

Summary
Mitochondrial fission and fusion cycles are integrated with cell cycle progression. In this paper, we demonstrate that the inhibition of mitochondrial fission protein Drp1 causes an unexpected delay in G2/M cell cycle progression and aneuploidy. In investigating the underlying molecular mechanism, we revealed that inhibiting Drp1 triggers replication stress, which is mediated by a hyperfused mitochondrial structure and unscheduled expression of cyclin E in the G2 phase. This persistent replication stress then induces an ATM-dependent activation of the G2 to M transition cell cycle checkpoint. Knockdown of ATR, an essential kinase in preventing replication stress, significantly enhanced DNA damage and cell death of Drp1-deficienct cells. Persistent mitochondrial hyperfusion also induces centrosomal overamplification and chromosomal instability, which are causes of aneuploidy. Analysis using cells depleted of mitochondrial DNA revealed that these events are not mediated by the defects in mitochondrial ATP production and reactive oxygen species (ROS) generation. Thus dysfunctional mitochondrial fission directly induces genome instability by replication stress, which then initiates the DNA damage response. Our findings provide a novel mechanism that contributes to the cellular dysfunction and diseases associated with altered mitochondrial dynamics.
Key words: Drp1, Cell cycle defects, Genome instability, Mitochondrial fission, Replication stress
Introduction
Mitochondria are dynamic organelles that constantly undergo fission and fusion events. Deficiencies in the proteins regulating mitochondrial dynamics are associated with a number of human pathologies including neurodegenerative diseases and newborn lethality (Westermann, 2010). Recently, mitochondria have been shown to undergo morphological remodeling as cells progress through the cell cycle (Mitra et al., 2009). At the G1/S boundary mitochondrial tubules form a highly fused network, which is associated with increased mitochondrial ATP production and high levels of cyclin E, in order to promote G1-to-S transition (Mitra et al., 2009). This hyperfused mitochondrial network is then disassembled and becomes increasingly fragmented through S, G2 and M phase of the cell cycle, with the greatest fragmentation evident during mitosis in order to allow the proper partitioning of mitochondria between two daughter cells during cytokinesis. Thus, mitochondrial remodeling throughout the cell cycle is considered to meet the cellular energy demands during the progression of specific stages of the cell cycle, and to ensure faithful inheritance of mitochondria during cell division. However, how deficiencies in the proteins that regulate mitochondrial dynamics impact cell cycle progression and hence directly contribute to the development of diseases is not clear.
The dynamic regulation of mitochondrial morphology is achieved by the coordination of mitochondrial fission and fusion events (Green and Van Houten, 2011). Dynamin-related protein 1 (Drp1), a large dynamin-related GTPase, is essential for mitochondrial fission (Smirnova et al., 2001). Loss of Drp1 results in elongated mitochondria, and Drp1 deficiencies have been identified in several human diseases (Cho et al., 2009; Wang et al., 2008; Waterham et al., 2007). Drp1 is directly regulated by the machinery that controls cell cycle progression. For example, Drp1 is phosphorylated at Ser585 by cdc2/cyclin B in order to promote mitochondrial fission during mitosis (Taguchi et al., 2007). Drp1 deficiency is generally thought to cause mitochondrial dysfunction due to a failure of a Drp1-dependent mechanism of mitophagy that removes damaged mitochondria within the cell (Twig et al., 2008). The resulting accumulation of damaged mitochondria has been suggested to cause a depletion of cellular ATP and an inhibition of cell proliferation (Parone et al., 2008). Such an energy depletion-related cell proliferation defect may be caused by a metabolic checkpoint that triggers an AMPK- and p53-dependent G1/S cell cycle arrest (Jones et al., 2005; Owusu-Ansah et al., 2008). Consistent with such a mechanism, overexpression of mutant Drp1 (K38A), results in a hyperfused mitochondrial network and a p53-dependent delay of S phase entry (Mitra et al., 2009). However, reduced cell proliferation has also been observed in the absence of cellular ATP depletion in non-immortalized Drp1-knockout mouse embryonic fibroblasts (MEFs) (Wakabayashi et al., 2009). This suggests that defective mitochondrial dynamics may affect cell proliferation through mechanisms that are not associated with mitochondrial energy metabolism.
In our present study we show that Drp1 deficiency-induced mitochondrial hyperfusion triggers replication stress and subsequent ATM/Chk2 and ATR/Chk1 DNA damage signaling, as well as the ATM kinase-dependent G2/M cell cycle checkpoint and aneuploidy in both p53 wild-type and p53 mutated cells. Significantly, we show that these phenotypes are not associated with defects in mitochondrial ATP production and reactive oxygen species (ROS) generation. Thus, we have dissociated mitochondrial hyperfusion-associated genome instability from mechanisms that mediate mitochondrial energy metabolism. These data identify a novel mechanism that connects Drp1 disruption-induced mitochondrial hyperfusion to the cell cycle regulation apparatus.
Results
Loss of the fission protein Drp1 causes mitochondrial hyperfusion and induces G2/M cell cycle arrest and aneuploidy
To investigate the functional consequences of defective mitochondrial dynamics on cell cycle progression, we knocked down the expression of the mitochondrial fission protein Drp1 using siRNA. Cell cycle analysis revealed that loss of Drp1 induced G2/M cell cycle arrest and aneuploidy (DNA content >4N) in a variety of cell lines independent of their p53 status (Fig. 1A; supplementary material Fig. S1A-C). This p53-independent effect was further confirmed by concurrent knocking down of both Drp1 and p53 in MCF7 p53 wild-type cells. Similar increase in G2/M and aneuploidy cell number was observed in p53 deficient as in p53 proficient MCF7 parental cells (supplementary material Fig. S1B,C). Since MDA-MB-231 cells showed the most severe phenotype, we selected this cell line for further investigations into the underlying mechanism. Drp1 deficiency in MDA-MB-231 cells induced a hyperfused mitochondrial network as expected (Fig. 1B), as well as a remarkable decrease in cell proliferation (Fig. 1C). The decrease in cell proliferation is a consequence of G2/M cell cycle arrest, as only a slight increase in the number of apoptotic cells was observed following Drp1 knockdown (Fig. 1D). To examine cell cycle progression over time, BrdU pulse-chase time course experiment was performed (Fig. 1E). In control cells, BrdU-labeled S phase cells progressed through S G2 and M phase and were detected in G1 phase at as early as 6 h (indicated by dashed circle). In contrast, not only the number of BrdU labeled S phase cells was reduced in Drp1-deficient cells (Fig. 1E; supplementary material Fig. S1A-C), but also their progression through S G2 and M phase was delayed. The appearance of BrdU-labeled Drp1-deficient cells in G1 phase was first detected after 10-hour chase (Fig. 1E). These data are consistent with the G2/M arrest in Drp1-deficient cells. To confirm that these phenotypes were the consequences of Drp1 deficiency and to exclude the possible off-target effects of the siRNA, we employed a selective small molecule inhibitor of Drp1, mdivi-1 (Cassidy-Stone et al., 2008). Mdivi-1 induced the similar G2/M cell cycle arrest and aneuploidy as Drp1 deficiency achieved by using siRNA (supplementary material Fig. S1D). Thus, Drp1 function is essential for proper cell cycle progression.
Fig. 1.
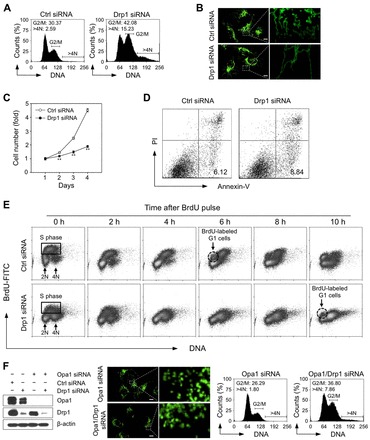
Loss of the fission protein Drp1 causes mitochondrial hyperfusion and induces G2/M cell cycle arrest and aneuploidy. (A) Loss of Drp1 induces G2/M cell cycle arrest and aneuploidy. MDA-MB-231 cells were examined four days after their transfection with control siRNA or Drp1 siRNA. Cell cycle distribution was determined by flow cytometric analysis of propidium iodide-stained cells. The graph indicates the percentage of cells containing the DNA content of 4N (G2 and M phase cells) and the DNA content greater than 4N (aneuploidy cells). These data represent three independent experiments. (B) Loss of Drp1 causes mitochondrial hyperfusion. Changes in mitochondrial morphology are visible in the control cells and in Drp1-deficient MDA-MB-231 cells that express pAcGFP1-Mito. The bars indicate 10 µm. (C) Loss of Drp1 causes decreased cell proliferation. Proliferation of the control cells and Drp1-deficient MDA-MB-231 cells was determined by using a CyQUANT assay. These data represent the mean ± s.d.; n = 4 wells. **P<0.01. (D) Loss of Drp1 does not enhance apoptotic cell death. Apoptosis was determined by annexin V assay four days after the cells were transfected with control siRNA or with Drp1 siRNA. The scatter plot graph indicates the percentage of annexin V-positive and PI-negative early apoptotic cells. (E) BrdU pulse-chase time course assay after the cells were transfected with the control siRNA or Drp1 siRNA for three days. Cells were incubated with 10 µM BrdU for 30 min (pulse) and the cell cycle progression of BrdU-labeled cells (S phase cells) was followed for the indicated time points (i.e. the chase). (F) Loss of the fusion protein Opa1 reverses the phenotypes observed in Drp1-deficient cells. Immunoblots show the knockdown efficiency of Opa1 and Drp1 in MDA-MB-231 cells. Changes in mitochondrial morphology are visible in Opa1 knockdown cells and Opa1/Drp1 double knockdown MDA-MB-231 cells that express pAcGFP1-Mito. The bars indicate 10 µm. Cell cycle distribution was determined as previously described.
To determine whether mitochondrial fission per se is directly responsible for the G2/M cell cycle arrest and aneuploidy observed in Drp1-deficient cells, we knocked down an essential mitochondrial fusion protein Opa1 (Cipolat et al., 2004) to counteract the Drp1 knockdown-induced mitochondrial hyperfusion. Even though mitochondrial outer membrane fusion occurs in Opa1-depleted cells, the fusion rate is reduced and Opa1-depleted cells contain fragmented mitochondria (Song et al., 2009). Consistent with this observation, knockdown of Opa1 induced extensive mitochondrial fragmentation in MDA-MB-231 cells, and prevented mitochondrial hyperfusion induced by Drp1 depletion (Fig. 1F). These Drp1 and Opa1 double knockdown cells showed lower G2/M accumulation and a twofold decrease in aneuploidy as compared to when Drp1 alone was knocked down (compare Fig. 1F and Fig. 1A). These results indicate that the G2/M cell cycle arrest and aneuploidy induced in Drp1-deficient cells required mitochondrial hyperfusion. Since efficient knockdown of Opa1 was not able to completely reverse G2/M arrest, some none mitochondrial role of Drp1 might also be involved in this process.
The G2/M cell cycle arrest and aneuploidy observed in Drp1-deficient cells are not caused by changes in mitochondrial energy metabolism
Cellular energy status has been recognized as important for cell cycle progression (Mandal et al., 2005), we therefore evaluated if the mitochondrial energy metabolism plays a role in regulating the G2/M cell cycle arrest and aneuploidy in Drp1-deficient MDA-MB-231 cells. Contrary to the previous report using HeLa cells (Parone et al., 2008), there is no depletion of total cellular ATP following Drp1 knockdown (Fig. 2A). Since changes in total intracellular ATP levels may not reflect the changes in mitochondrial metabolism in cancer cells because of the Warburg effect (Warburg et al., 1927), we undertook a detailed analysis to address whether mitochondrial function is altered in Drp1-deficient cells. We observed a slight decrease in mitochondrial membrane potential (Fig. 2B) and about a ∼25% decrease in the oxygen consumption rate (OCR) (Fig. 2C), which is accompanied with a concomitant increase in extracellular acidification rate (ECAR) (Fig. 2D), a marker of glycolysis, in Drp1-deficient cells. Consistent with these data, we observed a decreased mitochondrial contribution [indicated by lower ATP levels in the presence of 2-deoxyglucose (2DG)] (Fig. 2E), which is compensated by an increased glycolysis contribution (indicated by higher ATP levels in the presence of oligomycin) (Fig. 2F), in order to maintain total cellular ATP levels in Drp1-deficient cells. These observations indicate that loss of Drp1 in MDA-MB-231 cells reduces mitochondrial energy metabolism. ROS production due to mitochondrial dysfunction is frequently considered a cause of cellular damage and cell cycle arrest (Owusu-Ansah et al., 2008). However, we observed no increase in mitochondrial ROS generation in Drp1-deficient MDA-MB-231 cells using the mitochondrial superoxide indicator MitoSox (Fig. 2G). These results indicate that the G2/M cell cycle arrest and aneuploidy observed in Drp1-deficient cells cannot be attributed to changes in total ATP production or mitochondrially generated ROS.
Fig. 2.
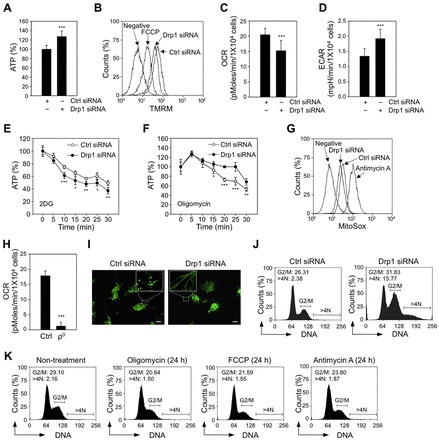
The G2/M cell cycle arrest and aneuploidy observed in Drp1-deficient cells are not caused by changes in mitochondrial energy metabolism. (A) Loss of Drp1 does not deplete the total intracellular ATP levels. The ATP levels were measured after transfection with siRNA for four days (in Fig. 2 all measurements were performed four days after transfection). Data are presented as the mean ± the standard deviation (s.d.); n = 3 wells. ***P<0.005. (B) Loss of Drp1 induces a slight decrease in the mitochondrial membrane potential, which was measured after incubating cells with 20 nM of TMRM for 20 min. Cells that were not incubated with TMRM were used as a negative control. Cells treated with 10 µM of FCCP for 20 min were used as a positive control to show depolarized mitochondrial membrane potential. (C,D) Loss of Drp1 impacts the oxygen consumption rate (OCR) (C) and the extracellular acidification rate (ECAR) (D). The OCR and ECAR were measured by using a Seahorse Extracellular Flux analyzer. These data represent the mean ± s.d.; n = 3 wells. ***P < 0.005. (E,F) Loss of Drp1 suppresses mitochondrial ATP generation. The contribution of mitochondria (E) and contribution of glycolysis (F) to total intracellular ATP levels were determined by measuring the changes in total intracellular ATP levels over time in the presence of 100 mM 2DG and 1 µg/ml of oligomycin, respectively. ATP levels were monitored at 5-minute intervals for 30 min. These data represent the mean ± s.d.; n = 3 wells. *P<0.05; **P<0.01; ***P<0.005 (G) Loss of Drp1 does not increase mitochondrial superoxide levels, which were measured after incubating cells with 2.5 µM of MitoSox for 20 min. Cells that were not incubated with MitoSox were used as a negative control. Cells treated with 20 µg/ml of antimycin A for 20 min were used as a positive control to show increased mitochondrial superoxide generation. (H) Oxygen consumption is dramatically decreased in MDA-MB-231 ρ0 cells. OCR was measured by using a Seahorse Extracellular Flux analyzer. These data represent the mean ± s.d.; n = 3 wells. ***P < 0.005. (I) Loss of Drp1 induces mitochondrial hyperfusion in MDA-MB-231 ρ0 cells. Changes in mitochondrial morphology were visualized by staining the control cells and the Drp1-deficient MDA-MB-231 ρ0 cells with 100 nM of MitoTracker green FM for 20 min. The bars represent 10 µm. (J) Loss of Drp1 induces G2/M cell cycle arrest and aneuploidy in MDA-MB-231 ρ0 cells. Cell cycle distribution was determined by flow cytometric analysis of propidium iodide stained cells. The percentage of cells containing DNA content of 4N and DNA content greater than 4N is indicated. (K) Pharmacological inhibition of mitochondrial respiration, depolarization of mitochondrial membrane potential, or stimulation of mitochondrial ROS production does not induce G2/M cell cycle arrest or aneuploidy. MDA-MB-231 cells were treated with 5 µg/ml oligomycin, 5 µM FCCP, or 10 µg/ml antimycin A for 24 h. Cell cycle distribution was determined as described above.
To further exclude the potential role for mitochondrial energy and ROS in mediating the cell cycle defects observed in Drp1-deficient cells, we examined cell cycle progression in MDA-MB-231 ρ0 cells following knockdown of Drp1. Mitochondria in ρ0 cells do not contribute to total cellular ATP generation and are unable to produce ROS (Weinberg et al., 2010), due to a deficiency in mitochondrial oxidative phosphorylation that is caused by the depletion of mitochondrial DNA (Qian and Van Houten, 2010) (Fig. 2H). Loss of Drp1 in MDA-MB-231 ρ0 cells resulted in elongated mitochondria (Fig. 2I), G2/M cell cycle arrest and aneuploidy (Fig. 2J), that are similar to what we observed in the parental MDA-MB-231 cells (Fig. 1A,B). Furthermore, treatment of MDA-MB-231 cells with either oligomycin, a complex V inhibitor that suppresses mitochondrial respiration, FCCP, an uncoupler that depolarizes mitochondrial membrane potential, or antimycin A, a complex III inhibitor that stimulates mitochondrial ROS production, did not induce G2/M cell cycle arrest and aneuploidy (Fig. 2K). These data further support our conclusion that the G2/M cell cycle arrest and aneuploidy observed in Drp1-deficient cells are not caused by defects in mitochondrial energy metabolism.
The G2/M cell cycle arrest observed in Drp1-deficient cells is not caused by disruptions in the molecular machinery that is essential for the G2/M cell cycle transition
To investigate the mechanisms underlying the G2/M cell cycle arrest and aneuploidy observed in Drp1-deficient cells, we synchronized MDA-MB-231 cells at G2/M phase using a single thymidine block followed by nocodazole treatment (henceforth referred to as a thymidine/nocodazole block), and then monitored the changes in G2/M phase-related molecular events following the release from G2/M cell cycle block (Fig. 3A). Both control and Drp1-deficient MDA-MB-231 cells were able to be blocked at G2/M phase as shown by their 4N DNA content immediately following the thymidine/nocodazole block (Fig. 3B, 0 h release). Phosphorylation of histone H3 at serine residue (Ser10) is associated with chromosome condensation and mitotic entry (Crosio et al., 2002); therefore, the phosphorylation of histone H3 is used as a marker to distinguish M phase cells from G2 phase cells, both of which contain 4N DNA content. Immediately following the thymidine/nocodazole block we observed that the levels of positive phospho-histone H3 in Drp1 knockdown cells was ∼10-fold lower than in control cells (Fig. 3C), even though both control and Drp1-deficient cells contained 4N DNA content. These data suggest that the accumulation of 4N DNA content in Drp1-deficient cells was largely due to the cell cycle arrest at G2 phase rather than M phase. Following the release from the G2/M block control cells rapidly entered the cell cycle such that the majority of cells were in G1 phase with 2N DNA content at 6 h (Fig. 3B). In contrast, the majority of Drp1-deficient cells were still at G2/M phase as indicated by the large fraction of cells with 4N DNA content at 6 h following release from the block (Fig. 3B). These data indicated that loss of Drp1 prevented G2 to M cell cycle transition.
Fig. 3.
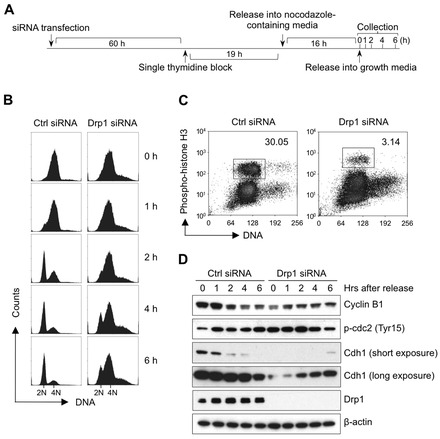
The G2/M cell cycle arrest observed in Drp1-deficient cells is not caused by disruptions in the molecular machinery that is essential for the G2/M cell cycle transition. (A) The schematic indicates the thymidine/nocodazole block protocol used to synchronize siRNA-transfected cells in the G2/M phase. (B) Loss of Drp1 prevents cell cycle progression after the release from the G2/M block. Control cells and Drp1-deficient cells were released from a thymidine/nocodazole block and cell cycle distribution was determined by flow cytometric analysis of propidium iodide stained cells collected at the indicated time points (right). (C) Loss of Drp1 decreases the number of cells in mitosis immediately after the thymidine/nocodazole block. Control cells and Drp1-deficient cells were synchronized and cells expressing phospho-histone H3 were labeled by using Alex Fluor 647-conjugated anti-phospho-histone H3 antibody and detected by flow cytometry. (D) Loss of Drp1 suppresses the factors that are essential for mitotic entry. Control cells and Drp1-deficient cells were synchronized and collected at the indicated time points after release. The changes in the proteins that are associated with mitotic entry were analyzed by western blot. These data represent three independent experiments.
To examine the integrity of molecular events associated with M phase entry, we examined the activity of maturation/mitosis-promoting factor (MPF). MPF is a heterodimeric protein composed of cyclin B and cdc2 serine/threonine kinase (Dorée and Hunt, 2002). Both the accumulation of cyclin B1 and dephosphorylation of cdc2 at Tyr15 are required for the initiation of mitosis (Norbury et al., 1991). Drp1-deficient MDA-MB-231 cells were defective in the accumulation of cyclin B1 and the dephosphorylation of cdc2 at Tyr15 in response to thymidine/nocodazole block (Fig. 3D). High levels of phosphorylated cdc2 have previously been associated with effective G2/M cell cycle checkpoint activation (Lew and Kornbluth, 1996). These data indicate that Drp1 deficiency causes suppression of MPF activity and subsequent defect in mitotic entry. Cdh1 is required for the degradation of cyclin B1 and the expression pattern of cdh1 through the cell cycle is similar to that of cyclin B1 (Listovsky et al., 2004). Immediately following the release from the thymidine/nocodazole block control cells showed high levels of cyclin B1 and cdh1, which rapidly declined during the next few hours. This is in stark contrast to the Drp1 knockdown cells, which showed initially low levels of cyclin B1 and very low levels of cdh1 with high levels of phosphorylated cdc2. These Drp1 knockdown cells once released from the thymidine/nocodazole block showed a slow increase in cyclin B1 and cdh1, with a decrease in phosphorylated cdc2 (Fig. 3D). This particular pattern observed in Drp1 knockdown cells represents the slow progression through G2/M phase, which is associated with subsequent aneuploidy. These data are consistent with an induction of a G2/M cell cycle checkpoint in Drp1-deficient cells rather than a disruption in the molecular machinery that is essential for the G2/M cell cycle transition.
Loss of Drp1 induces chromosomal instability and centrosome overamplification
Aneuploidy is frequently a consequence of the chromosomal instability that is associated with defects in mitotic segregation of chromosomes (Rajagopalan and Lengauer, 2004). We observed misaligned chromosomes in metaphase, and lagging chromosomes in anaphase as Drp1-deficient cells progressed through mitosis (Fig. 4A,B). The hyperfused mitochondrial network was maintained throughout the mitosis in Drp1-deficient cells, in contrast to the fragmented mitochondrial morphology observed in control cells (Fig. 4A). These data indicate that defects in mitochondrial fission lead to the defects in chromosome segregation during mitosis. Given that Drp1-deficient cells still retained the ability to progress through mitosis (albeit at a greatly reduced rate) (Fig. 3D), such defects in chromosome segregation could give rise to aneuploidy.
Fig. 4.
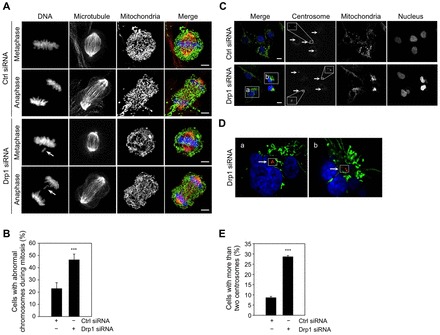
Loss of Drp1 induces chromosomal instability and centrosome overamplification. (A,B) Loss of Drp1 induces chromosome abnormalities in mitosis. (A) Mitotic chromosomes were visualized in the control cells and Drp1-deficient cells stably expressing pAcGFP1-Mito by DAPI staining. Microtubules were visualized by staining cells with Alex Fluor 555-conjugated anti-β-tubulin antibody. The images show representative cells in metaphase and anaphase. Arrows indicate lagging chromosomes. The bars indicate 5 µm. (B) The percentage of mitotic control cells and Drp1-deficient cells with abnormal chromosomes was determined by counting at least 30 mitotic cells from three independent slides. These data represent the mean ± the standard deviation (s.d.). *** P<0.005. (C-E) Loss of Drp1 induces centrosome overamplification. (C) Centrosomes in control and Drp1-deficient cells stably expressing pAcGFP1-Mito were visualized by staining cells with anti-γ-tubulin antibody, followed by secondary Alex Fluor 594 goat anti-mouse antibody. The nuclei were visualized by DAPI staining. Arrows indicate the centrosomes. The bars indicate 10 µm. (D) Enlarged images of box a and box b in panel C of a single focal plane from Drp1 knockdown cells. (E) The percentage of control and Drp1-deficient cells with more than two centrosomes was determined by counting at least 100 cells from three independent slides. These data represent the mean ± the standard deviation (s.d.). ***P<0.005.
Abnormal extra centrosomes are known to be associated with chromosome instability via formation of aberrant mitotic spindles (Ganem et al., 2009). Depending on the cell cycle phase, normal cells contain one or two centrosomes. We observed that Drp1-deficient cells frequently contained more than two centrosomes, relative to control cells (Fig. 4C-E). The Drp1-deficient cells containing extra centrosomes also showed abnormal nuclear morphology and micronuclei (Fig. 4C), a phenotype that is again indicative of chromosome instability. In images obtained from single focal plane we observed that over-amplified centrosomes were often surrounded by aggregated mitochondria, indicating that mitochondrial aggregation is associated with centrosomal abnormalities (Fig. 4D).
Loss of Drp1 induces mitochondrial aggregation around the microtubule organizing center (MTOC)
Since the changes in the mitochondrial morphology per se play a direct role in mediating Drp1 deficiency-induced cell cycle defects, as demonstrated by that knockdown of mitochondrial fusion protein Opa1 reversed the G2/M arrest and aneuploidy observed in Drp1-deficient cells, we undertook a detailed analysis of mitochondrial dynamics in Drp1-deficient cells. Microtubules originate at the centrosome (the main microtubule organizing center), and are tracks for mitochondrial transportation and distribution. Since we had observed a centrosome defect in Drp1-deficient cells, we reasoned that the morphological relationship between mitochondria and microtubules might be defective in these cells. In control MDA-MB-231 cells, both the mitochondria and microtubules were observed to be widespread in the cytoplasm, and the area that contained a high concentration of microtubules indirectly marked the location of the MTOC (Fig. 5A,B). In Drp1-deficient cells we observed that mitochondria aggregated around the MTOC, with few elongated mitochondria radiating along microtubule tracks from the mitochondrial aggregates toward the peripheral regions of the cells. We propose that this morphological remodeling results from the retraction and fusion of mitochondria dispersed in peripheral region of the cytoplasm to the MTOC, and this in turn lead to mitochondrial aggregation and a large region of cytoplasm that contains no mitochondria (for example see ‘cell b’ in Fig. 5B). It is also notable that ‘cell b’ shows an abnormal nuclear morphology, and the micronuclei were located inside such mitochondrial aggregates. Therefore the aggregation of mitochondria around the MTOC may affect intracellular homeostasis such as Ca2+ signaling that directly contributes to the defects of centrosome duplication and other cell cycle related events (Matsumoto and Maller, 2002).
Fig. 5.
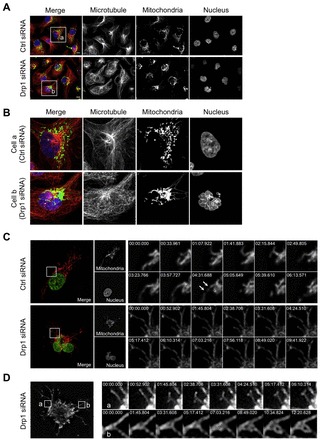
Loss of Drp1 induces mitochondrial aggregation around the microtubule organizing center (MTOC). (A,B) In Drp1-deficient cells mitochondria aggregate around the MTOC. (A) Microtubules in control cells and in Drp1-deficient cells stably expressing pAcGFP1-Mito were visualized by staining the cells with Alex Fluor 555-conjugated anti-β-tubulin antibody. Their nuclei were visualized by DAPI staining. Regions with concentrated microtubule staining indicate the locations of MTOC. The bars indicate 10 µm. (B) Enlarged images of box a and box b in panel A represent the control cells and Drp1-deficient cells, respectively. (C) Loss of Drp1 results in reduced mitochondrial motility and redistribution. Mitochondrial dynamics were recorded over time in the control cells and Drp1-deficient MDA-MB-231 cells stably expressing pDsRed2-Mito and histone H2B-GFP. Image sequences show representative mitochondrial movements in the indicated region. Arrows indicate fission events. (D) Mitochondrial remodeling in Drp1-deficient cells. Image sequences obtained in region a show a mitochondrial branching event and in region b show transformation of the mitochondrial structure from a single fork shape to a netlike morphology.
Mitochondrial aggregates have been described as clusters of tubules rather than a large mass of coalescing membrane (Smirnova et al., 1998). However, the exact process through which the highly interconnected mitochondrial aggregates are formed is not known. We generated time-lapse movies that revealed markedly reduced mitochondrial motility and redistribution due to lack of fission in Drp1-deficient cells (Fig. 5C; supplementary material Movies 1–4). Nevertheless, mitochondria were still able to undergo some remodeling in these cells, even with the limited frequency and within the limited spatial region. For example, a branching event in Drp1-deficient cells is indicated ‘a’ in Fig. 5D and supplementary material Movie 5. Furthermore, fusion is not limited to end-joining fusion between separate mitochondria, and can occur as a cross-fusion between branched mitochondrial tubules that make contact as shown in region ‘b’ in Fig. 5D and supplementary material Movie 6. In this example, mitochondria underwent a morphological change from a simple fork shape to a complicated netlike structure. These preserved branching and fusion abilities in Drp1-deficient cells eventually build up a complicated mitochondrial network, which appears as mitochondrial aggregates. We believe that these examples are the paradigms that demonstrate how a vast hyperfused mitochondrial network may be generated in the condition of lack of fission.
The G2/M cell cycle arrest and aneuploidy observed in Drp1-deficient cells are consequences of replication stress-initiated DNA damage signaling that involves ATM/Chk2 and ATR/Chk1 kinases
We sought to understand the molecular mechanism that causes the G2/M arrest and aneuploidy in Drp1-deficient cells. Mitochondrial hyperfusion induced either by overexpression of mutant Drp1 (K38A) or by Drp1 inhibitor mdivi-1 has previously been associated with the onset of DNA replication and cyclin E accumulation in HCT116 and NRK cells (Mitra et al., 2009). During a normal cell cycle, cyclin E levels accumulate at the G1/S phase boundary, decline during S phase and become low or undetectable when replication is complete (Fig. 6A). In contrast to the previous report (Mitra et al., 2009), we observed a decrease in cyclin E levels in Drp1-deficient MDA-MB-231 cells relative to control cells in a non-synchronized condition (Fig. 6B, lanes 1 and 2). To determine whether this reduction in cyclin E was a reflection of the accumulation of Drp1-deficient cells at G2/M phase, we used nocodazole to arrest both Drp1-deficient and control cells at the G2/M phase. While cyclin E was low in control cells arrested at G2/M by nocodazole block, the cyclin E levels were not changed in nocodazole-treated Drp1-deficient cells (Fig. 6B, lanes 3 and 4). Thus, cyclin E is maintained at high levels in G2/M phase in Drp1-deficient cells as compared to that in control cells. These results suggest that the control of cyclin E expression is uncoupled from the cell cycle in Drp1-deficient cells. Overexpression of cyclin E can induce G2/M cell cycle arrest, aneuploidy and genomic instability (Bartkova et al., 2005; Keck et al., 2007; Spruck et al., 1999). To determine whether cyclin E is required for the G2/M cell cycle arrest and aneuploidy observed in Drp1-deficient MDA-MB-231 cells, we disrupted cyclin E using siRNA (Fig. 6C). The concurrent knockdown of cyclin E and Drp1 greatly reduced the G2/M cell cycle arrest and aneuploidy as compared to cells in which only Drp1 was disrupted (Fig. 6D), suggesting that cyclin E plays an important role in mediating the cell cycle defects observed in Drp1-deficient cells.
Fig. 6.
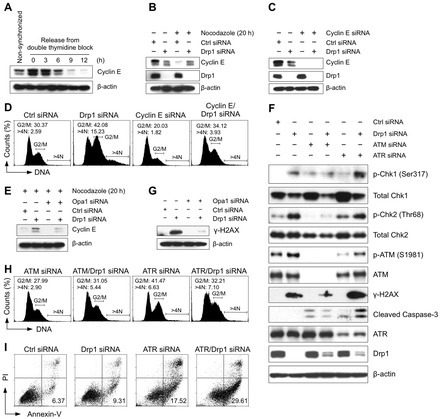
The G2/M cell cycle arrest and aneuploidy observed in Drp1-deficient cells result from replication stress-initiated DNA damage signaling that involves ATM/Chk2 and ATR/Chk1 kinases. (A) Cyclin E expression during the cell cycle. MDA-MB-231 cells were synchronized at G1/S border by a double thymidine block and then released in fresh media containing nocodazole. Cells were then harvested at the indicated time points, and the expression of cyclin E was detected by western blot. (B) Loss of Drp1 causes an accumulation of cyclin E in the G2/M phase. Cyclin E expression was assessed by western blot by using cell extracts generated from the control cells and from Drp1-deficient cells in the presence or absence of nocodazole for 20 h. (C) Knockdown efficiency of cyclin E and Drp1 was confirmed by western blot. (D) The loss of cyclin E reverses the G2/M cell cycle arrest and aneuploidy observed in Drp1-deficient cells. Four days after siRNA transfection, cell cycle distribution was determined by flow cytometric analysis of propidium iodide stained cells. The percentage of cells containing a DNA content of 4N and DNA content greater than 4N is indicated. (E) Loss of Opa1 reverses cyclin E accumulation in G2/M phase observed in Drp1-deficient cells. (F) Loss of Drp1 induces a DNA damage response. Cells were transfected with the indicated siRNA for four days and the changes in the proteins associated with the DNA damage response were assessed by western blot. (G) Loss of Opa1 prevented the accumulation of γ-H2AX in Drp1-deficient cells. (H) Loss of ATM reverses the G2/M cell cycle arrest and aneuploidy observed in Drp1-deficient cells, whereas the loss of ATR induces G2/M cell cycle arrest and aneuploidy. Four days after siRNA transfection, the cell cycle distribution was determined, as previously described. (I) ATR is essential for the survival of Drp1-deficient cells. Cells were transfected with the indicated siRNA for four days and apoptosis was assessed by annexin V and PI staining. The percentage of annexin V-positive and PI-negative early apoptotic cells is indicated. These data represent three independent experiments.
Dysregulated cyclin E expression and decreased cyclin B/cdc2 kinase activity have been previously associated with DNA damage response and G2/M cell cycle checkpoint activation (Bartkova et al., 2005; Kastan and Bartek, 2004). We therefore examined the activities of ATM, Chk1 and Chk2, three kinases that are integral to the DNA damage response and activation of cell cycle checkpoint. In control and Drp1-deficient MDA-MB-231 cells, we observed that the activities of ATM, Chk1 and Chk2 kinases as indicated by the levels of their phosphorylation were increased in Drp1-deficient cells compared to control cells (Fig. 6F). ATM phosphorylates Chk2 and activates G2/M cell cycle checkpoint through inhibiting cdc2 kinase activity. We observed that ATM knockdown impeded the phosphorylation of Chk2 and abrogated the G2/M cell cycle arrest and aneuploidy in Drp1-deficient cells (Fig. 6F,H), suggesting ATM/Chk2-mediated signaling is required for the cell cycle defects in cells that lack mitochondrial fission. We also observed an increase of γ-H2AX in Drp1 deficient cells indicating DNA damage (Fig. 6F). Knockdown of fusion protein Opa1 concurrently with Drp1 reversed the accumulation of cyclin E in G2/M phase (Fig. 6E) and also decreased the accumulation of γ-H2AX (Fig. 6G) as compared to cells in which only Drp1 was knocked down. These data indicate that cyclin E dysregulation and DNA damage are consequences of mitochondrial hyperfusion.
ATR kinase-dependent phosphorylation and activation of Chk1 are central to the signal transduction axis activated by replication stress (Toledo et al., 2008). The phosphorylation of Chk1 as well as the aberrant expression of cyclin E at G2 phase observed in Drp1-deficient cells suggested that replication stress is induced by the mitochondrial hyperfusion. ATR is essential for the stability of stalled DNA replication forks (Toledo et al., 2008). As such, ATR disruption induces replication stress (Murga et al., 2009) and causes DNA double-strand breaks (DSBs) to be generated at sites of stalled replication forks. We observed increased Chk2 phosphorylation and G2/M cell cycle arrest in ATR-deficient MDA-MB-231 cells, and as such ATR deficiency phenocopies Drp1 deficiency in this replication stress-mediated cell cycle arrest (Fig. 6F,H). In order to test the hypothesis that ATR deficiency would further increase the replication stress in Drp1-deficient cells we knocked down both proteins in MDA-MB-231 cells. Knockdown of both Drp1 and ATR dramatically increased the levels of γ-H2AX, the cleavage of caspase-3 and the number of annexin V-positive cells that both are associated with apoptosis (Fig. 6F,I). Thus, ATR is essential in preventing the replication stress and hence the survival of Drp1-deficient cells.
Discussion
The replication stress-mediated genome instability and cell cycle defects identified in this study revealed a novel mechanism underlying Drp1 deficiency-related cellular dysfunction. These cell cycle defects were not a result of loss of mitochondrial oxidative phosphorylation and ATP production, but required the hyperfused state of mitochondrial morphology, as G2/M arrest and aneuploidy were also observed in ρ0 cells which have diminished mitochondrial energy metabolism, and Opa1 knockdown was able to abrogate these cell cycle defects. Thus, we have separated a function for Drp1 in genome stability from its potential role in mitochondrial energy metabolism. We have identified that dysregulation of cyclin E expression in G2 phase is a direct consequence of Drp1 deficiency causing replication stress and a subsequent DNA damage response that is associated with activation of ATM kinase-dependent delay of mitotic entry. Preventing ATR-mediated signaling in response to replication stress in Drp1-defecient cells enhances DNA damage and cell death. Together these data indicate that the cycles of mitochondrial fission and fusion are integrated with the cell cycle apparatus that are essential for genome stability (Fig. 7).
Fig. 7.
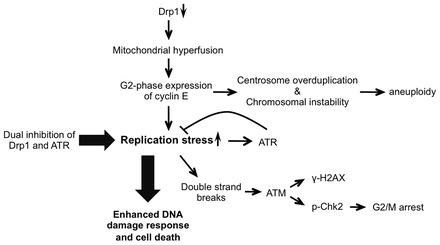
Loss of Drp1 induces replication stress-mediated genome instability. Our working model shows that mitochondrial hyperfusion that is induced by loss of the fission protein Drp1 leads to replication stress, centrosome overduplication, and chromosomal instability. These are mediated, at least in part, by the aberrant expression of cyclin E in the G2-phase. Persistent replication stress activates an ATM kinase signaling cascade that induces a G2/M cell cycle checkpoint. This is consistent with our data, which show that knockdown of the fusion protein Opa1, cyclin E or ATM reverses the G2/M cell cycle arrest and aneuploidy observed in Drp1-deficient cells. ATR kinase is essential for DNA damage responses to replication stress. Loss of Drp1 induces replication stress, which is further increased by the loss of ATR. This subsequently increases DNA damage and cell death.
Mitochondria form a highly interconnected network at the G1/S border, and the disassembly of this hyperfused network occurs when the G1/S transition process is completed (Mitra et al., 2009). This phenomenon suggests that mitochondrial fission, which is responsible for the disassembly of the hyperfused mitochondrial network, may play a significant role for the progression of following stages of the cell cycle after G1/S transition. In support of this idea, our data revealed that persistent mitochondrial hyperfusion due to loss of fission protein Drp1 is associated with aberrant accumulation of cyclin E in G2 phase. Cyclin E promotes cell cycle entry into S phase and is related with DNA replication associated functions (Ekholm and Reed, 2000). It has also been shown that cyclin E overexpression impacts DNA replication and leads to replication stress (Toledo et al., 2011). We observed a reduced rate of DNA replication (shown by the decrease in the number of BrdU-positive S-phase cells) in Drp1-deficient cells (supplementary material Fig. S1A–C), which may reflect either a reduced rate of replication or the activation of a G1/S or intra-S phase cell cycle checkpoint. These data are consistent with previous reports that described a similar reduction in BrdU incorporation (Mitra et al., 2009; Parone et al., 2008). However, our data indicate that the underlying mechanism is not dependent on the depletion of mitochondrial energy as suggested in these reports. We were not able to phenocopy Drp1 deficiency-induced cell cycle defects by pharmacological inhibition of mitochondrial energy metabolism (Fig. 2K). Rather, the reduction in the number of BrdU-positive S-phase cells in Drp1-deficient cells reflects replication stress that might be caused by either the accumulation of stalled replication forks, inefficient firing of replication origin (Liberal et al., 2012) and/or defects in pre-replication complex (preRC) assembly (Ekholm-Reed et al., 2004), phenotypes that have all been associated with cyclin E overexpression. Such cyclin E-mediated replication stress induces DNA damage and triggers the cell cycle checkpoint in G2 phase (Bartkova et al., 2005). It is also noteworthy that such cell cycle defects are not mediated by p53. Cells normally accumulate in G2 phase following replication stress, which is independent of p53, as demonstrated in ATR deficient and p53 wild-type MEF cells (Murga et al., 2009). Further, the accumulation of cyclin E is able to induce centrosome overduplication and chromosome instability (Nakayama et al., 2000; Rajagopalan et al., 2004; Spruck et al., 1999). Our results showing an increased number of centrosomes and chromosome instability in Drp1-deficient cells are consistent with these reports (Fig. 4). However, at present the mechanism underlying Drp1 deficiency-mediated alterations in mitochondrial dynamics and cyclin E dysregulation is unknown.
Our results revealed that as a consequence of replication stress in Drp1-deficient cells, ATR/Chk1 and ATM/Chk2 DNA damage signaling cascades are activated (Fig. 6F). A major cellular defense against DNA damage and control of cell cycle transition and cell death is a signaling network known as the DNA damage response (DDR) that is largely regulated by the ataxia telangiectasia mutated (ATM) and ATM- and Rad3-related (ATR). Cyclin E dysregulation-induced abnormalities in DNA replication are known inducers of the ATR/Chk1 cascade (Kastan and Bartek, 2004). ATR kinase activity is increased by single-stranded DNA (ssDNA). ssDNA accumulates at stalled replication forks that may arise as a consequence of replication stress (Cimprich and Cortez, 2008). Persistent replication stress can lead to the generation of DNA DSBs when stalled replication forks are subject to nucleolytic attack. ATM kinase activity is then increased by DNA DSBs and the subsequent G2/M cell cycle checkpoint is induced. It is noteworthy that knockdown of ATM prevented both G2/M arrest and aneuploidy in Drp1-deficienct cells (Fig. 6H), indicating that these cell cycle defects are dependent on ATM-mediated DNA damage signaling. ATM and ATR share many of the numerous substrates that promote cell cycle arrest and DNA repair. However, one of the best established roles of ATR that is not shared by ATM is preventing the formation DSBs by protecting the collapse of stalled replication forks (Cimprich and Cortez, 2008). Thus, ATR should attenuate DSBs and subsequent ATM kinase activity under conditions that induce replication stress. Our results showed that ATR kinase inhibition by knockdown of ATR in Drp1-deficient cells enhanced phosphorylation of H2AX and apoptosis (Fig. 6F,I). This increase in DNA damage and cell death is considered as the consequence of Drp1 deficiency-related replication stress under the condition of in the absence of ATR. This finding is reminiscent of the increased DNA damage and cell death when cells overexpressing cyclin E were treated with an ATR kinase inhibitor (Toledo et al., 2011). Since ATR activity is restricted to S and G2 phase (Toledo et al., 2011), these data further suggest that the direct impact of Drp1 disruption occurs in these phases. Moreover, centrosome overduplication requires functional G2/M checkpoint (Inanç et al., 2010), and ATM is also involved in initiating the signaling that regulates centrosome reduplication upon DNA damage (Fukasawa, 2007).
Mitochondrial fission is required for inheritance and partitioning of mitochondria during cell division (Westermann, 2010). Inhibiting Drp1-mediated mitochondrial fission has been reported to cause accumulation of mutant mtDNA (Malena et al., 2009). Our results provided the first evidence that mitochondrial dynamics are involved in initiating mitochondria-to-nucleus retrograde signaling, in order to ensure proper mitochondrial inheritance by enforcing cell cycle delay upon detection of abnormal mitochondrial morphology. One of the important roles of mitochondrial hyperfusion at G1/S border is postulated to allow homogenization of mitochondrial matrix and mtDNA. After G1/S transition is completed, mitochondrial fission has to take place during S G2 and M phase to ensure daughter cells inherit even and healthy mitochondria. When mitochondrial fission is impaired, the activation of G2/M cell cycle checkpoint thus allows more time for cells to fragment their mitochondria in preventing unequal segregation of mitochondria and mtDNA.
From a broader perspective, the genomic instability that we have identified in this study as a result of Drp1 deficiency may help explain why disruption of Drp1 induces lethality (Ishihara et al., 2009; Labrousse et al., 1999; Waterham et al., 2007) and cellular senescence (Yoon et al., 2006). Since replication stress has been observed in human precancerous lesions (Gorgoulis et al., 2005), alterations in mitochondrial dynamics may also play a significant role in cancer etiology.
Materials and Methods
Cell culture and transfection
The human breast carcinoma cell lines MDA-MB-231, MCF7 and MDA-MB-157, and the human lung carcinoma cell line A549 and H1299 were obtained from American Type Culture Collection (ATCC). Cells were cultured in RPMI 1640 supplemented with 10% heat-inactivated fetal calf serum and 1% penicillin-streptomycin in 5% CO2 at 37°C. MDA-MB-231 ρ0 cell line was established by culturing MDA-MB-231 cells in RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum, 1% penicillin-streptomycin, 1 mM sodium pyruvate, 50 µg/ml uridine, and 50 ng/ml ethidium bromide for at least four weeks (King and Attardi, 1996). DNA transfection was performed using FuGENE 6 (Roche) and siRNA transfection was performed using oligofectamine (Invitrogen) according to the manufacturer's instructions.
Expression vectors and RNA interference
pAcGFP1-Mito and pDsRed2-Mito vectors were purchased from Clontech. Histone H2B-GFP (Kanda et al., 1998) was purchased from Addgene (Addgene plasmid 11680). ATR siRNA was purchased from Dharmacon, while all other siRNAs including AllStars Negative Control siRNA were purchased from Qiagen. The siRNA sense strand sequences are as follows: Drp1, 5′-AACGCAGAGCAGCGGAAAGAG-3′ (Sugioka et al., 2004); Opa1, 5′-AAGTTATCAGTCTGAGCCAGGTT-3′; cyclin E, 5′-AACCAAACTTGAGGAAATCTA-3′ (Hemerly et al., 2009); ATM, 5′-AAGCGCCTGATTCGAGATCCT-3′ (White et al., 2008); ATR, 5′-AAGAGTTCTCAGAAGTCAACC-3′; p53, 5′-AACAGACCTATGGAAACTACT-3′.
Cell proliferation
Cells were transfected with either control or Drp1 siRNA and replated into 96-well plates at the following day. Cell proliferation was determined at 24 h intervals using a CyQUANT Direct Cell Proliferation Assay kit (Invitrogen), according to the manufacturer's instruction.
Apoptosis detection
Apoptosis was determined by staining cells with Annexin V-FITC and propidium iodide (PI) using an FITC Annexin V Apoptosis Detection Kit (BD PharMingen, San Diego, CA) followed by flow cytometric analysis using a CyAn ADP Analyzer (Beckman Coulter, Brea, CA). Data were analyzed using Summit software.
Cell synchronization
MDA-MB-231 cells were synchronized at G2/M phase by single thymidine (2 mM) block (19 h) followed by release into nocodazole (100 ng/ml)-containing media (16 h) (thymidine/nocodazole block). Double thymidine block was used to synchronize cells at G1/S border. Cells were blocked with thymidine (2 mM) for two overnights with 10 h release in fresh media between each thymidine block.
Cell cycle analysis
For DNA content analysis, cells were trypsinized and fixed in 70% ice-cold ethanol overnight at 4°C. After fixation, the cells were washed with 1% BSA/PBS, and permeabilized using 0.25% triton-X 100 in 1% BSA/PBS. Cells were then incubated in PI solution (PBS containing 50 µg/ml of PI and 40 µg/ml of RNase A) for 30 min at room temperature. S phase cells were detected using a bromodeoxyuridine (BrdU) incorporation assay. Cells were pulse-labeled with 10 µM BrdU for 30 min at 37°C. Cells were then trypsinized and fixed in 70% ice-cold ethanol overnight at 4°C. DNA was denatured in 2 N HCl containing 0.5% Triton X-100, and the cells were then neutralized with 0.1 M Na2B4O7. Cells were then stained with FITC-labeled anti-BrdU antibody (BD Biosciences, San Jose, CA). To determine the number of cells in mitosis, cells were fixed, permeabilized and stained with Alexa Fluor 647-conjugated phospho-Histone H3 (Ser 10) antibody (Cell Signaling Technology). Samples were then analyzed on a CyAn ADP Analyzer (Beckman Coulter, Brea, CA). 5×104 events per sample were acquired to ensure adequate mean fluorescence levels. Data were analyzed using Summit software.
ATP measurement
Total cellular ATP content was determined using a luminescent ATP detection kit, ATPlite (PerkinElmer Life Sciences, Boston, MA), according to the manufacturer's instructions. The luminescence intensity was measured using a microplate reader, Synergy 2 (BioTek Instruments, Winooski, VT).
Extracellular flux (XF) analysis
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured as we previously described (Qian and Van Houten, 2010). Cells were seeded in XF24 cell culture plates at 4×104 cells/well and incubated in 5% CO2 at 37°C. Prior to the analysis, cells were washed and growth medium was replaced with bicarbonate-free modified RPMI 1640 medium, the ‘assay medium’ (Molecular Devices, Sunnyvale, CA). Cells were then incubated for another 60 min in a 37°C incubator without CO2. OCR and ECAR measurements were then performed simultaneously using a Seahorse XF24 Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA).
Mitochondrial membrane potential and superoxide generation
To measure mitochondrial membrane potential and superoxide generation, cells were incubated in either 20 nM of TMRM (Invitrogen) or 2.5 µM of MitoSox (Invitrogen) for 20 min at 37°C, respectively. Cells were then trypsinized and suspended in HBSS containing 1% BSA. TMRM and MitoSox fluorescence intensity were analyzed using a CyAn ADP Analyzer (Beckman Coulter, Brea, CA). 5×104 events per sample were acquired and the results were analyzed using Summit software.
Western blot analysis
Cells were lysed in cell lysis buffer (Cell Signaling Technology) containing complete protease inhibitor (Roche). Cell lysates were cleared at 15,000 rpm for 15 min at 4°C. The protein content of the cleared cell lysate was quantified using a Bio-Rad Protein Assay kit (Bio-Rad Laboratories, Hercules, CA). The equal amount of protein was separated on Tris-glycine or Tris-acetate gels (Invitrogen). The separated proteins were blotted onto a polyvinylidene difluoride membrane and blocked overnight at 4°C in phosphate-buffered saline containing 0.1% Tween 20 and 10% nonfat dry milk (blocking buffer). Membranes were incubated with primary antibody in blocking buffer overnight at 4°C. Primary antibodies used were: Drp1 and Opa1 were from BD Biosciences, β-actin (AC-15) and ATM (MAT3-4G10/8) were from Sigma, p53 (DO-1), cyclin E (HE12), cyclin B1 (H-20) and ATR (N-19) were from Santa Cruz Biotechnology, phospho-cdc2 (Tyr15), Chk1 (2G1D5), phospho-Chk2 (Thr68) (C13C1), Chk2 (1C12) and cleaved caspase-3 were from Cell Signaling Technology, phospho-Chk1 (Ser317) was from R&D Systems, cdh1 was a gift from Dr Yong Wan, phospho-ATM (S1981) was from Epitomics, and phospho-Histone H2AX (Ser139) (JBW301) was from Millipore. Membranes were then washed and incubated in peroxidase conjugated anti-rabbit IgG (Sigma), anti-mouse IgG (Sigma), or anti-goat IgG (Santa Cruz Biotechnology) secondary antibody for 1 h at room temperature. Membranes were washed and developed using SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific).
Immunofluorescence
Cells were fixed in 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) in PBS for 15 min at 37°C and blocked using 3% BSA in PBS containing 0.3% Triton X-100 overnight at 4°C. For centrosome staining, cells were incubated for 1 h at room temperature with anti-γ-tubulin antibody (Sigma), followed by an incubation with the secondary Alex Fluor 594 goat anti-mouse antibody (Invitrogen) for 1 h at room temperature. β-tubulin was visualized by incubation with Alex Fluor 555-conjugated anti-β-tubulin antibody (Cell Signaling Technology). Slides were mounted with VECTASHIELD mounting medium containing DAPI (Vector Laboratories, Burlingame, CA). Confocal images were captured using a laser-scanning confocal microscope, Olympus FLUOVIEW FV-1000, with a PlanApo N 60×oil immersion objective, NA = 1.42 (Olympus).
Live cell confocal microscopy analysis
Cells were plated on 40 mm diameter coverglass and incubated for 24 h at 37°C. The coverglass was then assembled into an environmentally controlled closed chamber system, FCS2 live cell chamber (Bioptechs, Butler, PA). The chamber system was then mounted on an inverted confocal microscope (Nikon A1, Nikon), controlled by NIS-Elements software. Leibovitz's L-15 medium supplemented with 10% FBS and 1% penicillin-streptomycin was used, and the temperature of the chamber was maintained at 37°C during the imaging. The excitation wavelengths for GFP and DsRed2 were 488 and 543 nm, respectively. Signal was collected through a Plan Apo VC 60×oil immersion objective, NA = 1.40 (Nikon). The number of individual Z-stacks was set to cover the entire thickness of the cell, with the step size of 1 µm. Time-lapse images were captured at the interval of no time delay. The perfect focus system (PFS) was applied to automatically correct possible focus drift during the period of time-lapse imaging. Post-acquisition analysis of image files was performed using MetaMorph (Molecular Devices), ImageJ (National Institutes of Health) and Photoshop (Adobe).
Statistical analysis
Data are expressed as mean ± standard deviation (s.d.). A Student's t-test was used for the comparisons between two groups. P<0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Dr Stefan Duensing for his assistance with centrosome analysis, Dr Yong Wan for assistance with cell cycle synchronization, and Dr Geoffrey M. Wahl for developing the Histone H2B-GFP plasmid. We are also grateful to Dr Katsuri Mitra, the members of the Van Houten laboratory, and the Merrill Egorin Writing Group of the University of Pittsburgh Cancer Institute for their helpful discussions.
Wei Qian, Serah Choi, Gregory Gibson, Simon Watkins, Christopher Bakkenist and Bennett Van Houten conceived and designed the experiments. Wei Qian performed the experiments.Wei Qian, Serah Choi, Gregory Gibson, Christopher Bakkenist, and Bennett Van Houten analyzed the data. Wei Qian, Christopher Bakkenist and Bennett Van Houten wrote the paper.
Footnotes
Funding
This work was funded, in part, by a grant with the Pennsylvania Department of Health, PA CURE. This work was also supported by funding from the National Insitutes of Health [grant numbers R01CA148644 to C.J.B., P30CA047904, P50CA097190, P50CA121973]. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.109769/-/DC1
References
- Bartkova J., Horejsí Z., Koed K., Krämer A., Tort F., Zieger K., Guldberg P., Sehested M., Nesland J. M., Lukas C.et al. (2005). DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434, 864–870 10.1038/nature03482 [DOI] [PubMed] [Google Scholar]
- Cassidy–Stone A., Chipuk J. E., Ingerman E., Song C., Yoo C., Kuwana T., Kurth M. J., Shaw J. T., Hinshaw J. E., Green D. R.et al. (2008). Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 14, 193–204 10.1016/j.devcel.2007.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho D. H., Nakamura T., Fang J., Cieplak P., Godzik A., Gu Z., Lipton S. A. (2009). S-nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science 324, 102–105 10.1126/science.1171091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimprich K. A., Cortez D. (2008). ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 9, 616–627 10.1038/nrm2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolat S., Martins de Brito O., Dal Zilio B., Scorrano L. (2004). OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 101, 15927–15932 10.1073/pnas.0407043101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosio C., Fimia G. M., Loury R., Kimura M., Okano Y., Zhou H., Sen S., Allis C. D., Sassone–Corsi P. (2002). Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian Aurora kinases. Mol. Cell. Biol. 22, 874–885 10.1128/MCB.22.3.874-885.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorée M., Hunt T. (2002). From Cdc2 to Cdk1: when did the cell cycle kinase join its cyclin partner? J. Cell Sci. 115, 2461–2464 [DOI] [PubMed] [Google Scholar]
- Ekholm S. V., Reed S. I. (2000). Regulation of G1 cyclin-dependent kinases in the mammalian cell cycle. Curr. Opin. Cell Biol. 12, 676–684 10.1016/S0955-0674(00)00151-4 [DOI] [PubMed] [Google Scholar]
- Ekholm–Reed S., Méndez J., Tedesco D., Zetterberg A., Stillman B., Reed S. I. (2004). Deregulation of cyclin E in human cells interferes with prereplication complex assembly. J. Cell Biol. 165, 789–800 10.1083/jcb.200404092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukasawa K. (2007). Oncogenes and tumour suppressors take on centrosomes. Nat. Rev. Cancer 7, 911–924 10.1038/nrc2249 [DOI] [PubMed] [Google Scholar]
- Ganem N. J., Godinho S. A., Pellman D. (2009). A mechanism linking extra centrosomes to chromosomal instability. Nature 460, 278–282 10.1038/nature08136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoulis V. G., Vassiliou L. V., Karakaidos P., Zacharatos P., Kotsinas A., Liloglou T., Venere M., Ditullio R. A., Jr, Kastrinakis N. G., Levy B.et al. (2005). Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434, 907–913 10.1038/nature03485 [DOI] [PubMed] [Google Scholar]
- Green D. R., Van Houten B. (2011). SnapShot: Mitochondrial quality control. Cell 147, 950, e1 10.1016/j.cell.2011.10.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemerly A. S., Prasanth S. G., Siddiqui K., Stillman B. (2009). Orc1 controls centriole and centrosome copy number in human cells. Science 323, 789–793 10.1126/science.1166745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inanç B., Dodson H., Morrison C. G. (2010). A centrosome-autonomous signal that involves centriole disengagement permits centrosome duplication in G2 phase after DNA damage. Mol. Biol. Cell 21, 3866–3877 10.1091/mbc.E10-02-0124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N., Nomura M., Jofuku A., Kato H., Suzuki S. O., Masuda K., Otera H., Nakanishi Y., Nonaka I., Goto Y.et al. (2009). Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 11, 958–966 10.1038/ncb1907 [DOI] [PubMed] [Google Scholar]
- Jones R. G., Plas D. R., Kubek S., Buzzai M., Mu J., Xu Y., Birnbaum M. J., Thompson C. B. (2005). AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 18, 283–293 10.1016/j.molcel.2005.03.027 [DOI] [PubMed] [Google Scholar]
- Kanda T., Sullivan K. F., Wahl G. M. (1998). Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr. Biol. 8, 377–385 10.1016/S0960-9822(98)70156-3 [DOI] [PubMed] [Google Scholar]
- Kastan M. B., Bartek J. (2004). Cell-cycle checkpoints and cancer. Nature 432, 316–323 10.1038/nature03097 [DOI] [PubMed] [Google Scholar]
- Keck J. M., Summers M. K., Tedesco D., Ekholm–Reed S., Chuang L. C., Jackson P. K., Reed S. I. (2007). Cyclin E overexpression impairs progression through mitosis by inhibiting APCCdh1. J. Cell Biol. 178, 371–385 10.1083/jcb.200703202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King M. P., Attardi G. (1996). Isolation of human cell lines lacking mitochondrial DNA. Methods Enzymol. 264, 304–313 10.1016/S0076-6879(96)64029-4 [DOI] [PubMed] [Google Scholar]
- Labrousse A. M., Zappaterra M. D., Rube D. A., van der Bliek A. M. (1999). C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol. Cell 4, 815–826 10.1016/S1097-2765(00)80391-3 [DOI] [PubMed] [Google Scholar]
- Lew D. J., Kornbluth S. (1996). Regulatory roles of cyclin dependent kinase phosphorylation in cell cycle control. Curr. Opin. Cell Biol. 8, 795–804 10.1016/S0955-0674(96)80080-9 [DOI] [PubMed] [Google Scholar]
- Liberal V., Martinsson–Ahlzén H. S., Liberal J., Spruck C. H., Widschwendter M., McGowan C. H., Reed S. I. (2012). Cyclin-dependent kinase subunit (Cks) 1 or Cks2 overexpression overrides the DNA damage response barrier triggered by activated oncoproteins. Proc. Natl. Acad. Sci. USA 109, 2754–2759 10.1073/pnas.1102434108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listovsky T., Oren Y. S., Yudkovsky Y., Mahbubani H. M., Weiss A. M., Lebendiker M., Brandeis M. (2004). Mammalian Cdh1/Fzr mediates its own degradation. EMBO J. 23, 1619–1626 10.1038/sj.emboj.7600149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malena A., Loro E., Di Re M., Holt I. J., Vergani L. (2009). Inhibition of mitochondrial fission favours mutant over wild-type mitochondrial DNA. Hum. Mol. Genet. 18, 3407–3416 10.1093/hmg/ddp281 [DOI] [PubMed] [Google Scholar]
- Mandal S., Guptan P., Owusu–Ansah E., Banerjee U. (2005). Mitochondrial regulation of cell cycle progression during development as revealed by the tenured mutation in Drosophila. Dev. Cell 9, 843–854 10.1016/j.devcel.2005.11.006 [DOI] [PubMed] [Google Scholar]
- Matsumoto Y., Maller J. L. (2002). Calcium, calmodulin, and CaMKII requirement for initiation of centrosome duplication in Xenopus egg extracts. Science 295, 499–502 10.1126/science.1065693 [DOI] [PubMed] [Google Scholar]
- Mitra K., Wunder C., Roysam B., Lin G., Lippincott–Schwartz J. (2009). A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc. Natl. Acad. Sci. USA 106, 11960–11965 10.1073/pnas.0904875106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murga M., Bunting S., Montaña M. F., Soria R., Mulero F., Cañamero M., Lee Y., McKinnon P. J., Nussenzweig A., Fernandez–Capetillo O. (2009). A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat. Genet. 41, 891–898 10.1038/ng.420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K., Nagahama H., Minamishima Y. A., Matsumoto M., Nakamichi I., Kitagawa K., Shirane M., Tsunematsu R., Tsukiyama T., Ishida N.et al. (2000). Targeted disruption of Skp2 results in accumulation of cyclin E and p27Kip1, polyploidy and centrosome overduplication. EMBO J. 19, 2069–2081 10.1093/emboj/19.9.2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norbury C., Blow J., Nurse P. (1991). Regulatory phosphorylation of the p34cdc2 protein kinase in vertebrates. EMBO J. 10, 3321–3329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owusu–Ansah E., Yavari A., Mandal S., Banerjee U. (2008). Distinct mitochondrial retrograde signals control the G1-S cell cycle checkpoint. Nat. Genet. 40, 356–361 10.1038/ng.2007.50 [DOI] [PubMed] [Google Scholar]
- Parone P. A., Da Cruz S., Tondera D., Mattenberger Y., James D. I., Maechler P., Barja F., Martinou J. C. (2008). Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS ONE 3, e3257 10.1371/journal.pone.0003257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian W., Van Houten B. (2010). Alterations in bioenergetics due to changes in mitochondrial DNA copy number. Methods 51, 452–457 10.1016/j.ymeth.2010.03.006 [DOI] [PubMed] [Google Scholar]
- Rajagopalan H., Lengauer C. (2004). Aneuploidy and cancer. Nature 432, 338–341 10.1038/nature03099 [DOI] [PubMed] [Google Scholar]
- Rajagopalan H., Jallepalli P. V., Rago C., Velculescu V. E., Kinzler K. W., Vogelstein B., Lengauer C. (2004). Inactivation of hCDC4 can cause chromosomal instability. Nature 428, 77–81 10.1038/nature02313 [DOI] [PubMed] [Google Scholar]
- Smirnova E., Shurland D. L., Ryazantsev S. N., van der Bliek A. M. (1998). A human dynamin-related protein controls the distribution of mitochondria. J. Cell Biol. 143, 351–358 10.1083/jcb.143.2.351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova E., Griparic L., Shurland D. L., van der Bliek A. M. (2001). Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 12, 2245–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z., Ghochani M., McCaffery J. M., Frey T. G., Chan D. C. (2009). Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 20, 3525–3532 10.1091/mbc.E09-03-0252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruck C. H., Won K-A., Reed S. I. (1999). Deregulated cyclin E induces chromosome instability. Nature 401, 297–300 10.1038/45836 [DOI] [PubMed] [Google Scholar]
- Sugioka R., Shimizu S., Tsujimoto Y. (2004). Fzo1, a protein involved in mitochondrial fusion, inhibits apoptosis. J. Biol. Chem. 279, 52726–52734 10.1074/jbc.M408910200 [DOI] [PubMed] [Google Scholar]
- Taguchi N., Ishihara N., Jofuku A., Oka T., Mihara K. (2007). Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 282, 11521–11529 10.1074/jbc.M607279200 [DOI] [PubMed] [Google Scholar]
- Toledo L. I., Murga M., Gutierrez–Martinez P., Soria R., Fernandez–Capetillo O. (2008). ATR signaling can drive cells into senescence in the absence of DNA breaks. Genes Dev. 22, 297–302 10.1101/gad.452308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo L. I., Murga M., Zur R., Soria R., Rodriguez A., Martinez S., Oyarzabal J., Pastor J., Bischoff J. R., Fernandez–Capetillo O. (2011). A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. 18, 721–727 10.1038/nsmb.2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G., Elorza A., Molina A. J., Mohamed H., Wikstrom J. D., Walzer G., Stiles L., Haigh S. E., Katz S., Las G.et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446 10.1038/sj.emboj.7601963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi J., Zhang Z., Wakabayashi N., Tamura Y., Fukaya M., Kensler T. W., Iijima M., Sesaki H. (2009). The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J. Cell Biol. 186, 805–816 10.1083/jcb.200903065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Su B., Fujioka H., Zhu X. (2008). Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer's disease patients. Am. J. Pathol. 173, 470–482 10.2353/ajpath.2008.071208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O., Wind F., Negelein E. (1927). The metabolism of tumors in the body. J. Gen. Physiol. 8, 519–530 10.1085/jgp.8.6.519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterham H. R., Koster J., van Roermund C. W., Mooyer P. A., Wanders R. J., Leonard J. V. (2007). A lethal defect of mitochondrial and peroxisomal fission. N. Engl. J. Med. 356, 1736–1741 10.1056/NEJMoa064436 [DOI] [PubMed] [Google Scholar]
- Weinberg F., Hamanaka R., Wheaton W. W., Weinberg S., Joseph J., Lopez M., Kalyanaraman B., Mutlu G. M., Budinger G. R., Chandel N. S. (2010). Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 107, 8788–8793 10.1073/pnas.1003428107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann B. (2010). Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 11, 872–884 10.1038/nrm3013 [DOI] [PubMed] [Google Scholar]
- White J. S., Choi S., Bakkenist C. J. (2008). Irreversible chromosome damage accumulates rapidly in the absence of ATM kinase activity. Cell Cycle 7, 1277–1284 10.4161/cc.7.9.5961 [DOI] [PubMed] [Google Scholar]
- Yoon Y. S., Yoon D. S., Lim I. K., Yoon S. H., Chung H. Y., Rojo M., Malka F., Jou M. J., Martinou J. C., Yoon G. (2006). Formation of elongated giant mitochondria in DFO-induced cellular senescence: involvement of enhanced fusion process through modulation of Fis1. J. Cell. Physiol. 209, 468–480 10.1002/jcp.20753 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.