

Abstract
Gene targeting in mice (transgenic and knockout) has provided investigators with an unparalleled armamentarium in recent decades to dissect the cellular and molecular basis of critical pathophysiological states. Fruitful information has been derived from studies using these genetically engineered mice with significant impact on our understanding, not only of specific biological processes spanning cell proliferation to cell death, but also of critical molecular events involved in the pathogenesis of human disease. This review will focus on the use of gene-targeted mice to study various models of lung disease including airways diseases such as asthma and chronic obstructive pulmonary disease, and parenchymal lung diseases including idiopathic pulmonary fibrosis, pulmonary hypertension, pneumonia, and acute lung injury. We will attempt to review the current technological approaches of generating gene-targeted mice and the enormous dataset derived from these studies, providing a template for lung investigators.
Keywords: transgenic, knockout, asthma, chronic obstructive pulmonary disease, idiopathic pulmonary fibrosis, acute respiratory distress syndrome, pulmonary hypertension
the successful application of gene targeting to specifically induce genetic modifications in animals represents a tour de force approach to better understand regulation and function of specific genes. The sequencing of the human genome, and the subsequent massive high throughput profiling “omics” assays currently available in the functional genomics era such as genome-wide association studies, epigenetics, large-scale analysis of gene expression, microRNA targeting and expression, metabolomics, and proteomics have made it even more imperative that we validate the function of these candidate molecules in the context of human disease models. Gene targeting (i.e., overexpression, or gain of function, via transgenic mice; and knockdown, or loss of function, via null knockout mice) in animals plays critical roles in these functional studies. Moreover, current technology exists to generate gene-targeted modifications in animals ranging from mice and rats to large animals such as sheep, goats, chicken, cows, pigs, and nonhuman primates. The seminal discovery of principles for introducing specific gene modifications in mice by Drs. Mario R. Capecchi, Martin J. Evans, and Oliver Smithies [awarded the 2007 Nobel Prize in Physiology or Medicine (101)] has transformed biomedical research over the past 30 years and has generated intense investigations in the study of mammalian biology, and its application toward improved understanding of the pathophysiology and pathogenesis of human diseases.
Pulmonary investigators, as well as investigators in all fields of biomedical research and clinical disciplines, have been challenged to use gene-targeting approaches to unravel the function of specific gene(s), with the hope of translating the discoveries made in gene-targeted mice into new diagnostic and therapeutic modalities for human lung diseases. The lung community has experienced successes and has received priceless dividends from these gene-targeted approaches, but we are also reminded that this approach has limitations, as does any technology available currently in the armamentarium of research tools (see Table 1). The sheer number of cell types (∼60 cell types) in the lung, each with distinct and overlapping functions in disease processes, presents unique challenges compared with other organs. Furthermore, the unique physiological responses of the lung (e.g., hypoxic vasoconstriction in the lung in contrast to vasodilation as might occur in other organs) further highlight the complexity of the lung and the critical necessity to examine lung-specific molecular regulation and function and lung-specific gene targeting. Our goal in this review is to provide a basic update on gene-targeting approaches (with a primary focus on gene-targeted mice) that have been used by lung investigators in recent years, with specific application to the more “common” ailments chronic obstructive pulmonary disease (COPD), asthma, pneumonia, acute respiratory distress syndrome (ARDS), idiopathic pulmonary fibrosis (IPF), and pulmonary hypertension (see Supplemental Table S2).
Table 1.
Limitations of genetically modified mice in the study of lung disease
| Category of Limitation | Limitation | Possible Remedies |
|---|---|---|
| Mouse model effects | Variable biology based on disease model and mouse strain used | Test multiple models and strains and confirm biological findings mechanistically |
| Variable biology based on gender | Confine conclusions to 1 gender examined | |
| Integration of the genetically modified vector | It is possible that observed biology can be on the basis of disruption of another gene from random integration rather than the gene of interest | Test more than 1 line of genetically modified mice to confirm that the findings are related to the target gene |
| Variable biology might be observed based on the copy number of a transgenic insertion | Confirm biology with different lines of mice with variable copy numbers as is possible and confirm mechanism in vitro and/or in another in vivo system | |
| Off-target effects of tools used to generate genetically modified mice | Doxycycline (and tetracycline analogs) can have antibacterial effects, effects on bone, effects on the placenta in pregnant mice (fetoplacental toxicity) (114), effects on alveolar development (especially at high doses) (139) | Use lowest dose possible of doxycycline to achieve induction of expression; consider use of a more sensitive promoter that might allow induction with lower doses of doxycycline (114) |
| Air space enlargement observed in some CCSP-rtTA lines in absence of doxycycline and concerns raised that might also be the case for some SPC-rtTA lines (139) | As possible, use lines of mice that have been exhaustively tested for off-target effects and include controls to rule out independent effects of the tissue-specific promoter | |
| Toxicity to mammalian cells with prolonged exposure of Cre-recombinase (perhaps related to “illegitimate” recombination of nonloxP sites) (97, 137) | Limit intensity and duration of Cre exposure as much as possible; include controls in experiments to rule out off-target effects of Cre-recombinase [i.e., can consider conditional expression: more recently generated lines of SPC-rtTA and CCSP-rtTA have demonstrated less toxicity when combined with Cre-recombinase (120)] |
CCSP, Clara cell secretory protein; SPC, surfactant protein C; rtTA, reverse tetracycline transcriptional activator.
GENERATING KNOCKOUT, KNOCKIN, AND TRANSGENIC ANIMALS
The ability to insert exogenous DNA randomly into the genome through injection of pronuclei to produce transgenic mice (or expression of the DNA at a “trans” or nonendogenous location) was first reported over 30 years ago (55, 56). However, the fundamental concept of genetically engineered knockout or knockin mice rests in the ability to induce a specific genetic modification within the chromosome of an embryonic stem (ES) cell. These modified ES cells are then used to derive mice with this alteration that can be passed along to subsequent offspring. Creation of genetically engineered mice thus depends on two key concepts: homologous recombination and introduction of altered genetic material into ES cells (101). Homologous recombination was initially described in the context of sexual reproduction in which meiosis produces a haploid gamete from a diploid germ cell. With joining of two haploid gametes, a diploid zygote is formed that contains one set of chromosomes from the egg and one from the sperm. As the diploid zygote develops, chromosomes from each of the parents recombine at sites of homologous genes to produce unique genetic offspring. It was known that homologous recombination could occur within somatic cells in the context of DNA repair to allow the undamaged chromosome to serve as a template for repair of the damaged strand. However, it was Oliver Smithies (140) who discovered that homologous recombination within somatic cells could serve as a significant source of allelic variation and thus opened the door for considering introduction of new genetic material in a targeted fashion. In parallel, Mario R. Capecchi (152) reported microinjection of DNA into a somatic cell nucleus and noted homologous recombination of the introduced genetic material with the genetic machinery of the recipient cell. To propagate targeted genetic modification in mice, introduction of the new genetic material would be required in cells that contributed to germ cell formation. Both Smithies and Capecchi relied on Martin J. Evans's (39) development of a pluripotent ES cell line derived from mouse blastocysts, and their work led to development of the techniques that have been used widely to generate genetically engineered mice.
“Global” Knockout Mice (or Gene Knockout in All Tissues)
A general approach toward development of a “knockout” mouse relies on generating a vector that, through homologous recombination after introduction into ES cells (commonly through electroporation), will result in deletion of the desired portion of the target gene (Fig. 1). By including positive (and sometimes negative) selection markers within the targeting vector, recombinant ES cells harboring the knockout construct can be selected after screening, injected into mouse blastocysts, and transferred to surrogate mothers to produce a novel line of mice with targeted deletion of the desired gene or gene segment. Thus in vivo studies examining the effect of loss of gene expression of the target gene can be carried out. The initial offspring are chimeric (carrying cells derived both from host and ES cells, often initially from different strains), and mice carrying the germ line targeted deletion can be selected and bred to homozygosity. Similar techniques can be used to “knock in” a mutated version of the target gene in place of the endogenous gene, allowing study of changes in protein structure or function.
Fig. 1.
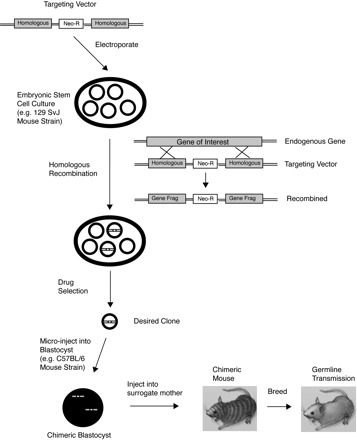
Generation of knockout mice. A vector is constructed containing homologous regions of the gene targeted for deletion (Homologous, in gray) that surround a drug selection marker such as the neomycin resistance cassette (Neo-R). This vector is then introduced into embryonic stem cells in culture (e.g., by electroporation), where homologous recombination with the endogenous gene of interest occurs, resulting in a subset of cells containing the recombined product containing the disrupted endogenous gene (gene fragments, or Gene Frag) and the drug selection marker. Drug selection is then performed to isolate the desired clone, which is then microinjected into a blastocyst that is then transferred to the uterus of a surrogate mother, with the goal of obtaining germ line transmission and offspring harboring the genetic modification. (Adapted from Ref. 101 with permission).
Pitfalls of global knockout mice.
Global knockout animals are now widely used and have revealed unexpected functions for targeted genes (see Supplemental Table S1), but generation of these mice still can be complicated and time consuming. Deletion of genes important for development can result in embryonic lethality. Thus study of these targets as well as the analysis of tissue-specific effects of gene targeting might require development of more complicated mice with conditional expression (see below). Conversely, deletion of genes for which there exist other genes with overlapping functions can result in the development of no novel phenotype at all (29). For example, there exists a single gene in humans that produces the protein α-1-antitrypsin (A1AT), Serpinea1; however, in mice there exist three to five genes with overlapping functions responsible for A1AT production, thus making generation of an A1AT knockout mouse extremely challenging (161).
Global Transgenic Mice (or Gene Overexpression in All Tissues)
Conversely, generation of a “transgenic” mouse that overexpresses a target gene requires microinjection of the gene sequence (often under control of a tissue-specific promoter, see below and Fig. 2) into a newly fertilized oocyte, where the gene integrates randomly into the genome, usually at a single chromosomal location, and with variable copy numbers of the gene inserted. The injected eggs are transferred to a surrogate mother, and offspring harboring germ line expression of the transgene are bred to produce a novel (or “founder”) line of mice with overexpression of the target gene. Compared with knockout animals, transgenic mice generally are easier to generate, can produce high levels of expression of the desired transgene, and, as a result, were more widely used before knockout animals.
Fig. 2.
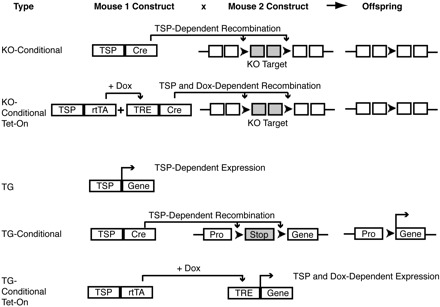
Approaches in generating knockout and transgenic mice. Shown here are some examples of strategies in generating knockout and transgenic mice with tissue-specific and conditional expression of genetic modifications. In 1 type of conditional expression system, a mouse containing a tissue-specific promoter (TSP) driving Cre-recombinase can be bred to mice harboring targeted regions for deletion flanked by loxP sites, with resultant offspring harboring tissue-specific expression of the recombined genetic material. This strategy can be used to conditionally delete a gene target in knockout mice (KO) or overexpress a gene target in a transgenic mouse (TG) by deleting a genetically engineered Stop codon, thus allowing the promoter (Pro) to transcribe the gene of interest (see KO-Conditional and TG-Conditional examples in figure). Another type of conditional expression system involves engineering a TSP to drive rtTA (reverse tetracycline-controlled transactivator) that, when combined with a tetracycline-response element (TRE) in the presence of doxycycline, permits TSP and doxycycline (Dox)-dependent gene expression (see TG-Conditional example in figure). Dox-dependent Cre-recombination can also be undertaken, as in the KO-Conditional Tet-on example in the figure, thus employing multiple levels of complexity in controlling gene expression. [Adapted with kind permission from Springer Science+Business Media: Aoki K, Taketo MM. Tissue-specific transgenic, conditional knockout and knock-in mice of genes in the canonical Wnt signaling pathway. In: Wnt Signaling Volume 1: Pathway Methods and Mammalian Models, edited by Vincan E. Totowa, NJ: Humana, 2008, vol. 468, chapt. 24.].
Pitfalls of global transgenic mice.
The site of integration of the transgene can affect expression of the transgene, such that copy number of the inserted transgene does not necessarily correlate with levels of transgene expression. Moreover, most transgenic mice are mosaic for the transgene, in that the transgene may not be expressed in every cell of the body. Mosaicism has been attributed to the fact that chromosomal replication is rapid in mice (in contrast to larger animals) and is already occurring at the time of microinjection of the embryo (168). Thus levels of transgene expression should be analyzed in each founder line. Some investigators have proposed using DNA insulator sequences to mitigate the integration effects of transgene expression, but this technique has not proven uniformly successful (155). Transgene integration also has the potential to disrupt unintended DNA sequences (in as many as 5–10% of transgenic mice) and thus can affect expression of other genes (105, 150). Thus at least two independent lines of mice from different founders should be analyzed for function of the transgene, to confirm that the observed effects result from overexpression of the intended target gene, rather than an unintended genetic mutation. Moreover, in global transgenic mice, tissue-specific expression of the transgene should be analyzed as well, as the efficacy of promoter expression can vary between tissues (especially in nonendogenous target tissues). Finally, transgene expression can produce nonphysiological (and perhaps even toxic levels) of gene expression, such that the observed effect of the transgene might not apply to endogenous levels of gene expression (29, 120).
To date, the majority of genetic manipulation has been carried out in mice, although clearly limitations in translatability of murine models to human disease have been extensively described in recent reviews, with some aspects reviewed briefly below (19, 51, 65, 102). The ability to routinely perform genetic modification in larger mammals will expand our capabilities of carrying out clinically relevant research. Interestingly, Rogers et al. (128, 129) used this rationale to generate pigs with targeted disruption of the cystic fibrosis transmembrane receptor gene (CFTR) and found that CFTR-deficient (CFTR−/−) pigs recapitulated human cystic fibrosis far more accurately than did CFTR−/− mice. More recently, Tong et al. (154) demonstrated the feasibility of gene targeting in rats through homologous recombination in pluripotent rat ES cells, thus expanding dramatically the future repertoire of possible experiments using rat-based systems.
A number of exciting and unexpected findings have arisen from examination of the lung in global knockout or transgenic mice, and many of these have furthered our basic mechanistic understanding of “common” diseases, such as COPD, IPF, pneumonia, ARDS, and pulmonary hypertension (see Supplemental Table S1). Although beyond the scope of this article, there also exist important examples of the contribution of global knockout mice to our understanding of other “less common” diseases, such as the discovery that granulocyte macrophage colony-stimulating factor (GMCSF)-deficient mice develop pulmonary alveolar proteinosis (PAP) as a result of ineffective macrophage degradation of ingested surfactant (141). These studies ultimately led to important further mechanistic understanding of the disease and to consideration of GMCSF repletion in a subset of humans with PAP (81). However, many genes are lethal in mice when globally disrupted, requiring more complex genetic manipulations to derive animals with tissue- and temporal-specific expression.
TISSUE AND TEMPORAL SPECIFICITY
The ability to localize expression within specific tissues (by using a tissue-specific promoter) and under specific times/conditions (by using conditional expression systems) represent powerful tools for dissecting the effects of altered gene expression. Moreover, this technology has facilitated the study of genes that when globally and/or constitutively overexpressed or knocked out result in embryonic lethality and have allowed examination of biological effects as a result of localized expression within the lung.
Lung Specificity
There exist a number of promoters that have been well-characterized and have allowed for localized expression within specific tissues, including central nervous system, liver, heart, immune system, mammary gland, reproductive system, skin, and bone (6, 54). Interestingly, a recent paper used the VE-cadherin-5 promoter to target endothelial-specific blockade of NF-κB activation during murine models of endotoxemia and sepsis (cecal ligation and puncture) (174). Within the lung, the specific location and timing of gene expression of Clara cell secretory protein (CCSP, also known as CC10) and surfactant protein C (SPC) that are expressed exclusively within the lung epithelium have led to widespread use of these promoters to direct gene expression to distinct lung sites (138, 145, 163). CCSP is synthesized and expressed primarily in the larger airway and proximal epithelial cells of the lung (∼40% of airway epithelial cells), whereas SPC is synthesized and secreted predominantly by the more distal type II alveolar epithelial cells. To target larger airway-specific expression in adult mice, a 2.3-kb rat CCSP promoter was engineered and has been widely used (127), and, similarly, to target more distal airway-specific expression a 3.7-kb human SPC promoter was generated and has also been widely used (165).
Pitfalls of lung-specific promoters.
SPC-targeted distal expression is observed only after the embryonic day 15.5–17.5 stage of embryonic development, such that SPC-driven transgene expression prior to this stage affects a more uniform population of precursor cells in the entire lung epithelium. Thus a number of investigators have used SPC-driven gene expression to study lung development and the influence of different targeted pathways on the differentiation of cells into type I or type II epithelial cells (96). Although CCSP and SPC have been the main promoters used to target gene expression to lung epithelial cells, there has been interest in promoters with more specific proximal or distal localization of epithelial expression. Interestingly, Flodby et al. (43) have recently described a new transgenic mouse line with selective expression of Cre-recombinase (see below) in the distal type I alveolar epithelial cells (ATI cells), using aquaporin-5 (Aqp5) to direct expression at this site. To do this, they knocked in Cre-recombinase (described in detail below) into exon 1 of the endogenous Aqp5 gene. Although, as hoped, there was no specific expression of Cre-recombinase in these mice in the distal type II alveolar epithelial cells (ATII cells), the authors observed variable expression in some other tissues with endogenous Aqp5 expression, such as submandibular salivary gland, tracheal epithelium, brain, and stomach. It should be noted that selective expression of Aqp5 in ATI cells exclusively without ATII expression has been observed in 129S6/SvEvTac mice, but not in C57BL6 mice, in which low-level Aqp5 expression can be detected in ATII cells.
Conditional-Expression Systems
Conditional expression systems have allowed for significant advancement in biological understanding with the possibility of turning genes “on” or “off” at specific times. This technology has allowed complex study of developmental biology and, conversely, has allowed for expression of genes during development that can later be modified postnatally and under specific injury conditions. Although there exist a number of conditional expression systems (6), two often-used systems in lung biology are 1) the tetracycline-responsive system and 2) the Cre-mediated recombination system, with both systems often used under the direction of a tissue-specific promoter (see Fig. 2). A system less often used in lung biology studies, the estrogen receptor-activated system, will also be briefly mentioned.
Tetracycline-Responsive System
The tetracycline-responsive system is based on the Escherichia coli tetracycline resistance operon (57, 58), in which the tetracycline repressor (tetR) binds to the operator sequences (tetO) within the promoter of the tetracycline resistance operon, thus inhibiting transcription. Tetracycline, when present, binds tetR, thus prohibiting binding of tetR to tetO, and gene transcription of the tetracycline resistance operon is permitted. Thus investigators fused tetR to VP16 (a transcriptional activator) to produce the tetracycline-dependent transactivating factor (tTA). Furthermore, the target gene of interest was placed under control of the tetO sequences and basic promoter elements, producing what has been termed a tetracycline-response element (TRE) (57, 150). Thus, when tetracycline (or the commonly used analog doxycycline) is present, tTA cannot bind the TRE, and gene transcription is prohibited. Thus this system has been termed the “tet-off” system, because transcription of the target gene is off in the presence of tetracycline. One approach toward generating conditional lung-specific expression involves mating a mouse with the tTA region under control of a lung-specific promoter, with a mouse that harbors the target gene under control of a TRE promoter. Thus, in mice carrying both the tTA and TRE elements, transcription of the gene of interest can be expressed the majority of the time, then turned off in the presence of doxycycline. This tet-off system is often used to allow for gene expression during development and subsequent modulation of its expression thereafter.
A “tet-on” system has been developed through engineering the reverse tetracycline transcriptional activator (rtTA) that binds tetO and activates gene transcription only in the presence of doxycycline (59, 105). Thus, in mice carrying both the rtTA under the control of a lung-specific promoter and TRE elements (as a result of a breeding strategy as described above for tet-off mice), lung-specific gene expression occurs only in the presence of administration of doxycycline (or another tetracycline analog). This tet-on system allows for a gene to be kept inactive the majority of the time, then activated as desired with the addition of doxycycline. It must be remembered that the kinetics of altered gene expression after administration (or withdrawal) of doxycycline can vary, and thus changes in gene expression (in addition to basal expression, see Pitfalls of conditional expression systems: promoter leakiness) have to be assessed over a time course after doxycycline administration. CCSP-rtTA mice and SPC-rtTA mice have been widely used to drive conditional proximal airway or distal airway expression, respectively, of target genes. Clearly, independent effects relating to use of the antibiotic doxycycline in murine models of lung injury (especially those induced by infectious causes) must be considered, and appropriate controls must be used [see Pitfalls of Cre-mediated recombination system (including doxycycline administration)]. However, a recent publication demonstrated no independent antibacterial or anti-inflammatory properties of doxycycline when used to induce transgene expression during a model of polymicrobial sepsis (149).
Pitfalls of conditional expression systems: off-target effects.
As has been reviewed (167), biological (and/or toxic) effects unrelated to expression of the gene target have been attributed to CCSP, rtTA, and doxycycline, and earlier lines of CCSP-rtTA mice exhibited air space enlargement not attributable to transgene expression. However, whereas newer lines of CCSP-rtTA and SPC-rtTA mice used in conjunction with the Cre-recombinase system have not been reported to exhibit these off-target effects (120), it remains important to test transgenic littermates not expressing the transgene (e.g., including CCSP or SPC-rtTA mice, TRE-Cre mice, etc.) to be certain that observed effects are attributable to transgene expression [see discussion regarding doxycycline administration under Pitfalls of Cre-mediated recombination system (including doxycycline administration)].
Pitfalls of conditional expression systems: promoter leakiness.
Concerns have been raised regarding “leakiness” of the TRE promoter (i.e., low levels of undesired gene expression when the gene is supposed to be “turned off”), and thus it is important to confirm that gene expression is truly inducible as intended with the conditional expression system. Many investigators have used newer generation TRE promoters with the goal of decreasing basal activity of the promoter and improving inducibility of the transgene. For example, the pTRE-Tight vector (Clontech) (142) is engineered with the 7-tetO sequences more closely approximated compared with the original pTRE vector, such that there are fewer potential binding sites available for nonspecific endogenous transcription factors. However, intense interest persists in developing “leak-free” doxycycline systems, and Duerr et al. (33) recently reported that a newer-generation rtTA driven by CCSP (termed CCSP-rtTA2S-M2) exhibits no basal activity in the absence of doxycycline, high levels of doxycycline inducibility with dose-dependent effects of doxycycline, increased sensitivity of the promoter to doxycycline, such that 10-fold lower doses of doxycycline are required for induction, and complete reversibility of expression of the conditionally expressed transgene in the absence of doxycycline.
Another more involved strategy to address promoter leakiness that has been employed alone, or in combination with vectors such as the pTRE-Tight vector, includes the combination of rtTA-mice with “silencer” mice, such as those that harbor the tetracycline transcriptional silencer (tTS) element. With the tTS-rtTA double transgenic mice in this system, in the absence of doxycycline, the tTS element binds and silences the TRE promoter element with elimination of basal transcription of the downstream target gene. In the presence of doxycycline, tTS is dissociated, and doxycycline binds the TRE element to activate transcription of the downstream transgene. The tTS strategy thus requires additional generation and breeding steps for double transgenic mice. Generation of these double transgenic mice usually includes a construct containing the desired promoter element, the rtTA sequence, the tTS sequence, and an internal ribosome entry site (IRES) sequence that allows for production of both the rtTA and tTS elements.
Cre-Mediated Recombination System
The Cre-mediated recombination system (or Cre-loxP system) depends on the enzyme Cre-recombinase (cyclization of recombination, a product of the bacteriophage P1 gene and a site-specific recombinase) cleaving at lox sites of bacteriophage P1 (locus of crossing over of P1, loxP) consensus sites, with recombination of the cleaved ends (97, 137). Thus mating one mouse with Cre-recombinase under the control of a tissue-specific promoter (produced by transgenic approaches, see above) to a second mouse engineered to contain a gene or gene segment flanked by two 34-base loxP sites (i.e., “floxed” sites placed within introns of the gene; mice produced by homologous recombination in ES cells, see above) results in excision and recombination of DNA around the gene of interest in mice inheriting both the Cre transgene and the floxed target gene (29). The Cre-loxP system has been used extensively to allow time- and tissue-specific expression of genes, and, moreover, once a floxed mouse has been generated, it can be bred to a variety of tissue-specific (or even global) Cre-recombinase-expressing mice to allow for the study of gene function specifically in different tissues. Given concern from toxicity of prolonged exposure of mammalian cells to Cre-recombinase (see below, and Table 1), Whitsett and colleagues (120) bred the CCSP-rtTA and SPC-rtTA mice, respectively, to TRE-Cre mice, thus enabling conditional expression of Cre-recombinase in localized sites within the respiratory epithelium (see Fig. 2). As described above, whereas initial lines of these rtTA-Cre mice exhibited off-target effects, more recent lines of mice have been made available to the research community that appear to exhibit fewer off-target effects and nonspecific expression.
Pitfalls of Cre-mediated recombination system (including doxycycline administration).
Long-term exposure to Cre-recombinase has been reported to produce toxicity in mammalian cells, as a result of unintended cleavage at “pseudo”-loxP sites and “inappropriate” recombination, with subsequent DNA damage (167). This toxicity is limited with shorter-term Cre-recombinase expression, and thus many investigators are using inducible systems as described above (120). As with any transgenic animal (see Pitfalls of global transgenic mice above), integration and expression levels of Cre-recombinase can be variable. Thus newly generated lines of Cre mice need to be tested as would any new transgenic animal, and efficacy of excision of the floxed target must be confirmed by examining tissue-specific levels of the gene of interest (150). Additionally, placement of the loxP sites can also affect target gene expression, even though these sites are targeted to intron segments, such that expression and function of the wild-type floxed gene prior to excision and recombination should be confirmed. Given the complexity of generating these mouse lines, it is important to include controls that allow for optimal interpretation of the effects of the gene target. Controls should include littermate mice from the floxed lines, Cre lines, and wild-type mice, and when inducible mice are being used (e.g., with doxycycline or 4-hydroxytamoxifen, as below), inducer-treated mice from all of these lines should be included as well as the untreated controls. Additionally, as prolonged administration of doxycycline can result in its deposition and later release from tissues, it has been recommended, especially for experiments involving developmental studies, that doxycycline be limited to as short a period as possible, with limited off-target effects observed with short-term administration during distinct developmental stages (120, 167).
Estrogen Receptor-Activated Systems
The estrogen receptor-activated system is based on a promoter (often tissue-specific) driving the ligand binding domain of a human estrogen receptor (ER) that is activated by administration and binding of 4-hydroxytamoxifen (4-HT) (132). The ER may be fused to proteins (including transcription factors or kinases) that are active in the cell nucleus and is often fused to Cre recombinase (Cre-ER), thus providing another system for time- and location-specific induction of genetic modifications in mice, similar to the concept underlying the rtTA-Cre mice described in the previous section. Basally, the ER fusion protein is sequestered in the cytoplasm; then, with addition of 4-HT and binding and stabilization of the ER, the fusion protein translocates to the nucleus where downstream effects then ensue. ER-driven expression effects depend on stabilization of an existing protein rather than transcription and translation of the target protein as in the tet system, and thus target gene expression may be realized more quickly than the tet system. Additionally, fewer crossings may be required to generate ER-driven conditionally expressing mice, compared with the need to mate lines of rtTA (or tTA) and TRE mice in the tet system.
Pitfalls of estrogen receptor-activated systems.
Concerns are heightened beyond those for the tet system for basal expression of the ER in the absence of 4-HT (i.e., “promoter leak”), given that the ER is endogenously expressed, whereas, in contrast, tetO is not expressed in mammalian cells. In response to these concerns, newer generation ER fusion proteins have been generated through amino acid mutations to decrease the likelihood of binding endogenous 17β-estradiol, while retaining sensitivity to 4-HT (48, 132). Finally, similar to concerns regarding non-gene-targeted effects of administering doxycycline in the tet system, administration of 4-HT can have nonspecific effects on responsive organs (e.g., breast, uterus, liver, bone), and experimental controls should include non-gene-targeted littermate mice treated with 4-HT as well as the untreated controls (29).
DISCOVERIES AND PITFALLS OF GENETICALLY MODIFIED MICE IN LUNG DISEASES
There exist numerous studies that have employed genetically modified mice in characterizing the biology and underlying mechanisms in lung disease pathogenesis. Although some lung biology findings have emerged from “unchallenged” mice at baseline (see example of GMCSF-null mouse above), many important observations in lung biology have emerged after subjecting genetically modified mice to models of lung disease. Because it is impossible to do justice to the extensive important findings in lung biology that have been identified through use of modeling genetically modified mice, we have reviewed some of the key findings that have contributed to our basic understanding of COPD, asthma, pneumonia, ARDS, IPF, and pulmonary hypertension (Supplemental Table S1). Although the list is certainly not exhaustive, it is apparent that: 1) critical discoveries have arisen as a result of use of these animal models; 2) the same genetically modified animal has yielded key information regarding disease pathogenesis for multiple illnesses and has contributed substantially to our understanding of human disease; and 3) despite the immense information of these animal models, there exist important limitations in direct translatability of the findings for each lung disease and have recently been reviewed in depth (11, 19, 65, 102, 109, 144). It is important to understand the limitations of the models themselves, since these factors are of enhanced importance in interpreting findings from genetically modified mice subjected to these models. We have therefore summarized a few key limitations below.
With regard to modeling COPD in mice, it must be considered that mice and humans exhibit different lung anatomy, with mice having few submucosal glands and only six to eight branches before reaching the terminal bronchiole, whereas humans have more than 20 airway branches. Moreover, mice do not possess the respiratory bronchiole structure that is an important initial site of inflammation and is believed to be a critical site in the development of centriacinar emphysema. In addition, genes that are important in the development of COPD in humans are not always expressed in mice, thus prompting some investigators to develop genetically modified mice that overexpress human genes (e.g., see MMP-1 mice in Supplemental Table S1). Although we have chosen to focus on murine models of COPD that arise from cigarette exposure, there are clearly other conditions (genetic and environmentally mediated) that lead to COPD, and thus translatability of findings in one model must be applied with caution to all forms of the disease (135).
Although many of the key biological findings in asthma have arisen from the study of airway hyperreactivity (AHR) as a result of ovalbumin sensitization and challenge in mice (OVA model), there exist important differences in human and murine aspects of asthma: 1) humans with asthma may have basal AHR to methacholine, whereas mice may not exhibit this finding; 2) humans might develop chronic allergic asthma, and mice develop tolerance; 3) IgE and mast cells are critical for the development of human asthma, whereas this is not necessarily the case in mice; 4) mice require short-term, high-level exposure to allergens to develop AHR, whereas humans more typically experience low-level chronic exposure; 5) although eosinophils are activated in some human asthma subjects, these cells may not be activated in the murine model of allergic inflammation (reviewed in Ref. 68). There also exist important strain-related findings with the OVA model, with BALB/c mice exhibiting the best reproducible features of AHR. However, generation of genetically modified mice has not been as straightforward with BALB/c as with other strains such as C57BL/6 strains, and C57BL/6 exhibit different findings in the OVA model (e.g., lateral granulocyte infiltration rather than the peribronchial eosinophil infiltration observed in BALB/c mice). Finally, although we have chosen to focus on the role of genetically modified mice in advancing our understanding of the inflammatory response in asthma, there exist a large number of critical studies that have focused on airway remodeling that has been observed without an accompanying significant inflammatory/eosinophilic response in a number of transgenic mice with CCSP-specific overexpression [e.g., CCSP-driven IL-6 expression (50)].
The study of pneumonia and ARDS has been extremely challenging (102). We have therefore chosen to focus on pneumonia that arises from bacterial infections (Supplemental Table S1). Challenges have included the following: 1) it is necessary to maintain mice in a uniform barrier facility, such that underlying murine infections or differences in microbial flora colonization do not influence the effects of the induced infections; 2) titration of the administered bacteria can often be difficult in achieving the desired infectious response without inducing unintended mortality during the experiment; 3) observed findings from one infectious organism or even a different serotype of the same organism might vary quite widely with another agent, and thus observed conclusions apply solely to the organism under study; 4) genetically modified mice that require administration of an antibiotic, such as doxycycline or tetracycline, to induce expression of a transgene clearly have the potential to influence the course of the experimental bacterial infection, and thus results from such experiments much be interpreted with caution. We have focused on ARDS that arises from induction of ventilator-induced lung injury (VILI) and hyperoxia (75), given that these models have many features that replicate human ARDS (Supplemental Table S1). In VILI, mice are ventilated for 2–8 h, with induction of lung injury as a result of alveolar stretch. Although it is possible to induce VILI with low-tidal-volume ventilation, as has been adopted in the support of patients with ARDS, the degree of injury is often not sufficient to allow perturbation in an experimental system, and thus an element of clinical relevance of the model must be sacrificed. Some studies of VILI have reported using tidal volumes as high as 20–35 ml/kg that would never be considered for human use. A limitation to use of the hyperoxia model is that humans do not appear to develop the profound lung injury that mice exhibit after short-term hyperoxic exposure (102). Increasingly, investigators are applying “two-hit” models, employing sequential lung injuries, which might more accurately reflect clinical disease (e.g., development of sepsis, followed by the need for mechanical ventilation). These dual-injury models, in addition to being time intensive and complicated to reproduce, require increased sophistication in interpreting the complex biological effects of diverse insults. The difficulty of linking a specific genetic modification of a transgenic or knockout animal with multiple different injuries might result in some loss of “elegance” of the mechanistic approaches to genetically modified mice. As with any illness, the ability to confirm key murine findings in human samples heightens the translatability and importance of the genetically modified mouse models.
The study of IPF has been hampered by the lack of murine models that recapitulate the histological features and the chronic, progressive nature of the human disease. We have chosen to focus on the bleomycin model, as intratracheal instillation of bleomycin into rodents has provided important insights into the molecular pathways leading to fibrotic responses to lung injury (Supplemental Table S1). Limitations of the bleomycin model include the following: 1) the findings in mice are strain dependent, and Haston et al. (67) have shown that many of these strain-related differences might be explained by differential expression of bleomycin hydrolase (67, 134); 2) the prominent proinflammatory response following bleomycin may not be present in human IPF (115, 136, 153); and 3) the time course of bleomycin-induced fibrosis is self-limited, whereas human IPF is often an inexorably progressive disease (121).
Pulmonary arterial hypertension (PAH) in humans is characterized by medial hypertrophy and intimal proliferation, and we have chosen to focus on models of hypoxia and exposure of rats to the pyrrole liver metabolite of seeds of Crotalaria spectabilis [monocrotaline, an alkaloid toxin that induces endothelial cell injury (62, 125, 130, 179)]. Limitations of these models include the following: 1) monocrotaline and hypoxia models lack plexogenic lesions seen in human PAH; 2) hypoxia may induce only modest increases in right ventricular pressure and vascular remodeling; 3) hypoxia may not induce significant endothelial cell proliferation; and 4) in contrast to human PAH, the hypoxia-induced muscularization of the precapillary pulmonary arterioles and vasoconstriction are slowly reversible when normal oxygen levels are restored (106). There has been recent interest in VEGFR-2 blockade with SU5416 (Sugen) combined with hypoxia as a model of PAH, since studies in rats have demonstrated formation of plexiform lesions that have been lacking in the other models described (1). A recent study of Sugen plus hypoxia in mice demonstrated development of pulmonary vascular remodeling, but the vessels lacked plexiform lesions, which is an important limitation of this mouse model (26).
General pitfalls in using genetically modified mice in murine models of lung disease.
Given that every mouse model has limitations in replicating the human disease, many investigators have replicated biological findings in at least two different models of disease, along with confirming relevant mechanistic findings in an in vitro system. Ideally, clinically relevant findings from mouse models of disease will be repeated in large animal models and ultimately in human samples and in human trials. In addition, findings can be quite different between males and females, and thus findings should be compared between sexes, and ideally both male and female mice should be examined. As described above with mouse models of asthma, observed findings can vary dramatically with the genetic background of the animal, and especially with generation and breeding of multiple lines of mice, offspring with mixed genetic backgrounds can emerge. Thus it is important to use littermate controls for all experiments, and genetically modified animals on mixed backgrounds should ideally be backcrossed to at least 99% purity (which may be as many as 10 generations). There exist genotyping strategies through commercial vendors to determine the percent purity of the genetic background and to facilitate faster achievement of genetic purity through selecting “purer” animals for the backcrossing matings (termed “speed congenics”) (171). Additional considerations include that phenotypes of genetically modified mice may not be apparent with one “stressor” (i.e., model) but might emerge on a different genetic background, by studying animals at different ages, or by using a different model of illness (29).
Finally, there exist important limitations to the study of any genetically modified mice, not only from limitations of each of the disease models as described above, but also as a result of the process of inducing genetic modification, and we have reviewed key points in previous sections and in Table 1. Importantly, any studies must be performed with the appropriate controls to rule out off-target effects of tissue-specific promoters, administered drugs to induce transgene expression, and expression of nonendogenous substances such as Cre-recombinase. Additionally, it is critical with any genetically modified mouse to make certain that the observed biology arises from altering expression of the gene of interest, rather than from disruption of another gene from random integration of the introduced vector, or compensatory response arising from either deficiency or overexpression of a gene other than the intended target that has been modified. Thus, although data gathered from genetically modified mice are incredibly valuable in determining gene function, mechanistic findings are ideally replicated and further explored by using both in vitro systems and, ultimately, translational systems that allow confirmation of relevant findings in human samples.
SUMMARY AND CONCLUSIONS
This review has outlined the major technological approaches to the utilization of gene-targeted and/or genetically modified mice to delineate the cellular and molecular basis of experimental lung disease. Excitingly, with the ongoing international effort of the Knockout Mouse Project (KOMP) to generate and characterize knockout mice for each protein-coding gene in the mouse genome, we can expect that a wealth of additional important information will be generated from the evaluation of gene-targeted mice (61). We have outlined the strengths and limitations of gene manipulated mice, focusing on the tremendous potential and pitfalls for investigators to consider when performing studies with these genetically engineered mice. We hope that this review serves as an effective and helpful template for pulmonologists in our lung community.
Supplementary Material
REFERENCES
- 1. Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, Voelkel NF, McMurtry IF, Oka M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation 121: 2747–2754, 2010. [DOI] [PubMed] [Google Scholar]
- 2. Akram A, Han B, Masoom H, Peng C, Lam E, Litvack ML, Bai X, Shan Y, Hai T, Batt J, Slutsky AS, Zhang H, Kuebler WM, Haitsma JJ, Liu M, dos Santos CC. Activating transcription factor 3 confers protection against ventilator-induced lung injury. Am J Respir Crit Care Med 182: 489–500, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Albaiceta GM, Gutierrez-Fernández A, García-Prieto E, Puente XS, Parra D, Astudillo A, Campestre C, Cabrera S, Gonzalez-Lopez A, Fueyo A, Taboada F, López-Otin C. Absence or inhibition of matrix metalloproteinase-8 decreases ventilator-induced lung injury. Am J Respir Cell Mol Biol 43: 555–563, 2010. [DOI] [PubMed] [Google Scholar]
- 4. Albaiceta GM, Gutiérrez-Fernández A, Parra D, Astudillo A, García-Prieto E, Taboada F, Fueyo A. Lack of matrix metalloproteinase-9 worsens ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol 294: L535–L543, 2008. [DOI] [PubMed] [Google Scholar]
- 5. Albiger B, Dahlberg S, Sandgren A, Wartha F, Beiter K, Katsuragi H, Akira S, Normark S, Henriques-Normark B. Toll-like receptor 9 acts at an early stage in host defence against pneumococcal infection. Cell Microbiol 9: 633–644, 2007. [DOI] [PubMed] [Google Scholar]
- 6. Aoki K, Taketo MM. Tissue-specific transgenic, conditional knockout and knock-in mice of genes in the canonical Wnt signaling pathway. In: Wnt Signaling Volume 1: Pathway Methods and Mammalian Models, edited by Vincan E. Totowa, NJ: Humana, 2008, vol. 468, chapt. 24, p. 307–331. [DOI] [PubMed] [Google Scholar]
- 7. Arredouani M, Yang Z, Ning Y, Qin G, Soininen R, Tryggvason K, Kobzik L. The scavenger receptor MARCO is required for lung defense against pneumococcal pneumonia and inhaled particles. J Exp Med 200: 267–272, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bai KJ, Spicer AP, Mascarenhas MM, Yu L, Ochoa CD, Garg HG, Quinn DA. The role of hyaluronan synthase 3 in ventilator-induced lung injury. Am J Respir Crit Care Med 172: 92–98, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Balamayooran T, Balamayooran G, Jeyaseelan S. Review: Toll-like receptors and NOD-like receptors in pulmonary antibacterial immunity. Innate Immun 16: 201–210, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baran CP, Opalek JM, McMaken S, Newland CA, O'Brien JM, Jr, Hunter MG, Bringardner BD, Monick MM, Brigstock DR, Stromberg PC, Hunninghake GW, Marsh CB. Important roles for macrophage colony-stimulating factor, CC chemokine ligand 2, and mononuclear phagocytes in the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med 176: 78–89, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bates JHT, Rincon M, Irvin CG. Animal models of asthma. Am J Physiol Lung Cell Mol Physiol 297: L401–L410, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Beller TC, Friend DS, Maekawa A, Lam BK, Austen KF, Kanaoka Y. Cysteinyl leukotriene 1 receptor controls the severity of chronic pulmonary inflammation and fibrosis. Proc Natl Acad Sci USA 101: 3047–3052, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Betsuyaku T, Fuke S, Nasuhara Y, Morikawa T, Kondo S, Nishimura M. Diverse expression of antioxidants and inflammatory chemokines in terminal bronchiolar epithelium in chronic obstructive pulmonary disease. Proc Am Thorac Soc 3: 471–472, 2006. [DOI] [PubMed] [Google Scholar]
- 14. Betsuyaku T, Fukuda Y, Parks WC, Shipley JM, Senior RM. Gelatinase B is required for alveolar bronchiolization after intratracheal bleomycin. Am J Pathol 157: 525–535, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bonniaud P, Kolb M, Galt T, Robertson J, Robbins C, Stampfli M, Lavery C, Margetts PJ, Roberts AB, Gauldie J. Smad3 null mice develop airspace enlargement and are resistant to TGF-beta-mediated pulmonary fibrosis. J Immunol 173: 2099–2108, 2004. [DOI] [PubMed] [Google Scholar]
- 16. Bostrom H, Willetts K, Pekny M, Levéen P, Lindahl P, Hedstrand H, Pekna M, Hellström M, Gebre-Medhin S, Schalling M, Nilsson M, Kurland S, Törnell J, Heath JK, Betsholtz C. PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell 85: 863–873, 1996. [DOI] [PubMed] [Google Scholar]
- 17. Budhiraja R, Tuder RM, Hassoun PM. Endothelial dysfunction in pulmonary hypertension. Circulation 109: 159–165, 2004. [DOI] [PubMed] [Google Scholar]
- 18. Caironi P, Ichinose F, Liu R, Jones RC, Bloch KD, Zapol WM. 5-Lipoxygenase deficiency prevents respiratory failure during ventilator-induced lung injury. Am J Respir Crit Care Med 172: 334–343, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chan YR, Liu JS, Pociask DA, Zheng M, Mietzner TA, Berger T, Mak TW, Strong RK, Ray P, Kolls JK. Lipocalin 2 is required for pulmonary host defense against Klebsiella infection. J Immunol 182: 4947–4956, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen Q, Rabach L, Noble P, Zheng T, Lee CG, Homer RJ, Elias JA. IL-11 receptor alpha in the pathogenesis of IL-13-induced inflammation and remodeling. J Immunol 174: 2305–2313, 2005. [DOI] [PubMed] [Google Scholar]
- 21. Chen SC, Mehrad B, Deng JC, Vassileva G, Manfra DJ, Cook DN, Wiekowski MT, Zlotnik A, Standiford TJ, Lira SA. Impaired pulmonary host defense in mice lacking expression of the CXC chemokine lungkine. J Immunol 166: 3362–3368, 2001. [DOI] [PubMed] [Google Scholar]
- 22. Chen ZH, Lam HC, Jin Y, Feghali-Bostwick C, Ryter SW, Kim HP, Cao J, Lee SJ, Ifedigbo E, Parameswaran H, Ryter SW, Choi AMK. Autophagy protein LC3B activates extrinsic apoptosis during cigarette smoke induced emphysema. Proc Natl Acad Sci USA 107: 18880–18885, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chua F, Dunsmore SE, Clingen PH, Mutsaers SE, Shapiro SD, Segal AW, Roes J, Laurent GJ. Mice lacking neutrophil elastase are resistant to bleomycin-induced pulmonary fibrosis. Am J Pathol 170: 65–74, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Churg A, Cosio M, Wright L. Mechanisms of cigarette smoke-induced COPD: insights from animal models. Am J Physiol Lung Cell Mol Physiol 294: L612–L631, 2008. [DOI] [PubMed] [Google Scholar]
- 25. Churg A, Wang RD, Tai H, Wang X, Xie C, Wright JL. Tumor necrosis factor-alpha drives 70% of cigarette smoke-induced emphysema in the mouse. Am J Respir Crit Care Med 170: 492–498, 2004. [DOI] [PubMed] [Google Scholar]
- 26. Ciuclan L, Bonneau O, Hussey M, Duggan N, Holmes AM, Good R, Stringer R, Jones P, Morrell NW, Jarai G, Walker C, Westwick J, Thomas M. A novel murine model of severe pulmonary arterial hypertension. Am J Respir Crit Care Med 184: 1171–1182, 2011. [DOI] [PubMed] [Google Scholar]
- 27. D'Armiento J, Dalal SS, Okada Y, Berg RA, Chada K. Collagenase expression in the lungs of transgenic mice causes pulmonary emphysema. Cell 71: 955–961, 1992. [DOI] [PubMed] [Google Scholar]
- 28. Damarla M, Hasan E, Boueiz A, Le A, Pae HH, Montouchet C, Kolb T, Simms T, Myers A, Kayyali US, Gaestel M, Peng X, Reddy SP, Damico R, Hassoun PM. Mitogen activated protein kinase activated protein kinase 2 regulates actin polymerization and vascular leak in ventilator associated lung injury. PLoS One 4: e4600, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Davey RA, MacLean HE. Current and future approaches using genetically modified mice in endocrine research. Am J Physiol Endocrinol Metab 291: E429–E438, 2006. [DOI] [PubMed] [Google Scholar]
- 30. Deng JC, Zeng X, Newstead M, Moore TA, Tsai WC, Thannickal VJ, Standiford TJ. STAT4 is a critical mediator of early innate immune responses against pulmonary Klebsiella infection. J Immunol 173: 4075–4083, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dessing MC, Hirst RA, de Vos AF, van der Poll T. Role of Toll-like receptors 2 and 4 in pulmonary inflammation and injury induced by pneumolysin in mice. PLoS One 4: e7993, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dolinay T, Wu W, Kaminski N, Ifedigbo E, Kaynar AM, Szilasi M, Watkins SC, Ryter SW, Hoetzel A, Choi AMK. Mitogen activated protein kinase regulates susceptibility to ventilator induced lung injury. PLoS One 3: e1601, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Duerr J, Gruner M, Schubert SC, Haberkorn U, Bujard H, Mall MA. Use of a new-generation reverse tetracycline transactivator system for quantitative control of conditional gene expression in the murine lung. Am J Respir Cell Mol Biol 44: 244–254, 2011. [DOI] [PubMed] [Google Scholar]
- 34. Eckle T, Grenz A, Laucher S, Eltzschig HK. A2B adenosine receptor signaling attenuates acute lung injury by enhancing alveolar fluid clearance in mice. J Clin Invest 18: 3301–3315, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Eddahibi S, Hanoun N, Lanfumey L, Lesch KP, Raffestin B, Hamon M, Adnot S. Attenuated hypoxic pulmonary hypertension in mice lacking the 5-hydroxytryptamine transporter gene. J Clin Invest 105: 1555–1562, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Eitzman DT, McCoy RD, Zheng X, Fay WP, Shen T, Ginsburg D, Simon RH. Bleomycin-induced pulmonary fibrosis in transgenic mice that either lack or overexpress the murine plasminogen activator inhibitor-1 gene. J Clin Invest 97: 232–237, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. El-Bizri N, Wang L, Merklinger SL, Guignabert C, Desai T, Urashima T, Sheikh AY, Knutsen RH, Mecham RP, Mishina Y, Rabinovitch M. Smooth muscle protein 22alpha-mediated patchy deletion of Bmpr1a impairs cardiac contractility but protects against pulmonary vascular remodeling. Circ Res 102: 380–388, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Elias JA, Zhu Z, Chupp G, Homer RJ. Airway remodeling in asthma. J Clin Invest 104: 1001–1006, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature 292: 154–156, 1981. [DOI] [PubMed] [Google Scholar]
- 40. Faul JL, Nishimura T, Berry GJ, Benson GV, Pearl RG, Kao PN. Triptolide attenuates pulmonary arterial hypertension and neointimal formation in rats. Am J Respir Crit Care Med 162: 2252–2258, 2000. [DOI] [PubMed] [Google Scholar]
- 41. Feuillet V, Medjane S, Mondor I, Demaria O, Pagni PP, Galán JE, Flavell RA, Alexopoulou L. Involvement of Toll-like receptor 5 in the recognition of flagellated bacteria. Proc Natl Acad Sci USA 103: 12487–12492, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Finotto S, Hausding M, Doganci A, Maxeiner JH, Lehr HA, Luft C, Galle PR, Glimcher LH. Asthmatic changes in mice lacking T-bet are mediated by IL-13. Int Immunol 17: 993–1007, 2005. [DOI] [PubMed] [Google Scholar]
- 43. Flodby P, Borok A, Banfalvi A, Zhou B, Gao D, Minoo P, Ann DK, Morrisey EE, Crandall ED. Directed expression of cre in alveolar epithelial type 1 cells. Am J Respir Cell Mol Biol 43: 173–178, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Foronjy RF, Mirochnitchenko O, Propokenko O, Lemaitre V, Jia Y, Inouye M, Okada Y, D'Armiento JM. Superoxide dismutase expression attenuates cigarette smoke- or elastase-generated emphysema in mice. Am J Respir Crit Care Med 173: 623–631, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Frank JA, Pittet JF, Wray C, Matthay MA. Protection from experimental ventilator-induced acute lung injury by IL-1 receptor blockade. Thorax 63: 147–153, 2008. [DOI] [PubMed] [Google Scholar]
- 46. Fredenburgh LE, Liang OD, Macias AA, Polte TR, Liu X, Riascos DF, Chung SW, Schissel SL, Ingber DE, Mitsialis SA, Kourembanas S, Perrella MA. Absence of cyclooxygenase-2 exacerbates hypoxia-induced pulmonary hypertension and enhances contractility of vascular smooth muscle cells. Circulation 117: 2114–2122, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fredenburgh LE, Ma J, Perrella MA. Cyclooxygenase-2 inhibition and hypoxia-induced pulmonary hypertension: effects on pulmonary vascular remodeling and contractility. Trends Cardiovasc Med 19: 31–37, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Friedel RH, Wurst W, Wefers B, Kuhn R. Generating conditional knockout mice. Methods Mol Biol 693: 205–231, 2011. [DOI] [PubMed] [Google Scholar]
- 49. Fujita M, Shannon JM, Morikawa O, Gauldie J, Hara N, Mason RJ. Overexpression of tumor necrosis factor-alpha diminishes pulmonary fibrosis induced by bleomycin or transforming growth factor-beta. Am J Respir Cell Mol Biol 29: 669–676, 2003. [DOI] [PubMed] [Google Scholar]
- 50. Geraci MW, Gao B, Hoshikawa Y, Yeager ME, Tuder RM, Voelkel NF. Genomic approaches to research in pulmonary hypertension. Respir Res 2: 210–215, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Geraci M, Gao B, Shepherd D, Allard J, Curiel D, Westcott J, Voelkel N. Pulmonary prostacyclin synthase overexpression by adenovirus transfection and in transgenic mice. Chest 114: 99S, 1998. [DOI] [PubMed] [Google Scholar]
- 52. Geraci MW, Gao B, Shepherd DC, Moore MD, Westcott JY, Fagan KA, Alger LA, Tuder RM, Voelkel NF. Pulmonary prostacyclin synthase overexpression in transgenic mice protects against development of hypoxic pulmonary hypertension. J Clin Invest 103: 1509–1515, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Glasser SW, Detmer EA, Ikegami M, Na CL, Stahlman MT, Whitsett JA. Pneumonitis and emphysema in Sp-C gene targeted mice. J Biol Chem 278: 14291–14298, 2003. [DOI] [PubMed] [Google Scholar]
- 54. Glasser SW, Korfhagen TR, Wert SE, Whitsett JA. Transgenic models for study of pulmonary development and disease. Am J Physiol Lung Cell Mol Physiol 267: L489–L497, 1994. [DOI] [PubMed] [Google Scholar]
- 55. Gordon JW, Ruddle FH. Integration and stable germ line transmission of genes injected into mouse pronuclei. Science 214: 1244–1246, 1981. [DOI] [PubMed] [Google Scholar]
- 56. Gordon JW, Sangos GA, Plotkin DJ, Barbosa JL, Ruddle FH. Genetic transformation of mouse embryos by microinjection of purified DNA. Proc Natl Acad Sci USA 77: 7380–7384, 1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gossen M, Bujard H. Efficacy of tetracycline-controlled gene expression is influenced by cell type: commentary. Biotechniques 19: 213–216, 1995. [PubMed] [Google Scholar]
- 58. Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA 89: 5547–5551, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gossen M, Freundlieb S, Bender B, Müller G, Hillen W, Bujard H. Transcriptional activation by tetracyclines in mammalian cells. Science 268: 1766–1769, 1995. [DOI] [PubMed] [Google Scholar]
- 60. Green MC, Sweet HO, Bunker LE. Tight-skin, a new mutation of the mouse causing excessive growth of connective tissue and skeleton. Am J Pathol 82: 493–512, 1976. [PMC free article] [PubMed] [Google Scholar]
- 61. Hampton T. Knockout science: massive mouse project to provide window into human diseases. JAMA 306: 1968, 2011. [DOI] [PubMed] [Google Scholar]
- 62. Hansmann G, Wagner RA, Schellong S, Perez VA, Urashima T, Wang L, Sheikh AY, Suen RS, Stewart DJ, Rabinovitch M. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma activation. Circulation 115: 1275–1284, 2007. [DOI] [PubMed] [Google Scholar]
- 63. Hansmann G, de Jesus Perez VA, Alastalo TP, Alvira CM, Guignabert C, Bekker JM, Schellong S, Urashima T, Wang L, Morrell NW, Rabinovitch M. An antiproliferative BMP-2/PPARgamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Invest 118: 1846–1857, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hardie WD, Glasser SW, Hagood JS. Emerging concepts in the pathogenesis of lung fibrosis. Am J Pathol 175: 3–16, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hardie WD, Hagood JS, Dave V, Perl AK, Whitsett JA, Korfhagen TR, Glasser S. Signaling pathways in the epithelial origins of pulmonary fibrosis. Cell Cycle 9: 2769–2776, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hardie WD, Le Cras TD, Jiang K, Tichelaar JW, Azhar M, Korfhagen TR. Conditional expression of transforming growth factor-alpha in adult mouse lung causes pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 286: L741–L749, 2004. [DOI] [PubMed] [Google Scholar]
- 67. Haston CK, Wang M, Dejournett RE, Zhou X, Ni D, Gu X, King TM, Weil MM, Newman RA, Amos CI, Travis EL. Bleomycin hydrolase and a genetic locus within the MHC affect risk for pulmonary fibrosis in mice. Hum Mol Genet 11: 1855–1863, 2002. [DOI] [PubMed] [Google Scholar]
- 68. Hausding M, Sauer K, Maxeiner JH, Finotto S. Transgenic models in allergic responses. Curr Drug Targets 9: 503–510, 2008. [DOI] [PubMed] [Google Scholar]
- 69. Hautamaki RD, Kobayashi DK, Senior RM, et al. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science 277: 2002–2004, 1997. [DOI] [PubMed] [Google Scholar]
- 70. Herbold W, Maus R, Hahn I, Ding N, Srivastava M, Christman JW, Mack M, Reutershan J, Briles DE, Paton JC, Winter C, Welte T, Maus UA. Importance of CXC chemokine receptor 2 in alveolar neutrophil and exudate macrophage recruitment in response to pneumococcal lung infection. Infect Immun 78: 2620–2630, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hodges RJ, Jenkins RG, Wheeler-Jones CP, Copeman DM, Bottoms SE, Bellingan GJ, Nanthakumar CB, Laurent GJ, Hart SL, Foster ML, McAnulty RJ. Severity of lung injury in cyclooxygenase-2-deficient mice is dependent on reduced prostaglandin E(2) production. Am J Pathol 165: 1663–1676, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hoetzel A, Dolinay T, Vallbracht S, Zhang Y, Kim HP, Ifedigbo E, Alber S, Kaynar AM, Schmidt R, Ryter SW, Choi AM. Carbon monoxide protects against ventilator-induced lung injury via PPAR-gamma and inhibition of Egr-1. Am J Respir Crit Care Med 177: 1223–1232, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hoetzel A, Schmidt R, Vallbracht S, Goebel U, Dolinay T, Kim HP, Ifedigbo E, Ryter SW, Choi AM. Carbon monoxide prevents ventilator-induced lung injury via caveolin-1. Crit Care Med 37: 1708–1715, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hong SB, Huang Y, Moreno-Vinasco L, Sammani S, Moitra J, Barnard JW, Ma SF, Mirzapoiazova T, Evenoski C, Reeves RR, Chiang ET, Lang GD, Husain AN, Dudek SM, Jacobson JR, Ye SQ, Lussier YA, Garcia JG. Essential role of pre-B-cell colony enhancing factor in ventilator-induced lung injury. Am J Respir Crit Care Med 78: 605–617, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hosford GE, Fang X, Olson DM. Hyperoxia decreases matrix metalloproteinase-9 and increases tissue inhibitor of matrix metalloproteinase-1 protein in the newborn rat lung: association with arrested alveolarization. Pediatr Res 56: 26–34, 2004. [DOI] [PubMed] [Google Scholar]
- 76. Hoshikawa Y, Voelkel NF, Gesell TL, Moore MD, Morris KG, Alger LA, Narumiya S, Geraci MW. Prostacyclin receptor-dependent modulation of pulmonary vascular remodeling. Am J Respir Crit Care Med 164: 314–318, 2001. [DOI] [PubMed] [Google Scholar]
- 77. Iizuka T, Ishii Y, Itoh K, Kiwamoto T, Kimura T, Matsuno Y, Morishima Y, Hegab AE, Homma S, Nomura A, Sakamoto T, Shimura M, Yoshida A, Yamamoto M, Sekizawa K. Nrf2-deficient mice are highly susceptible to cigarette smoke-induced emphysema. Genes Cells 10: 1113–1125, 2005. [DOI] [PubMed] [Google Scholar]
- 78. Janssens SP, Bloch KD, Nong Z, Gerard RD, Zoldhelyi P, Collen D. Adenoviral-mediated transfer of the human endothelial nitric oxide synthase gene reduces acute hypoxic pulmonary vasoconstriction in rats. J Clin Invest 98: 317–324, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jenkins RG, Su X, Su G, Scotton CJ, Camerer E, Laurent GJ, Davis GE, Chambers RC, Matthay MA, Sheppard D. Ligation of protease-activated receptor 1 enhances alpha(v)beta6 integrin-dependent TGF-beta activation and promotes acute lung injury. J Clin Invest 116: 1606–1614, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kang MJ, Homer RJ, Gallo A, Lee CG, Crothers KA, Cho SJ, Rochester C, Cain H, Chupp G, Yoon H, Elias JA. IL-18 is induced and IL-18 receptor α plays a critical role in the pathogenesis of cigarette smoke-induced pulmonary emphysema and inflammation. J Immunol 178: 1948–1959, 2007. [DOI] [PubMed] [Google Scholar]
- 81. Kavuru MS, Sullivan EJ, Piccin R, Thomassen MJ, Stoller JK. Exogenous granulocyte-macrophage colony-stimulating factor administration for pulmonary alveolar proteinosis. Am J Respir Crit Care Med 161: 1143–1148, 2000. [DOI] [PubMed] [Google Scholar]
- 82. Kaynar AM, Houghton AM, Lum EH, Pitt BR, Shapiro SD. Neutrophil elastase is needed for neutrophil emigration into lungs in ventilator-induced lung injury. Am J Respir Cell Mol Biol 39: 53–60, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Keerthisingam CB, Jenkins RG, Harrison NK, Hernandez-Rodriguez NA, Booth H, Laurent GJ, Hart SL, Foster ML, McAnulty RJ. Cyclooxygenase-2 deficiency results in a loss of the anti-proliferative response to transforming growth factor-beta in human fibrotic lung fibroblasts and promotes bleomycin-induced pulmonary fibrosis in mice. Am J Pathol 158: 1411–1422, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Koh BH, Hwang SS, Kim JY, Lee W, Kang MJ, Lee CG, Park JW, Flavell RA, Lee GR. Th2 LCR is essential for regulation of Th2 cytokine genes and for pathogenesis of allergic asthma. Proc Natl Acad Sci USA 107: 10614–10619, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Koltsova EK, Ley K. The mysterious ways of the chemokine CXCL5. Immunity 33: 7–9, 2010. [DOI] [PubMed] [Google Scholar]
- 86. Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390: 45–51, 1997. [DOI] [PubMed] [Google Scholar]
- 87. Lawson WE, Polosukhin VV, Stathopoulos GT, Zoia O, Han W, Lane KB, Li B, Donnelly EF, Holburn GE, Lewis KG, Collins RD, Hull WM, Glasser SW, Whitsett JA, Blackwell TS. Increased and prolonged pulmonary fibrosis in surfactant protein C-deficient mice following intratracheal bleomycin. Am J Pathol 167: 1267–1277, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Leco KJ, Waterhouse P, Sanchez OH, Gowing KL, Poole AR, Wakeham A, Mak TW, Khokha R. Spontaneous air space enlargement in the lungs of mice lacking tissue inhibitor of metalloproteinases-3 (TIMP-3). J Clin Invest 108: 817–829, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Le Cras TD, Abman SH, Weinberger HD, Huang PL, McMurtry IF, Rodman DM. The pulmonary circulation of homozygous or heterozygous eNOS-null mice is hyperresponsive to mild hypoxia. J Clin Invest 103: 291–299, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Lee CG, Cho SJ, Kang MJ, Chapoval SP, Lee PJ, Noble PW, Yehualaeshet T, Lu B, Flavell RA, Milbrandt J, Homer RJ, Elias JA. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor β1-induced pulmonary fibrosis. J Exp Med 200: 377–389, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lee CG, Hartl D, Lee GR, Koller B, Matsuura H, Da Silva CA, Sohn MH, Cohn L, Homer RJ, Kozhich AA, Humbles A, Kearley J, Coyle A, Chupp G, Reed J, Flavell RA, Elias JA. Role of breast regression protein 39 (BRP-39)/chitinase 3-like-1 in Th2 and IL-13-induced tissue responses and apoptosis. J Exp Med 206: 1149–1166, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lee CG, Link H, Baluk P, Homer RJ, Chapoval S, Bhandari V, Kang MJ, Cohn L, Kim YK, McDonald DM, Elias JA. Vascular endothelial growth factor (VEGF) induces remodeling and enhances TH2-mediated sensitization and inflammation in the lung. Nat Med 10: 1095–1103, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lee JJ, McGarry MP, Farmer SC, Denzler KL, Larson KA, Carrigan PE, Brenneise IE, Horton MA, Haczku A, Gelfand EW, Leikauf GD, Lee NA. Interleukin-5 expression in the lung epithelium of transgenic mice leads to pulmonary changes pathognomonic of asthma. J Exp Med 185: 2143–2156, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lee SJ, Smith A, Guo L, Alastalo TP, Li M, Sawada H, Liu X, Chen ZH, Ifedigbo E, Jin Y, Feghali-Bostwick C, Ryter SW, Kim HP, Rabinovitch M, Choi AMK. Autophagic protein LC3B confers resistance against hypoxia-induced pulmonary hypertension. Am J Respir Crit Care Med 183: 649–658, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Lindahl P, Karlsson L, Hellström M, Gebre-Medhin S, Willetts K, Heath JK, Betsholtz C. Alveogenesis failure in PDGF-A-deficient mice is coupled to lack of distal spreading of alveolar smooth muscle cell progenitors during lung development. Development 124: 3943–3953, 1997. [DOI] [PubMed] [Google Scholar]
- 96. Liu Y, Jiang H, Crawford HC, Hogan BLM. Role for ETS domain transcription factors Pea3/Erm in mouse lung development. Dev Biol 261: 10–24, 2003. [DOI] [PubMed] [Google Scholar]
- 97. Loonstra A, Vooijs M, Beverloo BH, Allak BA, Drunen EV, Kanaar R, Berns A, Jonkers J. Growth inhibition and DNA damage induced by Cre recombinase in mammalian cells. Proc Natl Acad Sci USA 98: 9209–9214, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Madtes DK, Elston AL, Hackman RC, Dunn AR, Clark JG. Transforming growth factor-alpha deficiency reduces pulmonary fibrosis in transgenic mice. Am J Respir Cell Mol Biol 20: 924–934, 1999. [DOI] [PubMed] [Google Scholar]
- 99. Mahadeva R, Shapiro SD. Chronic obstructive pulmonary disease: experimental animal models of pulmonary emphysema. Thorax 57: 908–914, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Maniatis NA, Harokopos V, Thanassopoulou A, Oikonomou N, Mersinias V, Witke W, Orfanos SE, Armaganidis A, Roussos C, Kotanidou A, Aidinis V. A critical role for gelsolin in ventilator-induced lung injury. Am J Respir Cell Mol Biol 41: 426–432, 2009. [DOI] [PubMed] [Google Scholar]
- 101. Manis JP. Knock out, knock in, knock down — genetically manipulated mice and the Nobel Prize. N Engl J Med 357: 2426–2429, 2007. [DOI] [PubMed] [Google Scholar]
- 102. Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol 295: L379–L399, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. McMillan SJ, Kearley J, Campbell JD, Zhu XW, Larbi KY, Shipley JM, Senior RM, Nourshargh S, Lloyd CM. Matrix metalloproteinase-9 deficiency results in enhanced allergen-induced airway inflammation. J Immunol 172: 2586–2594, 2004. [DOI] [PubMed] [Google Scholar]
- 104. Mei J, Liu Y, Dai N, Favara M, Greene T, Jeyaseelan S, Poncz M, Lee JS, Worthen GS. CXCL5 regulates chemokine scavenging and pulmonary host defense to bacterial infection. Immunity 33: 106–117, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Meisler MH. Insertional mutation of ”classical“ and novel genes in transgenic mice. Trends Genet 8: 341–344, 1992. [DOI] [PubMed] [Google Scholar]
- 106. Michelakis ED, Wilkins MR, Rabinovitch M. Emerging concepts and translational priorities in pulmonary arterial hypertension. Circulation 118: 1486–1495, 2008. [DOI] [PubMed] [Google Scholar]
- 107. Minamino T, Christou H, Hsieh CM, Liu Y, Dhawan V, Abraham NG, Perrella MA, Mitsialis SA, Kourembanas S. Targeted expression of heme oxygenase-1 prevents the pulmonary inflammatory and vascular responses to hypoxia. Proc Natl Acad Sci USA 98: 8798–8803, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Mishina Y, Suzuki A, Ueno N, Behringer RR. Bmpr encodes a type I bone morphogenetic protein receptor that is essential for gastrulation during mouse embryogenesis. Genes Dev 9: 3027–3023, 1995. [DOI] [PubMed] [Google Scholar]
- 109. Mizgerd JP. Acute lower respiratory tract infection. N Engl J Med 358: 716–727, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Mizgerd JP, Lupa MM, Kogan MS, Warren HB, Kobzik L, Topulos GP. NF-κB p50 limits inflammation and prevents lung injury during E. coli pneumonia. Am J Respir Crit Care Med 168: 810–817, 2003. [DOI] [PubMed] [Google Scholar]
- 111. Moeller A, Ask K, Warburton D, Gauldie J, Kolb M. The bleomycin animal model: a useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int J Biochem Cell Biol 40: 362–382, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Moore BB, Hogaboam CM. Murine models of pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 294: L152–L160, 2008. [DOI] [PubMed] [Google Scholar]
- 113. Morris DG, Huang X, Kaminski N, Wang Y, Shapiro SD, Dolganov G, Glick A, Sheppard D. Loss of integrin alpha(v)beta6-mediated TGF-beta activation causes Mmp12-dependent emphysema. Nature 422: 169–173, 2003. [DOI] [PubMed] [Google Scholar]
- 114. Moutier R, Tchang F, Caucheteux SM, Kanellopoulos-Langevin C. Placental anomalies and fetal loss in mice, after administration of doxycycline in food for tet-system activation. Transgenic Res 12: 369–373, 2003. [DOI] [PubMed] [Google Scholar]
- 115. Nagai T, Tanaka M, Hasui K, Shirahama H, Kitajima S, Yonezawa S, Xu B, Matsuyama T. Effect of an immunotoxin to folate receptor beta on bleomycin-induced experimental pulmonary fibrosis. Clin Exp Immunol 161: 348–356, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Nagaya N, Yokoyama C, Kyotani S, Shimonishi M, Morishita R, Uematsu M, Nishikimi T, Nakanishi N, Ogihara T, Yamagishi M, Miyatake K, Kaneda Y, Tanabe T. Gene transfer of human prostacyclin synthase ameliorates monocrotaline-induced pulmonary hypertension in rats. Circulation 102: 2005–2010, 2000. [DOI] [PubMed] [Google Scholar]
- 117. Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY, Dietz HC. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet 33: 407–411, 2003. [DOI] [PubMed] [Google Scholar]
- 118. Papaiahgari S, Yerrapureddy A, Reddy SR, Reddy NM, Dodd-O JM, Crow MT, Grigoryev DN, Barnes K, Tuder RM, Yamamoto M, Kensler TW, Biswal S, Mitzner W, Hassoun PM, Reddy SP. Genetic and pharmacologic evidence links oxidative stress to ventilator-induced lung injury in mice. Am J Respir Crit Care Med 176: 1222–1235, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Peng X, Abdulnour RE, Sammani S, Ma SF, Han EJ, Hasan EJ, Tuder R, Garcia JG, Hassoun PM. Inducible nitric oxide synthase contributes to ventilator-induced lung injury. Am J Respir Crit Care Med 172: 470–479, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Perl AK, Zhang L, Whitsett JA. Conditional expression of genes in the respiratory epithelium in transgenic mice: cautionary notes and toward building a better mouse trap. Am J Respir Cell Mol Biol 40: 1–3, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Phan SH, Armstrong G, Sulavik MC, Schrier D, Johnson KJ, Ward PA. A comparative study of pulmonary fibrosis induced by bleomycin and an O2 metabolite producing enzyme system. Chest 83 Suppl: 44S–45S, 1983. [DOI] [PubMed] [Google Scholar]
- 122. Quintero PA, Knolle MD, Cala LF, Zhuang Y, Owen CA. Matrix metalloproteinase-8 inactivates macrophage inflammatory protein-1 alpha to reduce acute lung inflammation and injury in mice. J Immunol 184: 1575–1588, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Quinton LJ, Jones MR, Robson BE, Simms BT, Whitsett JA, Mizgerd JP. Alveolar epithelial STAT3, IL-6 family cytokines, and host defense during Escherichia coli pneumonia. Am J Respir Cell Mol Biol 38: 699–706, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Quinton LJ, Jones MR, Simms BT, Kogan MS, Robson BE, Skerrett SJ, Mizgerd JP. Functions and regulation of NF-kappaB RelA during pneumococcal pneumonia. J Immunol 178: 1896–1903, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Rabinovitch M, Gamble W, Nadas AS, Miettinen OS, Reid L. Rat pulmonary circulation after chronic hypoxia: hemodynamic and structural features. Am J Physiol Heart Circ Physiol 236: H818–H827, 1979. [DOI] [PubMed] [Google Scholar]
- 126. Rankin JA, Picarella DE, Geba GP, Temann UA, Prasad B, DiCosmo B, Tarallo A, Stripp B, Whitsett J, Flavell RA. Phenotypic and physiologic characterization of transgenic mice expressing interleukin 4 in the lung: lymphocytic and eosinophilic inflammation without airway hyperreactivity. Proc Natl Acad Sci USA 93: 7821–7825, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Ray NB, Durairaj L, Chen BB, McVerry BJ, Ryan AJ, Donahoe M, Waltenbaugh AK, O'Donnell CP, Henderson FC, Etscheidt CA, McCoy DM, Agassandian M, Hayes-Rowan EC, Coon TA, Butler PL, Gakhar L, Mathur SN, Sieren JC, Tyurina YY, Kagan VE, McLennan G, Mallampalli RK. Dynamic regulation of cardiolipin by the lipid pump Atp8b1 determines the severity of lung injury in experimental pneumonia. Nat Med 16: 1120–1127, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Rogers CS, Hao Y, Rokhlina T, Samuel M, Stoltz DA, Li Y, Petroff E, Vermeer DW, Kabel AC, Yan Z, Spate L, Wax D, Murphy CN, Rieke A, Whitworth K, Linville ML, Korte SW, Engelhardt JF, Welsh MJ, Prather RS. Production of CFTR-null and CFTR-DeltaF508 heterozygous pigs by adeno-associated virus-mediated gene targeting and somatic cell nuclear transfer. J Clin Invest 118: 1571–1577, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Rogers CS, Stoltz DA, Meyerholz DK, Ostedgaard LS, Rokhlina T, Taft PJ, Rogan MP, Pezzulo AA, Karp PH, Itani OA, Kabel AC, Wohlford-Lenane CL, Davis GJ, Smith TL, Samuel M, Wax D, Murphy CN, Rieke A, Whitworth K, Uc A, Starner TD, Brogden KA, Shilyansky J, McCray PB, Jr, Zabner J, Prather RS, Welsh MJ. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science 321: 1837–1841, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Rosenberg HC, Rabinovitch M. Endothelial injury and vascular reactivity in monocrotaline pulmonary hypertension. Am J Physiol Heart Circ Physiol 255: H1484–H1491, 1988. [DOI] [PubMed] [Google Scholar]
- 131. Said SI, Hamidi SA, Dickman KG, Szema AM, Lyubsky S, Lin RZ, Jiang YP, Chen JJ, Waschek JA, Kort S. Moderate pulmonary arterial hypertension in male mice lacking the vasoactive intestinal peptide gene. Circulation 115: 1260–1268, 2007. [DOI] [PubMed] [Google Scholar]
- 132. Saunders TL. Inducible transgenic mouse models. Methods Mol Biol 693: 103–115, 2011. [DOI] [PubMed] [Google Scholar]
- 133. Schissel SL, Dunsmore SE, Liu X, Shine RW, Perrella MA, Layne MD. Aortic carboxypeptidase-like protein is expressed in fibrotic human lung and its absence protects against bleomycin-induced lung fibrosis. Am J Pathol 174: 818–828, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Schrier DJ, Kunkel RG, Phan SH. Role of strain variation in murine bleomycin-induced pulmonary fibrosis. Am Rev Respir Dis 127: 63–66, 1983. [DOI] [PubMed] [Google Scholar]
- 135. Shapiro SD. Transgenic and gene-targeted mice as models for chronic obstructive pulmonary disease. Eur Respir J 29: 75–78, 2007. [DOI] [PubMed] [Google Scholar]
- 136. Sharma SK, MacLean JA, Pinto C, Kradin RL. The effect of an anti-CD3 monoclonal antibody on bleomycin-induced lymphokine production and lung injury. Am J Respir Crit Care Med 154: 193–200, 1996. [DOI] [PubMed] [Google Scholar]
- 137. Silver DP, Livingston DM. Self-excising retroviral vectors encoding the Cre recombinase overcome Cre-mediated cellular toxicity. Mol Cell 8: 233–243, 2001. [DOI] [PubMed] [Google Scholar]
- 138. Singh G, Singh J, Katyal SL, Brown WE, Kramps JA, Paradis IL, Dauber JH, Macpherson TA, Squeglia N. Identification, cellular localization, isolation, and characterization of human Clara cell-specific 10 KD protein. J Histochem Cytochem 36: 73–80, 1988. [DOI] [PubMed] [Google Scholar]
- 139. Sisson TH, Hansen JM, Shah M, Hanson KE, Du M, Ling T, Simon RH, Christensen PJ. Expression of the reverse tetracycline-transactivator gene causes emphysema-like changes in mice. Am J Respir Cell Mol Biol 34: 552–560, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Smithies O, Gregg RG, Boggs SS, Koralewski MA, Kucherlapati RS. Insertion of DNA sequences into the human chromosomal beta-globin locus by homologous recombination. Nature 317: 230–234, 1985. [DOI] [PubMed] [Google Scholar]
- 141. Stanley E, Lieschke GJ, Grail D, Metcalf D, Hodgson G, Gall JA, Maher DW, Cebon J, Sinickas V, Dunn AR. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci USA 91: 5592–5596, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Stegmeier F, Hu G, Rickles RJ, Hannon GJ, Elledge SJ. A lentiviral microRNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells. Proc Natl Acad Sci USA 102: 13212–13217, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res 104: 236–244, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol 297: L1013–L1032, 2009. [DOI] [PubMed] [Google Scholar]
- 145. Stripp BR, Sawaya PL, Luse DS, Wikenheiser KA, Wert SE, Huffman JA, Lattier DL, Singh G, Katyal SL, Whitsett JA. Cis-acting elements that confer lung epithelial cell expression of the CC10 gene. J Biol Chem 267: 14703–14712, 1992. [PubMed] [Google Scholar]
- 146. Suga T, Kurabayashi M, Sando Y, Ohyama Y, Maeno T, Maeno Y, Aizawa H, Matsumura Y, Kuwaki T, Kuro-O M, Nabeshima Y, Nagai R. Disruption of the klotho gene causes pulmonary emphysema in mice. Defect in maintenance of pulmonary integrity during postnatal life. Am J Respir Cell Mol Biol 22: 26–33, 2000. [DOI] [PubMed] [Google Scholar]
- 147. Swaisgood CM, French EL, Noga C, Simon RH, Ploplis VA. The development of bleomycin-induced pulmonary fibrosis in mice deficient for components of the fibrinolytic system. Am J Pathol 157: 177–187, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Tager AM, Kradin RL, LaCamera P, Bercury SD, Campanella GS, Leary CP, Polosukhin V, Zhao LH, Sakamoto H, Blackwell TS, Luster AD. Inhibition of pulmonary fibrosis by the chemokine IP-10/CXCL10. Am J Respir Cell Mol Biol 31: 395–404, 2004. [DOI] [PubMed] [Google Scholar]
- 149. Takenaka K, Nishimura Y, Nishiuma T, Sakashita A, Yamashita T, Kobayashi K, Satouchi M, Ishida T, Kawashima S, Yokoyama M. Ventilator-induced lung injury is reduced in transgenic mice that overexpress endothelial nitric oxide synthase. Am J Physiol Lung Cell Mol Physiol 290: L1078–L1086, 2006. [DOI] [PubMed] [Google Scholar]
- 150. Tecott LH, Johnson DS. Gene targeting and transgenic technologies. In: Neuropshychopharmacology: The Fifth Generation of Progress, edited by Davis KL, Charney D, Coyle JT, Nemeroff C. Philadelphia, PA: Lippincott, Williams & Wilkins, 2002, chapt. 19, p. 241–251. [Google Scholar]
- 151. Temann UA, Geba GP, Rankin JA, Flavell RA. Expression of interleukin 9 in the lungs of transgenic mice causes airway inflammation, mast cell hyperplasia, and bronchial hyperresponsiveness. J Exp Med 188: 1307–1320, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Thomas KR, Folger KR, Capecchi MR. High frequency targeting of genes to specific sites in the mammalian genome. Cell 44: 419–428, 1986. [DOI] [PubMed] [Google Scholar]
- 153. Tokuda A, Itakura M, Onai N, Kimura H, Kuriyama T, Matsushima K. Pivotal role of CCR1-positive leukocytes in bleomycin-induced lung fibrosis in mice. J Immunol 164: 2745–2751, 2000. [DOI] [PubMed] [Google Scholar]
- 154. Tong C, Li P, Wu NL, Yan Y, Ying QL. Production of p53 gene knockout rats by homologous recombination in embryonic stem cells. Nature 467: 211–214, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Truffinet V, Guglielmi L, Cogne M, Denizot Y. The chicken beta-globin HS4 insulator is not a silver bullet to obtain copy-number dependent expression of transgenes in stable B cell transfectants. Immunol Lett 96: 303–304, 2005. [DOI] [PubMed] [Google Scholar]
- 156. Tsai WC, Strieter RM, Wilkowski JM, Bucknell KA, Burdick MD, Lira SA, Standiford TJ. Lung-specific transgenic expression of KC enhances resistance to Klebsiella pneumoniae in mice. J Immunol 161: 2435–2440, 1998. [PubMed] [Google Scholar]
- 157. Vannella KM, McMillan TR, Charbeneau RP, Wilke CA, Thomas PE, Toews GB, Peters-Golden M, Moore BB. Cysteinyl leukotrienes are autocrine and paracrine regulators of fibrocyte function. J Immunol 179: 7883–7890, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Voelkel NF, Agusti A. COPD at the 2004 9th European Pulmonary Summit. Pulm Pharmcol Ther 17: 249–251, 2004. [DOI] [PubMed] [Google Scholar]
- 159. Voelkel NF, Tuder RM, Bridges J, Arend WP. Interleukin-1 receptor antagonist treatment reduces pulmonary hypertension generated in rats by monocrotaline. Am J Respir Cell Mol Biol 11: 664–675, 1994. [DOI] [PubMed] [Google Scholar]
- 160. Voelkel NF, Tuder RM, Wade K, Hoper M, Lepley RA, Goulet JL, Koller BH, Fitzpatrick F. Inhibition of 5-lipoxygenase-activating protein (FLAP) reduces pulmonary vascular reactivity and pulmonary hypertension in hypoxic rats. J Clin Invest 97: 2491–2498, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Wang D, Wang W, Dawkins P, Paterson T, Kalsheker N, Sallenave JM, Houghton AM. Deletion of Serpina1a, a murine α1-antitrypsin ortholog, results in embryonic lethality. Exp Lung Res 37: 291–300, 2011. [DOI] [PubMed] [Google Scholar]
- 162. Wang Z, Zheng T, Zhu Z, Homer RJ, Riese RJ, Chapman HA, Jr, Shapiro SD, Elias JA. Interferon gamma induction of pulmonary emphysema in the adult murine lung. J Exp Med 192: 1587–1600, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Weinstein M, Xu X, Ohyama K, Deng CX. FGFR-3 and FGFR-4 function cooperatively to direct alveogenesis in the murine lung. Development 125: 3615–3623, 1998. [DOI] [PubMed] [Google Scholar]
- 164. Wendel DP, Taylor DG, Albertine KH, Keating MT, Li DY. Impaired distal airway development in mice lacking elastin. Am J Respir Cell Mol Biol 23: 320–326, 2000. [DOI] [PubMed] [Google Scholar]
- 165. Wert SE, Glasser SW, Korfhagen TR, Whitsett JA. Transcriptional elements from the human SP-C gene direct expression in the primordial respiratory epithelium of transgenic mice. Dev Biol 156: 426–443, 1993. [DOI] [PubMed] [Google Scholar]
- 166. West J, Fagan K, Steudel W, Fouty B, Lane K, Harral J, Hoedt-Miller M, Tada Y, Ozimek J, Tuder R, Rodman DM. Pulmonary hypertension in transgenic mice expressing a dominant-negative BMPRII gene in smooth muscle. Circ Res 94: 1109–1114, 2004. [DOI] [PubMed] [Google Scholar]
- 167. Whitsett JA, Perl AK. Conditional control of gene expression in the respiratory epithelium: a cautionary note. Am J Respir Cell Mol Biol 34: 519–520, 2006. [DOI] [PubMed] [Google Scholar]
- 168. Wilkie TM, Brinster RL, Palmiter RD. Germline and somatic mosaicism in transgenic mice. Dev Biol 118: 9–18, 1986. [DOI] [PubMed] [Google Scholar]
- 169. Willingham SB, Allen IC, Bergstralh DT, Brickey WJ, Huang MT, Taxman DJ, Duncan JA, Ting JP. NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J Immunol 183: 2008–2015, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Wolters PJ, Wray C, Sutherland RE, Kim SS, Koff J, Mao Y, Frank JA. Neutrophil-derived IL-6 limits alveolar barrier disruption in experimental ventilator-induced lung injury. J Immunol 182: 8056–8062, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Wong GT. Speed congenics: applications for transgenic and knock-out mouse strains. Neuropeptides 36: 230–236, 2002. [DOI] [PubMed] [Google Scholar]
- 172. Wu H, Santoni-Rugiu E, Ralfkiaer E, Porse BT, Moser C, Høiby N, Borregaard N, Cowland JB. Lipocalin 2 is protective against E. coli pneumonia. Respir Res 11: 96, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Xia H, Diebold D, Nho R, Perlman D, Kleidon J, Kahm J, Avdulov S, Peterson M, Nerva J, Bitterman P, Henke C. Pathological integrin signaling enhances proliferation of primary lung fibroblasts from patients with idiopathic pulmonary fibrosis. J Exp Med 205: 1659–1672, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Ye X, Ding J, Zhou X, Chen G, Liu SF. Divergent roles of endothelial NF-kappaB in multiple organ injury and bacterial clearance in mouse models of sepsis. J Exp Med 205: 1303–1315, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, Shellito JE, Bagby GJ, Nelson S, Charrier K, Peschon JJ, Kolls JK. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med 194: 519–527, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Yi ES, Kim H, Ahn H, Strother J, Morris T, Masliah E, Hansen LA, Park K, Friedman PJ. Distribution of obstructive intimal lesions and their cellular phenotypes in chronic pulmonary hypertension. A morphometric and immunohistochemical study. Am J Respir Crit Care Med 162: 1577–1586, 2000. [DOI] [PubMed] [Google Scholar]
- 177. Yoshida M, Korfhagen TR, Whitsett JA. Surfactant protein D regulates NF-kappa B and matrix metalloproteinase production in alveolar macrophages via oxidant-sensitive pathways. J Immunol 166: 7514–7519, 2001. [DOI] [PubMed] [Google Scholar]
- 178. Zaidi SH, You XM, Ciura S, Husain M, Rabinovitch M. Overexpression of the serine elastase inhibitor elafin protects transgenic mice from hypoxic pulmonary hypertension. Circulation 105: 516–521, 2002. [DOI] [PubMed] [Google Scholar]
- 179. Zaiman A, Fijalkowska I, Hassoun PM, Tuder RM. One hundred years of research in the pathogenesis of pulmonary hypertension. Am J Respir Cell Mol Biol 33: 425–431, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Zhang DH, Yang L, Cohn L, Parkyn L, Homer R, Ray P, Ray A. Inhibition of allergic inflammation in a murine model of asthma by expression of a dominant-negative mutant of GATA-3. Immunity 11: 473–482, 1999. [DOI] [PubMed] [Google Scholar]
- 181. Zheng T, Zhu Z, Wang Z, Homer RJ, Ma B, Riese RJ, Jr, Chapman HA, Jr, Shapiro SD, Elias JA. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J Clin Invest 106: 1081–1093, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Zhou L, Dey CR, Wert SE, Whitsett JA. Arrested lung morphogenesis in transgenic mice bearing an SP-C-TGF-β1 chimeric gene. Dev Biol 175: 227–228, 1996. [DOI] [PubMed] [Google Scholar]
- 183. Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, Zhang Y, Elias JA. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest 103: 779–788, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Zhu Z, Ma B, Zheng T, Homer RJ, Lee CG, Charo IF, Noble P, Elias JA. IL-13-induced chemokine responses in the lung: role of CCR2 in the pathogenesis of IL-13-induced inflammation and remodeling. J Immunol 168: 2953–2962, 2002. [DOI] [PubMed] [Google Scholar]
- 185. Zuo F, Kaminski N, Eugui E, Allard J, Yakhini Z, Ben-Dor A, Lollini L, Morris D, Kim Y, DeLustro B, Sheppard D, Pardo A, Selman M, Heller RA. Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc Natl Acad Sci USA 99: 6292–6297, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.