

Cancer cell invasion is a fundamental aspect of metastasis and a major cause of mortality in patients[1]. The invasive potential of cancer cells is largely dependent on their ability to degrade and migrate through extracellular matrix (ECM) barriers. During invasion, cancer cells form asymmetrical morphology to escape from their original location and invade into surrounding microenvironment. Various migration types can be broadly categorized into single-cell migration, multicellular streaming and collective migration, regulated by physical and molecular determinants of the cells and from stromal tissues[2]. Well-defined in vitro experiments have allowed precise delineation of the various cell migration mechanisms involving receptor-ligand interactions, but their complexity and synergistic effects of physical and molecular guiding principles make it still challenging to apparently identify the mechanisms[2, 3]. Furthermore, tumour cells have plasticity adaptively changing structure of ECM, invasion mode, and interaction with other cells by various extra- and intra-cellular mechanisms[2, 3]. Previous in vitro studies have failed revealing the complexity and plasticity, because they have regarded invaded tissue as a homogeneous and passive scaffold[2]. In the present study, we have considered the invaded tissue as heterogeneous ECM, a regulator of tumor cell behavior simultaneously allowing cancer cells to lead either proteolytic or non-proteolytic invasion[4].
The ECM is generally classified into two main types: basement membrane and stromal/interstitial matrix[5, 6]. Basement membrane is normally deposited beneath the epithelium as a thin and dense polymeric network with high tensile strength[6, 7]. As a reconstituted basement membrane, Matrigel extracted from the murine Engelbreth-Holm-Swarm (EHS) tumor has been widely used to study cancer cell invasion[8]. Stromal/interstitial matrices form the majority of connective tissues in the body and primarily comprise cross-linked collagen type I to impart 3-dimensional (3D) structural support[6, 9]. As a critical regulator of tumor progression[10], ECMs, such as collagen type 1 and Matrigel, have been revealed to mediate malignant phenotype of breast cancer cells[11]. Several researchers have used the ECMs for migration studies of human breast cancer cells[12] and analyzed their interactions with cancer cell phenotypes[13]. However, there is an urgent requirement for technologies that permit quantified analysis and controlled perturbation of local cellular morphology and plasticity in various heterogeneous combinations of the ECMs in situ and in 3D while also allowing for chemotactic effects[14].
In the present study, a novel and powerful microfluidic in vitro platform was applied to analyze the ability of a highly invasive human breast cancer cell line, MDA-MB-231, to invade 3D heterogeneous ECMs. The hydrogel-incorporating microfluidic platform includes the 3D ECM microenvironment of collagen type I, Matrigel, or their alignments and mixtures under gradient of fetal bovine serum (FBS) to compare the phenotypes of MDA-MB-231 cells, including morphology change, migration potential and plasticity. This platform enables high-resolution real-time and quantitative in situ analysis of cells cultured with ECM, facilitating interactions of these cells with 3D ECMs and the capture of complex behaviors occurring within the in vivo microenvironment[14].
During the metastasis of breast cancer cells, they disrupt and interact with both ECMs (basement membrane and stromal matrix) in the early steps, escape from primary tumor aggregates and intravasate into the blood or lymphatic systems. We thought that ECMs additionally play an important role in cancer cell survival during and after extravasation (Figure 1a). Here, we cultured MDA-MB-231 cells in a microfluidic platform (Figure 1b) in which channels are connected by ECMs to mimic the ECM microenvironments surrounding tumor cells (pink, Figure 1c)[15]. Collagen type I (COL) or Matrigel (MAT) was injected and gelled in the ECM region (pink). MDA-MB-231 cells (green beads) were seeded onto the ECMs in both center and bottom microfluidic channels in medium containing 1% FBS. After seeding, cell culture medium containing 5% FBS (blue) was introduced into the top channel, applying the FBS gradient on cells in the center channel, while cells in the channel below remained free from the gradient. The established gradient was simulated (Figure 1d) considering diffusion of serum albumin protein in FBS in ECMs (D=6.10×10-11 m2/s in collagen, D=6.41×10-11 m2/s in matrigel; assuming molecular weight of serum albumin protein in FBS is 67 kDa[16] and diffusion through cells (D=10x10-13 m2/s), with accuracy confirmed in our previous study[17]. In the simulated gradient profile, a steep FBS gradient (>3% per 50 microns) within hydrogel between the top and center channels was generated after 4 h and maintained over 24 h, while gradients across the hydrogel between the center and bottom channels were much smaller (control: <2% per 50 microns). Details of assay preparation and the molecular gradient presented in the legend to Supporting Figure 1.
Figure 1.
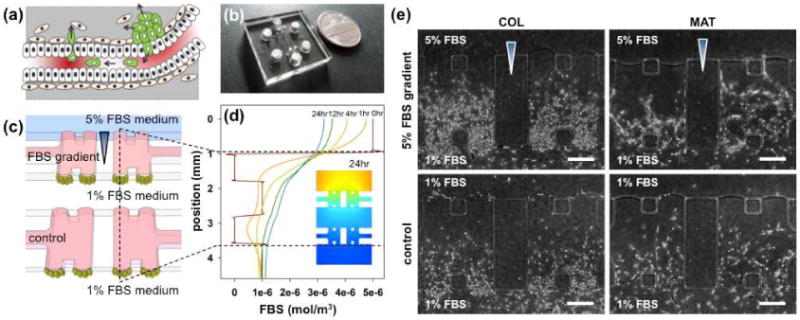
(a) Depiction of the metastasis process, (b) image of the developed microfluidic assay, and (c) its schematic depiction. The assay comprises three microfluidic channels and four gel regions (pink). Cancer cells (green beads) are seeded in the center and bottom channels and 5% FBS medium (blue) is supplied to the top channel. The two other channels are filled with 1% FBS medium as control. (d) Simulation of diffusion of 67KDa of serum albumin protein in 5% FBS through the hydrogel scaffolds. Lines signify the concentration of a molecule (67kDa) mimicking main protein in FBS over 24 hours, based on the line indicated in (c). (e) Morphological variations in MDA-MB-231 cells invading collagen type 1 (COL) and matrigel (MAT) hydrogels in the presence or absence (control) of the FBS gradient under our experimental conditions. Scale bar: 250 μm.
MDA-MB-231 cells invaded into the hydrogel under both FBS and negligible gradients (control) due to their highly invasive nature (Figure 1e, control & Supporting Figure 2). Interestingly, the invasion morphology was ECM-dependent, with cells displaying multicellular invasion in MAT and individual invasion in COL (Figure 1e). Confocal images confirmed the 3D morphologies of cancer cells. In MAT, cells were spread from the bottom (purple) to the top (red) plane of scaffolds, with long multicellular structures of leaders and connected followers by proteolytically breaking down ECM (Figure 2a & Supporting Figure 3). In COL, each cell extended multiple pseudopodia towards the FBS gradient and individually invaded the matrix. The invasion was quantified based on the projected area of cells that invaded into the hydrogel (details in Supporting Figure 4a). Interestingly, the FBS gradients facilitated proliferation of invading cells, only leading to increased cell numbers in both COL and MAT (the FBS gradient did not significantly affect other values of projected area and stalk cell length) (Figure 2b). Evaluation of the length of connected stalks (definition in Supporting Figure 4b) revealed a dramatic increase in connectivity of MDA-MB-231 cells in MAT, with increased proliferation of cell numbers per unit stalk length under the FBS gradient (Figure 2c), as depicted in Figure 2d &2e.
Figure 2.
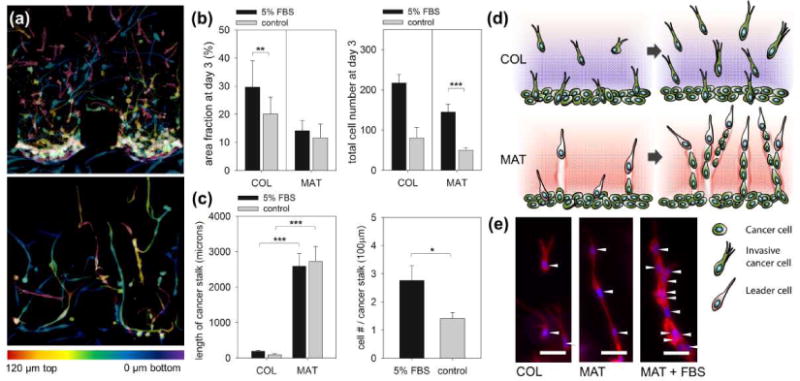
(a) Confocal images of MDA-MB-231 invading COL (top) and MAT (bottom). In total, 135 stacks of confocal images with 1 μm z-spacing were merged into one image, with color codes indicating the actual z-position in hydrogel (red, 120 μm from the bottom substrate; purple, 0 μm). Scale bars: 100 μm. (b) Quantification of the area fraction occupied by invading MDA-MB-231 (left) and total number of MDA-MB-231 in hydrogels (right) at day 3 under FBS gradient (5% FBS) or no gradient (control). (c) Quantification of the cancer stalk length in COL and MAT (left) and number of cancer cells per unit length of cancer stalk only in MAT (right). Total n=44, and error bars indicate mean values ± standard error (*P < 0.05; **P < 0.01; ***P < 0.001). Details of quantification are presented in Supporting information 4. (d) Simple depiction and (e) images of morphological variations in MDA-MB-231 in COL under FBS gradient (left), MAT under control conditions (center) and FBS gradient (right). Cancer cells were cultured for 3 days after seeding, and stained with rhodamine-phalloidin (red) for actin filament and DAPI (blue) for nuclei. White arrowheads indicate the position of nuclei in each cell. Scale bars: 50 μm
The ECM dependency of MDA-MB-231 in 3D morphology of invasion was supposed to be inherent, with a good consistency when they passed the interface between different ECMs. It was confirmed by heterogeneous alignment of COL and MAT in a microfluidic assay (Figure 3a; details in Supporting Figure 5). MDA-MB-231 cells (green beads) seeded in a bottom channel onto hydrogel 1 (pink) invaded and passed through hydrogel 1 (Figure 3b) and finally invaded into hydrogel 2 (green). Interestingly, MDA-MB-231 cells showed dramatic morphological changes during passage through the interface of hydrogels, both from MAT (multicellualr) to COL (single) and COL (single) to MAT (multicellular) (Figure 3c). The findings validated the inherent plasticity of MDA-MB-231 morphology of invasion dependent on heterogeneous alignment of ECMs.
Figure 3.
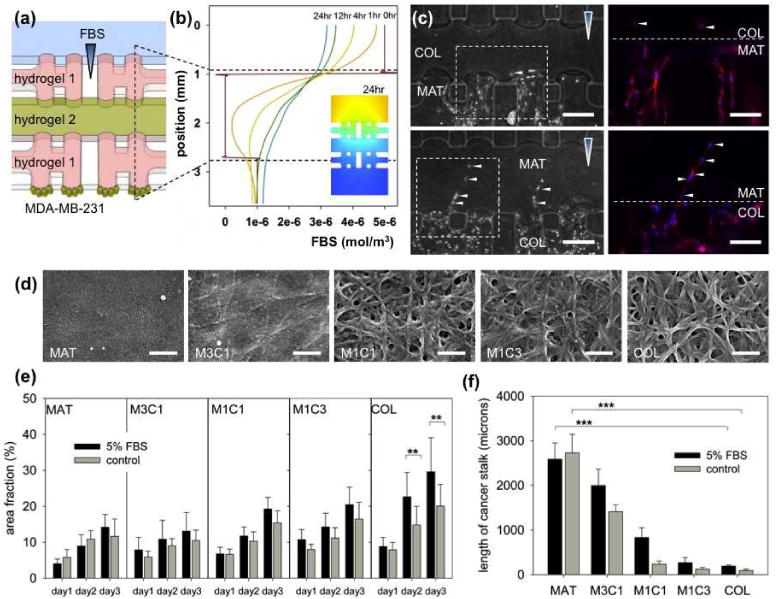
(a) Schematic of the microfluidic assay to incorporate heterogeneously aligned hydrogel scaffolds and (b) simulation results of the transient diffusion of FBS. (c) Morphological transition of MDA-MB-231 from MAT to COL (top) and COL to MAT (bottom) was observed at the ECM interface. White arrowheads indicate morphological transition of MDA-MB-231 cells stained with rhodamine-phalloidin (red) for actin filament and DAPI (blue) for nuclei. Scale bar: 250 μm. (d) SEM images of MAT, M3C1, M1C1, M1C3 and COL. Scale bar: 500nm. Quantification graphs of (e) area fraction and (f) total length of cancer stalks of MDA-MB-231 invading hydrogel generated with various mixing ratios of MAT and COL under 5% FBS and control. Total n=44, and error bars indicate mean values ± standard error (*P < 0.05; **P < 0.01; ***P < 0.001).
It suggested that cancer metastasis can be regulated not only by chemical stimuli but also by ECM compositions. In experiment with heterogeneous mixture of ECMs, we found that MDA-MB-231 cell morphology was regulated by a fraction of MAT in the MAT/COL mixture. SEM images of MAT, COL and MAT/COL mixtures are shown in Figure 3d. COL has fibril structure with pores through which MDA-MB-231 cells invade individually (Supporting Figure 6). In dense mat-like MAT, MDA-MB-231 cells appear to invade by proteolytic ECM degradation, followed by microtracks[2], which can easily be filled by followers (Supporting Figure 6). The microtracks explains multicellular invasion in MAT. Under the FBS gradient, the follower cells prefer proliferation in the microtrack to exploring a new route, as confirmed by the increased number of cells under the FBS gradient with no changes in stalk length (Figure 2c). In correlation with the SEM images, the increased invasion area (Figure 3e) and the sudden increase in cancer stalk length (Figure 3f) suggest that proteolytic invasion of MAT and M3C1 induces multicellular invasion of MDA-MB-231. It shows that in a heterogeneous ECM condition, cancer cells can invade by various ways of morphological alteration and plasticity.
Can the heterogeneity of ECMs effectively affect metastasis? As shown in Figure 3e, ECM heterogeneity regulated morphological changes of cancer cells, but not the amount of invasion and metastatic potential. We supposed that additional factor of heterogeneous cell populations (mixed invasive and less (or non)-invasive cacner cells) would be required as an effective strategy for metastasis into a proteolytic matrix, because breast cancer is a heterogeneous disease, consisting of morphologically evident subtypes with variable molecular genetic signatures[18]. Figure 4a shows that the highly invasive MDA-MB-231 leaded the multicellular invasion of non-invasive MCF-7 into MAT. The non-invasive MCF-7 cells invaded MAT through microtracks made by invasive MDA-MB-231 cells (details in Supporting Figure 7), while without the proteolytic leaders, MCF-7 cells did not invade MAT (Figure 4b). E-cadherin, a transmembrane protein in cell-cell junctions, was found only on the junctions of MCF-7 cells in MAT, but not on the juction of MDA-MB-231 cells and between MDA-MB-231 and MCF-7 cells. It showed that the invasion of non-invasive MCF-7 was induced by physical guidance of microtracks. The role of MDA-MB-231 leaders can also be similized as the role of cancer stem cells in heterogeneous breast tumors, expressing CD44 cancer stem cell marker (Figure 4c).
Figure 4.
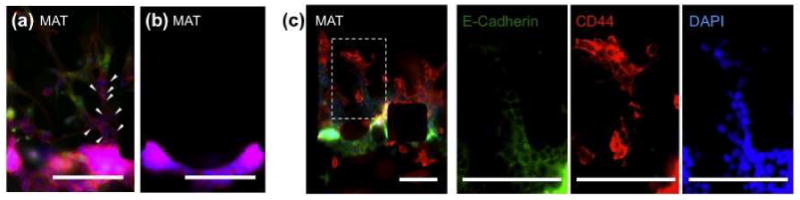
Fluorescence images of heterogeneous cancer cell population. (a) 3D multicellular invasion of highly invasive MDA-MB-231 (green) and non-invasive MCF-7 (white arrowheads) stained with rhodamine-phalloidin (red) for actin filament, DAPI (blue) for nuclei and CFDA-SE for MDA-MB-231 (green). Scale bar: 250 μm. (b) Without highly invasive MDA-MB-231, MCF-7 cannot invade into MAT. Scale bar: 250 μm. (c) Innunofluorescence staining of E-cadherin (green), CD44 (red) and DAPI (blue) in MCF-7 and MDA-MB-231 under co-culture condition. Scale bar: 150 μm.
The invasion of non-invasive cancer cells into a proteolytic matrix was also proved by metastasis–suppressed cancer cells. Metalloproteinases (MMPs) are possible candidate factors for degradation of ECMs and might therefore play a critical role in the formation of microtracks by leading cells in MAT. It is widely accepted that the pleiotropic effect of MMPs has a significant effect on cancer cell growth in a secondary site23. However in 3D invasion into ECMs, MMP-inhibited MDA-MB-231 cells showed limited reduction of invasion area in COL, and no reduction in MAT. MMP-inhibited cells invading MAT still maintained multicellular stalk structures with leaders and microtracks (Supporting Figure 8). Similar to the result of figure 4, we concluded that very limited population of the leaders (less-treated survivors or proliferated cancer cells) can induce invasion of MMP-inhibited (therefore non- or less-invasive) cells when they meet heterogeneous, proteolytically degrading ECMs.
The results demonstrates that plasticity of cancer cells is a very effective strategy for invasion into 3D proteolytic ECMs or ECMs with heterogeneous combinations. This might possibly explain the disappointing results of therapeutic approaches with inhibitors of MMPs in vivo24, but requires further investigation. We are additionally attempting to determine the internal mechanisms used by breast cancer cells to facilitate plasticity, in disrupting the basement membrane and migrating into the stromal matrix. Various molecular factors are currently under investigation with the developed method, including anti-angiogenic and anti-metastatic factors. Cell-cell interactions that are tightly related to initial cell density and number of cells on/in hydrogels may be another key regulator of cancer cell morphology of invasion. Plasticity and reciprocity of breast cancer cells to various ECMs were quantitatively and 3-dimensionally analyzed in vitro and proved to be an effective strategy of metastasis with an invasion routes (i.e. microtracks) in heterogeneous ECMs.
Experimental
Microfluidic device fabrication and preparation
Two of the microfluidic platforms used in this study were composed of 140 μm height channel-patterned PDMS (polydimethylsiloxane; Sylgard 184; Dow Chemical, MI) prepared on a SU-8 photoresist-patterned wafer (MicroChem, MA) using a conventional soft lithography process. Sterilized PDMS devices were bonded to glass coverslips with oxygen plasma (Femto Science, Korea) and coated with a PDL (poly-D-lysine hydrobromide; 1 mg/ml; Sigma-Aldrich, St Louis, MO) solution. Collagen type I (rat tail; cat. no. 40236; BD Bioscience), matrigel (Growth Factor Reduced (GFR) Matrigel™ Matrix, Phenol Red-free; BD Bioscience), and mixed hydrogel (collagen type I and matrigel at ratios of 1:1, 3:1 and 1:3) were used as the ECM scaffold material. Collagen type I was diluted to 2 mg/ml in 10× phosphate-buffered saline (PBS; Gibco) and gel stiffness controlled by altering the pH of collagen solution to 11 by adjusting the amount of 0.5N NaOH. The hydrogel scaffold-introduced into the microfluidic channel was allowed to gel in a 5% CO2 incubator at 37°C for 30 minutes. In the 3-gel device, hydrogel 1 was first injected into right and left gel scaffold regions, and after 30 minutes of incubation, hydrogel 2 or cells embedded in hydrogel 2 introduced into middle gel scaffold region, followed by incubation for 30 minutes. After gelation, all channels were filled with culture medium and plated in a humidified incubator before cell seeding. The hydrogel pH was irrelevant to cell morphology, since it was naturally neutralized by culture medium in the channels following gelation.
MDA-MB-231 breast cancer cell culture in microfluidic systems
The commercially available MDA-MB-231 human breast adenocarcinoma cell line was cultured in RPMI-1640 containing 10% fetal bovine serum (FBS). Cancer cells were seeded into cell culture channels of the microfluidic device by preparing suspensions at a density of 1×106 cells/ml, and the device maintained at 37°C in a 5% CO2 incubator for 2 h in a tilted position to facilitate attachment of cells onto collagen and matrigel scaffold by gravity. RPMI-1640 containing 1% FBS (basic nutrient amount for cell culture in a microfluidic channel) was filled in the cell culture channels, and medium containing 5% FBS introduced into the condition channel to generate a gradient within the gel scaffold. For cells embedded in hydrogel, the cancer suspension was prepared at a concentration of 1×107cells/ml diluted 1/10 times to obtain a desired concentration of 1×106 cells/ml. An aliquot of cell suspension (20 μl) was mixed in 200 μl gel and filled into the middle gel scaffold region. All microfluidic channels were filled with medium containing 5% FBS. Media in all channels were exchanged with fresh medium every day to maintain the linear concentration gradient. Approximately 50∼60 μl of medium was needed in each reservoir (100∼120 μl per channel) at the inlets and outlets to minimize medium evaporation from the channels.
MDA-MB-231 and MCF-7 cell co-culture in microfluidic systems
MCF-7 breast cancer cell line was cultured in 1640 RPMI containing 10% FBS and seeded into the cell culture channels by preparing suspensions at a density of 1×106 cells/ml, and the device maintained at 37°C in a 5% CO2 incubator for 2 h in a tilted position. Then, 1×106 cells/ml of MDA-MB-231 cells were additionally seeded into the same cell culture channels and cultured in a tilted position for 2 h. RPMI-1640 containing 1% FBS and 5% FBS was filled in the cell culture channels in the same way described in “MDA-MB-231 breast cancer cell culture in microfluidic systems”.
COMSOL simulation of the FBS diffusion gradient profile
The chemical gradient generated within hydrogel was simulated using COMSOL Multiphysics 4.0 (COMSOL Inc., MA, USA). We assumed that the diffusion coefficient of FBS in culture medium was 4.28×10-11 m2/s, and 6.10×10-11 m2/s and 6.41×10-11 m2/s in collagen and matrigel scaffold, respectively. Moreover, the concentrations of 5% FBS and 1% FBS in media were taken as 5×10-3mol/m3 and 1×10-2mol/m3, respectively.
Quantification of 3D cell invasion and migration
The morphologies of migrating and invading cancer cells were monitored using phase-contrast microscopy (Olympus) every day before medium exchange. Fluorescent images were obtained by fixing cells with 4% paraformaldehyde for 15 minutes and permeabilizing with 0.1% Triton X-100 for 5 minutes at room temperature. Actin filaments and nuclei were stained with rhodamine-phalloidin (Sigma-Aldrich) and DAPI (40,6-diamidino-2-phenylindole; Sigma-Aldrich) for 1 hour, respectively. Also, MCF-7 and MDA-MB-231 cells were blocked 4% (wt/vol) bovine serum albumin (BSA) and incubated with anti-E-cadherin (1:100, Santa Cruz Biotechnology, inc.) and CD44 (5 μl antibody/1×106 cells, Invitrogen) overnight at 4°C. Cells were washed with PBS and incubated with Alexa 488-conjugated goat anti-rabbit antibody (1:200, Molecular Probes®).
Supplementary Material
Footnotes
Supporting Information is available online from Wiley InterScience or from the author.
Contributor Information
Yoojin Shin, School of Mechanical Engineering, Korea University, Anam-Dong, Seongbuk-Gu, Seoul 136-713 (Korea).
Hyunju Kim, College of Like Science and Biotechnology, Korea University, Anam-Dong, Seongbuk-Gu, Seoul 136-701 (Korea).
Sewoon Han, School of Mechanical Engineering, Korea University, Anam-Dong, Seongbuk-Gu, Seoul 136-713 (Korea).
Jihee Won, School of Mechanical Engineering, Korea University, Anam-Dong, Seongbuk-Gu, Seoul 136-713 (Korea).
Prof. Eun-Sook Lee, National Cancer Center, Madu 1-dong, Ilsandong-gu, Goyang-si, Gyeonggi-do 410-769 (Korea)
Prof. Roger D. Kamm, Department of Mechanical Engineering and Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139 (USA)
Prof. Jae-Hong Kim, Email: jhongkim@korea.ac.kr, College of Like Science and Biotechnology, Korea University, Anam-Dong, Seongbuk-Gu, Seoul 136-701 (Korea).
Prof. Seok Chung, Email: sidchung@korea.ac.kr, School of Mechanical Engineering, Korea University, Anam-Dong, Seongbuk-Gu, Seoul 136-713 (Korea).
References
- 1.Fidler IJ. Nat Rev Cancer. 2003;3:453. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 2.Friedl P, Alexander S. Cell. 2011;147:992. doi: 10.1016/j.cell.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 3.Friedl P. Current Opinion in Cell Biology. 2004;16:14. doi: 10.1016/j.ceb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 4.Wolf K, Friedl P. Trends in Cell Biology. 2011;21:736. doi: 10.1016/j.tcb.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 5.Rowe RG, Weiss SJ. Annu Rev Cell Dev Biol. 2009;25:567. doi: 10.1146/annurev.cellbio.24.110707.175315. [DOI] [PubMed] [Google Scholar]
- 6.Katharine S, Maurice R. BMC Cancer. 8 [Google Scholar]
- 7.LeBleu VS, MacDonald B, Kalluri R. Experimental biology and medicine. 2007;232:1121. doi: 10.3181/0703-MR-72. [DOI] [PubMed] [Google Scholar]
- 8.Ma L, Teruya-Feldstein J, Weinberg RA. Nature. 2007;449:682. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]; Kleinman HK, Martin GR. Matrigel: basement membrane matrix with biological activity. 2005 doi: 10.1016/j.semcancer.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 9.Hotary KB, Allen ED, Brooks PC, Datta NS, Long MW, Weiss SJ. Cell. 2003;114:33. doi: 10.1016/s0092-8674(03)00513-0. [DOI] [PubMed] [Google Scholar]
- 10.Radisky D, Muschler J, Bissell M. Cancer investigation. 2002;20:139. doi: 10.1081/cnv-120000374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mi K, Wang G, Liu Z, Feng Z, Huang B, Zhao X. Macromolecular bioscience. 2009;9:437. doi: 10.1002/mabi.200800262. [DOI] [PubMed] [Google Scholar]
- 12.Wolf K, Mazo I, Leung H, Engelke K, von Andrian UH, Deryugina EI, Strongin AY, Brocker EB, Friedl P. J Cell Biol. 2003;160:267. doi: 10.1083/jcb.200209006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Demou ZN, Awad M, McKee T, Perentes JY, Wang X, Munn LL, Jain RK, Boucher Y. Cancer Res. 2005;65:5674. doi: 10.1158/0008-5472.CAN-04-1682. [DOI] [PubMed] [Google Scholar]; Hurst RE, Kyker KD, Bonner RB, Bowditch RD, Hemstreet GP., 3rd Anticancer Res. 2003;23:3119. [PMC free article] [PubMed] [Google Scholar]; Dozmorov MG, Kyker KD, Saban R, Knowlton N, Dozmorov I, Centola MB, Hurst RE. BMC Cancer. 2006;6:12. doi: 10.1186/1471-2407-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wozniak MA, Keely PJ. Biol Proced Online. 2005;7:144. doi: 10.1251/bpo112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chung S, Sudo R, Vickerman V, Zervantonakis I, Kamm R. Annals of Biomedical Engineering. 2010;38:1164. doi: 10.1007/s10439-010-9899-3. [DOI] [PubMed] [Google Scholar]
- 15.Chung S, Sudo R, Mack P, Wan C, Vickerman V, Kamm R. Lab on a Chip. 2009;9:269. doi: 10.1039/b807585a. [DOI] [PubMed] [Google Scholar]; Chung S, Sudo R, Zervantonakis I, Rimchala T, Kamm R. Advanced Materials. 2009;21:4863. doi: 10.1002/adma.200901727. [DOI] [PubMed] [Google Scholar]; Sudo R, Chung S, Zervantonakis I, Vickerman V, Toshimitsu Y, Griffith L, Kamm R. The FASEB Journal. 2009;23:2155. doi: 10.1096/fj.08-122820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamasaki Y, Sumimoto K, Nishikawa M, Yamashita F, Yamaoka K, Hashida M, Takakura Y. Journal of Pharmacology and Experimental Therapeutics. 2002;301:467. doi: 10.1124/jpet.301.2.467. [DOI] [PubMed] [Google Scholar]
- 17.Jeong SHGS, Shin Y, Kwon GH, Kamm RD, Lee SH, Chung S. Analytical Chemistry. 2011 doi: 10.1021/ac202170e. [DOI] [PubMed] [Google Scholar]
- 18.Hsiao YH, Chou MC, Fowler C, Mason JT, Man Y. Journal of Cancer. 2010;1:6. doi: 10.7150/jca.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson WF, Matsuno R. Journal of the National Cancer Institute. 2006;98:948. doi: 10.1093/jnci/djj295. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.