

Abstract
The microsomal prostaglandin E2 synthase (mPGES)-1 is the terminal enzyme in the biosynthesis of prostaglandin (PG)E2 from cyclooxygenase (COX)-derived PGH2. We previously found that mPGES-1 is inhibited by boswellic acids (IC50 = 3–30 μM), which are bioactive triterpene acids present in the anti-inflammatory remedy frankincense. Here we show that besides boswellic acids, additional known triterpene acids (i.e., tircuallic, lupeolic, and roburic acids) isolated from frankincense suppress mPGES-1 with increased potencies. In particular, 3α-acetoxy-8,24-dienetirucallic acid (6) and 3α-acetoxy-7,24-dienetirucallic acid (10) inhibited mPGES-1 activity in a cell-free assay with IC50 = 0.4 μM, each. Structure–activity relationship studies and docking simulations revealed concrete structure-related interactions with mPGES-1 and its cosubstrate glutathione. COX-1 and -2 were hardly affected by the triterpene acids (IC50 > 10 μM). Given the crucial role of mPGES-1 in inflammation and the abundance of highly active triterpene acids in frankincence extracts, our findings provide further evidence of the anti-inflammatory potential of frankincense preparations and reveal novel, potent bioactivities of tirucallic acids, roburic acids, and lupeolic acids.
The genus Boswellia comprises about 20 species, and among those Boswellia sacra Flück, B. carterii Birdw., B. frereana Birdw., B. papyrifera Hochst., and B. serrata Roxb. are commonly used as remedies in folk medicine. The gum resin from Boswellia spp. is composed of an essential oil fraction (5–10%), a mucilage fraction (up to 30%), and a pure resin fraction (up to 60%).1 The resin fraction has been intensively studied, and many triterpene acids with pentacyclic ursane, oleanane, and lupine scaffolds or tetracyclic tirucallane scaffolds have been isolated and characterized.2−5 Triterpene acids usually represent about 50% (m/m) of the resin fraction.1 However, depending on environmental fluctuations and the species, the amounts of triterpene acids may strongly differ, and resins from B. frereana, for instance, contain diminutive amounts of triterpene acids.6
β-Boswellic acid (1), 11-keto-β-boswellic acid (2), 3-O-acteyl-β-boswellic acid (3), and 3-O-acteyl-11-keto-β-boswellic acid (4) are pentacyclic triterpene acids that represent major ingredients in Boswellia spp. gum resins, reaching 14% to 25% (m/m) of the lipophilic extract from B. serrata gum resin.2,7 Many pharmacological activities and targets of boswellic acids have been identified.5 Boswellic acids are thus considered as the major bioactive principles of gum resins of Boswellia spp. The tetracyclic tirucallic acids, which are also part of further resinous remedies such as from Canarium,8Protium,9 and Pistacia spp.,10 may carry a hydroxy or a keto moiety at the 3 position and differ in the configuration of the hydroxy group and the acetylation of this residue. Further derivatives arise from the positioning of the cyclic double bond located at position 7 or 8, yielding 3-α-hydroxy-8,24-dienetirucallic acid (5), 3α-acetoxy-8,24-dienetirucallic acid (6), 3-β-hydroxy-8,24-dienetirucallic acid (7), 3-oxo-8,24-dienetirucallic acid (8), 3-α-hydroxy-7,24-dienetirucallic acid (9), and 3α-acetoxy-7,24-dienetirucallic acid (10).2,11−13 Nyctanthic acids and roburic acids represent seco-derivatives of boswellic acids that exhibit an open A-ring (e.g., roburic acid (11), 4,(23)-dihydroroburic acid (12), 4,(23)-dihydro-11-keto-roburic acid (13), and 4,(23)-dihydronyctanthic acid (14)) and are sparsely contained in gum resins of Boswellia spp.14 Lupeolic acid (15) and 3-O-acetyllupeolic acid (16) as well as the recently discovered 3-O-acetyl-28-hydroxylupeolic acid (17)15 also represent minor components (<1% (m/m)), respectively.2
Recently, the boswellic acids 1–4 were identified as inhibitors of microsomal prostaglandin E2 synthase (mPGES)-1 in cell-free, cellular, and in vivo studies as a molecular basis for the anti-inflammatory actions of frankincense.16 mPGES-1 is an inducible enzyme belonging to the three isoforms of PGE2 synthases that convert PGH2, formed by cyclooxygenases (COX)-1/2 from arachidonic acid (AA), to the pro-inflammatory PGE2. Inhibitors of mPGES-1 are considered as promising therapeutics for intervention with inflammatory disorders and cancer.17 In the present study we expand our investigations on triterpene acids derived from frankincense that may interfere with the enzymatic activity of mPGES-1.

Results and Discussion
Triterpene Acids from Gum Resins of Boswellia Species Inhibit mPGES-1 Activity
Previous studies showed that numerous mPGES-1 inhibitors are lipophilic acidic molecules.17,18 Therefore, special attention was paid to the acidic fraction of the gum resin extracts derived from different Boswellia spp. The acidic fractions (containing lipophilic acidic ingredients) of gum resins derived from different Boswellia spp. were separated from the neutral components (i.e., the essential oil and mucilage fraction); see the Supporting Information. First, aliquots of the neutral and acidic fractions were analyzed for inhibition of mPGES-1 activity in a cell-free assay using microsomes of IL-1β-stimulated A549 cells as enzyme source and 20 μM PGH2 as mPGES-1 substrate; MK-886 (10 μM; IC50 = 2.4 μM) was used as reference compound.19 The acidic fraction of all four tested species potently inhibited mPGES-1 activity. Thus, concentration–response analysis revealed IC50 values of 1.9, 2.8, 1.6, and 0.4 μg/mL for the acidic fraction of gum resins from B. serrata, B. sacra, B. carterii, and B. papyrifera, respectively (Figure 1B). In contrast, the neutral fraction (10 μg/mL) did not significantly inhibit mPGES-1 activity, regardless from which species it originated (not shown). In particular, the acidic fraction of B. papyrifera gum potently suppressed mPGES-1 activity with a maximal inhibition of 92% at 30 μg/mL, which was superior to the control inhibitor MK-886 (10 μM = 0.49 μg/mL, 79% inhibition) under the same assay conditions. Therefore, the remarkable potency of the acidic fraction of B. papyrifera gums suggested the presence of highly active constituents. It should be noted that the nature of the ingredients and their contents do not substantially differ between lipophilic extracts of gum resins from these four Boswellia spp.,7 indicating that defined mixtures or compositions of the bioactive components may result in efficient mPGES-1 inhibition.
Figure 1.

Microsomal preparations of IL-1β-stimulated A549 cells were preincubated with the indicated concentrations of acidic fractions derived from gums of B. papyrifera, B. serrata, B. sacra, and B. carterii, or vehicle (DMSO), for 15 min at 4 °C. The reaction was started by addition of 20 μM PGH2, and after 60 s at 4 °C, the reaction was terminated and PGE2 was analyzed. Data are given as mean + SEM, n = 3 or 4.
Besides the four boswellic acids 1–4 that were recently shown to inhibit mPGES-1,16 the acidic fractions of frankincense gum resins may contain additional triterpene acids that could interfere with mPGES-1 activity as well. We isolated 17 known triterpene acids (Table 1) from various gum resins of different Boswellia spp. (see Supporting Information), that is, the four boswellic acids 1–4, the six tirucallic acids 5–10, the three roburic acids 11–13, the nyctanthic acid 14, and the three lupeolic acids 15–17, by preparative HPLC. The chemical structures of the isolated compounds were analyzed by MS and NMR and compared to literature data (see Supporting Information). The four boswellic acids 1–4 were not retested for mPGES-1 inhibition. The 13 remaining triterpene acids 5–17 were tested at a fixed concentration of 10 μM (Table 1). The roburic acids 11 and 12 as well as the nyctanthic acid 14 failed to significantly inhibit mPGES-1 activity, whereas all tirucallic acids (5–10), the roburic acid 13, and the three lupeolic acids 15–17 markedly suppressed PGE2 production (Table 1). Just like MK-886 (at 10 μM), all these active compounds exerted a maximal inhibition of about 70% to 80%, except 5, which was able to suppress PGE2 formation by 96% (Table 1). Those triterpene acids that caused more than 60% inhibition at 10 μM were subjected to concentration–response analysis.
Table 1. Inhibition of mPGES-1 Activity by Triterpene Acids from Gum Resins of Boswellia spp. (mean ± SEM, n = 3–6).
| compound | residual activity at 10 μM | IC50 (μM) |
|---|---|---|
| 5 | 3.5 ± 0.5 | 1.1 |
| 6 | 36.8 ± 4.5 | 0.4 |
| 7 | 29.2 ± 3.7 | 1.2 |
| 8 | 16.7 ± 3.7 | 0.9 |
| 9 | 29.6 ± 0.5 | 3.0 |
| 10 | 20.6 ± 2.7 | 0.4 |
| 11 | 83.9 ± 5.8 | >10.0 |
| 12 | 92.4 ± 4.3 | >10.0 |
| 13 | 21.6 ± 3.1 | 1.0 |
| 14 | 74.1 ± 6.5 | >10.0 |
| 15 | 43.1 ± 3.6 | 8.5 |
| 16 | 51.4 ± 7.9 | 10.0 |
| 17 | 31.1 ± 9.6 | 0.9 |
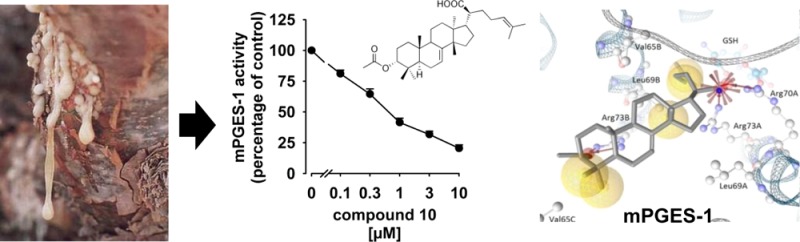
As depicted from Table 1, the six tirucallic acids 5–10, the roburic acid 13, and the lupeolic acid 17 potently suppressed mPGES-1 activity with IC50 values of 0.4 to 3 μM. In comparison to the boswellic acids 1–4 as inhibitors of mPGES-1 (IC50 = 3–30 μM16), the potency of the above-mentioned triterpene acids is indeed remarkable. The two acetylated tirucallic acids 6 and 10 with IC50 = 0.4 μM, each, were most potent, whereas the corresponding deacetylated analogues 5 and 9 were less efficient (IC50 = 1.1 and 3.0 μM, respectively) (Figure 2).
Figure 2.

Concentration–response analysis of triterpene acids and mPGES-1 activity. Microsomal preparations of IL-1β-stimulated A549 cells were preincubated with (A) 3-α-hydroxy-8,24-dienetirucallic acid (5), (B) 3α-acetoxy-8,24-dienetirucallic acid (6), (C) 3-α-hydroxy-7,24-dienetirucallic acid (9), and (D) 3α-acetoxy-7,24-dienetirucallic acid (10) at the indicated concentrations for 15 min at 4 °C. The reaction was started by addition of 20 μM PGH2, and after 60 s at 4 °C the reaction was terminated and PGE2 was analyzed Data are given as mean + SEM, n = 4–7.
From this structure–activity-relationship (SAR) study, we conclude that the acetylation of the free hydroxyl moiety at the 3 position of 6 and 10 is seemingly beneficial. On the other hand, replacement of the 3-hydroxy group of 5 (IC50 = 1.1 μM) by a keto group yielding 8 exhibited no significantly improved potency (IC50 = 0.9 μM). Moreover, 3β-configured 7 showed almost the same inhibitory potency (IC50 = 1.2 μM) as its 3α-isomer 5, indicating that the steric positioning of the 3-hydroxy group has negligible impact on the bioactivity. Among the roburic acids, the derivative 13, carrying a keto moiety, potently inhibited mPGES-1 activity (IC50 = 1.0 μM), whereas 11 and 12 (without the keto group) were not active up to 10 μM. Along these lines, the nyctanthic acid 14, also lacking a keto moiety, which is structurally related to the roburic acid 12, was also inactive. Inhibition of mPGES-1 by lupeolic acids was evident for all tested derivatives 15–17, but only the acetylated derivative 17 reached an IC50 (i.e., 0.9 μM) lower than 10 μM, suggesting that the additional hydroxyl moiety at position C(28) is responsible for this molecule’s bioactivity.
In contrast to boswellic acids, few studies have addressed the bioactivities and pharmacological properties of tirucallic acids and lupeolic acids. Thus, tirucallic acids were found to modulate 5-lipoxygenase product synthesis12 and to inhibit the serine/threonine kinase Akt in prostate cancer cells in association with apoptotic cell death.20 Cancer chemopreventive and cytotoxic activities in neuroblastoma cell lines were reported for the lupeolic acids 15 and 16, albeit at high concentrations (IC50 = 4.1–86.7 μM),21 and both compounds (applied topically) inhibited phorbol ester-induced ear inflammation in mice.11 For roburic acids, only one study on bioactivity has been published that describes moderate inhibitory effects on isolated COX enzymes (IC50 > 10 μM).22 Therefore, the identification of mPGES-1 as a target for the tirucallic acids 5–10, the roburic acid 13, and the luepolic acid 17 is a substantial insight into the pharmacology of these triterpene acids and underlines their anti-inflammatory and anticancer potential, which was proposed for other mPGES-1 inhibitors.17,23
Prediction of Binding Modes by Molecular Docking
The fact that several different structural classes of triterpene acids inhibit mPGES-1, while other derivatives failed, led us to deduce SARs in order to speculate about a common binding pattern of active compounds. In silico docking studies were performed to surmise binding modes of triterpene acids in mPGES-1. The tirucallic acids 5 and 10, the lupeolic acid 17, and the boswellic acid 1 were analyzed for common chemical features relevant to potent mPGES-1 inhibition and to compare the binding mode of these triterpene acids with less active derivatives. The tirucallic acid 10, one of the most potent derivatives (IC50 = 0.4 μM), exhibited various molecular interactions, which were subjected to subtle modifications during the MMFF94-based minimization within LigandScout (Figure 3A). For instance, the Arg73 of subunit A, which we refer to as Arg73A, extended as a solvent-exposed residue into the central pore of the homotrimeric mPGES-1. During the minimization, Arg73A shifted toward the acidic moiety of 10, making an ionic interaction feasible. Additionally, the acidic group of 10 formed an ionic interaction with Arg70A, a residue near the cofactor glutathione (GSH). Furthermore, the acetoxy group of 10 was involved in hydrogen bonding to Arg73B. We additionally observed various hydrophobic interactions between 10 and residues embedded in the central pore (e.g., Leu69A, Val65B, Leu69B, and Val65C). For comparison, the tirucallic acid 5 (IC50 = 1.1 μM) (Figure 3B) and the lupeolic acid 17 (IC50 = 0.9 μM) (Figure 3C) formed an ionic interaction or a hydrogen bond to Arg73C or Arg73A, respectively. Additionally, in the case of 5, a hydrogen bond was formed between the hydroxyl group of the triterpene acid and GSH, while, in the case of 17, a hydrogen bond was formed between the acetoxy group and the backbone amide of His53A.
Figure 3.

Predicted binding modes for representative triterpene acids using molecular docking. Predicted binding modes are shown for (A) 3α-acetoxy-7,24-dienetirucallic acid (10) (B), 3-α-hydroxy-8,24-dienetirucallic acid (5), (C) 3-O-acetyl-28-hydroxylupeolic acid (17), and (D) β-boswellic acid (1). Protein–ligand interactions are color-coded: red arrow, hydrogen-bond acceptor; green arrow, hydrogen-bond donor; yellow sphere, hydrophobic interaction; red star, negatively ionizable.
Interestingly, the sec-propenyl group of 17 protruded into the region adjacent to the cofactor GSH and formed a hydrophobic interaction with Leu69B. Finally, the boswellic acid 1, with less pronounced potency toward mPGES-1 (IC50 = 5 μM),16 was docked into the 3D structure of mPGES-1 (Figure 3D). This boswellic acid was predicted to form a hydrogen bond to Arg73B, similar to the other triterpene acids. Although hydrophobic interactions were formed (e.g., with Val65B and Leu69B), similar to other triterpene acids, these molecular interactions were less frequently observed for 1 compared to the other more potent compounds.
Together, a hydrogen bond or an ionic interaction of the carboxylic group to Arg73, which extends into the central pore of the homotrimeric enzyme, is observed in all docking poses. Thus, the acidic group of 5–10, 13, and 17 may essentially contribute to the potent interference of these triterpene acids with mPGES-1. However, additional interactions involving oxygen-containing substituents determine the potency, since 11, 12, and 14, lacking an additional oxygen substituent, failed to inhibit mPGES-1, and also 15 and 16, lacking the C(28)–OH, were less active than 17. In contrast, all six tirucallic acids (5–10) that carry an oxygen (hydroxyl, keto, or acetoxy group) distant from the carboxylic acid moiety potently suppressed mPGES-1 activity with IC50 values of 0.4 to 3 μM. Also, 13 and 17 substituted with a hydroxyl or keto group potently suppressed mPGES-1 activity. Therefore, the presence of oxygen distant from the carboxylic moiety is seemingly important for molecular interactions with mPGES-1. Moreover, the docking studies imply that hydrophobic interactions, which were frequently formed to residues embedded in the central pore (e.g., Val65, Leu69, and Met76), seem to contribute to mPGES-1 interference. Furthermore, among the most potent triterpene acids (e.g., 5 and 10), an ionic interaction to a residue near GSH (e.g., Arg70) or a hydrogen bond formed directly with GSH could be observed in the molecular docking.
Effects of Triterpene Acids on Cyclooxygenase-1 and -2 Activities
For generation of the proinflammatory PGE2, the release of AA as substrate and its conversion by COX enzymes are essential upstream processes leading to the formation of PGH2 as substrate for various prostanoid synthases.24 As mPGES-1 is considered to be a valuable drug target for selective inhibition of PGE2 biosynthesis, a concomitant suppression of COX enzymes would obliterate the selectivity of such compounds. In fact, the boswellic acids 1–4,25,22 the tirucallic acid 6,26 and the roburic acid 11(22) were reported to inhibit COX enzymes, though at higher concentrations, in the two-digit micromolar range. To estimate the selectivity of the triterpene acids that potently inhibited mPGES-1 activity with IC50 < 3 μM (i.e., 5–10, 13, and 17), the impact on COX enzymes was analyzed. In cell-free assays using purified ovine COX-1 or purified human recombinant COX-2, the triterpene acids (10 μM) elicited only moderate inhibitory effects without statistical significance (p > 0.05; Table 2).
Table 2. Effects of Triterpene Acids from Gum Resins of Boswellia spp. on COX-1 and COX-2 Activity in a Cell-Free Assay (mean ± SEM, n = 3, 4).
| compound | COX-1 residual activity at 10 μM | COX-2 residual activity at 10 μM |
|---|---|---|
| 5 | 81.9 ± 8.3 | n.d.c |
| 6 | 63.2 ± 4.1 | 81.8 ± 3.1 |
| 7 | 82.3 ± 1.2 | 83.4 ± 8.6 |
| 8 | 81.1 ± 6.5 | 70.1 ± 7.0 |
| 9 | 69.6 ± 3.7 | 83.4 ± 3.7 |
| 13 | 64.8 ± 9.5 | 66.3 ± 7.6 |
| 17 | 90.7 ± 7.7 | 75.8 ± 6.4 |
| indomethacina | 41.3 ± 5.2 | n.d. |
| celecoxibb | n.d. | 22.4 ± 3.8 |
Indomethacin, 10 μM, reference compound for COX-1.
Celecoxib, 5 μM, reference compound for COX-2.
n.d. = not determined.
In conclusion, we analyzed the major triterpene acids present in gum resins of Boswellia spp. for their ability to interfere with mPGES-1, and we identified six tirucallic acids (5–10), one lupeolic acid (17), and one roburic acid (13) as potent inhibitors of this enzyme. Of interest, these triterpene acids, except 9, inhibit mPGES-1 activity with improved potency (IC50 = 0.4–1.2 μM) as compared to boswellic acids (IC50 = 3–30 μM16). Molecular docking simulations confirm the improved potencies of tirucallic acids and lupeolic acids, as hydrogen bonds to or ionic interactions with the cofactor GSH, the neighboring Arg70, or Arg73 mediate tight binding, whereas the boswellic acid 1 forms hydrophobic interactions at a greater distance to the cofactor GSH, implying weaker binding. We suggest that mPGES-1 is a major target of several triterpene acids that are contained in gum resins of certain Boswellia spp., and suppression of mPGES-1 by these compounds may underlie their beneficial properties in the treatment of painful inflammatory disorders.
Experimental Section
General Experimental Procedures
The structures of the triterpene acids used in this study have been described previously and were isolated from the gum resin of B. serrata, B. papyrifera, B. carterii, and B. socotrana, respectively. The gum resins from B. serrata, B. papyrifera, B. carterii, and B. sacra were purchased from Gerhard Eggebrecht Vegetabilien & Harze, Süderau, Germany, whereas the gum resin of B. socotrana was a gift by the Botanical Garden and Botanical Museum (BGBM) in Berlin-Dahlem, Germany. All solvents were distilled. Fractions obtained by extraction and/or chromatography were stored in a refrigerator at −30 °C until they were used.
Structure elucidation was done by 1D- and 2D-NMR spectroscopy on a Bruker AV II 400 and/or a Bruker AV 500 NMR spectrometer. As 1D-NMR spectra, 1H, 13C, DEPT90, and DEPT135 spectra were recorded. As 2D experiments, H,H-gs-DQF-COSY, HMQC, HMQC-COSY, HMBC, and NOESY spectra were recorded. The molecular formula was calculated from HRMS spectra obtained with MAT95S (HRMS with CI) from Bruker. ESI mass spectra were measured with ZQ4000 from Waters (ESI in the negative ion mode). For more details, see the Supporting Information.
General Procedure for Extraction of the Resins
The resin was frozen at −30 °C overnight. Then, it was finely ground in a laboratory mill and extracted in a Soxhlet extractor with distilled dichloromethane for 16 h. A 180 g amount of the finely ground resin was extracted with ca. 1.5 L of solvent. The solvent was evaporated with the aid of a rotary evaporator at 40 °C under vacuum (ca. 10 mbar). The residue (raw extract) was dissolved in 200 mL of diethyl ether and extracted with 200 mL of 5% (m/v) aqueous KOH in a separatory funnel. After separating the phases, the alkaline aqueous phase was extracted three times with 50 mL of diethyl ether each. The combined ethereal phases were washed with brine (20 mL) and dried with MgSO4. After filtration of the drying agent, the diethyl ether was evaporated under vacuum in a rotary evaporator. The remaining oily residue is the neutral fraction of the extract. The alkaline aqueous phase from above was cooled in an ice bath and carefully acidified with ice cold, concentrated aqueous hydrochloric acid. The mixture became turbid milky through separation of insoluble acidic compounds. These were each extracted three times with 50 mL of distilled diethyl ether. The combined extracts were washed with brine (20 mL) and dried over MgSO4. After filtration of the drying agent, the solvent was evaporated under vacuum. The remaining yellow to orange foam is the acidic fraction of the resin and contains all lipophilic acids of the particular resin. The amounts of the neutral fraction and the acidic fraction depend strongly on the particular resin.
Separation of the Acidic Fraction through Flash Chromatography
The column was packed with a slurry of silica gel (normal phase NP, Merck AG, Darmstadt, Germany) with particle size from 40 to 63 μm in the appropriate mobile phase. After covering the silica gel with a small pad of purified sand, the sample, dissolved in a small amount of mobile phase, was applied on top of the column. Elution of the compounds was done with 500 mL portions of mobile phase (usually pentane/diethyl ether +1% (v/v) of acetic acid) with stepwise increasing polarity (starting with pentane/diethyl ether, 8:1, then 7:1, then 6:1, and so on up to pentane/diethyl ether, 1:2). The fractions (ca. 20 mL each) were collected in test tubes with the aid of a fraction collector. Analysis of the fractions was done with TLC (Merck glass plates) with a suitable solvent. Fractions with similar composition were combined, and the solvent was evaporated to dryness. Usually, a bright yellow to orange foam is obtained. These combined fractions were analyzed by analytical HPLC and further separated by preparative HPLC.
Preparative HPLC was done with a preparative HPLC pump model SYKAM S1521 (Sykam, Fürstenfeldbruck, Germany), a Rheodyne injection valve type 7725i, a diode array detector model Sykam S3210 (Sykam, Fürstenfeldbruck, Germany), and Chromstar 6.0 software (SCPA, Weyhe-Leeste, Germany). As a column, a semipreparative Nucleodur 100-5 C18 ec, 5 μm, 250 × 20, from Machery & Nagel, Düren, Germany, was used. The purity of the isolated compounds used for biological evaluation was at least 95%.
Materials for Bioassays
DMEM/high glucose (4.5 g/L) medium, penicillin, streptomycin, trypsin/EDTA solution, PAA (Coelbe, Germany), PGH2 (Larodan, Malmö, Sweden), 11β-PGE2 and MK-886 (BioTrend Chemicals GmbH, Cologne, Germany), arachidonic acid, and fetal calf serum were used, and all other chemicals were obtained from Sigma-Aldrich (Deisenhofen, Germany) unless stated otherwise.
Stimulation of A549 Cells and Isolation of Microsomes
Induction of mPGES-1 expression in A549 cells and isolation of microsomes were performed as described.19 In brief, cells were incubated for 16 h at 37 °C and 5% CO2, and after changing the medium, mPGES-1 expression was induced by IL-1β (1 ng/mL). After 72 h, cells were frozen in liquid nitrogen, ice-cold homogenization buffer (0.1 M potassium phosphate buffer pH 7.4, 1 mM phenylmethanesulfonyl fluoride, 60 μg/mL soybean trypsin inhibitor, 1 μg/mL leupeptin, 2.5 mM GSH, and 250 mM sucrose) was added, and after 15 min cells were resuspended and sonicated on ice (3 × 20 s). The homogenate was then subjected to differential centrifugation at 10000g for 10 min and at 174000g for 1 h at 4 °C. The pellet (microsomal fraction) was finally resuspended in 1 mL of homogenization buffer, and the protein concentration was determined by the Coomassie protein assay.
Determination of mPGES-1 Activity in Microsomes of A549 Cells
Microsomal membranes of A549 cells were diluted in potassium phosphate buffer (0.1 M, pH 7.4) containing 2.5 mM GSH (100 μL total volume), and PGE2 formation was initiated by addition of PGH2 (20 μM, final concentration). After 1 min at 4 °C, the reaction was terminated with 100 μL of stop solution (40 mM FeCl2, 80 mM citric acid, and 10 μM 11β-PGE2), and PGE2 was separated by solid-phase extraction and analyzed by RP-HPLC as described.19
Activity Assays of Isolated COX-1 and -2
Inhibition of the activities of isolated COX-1 and COX-2 was performed as described.25 Briefly, purified COX-1 (ovine, 50 units) or COX-2 (human recombinant, 20 units) was diluted in 1 mL of reaction mixture containing 100 mM Tris buffer pH 8, 5 mM glutathione, 5 μM hemoglobin, and 100 μM EDTA at 4 °C and preincubated with the test compounds for 5 min. Samples were prewarmed for 60 s at 37 °C, and AA (5 μM for COX-1, 2 μM for COX-2) was added to start the reaction. After 5 min at 37 °C, 12(S)-hydroxy-5Z,8E,10E-heptadecatrienoic acid was extracted and then analyzed by HPLC.
Molecular Docking
The binding modes of the investigated triterpene acids were analyzed using the quantum mechanics-polarized ligand docking (QPLD) workflow,27,28 which is available in the Maestro suite version 9.2.112.29 For this purpose, the X-ray crystal structure of mPGES-1 with bound cofactor GSH was used, which is deposited in the Protein Data Bank (PDB, http://rcsb.org/pdb/),30 entry 3dww.31
For the docking procedure, 2D structures of the triterpene acids were converted into 3D coordinates employing Maestro’s module Ligprep. This included the ionization of acidic moieties of the ligands with the module Ligprep (Epik/OPLS-2005). The protein was prepared with the Protein Preparation Wizard. The hydrogen atoms were added, and atom and bond types were assigned, which was followed by exploration of the hydrogen bond assignment in “extensive” mode. Within this procedure, the protonated form of His72 was assigned. Furthermore, the protein was refined by a minimization as a final step of the protein preparation within the respective assistant (OPLS-AA 2005/RMSD threshold: 0.3 Å). The molecular docking was performed with Glide in extra precision (XP) mode and a scaling of the receptor van der Waals radius by a factor of 0.9. In the QPLD workflow, the proposed orientation of the ligands within the binding site of the macromolecule target is used to calculate atomic (partial) charges of the ligands employing the quantum mechanical/molecular mechanical (QM/MM) approach performed with the module QSite (semiempirical method/Mulliken charges). The initial docking poses were submitted to a second docking procedure with Glide in XP mode, involving atomic (partial) charges of ligands from the QM/MM approach. The final docking poses were ranked according to the calculations from the GlideScore scoring function. The analysis of the docking poses, which were retrieved from the QPLD workflow, was performed within LigandScout version 3.132,33 following an MMFF94-based minimization of the investigated triterpene acid and of the binding site residue side chains within LigandScout, which was also used for visualization purposes.
Statistics
Data are expressed as mean ± SEM. The program Graphpad Instat (Graphpad Software Inc., San Diego, CA, USA) was used for statistical comparisons of the data by one-way ANOVAs for independent or correlated samples followed by Tukey HSD post hoc tests. Where appropriate, Student’s t test for paired and correlated samples was applied. A p value of <0.05 (*) was considered significant. IC50 values of compounds are approximations determined by graphical analysis (linear interpolation between the points between 50% activity).
Acknowledgments
This work was supported by the Austrian Science Fund (FWF, project S10711), the Tyrolean Science Foundation (TWF), and Aureliasan GmbH, Tuebingen, Germany. We thank D. Müller for expert technical assistance.
Supporting Information Available
Isolation and structure elucidation of the triterpene acids used in this study. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Tucker A. O. Econ. Bot. 1986, 40, 425–433. [Google Scholar]
- Buchele B.; Zugmaier W.; Simmet T. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2003, 791, 21–30. [DOI] [PubMed] [Google Scholar]
- Schweizer S.; von Brocke A. F.; Boden S. E.; Bayer E.; Ammon H. P.; Safayhi H. J. Nat. Prod. 2000, 63, 1058–1061. [DOI] [PubMed] [Google Scholar]
- Atta-ur-Raman; Naz H.; Fadimatou; Makhmoor T.; Yasin A.; Fatima N.; Ngounou F. N.; Kimbu S. F.; Sondengam B. L.; Choudhary M. I. J. Nat. Prod. 2005, 68, 189–193. [DOI] [PubMed] [Google Scholar]
- Abdel-Tawab M.; Werz O.; Schubert-Zsilavecz M. Clin. Pharmacokinet. 2011, 50, 349–369. [DOI] [PubMed] [Google Scholar]
- Mathe C.; Culioli G.; Archier P.; Vieillescazes C. Chromatographia 2004, 60, 493–499. [Google Scholar]
- Paul M.; Jauch J. Nat. Prod. Commun. 2012, 7, 283–288. [PubMed] [Google Scholar]
- Cotterre G.; Wriglesw M. J. Chem. Soc. C: Org. 1970, 5, 739. [Google Scholar]
- Mora A. J.; Delgado G.; de Delgado G. D.; Usubillaga A.; Khouri N.; Bahsas A. Acta Crystallogr. Sect. C: Cryst. Struct. Commun. 2001, 57, 638–640. [DOI] [PubMed] [Google Scholar]
- Assimopoulou A. N.; Papageorgiou V. P. Biomed. Chromatogr. 2005, 19, 285–311. [DOI] [PubMed] [Google Scholar]
- Banno N.; Akihisa T.; Yasukawa K.; Tokuda H.; Tabata K.; Nakamura Y.; Nishimura R.; Kimura Y.; Suzuki T. J. Ethnopharmacol. 2006, 107, 249–253. [DOI] [PubMed] [Google Scholar]
- Boden S. E.; Schweizer S.; Bertsche T.; Dufer M.; Drews G.; Safayhi H. Mol. Pharmacol. 2001, 60, 267–273. [DOI] [PubMed] [Google Scholar]
- Pardhy R. S.; Bhattacharyya S. C. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 1978, 16, 174–175. [Google Scholar]
- Fattorusso E.; Santacroce C.; Xaasan C. F. Phytochemistry 1983, 22, 2868–2869. [Google Scholar]
- Verhoff M.; Seitz S.; Northoff H.; Jauch J.; Schaible A. M.; Werz O. Biochem. Pharmacol. 2012, 84, 681–691. [DOI] [PubMed] [Google Scholar]
- Siemoneit U.; Koeberle A.; Rossi A.; Dehm F.; Verhoff M.; Reckel S.; Maier T. J.; Jauch J.; Northoff H.; Bernhard F.; Doetsch V.; Sautebin L.; Werz O. Br. J. Pharmacol. 2011, 162, 147–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeberle A.; Werz O. Curr. Med. Chem. 2009, 16, 4274–4296. [DOI] [PubMed] [Google Scholar]
- Waltenberger B.; Wiechmann K.; Bauer J.; Markt P.; Noha S. M.; Wolber G.; Rollinger J. M.; Werz O.; Schuster D.; Stuppner H. J. Med. Chem. 2011, 54, 3163–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeberle A.; Siemoneit U.; Buhring U.; Northoff H.; Laufer S.; Albrecht W.; Werz O. J. Pharmacol. Exp. Ther. 2008, 326, 975–982. [DOI] [PubMed] [Google Scholar]
- Estrada A. C.; Syrovets T.; Pitterle K.; Lunov O.; Buchele B.; Schimana-Pfeifer J.; Schmidt T.; Morad S. A.; Simmet T. Mol. Pharmacol. 2010, 77, 378–387. [DOI] [PubMed] [Google Scholar]
- Akihisa T.; Tabata K.; Banno N.; Tokuda H.; Nishimura R.; Nakamura Y.; Kimura Y.; Yasukawa K.; Suzuki T. Biol. Pharm. Bull. 2006, 29, 1976–1979. [DOI] [PubMed] [Google Scholar]
- Cao H.; Yu R.; Choi Y.; Ma Z. Z.; Zhang H.; Xiang W.; Lee D. Y.; Berman B. M.; Moudgil K. D.; Fong H. H.; van Breemen R. B. Pharmacol. Res. 2010, 61, 519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radmark O.; Samuelsson B. J. Int. Med. 2010, 268, 5–14. [DOI] [PubMed] [Google Scholar]
- Funk C. D. Science 2001, 294, 1871–1875. [DOI] [PubMed] [Google Scholar]
- Siemoneit U.; Hofmann B.; Kather N.; Lamkemeyer T.; Madlung J.; Franke L.; Schneider G.; Jauch J.; Poeckel D.; Werz O. Biochem. Pharmacol. 2008, 75, 503–513. [DOI] [PubMed] [Google Scholar]
- Ali S. I.; Zhang C. R.; Mohamed A. A.; El-Baz F. K.; Hegazy A. K.; Kord M. A.; Nair M. G. Nat. Prod. Commun. 2013, 8, 1365–1366. [PubMed] [Google Scholar]
- Raha K.; Peters M. B.; Wang B.; Yu N.; Wollacott A. M.; Westerhoff L. M.; Merz K. M. Jr. Drug Discovery Today 2007, 12, 725–731. [DOI] [PubMed] [Google Scholar]
- Chung J. Y.; Hah J. M.; Cho A. E. J. Chem. Inf. Model. 2009, 49, 2382–2387. [DOI] [PubMed] [Google Scholar]
- Maestro suite version 9.2.112; Schrödinger, LLC: New York, NY, 2011.
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jegerschold C.; Pawelzik S. C.; Purhonen P.; Bhakat P.; Gheorghe K. R.; Gyobu N.; Mitsuoka K.; Morgenstern R.; Jakobsson P. J.; Hebert H. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 11110–11115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolber G.; Langer T. J. Chem. Inf. Model. 2005, 45, 160–169. [DOI] [PubMed] [Google Scholar]
- LigandScout version 3.1; Inte:Ligand GmbH: Maria Enzersdorf, Austria, 1999. –2013.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.