and old (24 mo old) WT and MKO mice to determine whether Pgc-1 gene expression was impacted by age. Interestingly, expression of both Pgc-1α and Pgc-1β mRNA decreased in aged WT skeletal muscle (Fig. 1C), and the compensatory increase in Pgc-1β seen in young MKO muscle was lost in all skeletal muscle groups with aging [quadriceps; Fig. 1C (other muscles not shown)].
Fig. 1.
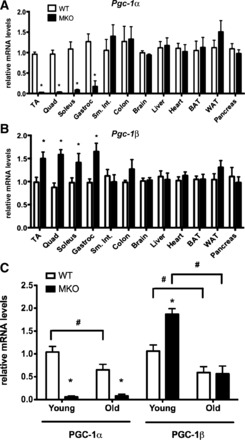
Aging promotes loss of both peroxisome proliferator-activated receptor-γ coactivator-1α (Pgc-1α) and Pgc-1β in skeletal muscle. Relative mRNA expression of Pgc-1α (A) or Pgc-1β (B) in tissues from young wild-type (WT) or muscle-specific Pgc-1α knockout (MKO) mice (6 mo; n = 8). Expression is normalized to levels in tissues of age-matched WT mice. C: expression of Pgc-1 coactivators in young (6 mo; n = 8) vs. aged quadriceps muscle (24 mo; n = 5) in WT and MKO mice expressed relative to mRNA levels in young WT mice. Data are means ± SE (*P < 0.05 WT vs. MKO of similar age; #P < 0.05 young vs. old mice of similar genotype).
Decreased mitochondrial oxidative capacity is a major effector pathway shared by aged muscle and muscle lacking PGC-1α.
To determine whether biological processes impacted by loss of Pgc-1α intersect with those altered during aging, global changes in muscle mRNA expression in gastrocnemius muscle from young (10 wk old) and old (24 mo old) WT and MKO mice were compared using genome-wide Affymetrix GeneChip arrays.
Although >2,500 genes were differentially expressed in young vs. old muscle samples (FDR <25%; Fig. 2A and Supplemental Data Set S1), only 11 molecular pathways were enriched following GSEA (FDR <25%; Fig. 2B and Supplemental Data Set S3). In comparison, 475 individual genes were different between WT and MKO mice regardless of age (FDR <25%; Fig. 2A), with GSEA identifying 95 gene sets (FDR <25%; Fig. 2B). Strikingly, 43% of the genes affected by loss of Pgc-1α were also changed in aging muscle (8% of total age-affected genes). Within this pool of 206 genes shared by both Pgc-1α loss and aging (Fig. 2A), 80% changed in the same direction (Supplemental Data Set S4), suggesting that a significant proportion of genes regulated by Pgc-1α (∼35%) may be involved in the molecular program of muscle aging.
Fig. 2.
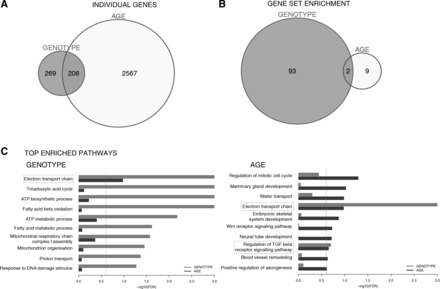
mRNA expression and gene set enrichment analysis in MKO vs. aging muscle. A and B: unique and overlapping transcript no. (A) and enriched gene sets (B) identified by multivariant analysis of expression arrays (n = 3; P < 0.05, FDR = 25%) of gastrocnemius muscle RNA from WT vs. MKO (genotype) or 10-wk-old vs. 24-mo-old (age) mice. C: the top 10 significantly enriched gene sets [ranked by false discovery rate (FDR)] for genotype or age. Gray (genotype) or black (age) bars represent relative strengths of gene set enrichment (FDR, x-axis). Gray dashed line represents cutoff of significance (FDR = 25%), and light gray boxes denote pathways shared across genotype and age data.
Although PGC-1α has been implicated in multiple aspects of muscle biology, the top eight gene sets affected by Pgc-1α loss were pathways of mitochondrial metabolism (Fig. 2C). Of note, only two gene sets were shared by genotype and age, “electron transport chain” function and “regulation of TGFβ signaling” (Fig. 2C), illustrating that altered oxidative phosphorylation is indeed a hallmark of both reduced Pgc-1α and aging muscle in mice.
Aging exaggerates decreased oxidative capacity in muscle.
As predicted by expression array data, mitochondrial genes were sensitive to both aging and loss of Pgc-1α (Fig. 3A). It is already established that PGC-1α is an important regulator of nuclear-encoded mitochondrial gene and protein expression (12, 55). To illustrate whether aging exaggerated decreases in MKO mice, we directly compared young vs. old mice by normalizing data to age-matched controls (Fig. 3B). This demonstrated that aging further lowered expression of many nuclear-encoded mitochondrial genes. Although Tfam expression was not significantly affected by Pgc-1α loss, mtDNA-encoded Nd1 and Atp6 were decreased in young MKO muscle, suggesting an early decrease in mitochondrial mass (Fig. 3A). Aging alone decreased mtDNA transcripts (Fig. 3A); however, unlike nuclear-encoded mitochondrial genes, aging did not further exacerbate mtDNA decreases caused by Pgc-1α ablation (Fig. 3B).
Fig. 3.
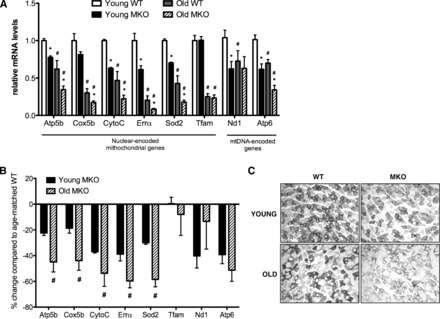
Reduced Pgc-1α expression exacerbated, decreasing mitochondrial gene expression, function, and oxidative fibers in aging muscle. A: relative mRNA expression of nuclear- and mitochondrial (mt)-encoded modulators of mitochondrial metabolism in gastronemius muscle of young (10 wk old; n = 8) vs. aged (24 mo old; n = 5) mice. Data are means ± SE normalized to young WT mice. B: %decreases in mitochondrial gene expression in young vs. old MKO mice (data are means ± SE normalized to relative age-matched WT controls). *P < 0.05, WT vs. MKO of similar age; #P < 0.05, young vs. old mice of similar genotype. C: succinate dehydrogenase activity in muscle sections (gastrocnemius) visualized by nitro blue tetrazolium staining.
SDH activity (an indicator of mitochondrial oxidative capacity) in muscle was decreased by age alone and loss of Pgc-1α in both young and old gastrocnemius muscle (Fig. 3C).
The combination of reduced muscle Pgc-1 and aging worsened glucose intolerance.
Because advanced age is a significant risk factor for diabetes, and low muscle ETC activity is linked to insulin resistance, we tested whether long-term loss of muscle Pgc-1α aggravated glucose intolerance. Chow-fed, WT, and MKO mice were followed over a period of 2 yr. At 3 mo of age, MKO mice had significantly decreased fasting glucose and circulating insulin (Table 1), and blood glucose was significantly lower 20 min following an ip glucose challenge (Fig. 4A). At 12 mo of age, although glycemia remained lower in MKO mice at fasting and 20 min post-glucose challenge, overall glucose clearance tended to worsen (Fig. 4B). For both 3- and 12-mo-old mice, despite similar AUCs (Fig. 4, A and B), the shapes of the MKO glucose disposal curves were different compared with WT controls (significant interaction between time and genotype, 2-way ANOVA, P < 0.05), suggesting that loss of skeletal muscle Pgc-1α impacted glucose excursion kinetics. After 24 mo, MKO mice were significantly less glucose tolerant than controls (Fig. 4C), and total AUC was increased. Thus, despite reduced mitochondrial gene expression and oxidative capacity at all ages, significant glucose intolerance manifested only after prolonged aging.
Table 1.
Metabolic parameters in WT vs. MKO mice at different ages
| 3 Mo |
12 Mo |
24 Mo |
||||
|---|---|---|---|---|---|---|
| WT | KO | WT | KO | WT | KO | |
| Weight, g | 23.4 ± 0.9 | 25.7 ± 0.7 | 36.6 ± 1.2 | 39.3 ± 2.2 | 33.9 ± 1.1 | 36.4 ± 2.1 |
| Fasting glucose, mg/dl | 96.7 ± 5.7 | 79.3 ± 6.8* | 97.6 ± 4.3 | 83.8 ± 2.8* | 91.0 ± 6.0 | 76.6 ± 9.5 |
| Insulin, ng/ml | 0.49 ± 0.05 | 0.37 ± 0.02* | 1.06 ± 0.1 | 0.51 ± 0.1* | ||
Data are expressed as means ± SE (n = 9–12 mice for 3- to 12-mo-old groups, n = 5 for 24-mo-old group).
WT, wild type; MKO, muscle-specific Pgc-1α knockout.
P < 0.05, comparing WT and MKO of similar age.
Fig. 4.
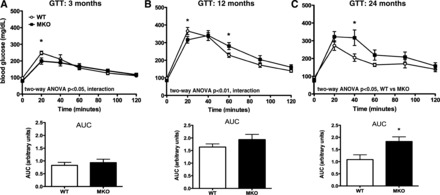
MKOs develop glucose intolerance with advanced age. A–C: glucose tolerance tests (GTT) performed in WT vs. MKO mice over 24 mo (n = 8–9). Area under the curve (AUC) calculated from baseline glucose represented under each curve. Data are means ± SE. *P < 0.05, WT vs. MKO.
MKO mice developed insulin resistance with advanced age.
We next assessed peripheral insulin sensitivity by insulin tolerance test. Similar to published reports (20, 57), younger MKOs (6 mo old) showed no differences in response to exogenous insulin (Fig. 5A). In contrast to young mice, 24-mo-old MKOs could not lower glucose as effectively in response to exogenous insulin (Fig. 5B). Moreover, although body composition was not different in young mice, old MKO mice had increased fat and decreased lean masses (Fig. 5C), with no significant difference in circulating triglycerides or free fatty acids in serum (Fig. 5, D and E). Despite decreased fasting insulin (Table 1) in MKOs, fed levels of serum insulin were similar (Fig. 5F), suggesting no defects in postprandial insulin secretion. Despite decreased oxidative capacity in MKO muscle, intramuscular triglyceride levels trended lower in old MKOs (Fig. 5G), arguing against insulin resistance due to high intramuscular lipids. The GLUT4 insulin-sensitive glucose transporter is a target of PGC-1α (32); however, Glut4 mRNA levels were unchanged in young mice and significantly lower only in aged MKO soleus, not gastrocnemius or other metabolically active tissues (Fig. 5, H and I). No significant differences were noted in total body weight between WT and MKO mice (Table 1), although the MKOs tended to be heavier.
Fig. 5.
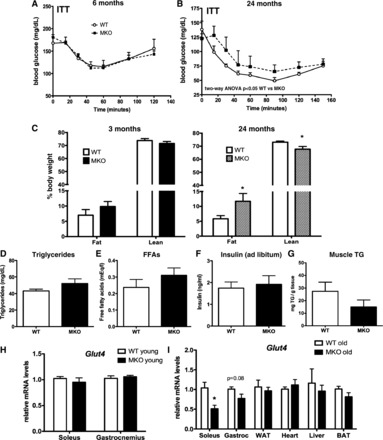
Aging promoted insulin resistance, increased adipose tissue mass, and lowered respiratory exchange ratio (RER) in MKO mice. A and B: insulin tolerance tests (ITT) in 6- (A) and 24-mo-old (B) WT and MKO mice (n = 5). C: body composition of mice by NMR (n = 6–8). D–G: triglyceride (D), free fatty acid (FFA; E), insulin (F), and intramuscular triglyceride (TG) content (G) measured by colorimetric assay using serum from fed WT and MKO mice at 24 mo of age (n = 5–9). G and I: Glut4 mRNA expression in tissues from young (3 mo, n = 8–9; H) and aged (24 mo, n = 5; I) WT vs. MKO mice. Data points represent means ± SE. *P < 0.05 WT vs. MKO. Gene expression is normalized to mean expression of corresponding tissue in age-matched WT mice. WAT, white adipose tissue; BAT, brown adipose tissue.
There were also no significant differences between genotypes in V̇o2, V̇co2, heat production, activity, or food intake by indirect calorimetry, regardless of whether data were corrected for total body weight (Fig. 6, A–E) or lean body mass (data not shown). However, aged MKO mice exhibited significantly lower RERs during the dark phase only (Fig. 6F), indicating a shift toward lipid metabolism. Because mice generally eat during the dark phase, increased catabolism of lipids when circulating insulin levels are highest can support insulin resistance (4, 15). And while defects in postprandial insulin secretion could also contribute to the shift in fuel utilization, fed insulin levels in 6-mo-old MKOs were similar to WT (Fig. 5F).
Fig. 6.
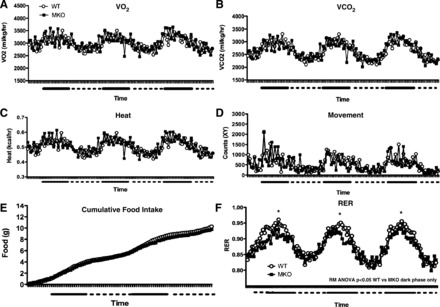
MKO mice had decreased RERs during dark phase only. Indirect calorimetry by Comprehensive Laboratory Animal Monitoring System measuring oxygen consumption (V̇o2; A), carbon dioxide production (V̇co2; B), heat (C), activity (laser breaks; D), cumulative food intake (E), and RER (F) in WT and MKO mice (24 mo old) over the course of 3 days. Solid bars represent dark phase (6 PM to 6 AM), and dashed lines represent light phase (6 AM to 6 PM). Data points represent means ± SE (n = 8). For RER: *P < 0.05 by repeated-measures (RM) ANOVA of values in dark phase only.
Loss of muscle Pgc-1α causes age-dependent inflammation in white adipose and liver tissue.
Low-grade, chronic inflammation is associated with aging and insulin resistance. Because loss of muscle Pgc-1α may increase circulating IL-6 (19), we hypothesized that increased inflammatory signaling in muscle contributes to glucose intolerance in old MKO mice. However, there was no significant increase in mRNA levels of Il-6, Tnfα, Socs3, or Mcp-1 (Ccl2) in sedentary young or aged MKO muscle (Fig. 7A). Cd68 expression (a marker of macrophage infiltration) and circulating TNFα were also similar between genotypes (Fig. 7, A and B).
Fig. 7.
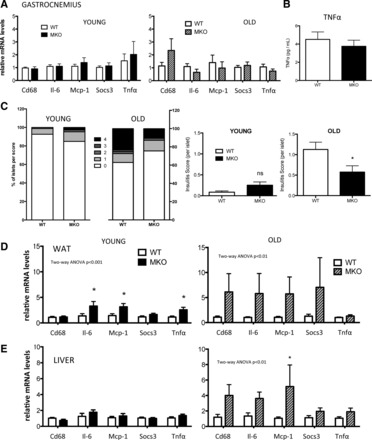
Aged, sedentary MKO mice have increased inflammation in liver and WAT. mRNA expression of inflammatory markers in gastrocnemius muscle (A), WAT (D), and liver (E) from young (10 wk old; n = 8) or old (24 mo old; n = 5) WT or MKO mice. Data are normalized to age-matched WT mice and are means ± SE. *P < 0.05. B: circulating TNFα in 6-mo-old mice. C: %islets scored for severity of lymphocyte inflammation and insulitus score per age and genotype. NS, not significant.
Since it was hypothesized that loss of muscle Pgc-1α leads to inflammatory signaling in islets (19), we assessed lymphocytic infiltration. The number of inflamed islets, the severity of lymphocyte infiltration, and the insulitus score of young MKO islets trended higher in younger mice but was not significant (Fig. 7C). Unexpectedly, quantitative histological analysis demonstrated that old MKO islets were actually significantly less inflamed than old WT littermates. No overt differences in islet size were noted (not shown). Thus, to investigate other possible mechanisms contributing to glucose intolerance and insulin resistance in aged MKO mice, we investigated inflammatory gene expression in other tissues that play a role in glucose clearance. In contrast to muscle, there was a significant increase in Il-6 and Mcp-1 expression in the white adipose tissue of young MKOs that became more pronounced with age (Fig. 7D). Similarly, inflammatory markers increased with age in livers of mice lacking Pgc-1α in skeletal muscle (Fig. 7E).
DISCUSSION
Reduced expression of Pgc-1 coactivators in muscle leads to significant defects in mitochondrial oxidative capacity (45, 55, 57). We show that this is not sufficient to worsen insulin sensitivity, but when combined with advanced age, it promotes glucose intolerance, insulin resistance, and inflammation in white adipose and liver. Thus, both genotype and chronological age played a role, highlighting an interaction between the two variables. Defects in mitochondrial function in muscle as a cause of insulin resistance are widely debated (14, 22). In fact, we show that young MKO mice had modestly improved metabolic parameters (fasting glucose, insulin, and glycemia immediately following glucose challenge) despite lower mitochondrial gene expression and function. This is consistent with other mouse models of disrupted mitochondrial function, including muscle-specific knockout of the Tfam and Aif genes (43, 53). The mechanism for improved glucose homeostasis in young MKOs and these other mouse models remains unclear but may involve increased glucose uptake or AMP-activated protein kinase activity.
It is important to note that most studies are performed in young mice (2–4 mo), whereas we investigated the long-term effects of modestly reduced muscle mitochondrial function. Our model provides mechanistic evidence that decreased Pgc-1α and deficiencies in electron transport chain function can indeed precede and lead to insulin resistance and impaired glucose clearance over time, which is particularly relevant, as age is a significant risk factor for diabetes. Consistent with our observations, overexpression of Pgc-1α in muscle improves glucose handling in sedentary aged mice (52) but causes insulin resistance in young high-fat-fed mice (6) that can be reversed by exercise (50). Although these studies support targeting muscle PGC-1s and mitochondria to improve insulin sensitivity, they highlight the importance of considering confounding variables such as physical activity, diet, and age. In fact, detrimental effects of age on metabolism can be improved by regular exercise (25, 31). In our current study, we limited the ability of the mice to exercise to better mimic the long-term sedentary lifestyle associated with metabolic disease. However, since muscle PGC-1α is intimately linked to exercise physiology, it would be interesting to evaluate how increased physical activity impacts the metabolic homeostasis of aging MKOs.
Our results in young MKOs contrast those of Handschin et al. (19), who reported decreased body weight and fat mass, increased energy expenditure, and resistance to diet-induced obesity despite glucose intolerance. One potential explanation for the discrepancy is their use of control mice lacking one allele of Pgc-1α in all tissues (due to breeding problems of their floxed line). Reduced Pgc-1α in metabolically active tissues such as liver, adipose, and brain has multiple confounding effects on baseline whole body glucose metabolism (11, 24, 26, 29, 30). In fact, aged whole body Pgc-1 knockout mice have increased circulating TNFα and IL-6 with no appreciable increase in muscle inflammatory signaling (36). We do note a trend toward increased islet lymphocyte infiltration in our young MKOs, consistent with evidence of pancreatic inflammation. However, we did not observe altered islet size or morphology, and interestingly, old MKO islets had less age-induced lymphocyte infiltration. Although these data suggeststhat MKO islets are protected from increased systemic inflammatory signaling associated with aging, we did not assess β-cell function in aged MKOs, and it is possible that secretory capacity was also affected.
mRNA expression analysis in muscle uncovered a potentially cooperative role for PGC-1β in muscle glucose homeostasis. Pgc-1β mRNA levels were higher in all skeletal muscle groups of young MKO mice. Although the mechanism for this seemingly compensatory increase is not known, increased Pgc-1β expression in young mice may limit or delay the consequences of losing Pgc-1α. Since complete loss of both Pgc-1α and -1β decreases the oxidative capacity of muscle but does not in itself worsen glucose tolerance in young mice (45, 57), our data suggest that a combination of advanced age and reduction of both Pgc-1 family members is required for impairment of glucose handling. This is consistent with a growing body of evidence suggesting that reduced mitochondrial function or content in muscle does not directly impair glucose tolerance but may intersect with other genetic or environmental factors to worsen disease (25). It is also possible that loss of muscle Pgc-1β impacts unique gene targets not shared with Pgc-1α and not involved in mitochondrial oxidation. Thus, it will be important to evaluate the individual and/or synergistic contributions of each family member to the observed phenotype.
Our model suggests inflammation in white adipose and liver tissues as an underlying mechanism of insulin resistance related to decreased mitochondrial oxidative capacity in aging muscle. Increased inflammation in these tissues is believed to contribute to the pathogenesis of type 2 diabetes and the metabolic syndrome (9, 16, 39, 48) and provides a plausible explanation for the age-dependent development of glucose intolerance, insulin resistance, and decreased circulating insulin in aged MKO mice. Important unanswered questions are how loss of Pgc-1α in muscle impacts inflammatory signaling in other tissues and whether these effects are due to defects in mitochondrial metabolism or nonmitochondrial actions of PGC-1. Overexpressed Pgc-1α or -1β inhibits NF-κB signaling in muscle cells (1, 10), and forced exercise potentiates inflammation in muscle lacking muscle Pgc-1α (18, 54). However, lack of increased inflammatory markers in muscle of sedentary MKO mice suggests that muscle may not be the primary site of inflammation and implicates alternative mechanisms of interorgan cross-talk. Recently, PGC-1α was shown to regulate the expression of myokines such as FNDC5, myostatin, and IGF-I (5, 35, 46), and our GSEA analysis identifies altered muscle TGFβ signaling in both aged and MKO muscle, indicating that alterations in hormone expression, secretion, or action may mediate the non-cell-autonomous effects of losing muscle Pgc-1α on whole body glucose homeostasis.
Our gene expression analysis also illustrated that a significant proportion of muscle Pgc-1α activity (∼35%) may be involved in molecular mechanisms underlying aging. However, only 6% of total age-related gene changes were mimicked by loss of Pgc-1α. Thus, although decreased Pgc-1α may not be sufficient to drive muscle aging, it appears to play a significant role in age-associated loss of muscle oxidative capacity and glucose intolerance. Of note, GSEA identified only altered ETC function within aging muscle, whereas other pathways of mitochondrial biology (lipid oxidation, ATP metabolism, or tricarboxylic acid cycling) were not significantly enriched. Although this may be a function of the large aging data set and limits of our analysis, it is consistent with studies specifically identifying oxidative phosphorylation as a major player in deregulated muscle metabolism during aging (40, 56).
Diabetes is a progressive and chronic disorder in humans, with diagnosis commonly made in midlife to late life. Evidence suggests that decreased physical activity, increased adiposity, and gradually declining mitochondrial function act synergistically to regulate insulin sensitivity during aging. Importantly, reduced mitochondrial function in MKO mice preceded increased adiposity, glucose intolerance, and insulin resistance. Our study highlights that preexisting, modest disruption of mitochondrial gene expression and function early in life, when combined with a sedentary lifestyle, can disrupt glucose homeostasis in later years. Identification of mitochondrial deficiencies in young adults may help predict diabetes risk, and augmentation of mitochondrial function could be a valid approach to delay or prevent disease progression.
GRANTS
This research was funded by grants from the National Research Council of Canada (H. L. Holmes Award) and Canadian Diabetes Association to J. L. Estall and the Institut de Recherches Cliniques de Montréal to J. L. Estall and B. Haibe-Kains.
DISCLOSURES
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
S.S., A.A., S.K., D.L.-B., C.D.W., and J.L.E. performed the experiments; S.S., A.B.-P., A.A., S.K., D.L.-B., C.D.W., J.L.R., B.H.-K., and J.L.E. analyzed the data; S.S., A.B.-P., J.L.R., B.H.-K., and J.L.E. interpreted the results of the experiments; S.S., A.B.-P., S.K., C.D.W., J.L.R., B.H.-K., and J.L.E. edited and revised the manuscript; S.S., A.B.-P., A.A., S.K., D.L.-B., C.D.W., J.L.R., B.H.-K., and J.L.E. approved the final version of the manuscript; J.L.R. and J.L.E. conceived and designed the research; B.H.-K. and J.L.E. prepared the figures; J.L.E. drafted the manuscript.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Bruce Spiegelman for critical analysis, helpful discussion, and guidance and the Specialized Assay Core at Joslin Diabetes Center, Boston, MA (5-P30-DK-36836), for hormone and lipid analysis.
REFERENCES
- 1.Alvarez-Guardia D, Palomer X, Coll T, Davidson MM, Chan TO, Feldman AM, Laguna JC, Vázquez-Carrera M. The p65 subunit of NF-kappaB binds to PGC-1alpha, linking inflammation and metabolic disturbances in cardiac cells. Cardiovasc Res 87: 449–458, 2010. [DOI] [PubMed] [Google Scholar]
- 2.Barres R, Osler ME, Yan J, Rune A, Fritz T, Caidahl K, Krook A, Zierath JR. Non-CpG methylation of the PGC-1alpha promoter through DNMT3B controls mitochondrial density. Cell Metab 10: 189–198, 2009. [DOI] [PubMed] [Google Scholar]
- 3.Barres R, Yan J, Egan B, Treebak JT, Rasmussen M, Fritz T, Caidahl K, Krook A, O'Gorman DJ, Zierath JR. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab 15: 405–411, 2012. [DOI] [PubMed] [Google Scholar]
- 4.Bergman BC, Howard D, Schauer IE, Maahs DM, Snell-Bergeon JK, Eckel RH, Perreault L, Rewers M. Features of hepatic and skeletal muscle insulin resistance unique to type 1 diabetes. J Clin Endocrinol Metab 97: 1663–1672, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bostrom P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, Rasbach KA, Bostrom EA, Choi JH, Long JZ, Kajimura S, Zingaretti MC, Vind BF, Tu H, Cinti S, Hojlund K, Gygi SP, Spiegelman BM. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 481: 463–468, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi CS, Befroy DE, Codella R, Kim S, Reznick RM, Hwang YJ, Liu ZX, Lee HY, Distefano A, Samuel VT, Zhang D, Cline GW, Handschin C, Lin J, Petersen KF, Spiegelman BM, Shulman GI. Paradoxical effects of increased expression of PGC-1alpha on muscle mitochondrial function and insulin-stimulated muscle glucose metabolism. Proc Natl Acad Sci USA 105: 19926–19931, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crane JD, Devries MC, Safdar A, Hamadeh MJ, Tarnopolsky MA. The effect of aging on human skeletal muscle mitochondrial and intramyocellular lipid ultrastructure. J Gerontol A Biol Sci Med Sci 65: 119–128, 2010. [DOI] [PubMed] [Google Scholar]
- 8.Dillon LM, Rebelo AP, Moraes CT. The role of PGC-1 coactivators in aging skeletal muscle and heart. IUBMB Life 64: 231–241, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol 11: 98–107, 2011. [DOI] [PubMed] [Google Scholar]
- 10.Eisele PS, Salatino S, Sobek J, Hottiger MO, Handschin C. The peroxisome proliferator-activated receptor γ coactivator 1α/β (PGC-1) coactivators repress the transcriptional activity of NF-κB in skeletal muscle cells. J Biol Chem 288: 2246–2260, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Estall JL, Kahn M, Cooper MP, Fisher FM, Wu MK, Laznik D, Qu L, Cohen DE, Shulman GI, Spiegelman BM. Sensitivity of lipid metabolism and insulin signaling to genetic alterations in hepatic peroxisome proliferator-activated receptor-gamma coactivator-1alpha expression. Diabetes 58: 1499–1508, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest 116: 615–622, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghosh S, Lertwattanarak R, Lefort N, Molina-Carrion M, Joya-Galeana J, Bowen BP, Garduno-Garcia Jde J, Abdul-Ghani M, Richardson A, DeFronzo RA, Mandarino L, Van Remmen H, Musi N. Reduction in reactive oxygen species production by mitochondria from elderly subjects with normal and impaired glucose tolerance. Diabetes 60: 2051–2060, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goodpaster BH. Mitochondrial deficiency is associated with insulin resistance. Diabetes 62: 1032–1035, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodpaster BH, Wolfe RR, Kelley DE. Effects of obesity on substrate utilization during exercise. Obes Res 10: 575–584, 2002. [DOI] [PubMed] [Google Scholar]
- 16.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol 29: 415–445, 2011. [DOI] [PubMed] [Google Scholar]
- 17.Hadjiyanni I, Baggio LL, Poussier P, Drucker DJ. Exendin-4 modulates diabetes onset in nonobese diabetic mice. Endocrinology 149: 1338–1349, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Handschin C, Chin S, Li P, Liu F, Maratos-Flier E, Lebrasseur NK, Yan Z, Spiegelman BM. Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1alpha muscle-specific knock-out animals. J Biol Chem 282: 30014–30021, 2007. [DOI] [PubMed] [Google Scholar]
- 19.Handschin C, Choi CS, Chin S, Kim S, Kawamori D, Kurpad AJ, Neubauer N, Hu J, Mootha VK, Kim YB, Kulkarni RN, Shulman GI, Spiegelman BM. Abnormal glucose homeostasis in skeletal muscle-specific PGC-1alpha knockout mice reveals skeletal muscle-pancreatic beta cell crosstalk. J Clin Invest 117: 3463–3474, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Handschin C, Kobayashi YM, Chin S, Seale P, Campbell KP, Spiegelman BM. PGC-1alpha regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev 21: 770–783, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hernández-Alvarez MI, Thabit H, Burns N, Shah S, Brema I, Hatunic M, Finucane F, Liesa M, Chiellini C, Naon D, Zorzano A, Nolan JJ. Subjects with early-onset type 2 diabetes show defective activation of the skeletal muscle PGC-1{alpha}/Mitofusin-2 regulatory pathway in response to physical activity. Diabetes Care 33: 645–651, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holloszy JO. “Deficiency” of mitochondria in muscle does not cause insulin resistance. Diabetes 62: 1036–1040, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson ML, Robinson MM, Nair KS. Skeletal muscle aging and the mitochondrion. Trends Endocrinol Metab 24: 247–256, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kleiner S, Mepani RJ, Laznik D, Ye L, Jurczak MJ, Jornayvaz FR, Estall JL, Chatterjee Bhowmick D, Shulman GI, Spiegelman BM. Development of insulin resistance in mice lacking PGC-1α in adipose tissues. Proc Natl Acad Sci USA 109: 9635–9640, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lanza IR, Nair KS. Muscle mitochondrial changes with aging and exercise. Am J Clin Nutr 89: 467S–471S, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leone T, Lehman J, Finck B, Schaeffer PJ, Boudina S, Courtois M, Wozniak DF, Sambandam N, Bernal-Mizrachi C, Chen Z, Holloszy JO, Medeiros DM, Schmidt RE, Saffitz JE, Abel ED, Semenkovich CF, Kelly DP. PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol 3: e101, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, Courtois M, Wozniak DF, Sambandam N, Bernal-Mizrachi C, Chen Z, Holloszy JO, Medeiros DM, Schmidt RE, Saffitz JE, Abel ED, Semenkovich CF, Kelly DP. PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol 3: e101, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li S, Czubryt MP, McAnally J, Bassel-Duby R, Richardson JA, Wiebel FF, Nordheim A, Olson EN. Requirement for serum response factor for skeletal muscle growth and maturation revealed by tissue-specific gene deletion in mice. Proc Natl Acad Sci USA 102: 1082–1087, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jager S, Vianna CR, Reznick RM, Cui L, Manieri M, Donovan MX, Wu Z, Cooper MP, Fan MC, Rohas LM, Zavacki AM, Cinti S, Shulman GI, Lowell BB, Krainc D, Spiegelman BM. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 119: 121–135, 2004. [DOI] [PubMed] [Google Scholar]
- 30.Ma D, Li S, Lucas EK, Cowell RM, Lin JD. Neuronal inactivation of peroxisome proliferator-activated receptor gamma coactivator 1alpha (PGC-1alpha) protects mice from diet-induced obesity and leads to degenerative lesions. J Biol Chem 285: 39087–39095, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mercken EM, Carboneau BA, Krzysik-Walker SM, de Cabo R. Of mice and men: the benefits of caloric restriction, exercise, and mimetics. Ageing Res Rev 11: 390–398, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Michael LF, Wu Z, Cheatham RB, Puigserver P, Adelmant G, Lehman JJ, Kelly DP, Spiegelman BM. Restoration of insulin-sensitive glucose transporter (GLUT4) gene expression in muscle cells by the transcriptional coactivator PGC-1. Proc Natl Acad Sci USA 98: 3820–3825, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mootha VK, Handschin C, Arlow D, Xie X, St Pierre J, Sihag S, Yang W, Altshuler D, Puigserver P, Patterson N, Willy PJ, Schulman IG, Heyman RA, Lander ES, Spiegelman BM. Erralpha and Gabpa/b specify PGC-1alpha-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc Natl Acad Sci USA 101: 6570–6575, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34: 267–273, 2003. [DOI] [PubMed] [Google Scholar]
- 35.Mormeneo E, Jimenez-Mallebrera C, Palomer X, De Nigris V, Vázquez-Carrera M, Orozco A, Nascimento A, Colomer J, Lerín C, Gómez-Foix AM. PGC-1α induces mitochondrial and myokine transcriptional programs and lipid droplet and glycogen accumulation in cultured human skeletal muscle cells. PLoS One 7: e29985, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Olesen J, Ringholm S, Nielsen MM, Brandt CT, Pedersen JT, Halling JF, Goodyear LJ, Pilegaard H. Role of PGC-1alpha in exercise training- and resveratrol-induced prevention of age-associated inflammation. Exp Gerontol 48: 1274–1284, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park SW, Goodpaster BH, Lee JS, Kuller LH, Boudreau R, de Rekeneire N, Harris TB, Kritchevsky S, Tylavsky FA, Nevitt M, Cho YW, Newman AB. Excessive loss of skeletal muscle mass in older adults with type 2 diabetes. Diabetes Care 32: 1993–1997, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci USA 100: 8466–8471, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pedersen BK, Febbraio MA. Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev Endocrinol 8: 457–465, 2012. [DOI] [PubMed] [Google Scholar]
- 40.Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science 300: 1140–1142, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peterson CM, Johannsen DL, Ravussin E. Skeletal muscle mitochondria and aging: a review. J Aging Res 2012: 194821, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Phielix E, Szendroedi J, Roden M. Mitochondrial function and insulin resistance during aging: a mini-review. Gerontology 57: 387–396, 2011. [DOI] [PubMed] [Google Scholar]
- 43.Pospisilik JA, Knauf C, Joza N, Benit P, Orthofer M, Cani PD, Ebersberger I, Nakashima T, Sarao R, Neely G, Esterbauer H, Kozlov A, Kahn CR, Kroemer G, Rustin P, Burcelin R, Penninger JM. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell 131: 476–491, 2007. [DOI] [PubMed] [Google Scholar]
- 44.Ringholm S, Olesen J, Pedersen JT, Brandt CT, Halling JF, Hellsten Y, Prats C, Pilegaard H. Effect of lifelong resveratrol supplementation and exercise training on skeletal muscle oxidative capacity in aging mice; impact of PGC-1alpha. Exp Gerontol 48: 1311–1318, 2013. [DOI] [PubMed] [Google Scholar]
- 45.Rowe GC, Patten IS, Zsengeller ZK, El-Khoury R, Okutsu M, Bampoh S, Koulisis N, Farrell C, Hirshman MF, Yan Z, Goodyear LJ, Rustin P, Arany Z. Disconnecting mitochondrial content from respiratory chain capacity in PGC-1-deficient skeletal muscle. Cell Rep 3: 1449–1456, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruas JL, White JP, Rao RR, Kleiner S, Brannan KT, Harrison BC, Greene NP, Wu J, Estall JL, Irving BA, Lanza IR, Rasbach KA, Okutsu M, Nair KS, Yan Z, Leinwand LA, Spiegelman BM. A PGC-1alpha isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell 151: 1319–1331, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci USA 91: 10771–10778, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shu CJ, Benoist C, Mathis D. The immune system's involvement in obesity-driven type 2 diabetes. Semin Immunol 24: 436–442, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Summermatter S, Handschin C. PGC-1alpha and exercise in the control of body weight. Int J Obes 36: 1428–1435, 2012. [DOI] [PubMed] [Google Scholar]
- 50.Summermatter S, Shui G, Maag D, Santos G, Wenk MR, Handschin C. PGC-1alpha improves glucose homeostasis in skeletal muscle in an activity-dependent manner. Diabetes 62: 85–95, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Summermatter S, Troxler H, Santos G, Handschin C. Coordinated balancing of muscle oxidative metabolism through PGC-1alpha increases metabolic flexibility and preserves insulin sensitivity. Biochem Biophys Res Commun 408: 180–185, 2011. [DOI] [PubMed] [Google Scholar]
- 52.Wenz T, Rossi SG, Rotundo RL, Spiegelman BM, Moraes CT. Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging. Proc Natl Acad Sci USA 106: 20405–20410, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 53.Wredenberg A, Freyer C, Sandstrom ME, Katz A, Wibom R, Westerblad H, Larsson NG. Respiratory chain dysfunction in skeletal muscle does not cause insulin resistance. Biochem Biophys Res Commun 350: 202–207, 2006. [DOI] [PubMed] [Google Scholar]
- 54.Wu J, Ruas JL, Estall JL, Rasbach KA, Choi JH, Ye L, Bostrom P, Tyra HM, Crawford RW, Campbell KP, Rutkowski DT, Kaufman RJ, Spiegelman BM. The unfolded protein response mediates adaptation to exercise in skeletal muscle through a PGC-1alpha/ATF6alpha complex. Cell Metab 13: 160–169, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98: 115–124, 1999. [DOI] [PubMed] [Google Scholar]
- 56.Zahn JM, Sonu R, Vogel H, Crane E, Mazan-Mamczarz K, Rabkin R, Davis RW, Becker KG, Owen AB, Kim SK. Transcriptional profiling of aging in human muscle reveals a common aging signature. PLoS Genet 2: e115, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zechner C, Lai L, Zechner JF, Geng T, Yan Z, Rumsey JW, Collia D, Chen Z, Wozniak DF, Leone TC, Kelly DP. Total skeletal muscle PGC-1 deficiency uncouples mitochondrial derangements from fiber type determination and insulin sensitivity. Cell Metab 12: 633–642, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.