

Abstract
CORL proteins (FUSSEL/SKOR proteins in humans) are related to Sno/Ski oncogenes but their developmental roles are unknown. We have cloned Drosophila CORL and show that its expression is restricted to distinct subsets of cells in the central nervous system. We generated a deletion of CORL and noted that homozygous individuals rarely survive to adulthood. Df(4)dCORL adult escapers display mushroom body (MB) defects and Df(4)dCORL larvae are lacking Ecdysone Receptor (EcR-B1) expression in MB neurons. This is phenocopied in CORL-RNAi and Smad2-RNAi clones in wild-type larvae. Furthermore, constitutively active Baboon (type I receptor upstream of Smad2) cannot stimulate EcR-B1 MB expression in Df(4)dCORL larvae, which demonstrates a formal requirement for CORL in Smad2 signaling. Studies of mouse Corl1 (Skor1) revealed that it binds specifically to Smad3. Overall, the data suggest that CORL facilitates Smad2 activity upstream of EcR-B1 in the MB. The conservation of neural expression and strong sequence homology of all CORL proteins suggests that this is a new family of Smad co-factors.
Keywords: Drosophila, Brain, Neurons, TGFβ, Signal transduction, Ecdysone receptor
INTRODUCTION
During embryonic development in animals, secreted Transforming Growth Factor β (TGFβ) proteins perform a multitude of tasks. Later in life, mutations that disrupt TGFβ signaling often lead to tumor growth. One group of TGFβ pathway regulators is oncogenic Sno/Ski proteins. These bind Smad signal transducers downstream of TGFβ/Activin subfamily members. The initial model for Sno/Ski function was based on data from gain-of-function studies in mammalian cells and states that they are obligate antagonists of TGFβ signaling (Jahchan and Luo, 2010). This model has evolved to accommodate loss-of-function data from flies and nematodes, and RNAi studies in mammalian cells. In Drosophila, data from Sno (Snoo – FlyBase) mutants suggests that Sno acts as a pathway switch: Sno facilitates TGFβ/Activin signaling via a molecular mechanism that simultaneously antagonizes Dpp/BMP signaling (Takaesu et al., 2006). The fly data are supported by evidence that DAF-5 facilitates Smad signaling in nematodes (da Graca et al., 2003) and that Sno facilitates activin signaling in two mammalian cell types (Sarker et al., 2005; Sarker et al., 2008). Consistent with this dual role in TGFβ signaling (negative when overexpressed but positive at physiological levels), human SNO (SKIL – Human Gene Nomenclature Database) can function as both an oncogene and a tumor suppressor gene (Jahchan and Luo, 2010).
Previous analysis of known Sno/Ski-related proteins clustered them into three subfamilies (Sno/Ski, Dachsund and CORL) (Takaesu et al., 2006) the defining feature of which is a Sno homology domain that interacts with Smads. The CORL subfamily contained two members: the Drosophila predicted protein CG11093 and mouse Corl1. At that time, only mouse Corl1 had been cloned and it displayed neural-specific expression during development (in a region of the hindbrain that will become Purkinje cells of the cerebellum). In cell culture, mouse Corl1 functioned as a co-factor for the LBX1 homeodomain protein (Mizuhara et al., 2005). Mouse Corl2 was subsequently cloned and found to have neural-specific expression similar to mouse Corl1 – starting at embryonic day 10.5 in the cell bodies of Purkinje cell progenitors and in the Purkinje cells of adults (Minaki et al., 2008; Miyata et al., 2010). Interestingly, the cerebellum is the site of motor coordination and cerebellar defects are associated with movement disorders known as ataxias (Orr, 2010).
Human CORL proteins FUSSEL15 (SKOR1 in human, Corl1 in mouse) and FUSSEL18 (SKOR2 in human, Corl2 in mouse) display conserved expression in adult Purkinje cells. Both bind Smads non-specifically in a melanoma cell assay and luciferase assays suggest human SKOR2 inhibits while human SKOR1 has no effect on TGFβ signaling (Arndt et al., 2005; Arndt et al., 2007). A genome-wide association study suggested that mutations in the chromosomal region containing SKOR1 were linked to an ataxia known as restless leg syndrome (Kemlink et al., 2009).
The possibility that CORL plays a role in Drosophila TGFβ signaling intrigued us. We cloned CORL and noted that it had neural-specific expression. We generated a null mutant and found that EcR-B1 expression was absent from the MB of larval brains, a phenotype we mimicked with CORL-RNAi and Smad2-RNAi in wild type. In an epistasis study, constitutively active Baboon could not stimulate EcR-B1 expression in CORL mutant brains placing CORL in the TGFβ pathway. Together with evidence that mouse Corl1 specifically binds Smad3, we propose that CORL proteins are a new family of co-factors for Smad signaling.
MATERIALS AND METHODS
Molecular biology
Genomic information refers to the D. melanogaster chromosome 4 complete sequence (Release 5.1; GenBank Accession Number AE014135). cDNA LD43973 for the predicted CG11093 was from Drosophila Genomics Research Center and BAC BACR13D24 (GenBank Accession Number AC010838) was from Children’s Hospital Oakland Research Institute. 5′ RACE was carried out using the GeneRacer kit (Invitrogen). The complete cDNA sequence of CORL (clone p1.10LD43973) is available at GenBank (Accession Number JX126878). Northern and Southern blots were analyzed as described previously (Newfeld and Gelbart, 1995). Two-sided PCR to verify the FLP-FRT-generated deletion in Df(4)dCORL was conducted as described previously (Parks et al., 2004). We employed a 3′ flanking primer (ending at 956,124 bp) with the Piggy-bac WH3′minus primer plus a 5′ flanking primer (ending at 1,002,248 bp) with the Piggy-bac WH5′minus primer. UAS.dCORL was generated from LD43973 using the BglII and XhoI sites of pUAST. CORL-RNAi was constructed from a 799 bp PCR fragment from within exon 3 cloned into pWiz. CORL-scrambled-RNAi was constructed from an 800 bp PCR fragment spanning exon 3 and exon 4 that included a 54 bp intron cloned into pWiz. The intron creates a small mismatch between CORL-scrambled-RNAi and the CORL mRNA that prevents knock-down of CORL but demonstrates the absence of off-target effects for the CORL-RNAi construct with which it shares 93.25% sequence identity.
Drosophila genetics
The FRT-bearing strains Pbac{WH}f07015 (956,754 bp) and Pbac{WH}f06253 (1,000,397 bp) were obtained from the Exelixis Collection. An FLP-FRT-based scheme that generates an exact interstitial deletion between two syntenic FRT elements using intrachromosomal recombination (Parks et al., 2004) was employed to create two independent strains of Df(4)dCORL. Precise deletion of four genes and the exclusion of toy from Df(4)dCORL was molecularly confirmed by two-sided PCR on single flies, as well as by genomic Southern and RNA in situ. Df(4)dCORL was characterized genetically in complementation tests with sphinx720RW (Dai et al., 2008), toyhdl (Pbac{XP}d0719) (Kronhamn et al., 2002) and Pbac{RB}e02096 (disrupts all predicted transcripts of CG32016). The stages of lethality tests were as described previously (Takaesu et al., 2006). Flip-out clones were as described previously (Struhl and Basler, 1993). Additional strains are: 238y.Gal4 (Aso et al., 2009), T80.Gal4 (Marquez et al., 2001), CA-Tkv (Haerry et al., 1998), CA-Babo (Brummel et al., 1999), UAS.dSno (Takaesu et al., 2006), UAS.Mad1 (Takaesu et al., 2005), GFP-Sphinx61 (M. Long, University of Chicago, IL, USA), dSmad2-RNAi and Medea-RNAi (M. O’Connor, University of Minnesota, MN, USA).
RNA and protein expression
RNA in situ of embryos and larval brains with riboprobes of CORL (cDNA LD43973) and toy (cDNA GH14454) were as described previously (Takaesu et al., 2006). Embryo VC fillets were as described previously (Broadie and Bate, 1993). Antibody staining of larval brains was as described previously (Shimizu et al., 2011). Hybridoma Bank antibodies were: Mabdac1-1 (1:50), EcR-B1 (AD4.4 1:50), lacZ (40-1A 1:1000), Pros (MR1A 1:10) and Fas2 (1D4 1:5). Additional antibodies were: Drosophila Act (rabbit 1:50; Santa Cruz), Tll (rabbit 1:500; Kosman et al., 1998), Trio (rabbit 1:1000; Awasaki et al., 2000), GFP (mouse 1:10, rabbit 1:1000, rat 1:1000; Abcam) and pH3 (rabbit 1:500; Abcam). Secondary antibodies were goat anti-mouse, anti-rabbit, anti-rat and anti-guinea pig Alexa Fluor 488, 546 and 633 (Molecular Probes), and biotinylated goat anti-rabbit (Vector Labs).
Biochemistry
Immunoprecipitation and immunoblotting were as described previously (Kawabata et al., 1998). 293T cells were transfected using FuGene6. Lysates were subject to immunoprecipitation using anti-FLAG M2 (Sigma). For mouse Corl1-Smad interactions, immunoprecipitation was followed by immunoblotting with anti-Myc 9E10 antibody (Santa Cruz) or lysates were directly blotted with anti-Myc, anti-FLAG or anti-HA.
Bioinformatics
All full-length Ski/Sno, Dac and CORL proteins from C. elegans, D. melanogaster, S. purpuratus, M. musculus and H. sapiens were included in an alignment of 18 sequences encompassing 2011 amino acids and a Maximum Likelihood tree was generated as described previously (Konikoff et al., 2010). Identification of conserved domains, coiled-coil motifs and alignment highlighting were conducted as described previously (Takaesu et al., 2006).
RESULTS
CORL family proteins are highly conserved
We identified a cDNA corresponding to CG11093, the only CORL family member in flies (Takaesu et al., 2006). Analysis of dCORL transcription by 5′RACE identified two initiation sites: a proximal start site that corresponds to the 5′ end of the cDNA; and a distal start site 10 kb upstream on the far side of the adjacent gene sphinx (Fig. 1A). These two transcripts contain distinct exons 1 and 2 with initiator methionines in exon 2. Both splice in-frame to the common exon 3 and are identical thereafter (Fig. 1B). Analysis of CORL reporter genes covering the region (N.T.T., unpublished observations) indicates that only the distal start site is meaningful.
Fig. 1.
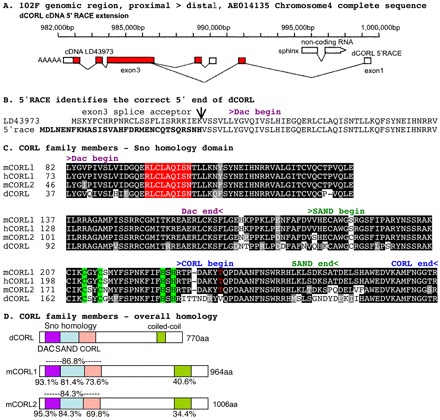
The Drosophila CORL locus, 5′RACE and alignments. (A) The CORL cDNA LD43973 and its extension by 5′RACE with exons shown as boxes (white, untranslated; red, protein coding). CORL transcription direction is shown by a poly-A tail and sphinx transcription direction (opposite from CORL) is indicated by an arrowhead. The CORL proximal start site at 990,000 bp corresponds to the 5′ end of LD43973 and the CORL start site identified by 5′RACE at 986,265 bp is distal to sphinx. The 5′RACE product is consistent with the prediction for CG11093 isoform B (GenBank Accession Number ACZ95098). (B) The open reading frames contained within exon 2 of both transcripts (5′RACE in bold) and their splice junctions with the common exon 3 are shown. (C) Sno homology domain of CORL aligned with mouse Corl1, mouse Corl2 and human CORL1. An amino acid is shaded if the residue is identical (black) or similar (gray), with coloring indicating the APC recognition site (red) and Cys2-His2 zinc finger (green). The red T (mCorl1 Thr235) absent in CORL is homologous to Smad4-binding Thr271 in human Ski. Conserved motifs within the Sno homology domain are Dac, SAND and CORL. (D) CORL proteins from fly and mouse with the locations of five distinct domains shown. The level of amino acid similarity between the indicated protein and Drosophila CORL is shown for all five domains.
Employing the extended ORF we conducted a new phylogenetic analysis (supplementary material Fig. S1A). Overall conservation between mammalian and fly CORL is notable (37.7% similarity) but conservation in the Sno homology domain is astonishingly high (Fig. 1C,D). There is an average of 85% similarity between Drosophila CORL and mouse Corl1/mouse Corl2 in the 197-residue Sno homology domain. Conservation between Drosophila CORL and mouse Corl1/2 in the Sno homology domain easily exceeds the similarity of the 188-residue ligand domain of fly Dpp and human BMP4 (77%) (Newfeld and Gelbart, 1995), two proteins that can function across species. Thus, members of the CORL subfamily probably have conserved functions and perhaps conserved expression patterns.
The similarity of CORL to its fly paralogs in the Sno and Dac family is less robust (supplementary material Fig. S1B,C). Comparing CORL to Sno, both the Dac and SAND domains are roughly 45% similar with the CORL domain not as well conserved. Alignments highlight similarities and differences between these proteins. Similarity in the N-terminal region of the Sno homology domain includes the APC site and Cys2-His2 motif. Differences in the C-terminal region of the Sno homology domain include the absence of the Smad4-interacting I-loop and its TCHW motif, suggesting that CORL is unlikely to bind Smad4. The last 20 residues are similar to a helix-turn motif in the dimerization domain of Siah-interacting protein (Sina in flies) (Santelli et al., 2005), suggesting a role in protein-protein interactions.
Sequence alignments identified additional similarities. Upstream of the Sno homology domain, Sno/Ski proteins contain a hydrophobic region that binds Smad2/3 (mSno 83-92; Lx3Lx4L). A similar region is found in CORL at a similar location (CORL 32-42; Vx3LLx4I; supplementary material Fig. S1D). Downstream of the Sno homology domain, Sno/Ski and CORL proteins contain a coiled-coil region that, for Sno/Ski, is involved in recruiting the co-repressor Sin3a. These conserved features suggest that CORL proteins can bind Smad2/3 and recruit co-factors.
CORL embryonic expression is restricted to the central nervous system
At stage 12, CORL transcripts become detectable in the ventral cord (VC) of the central nervous system (CNS) in a single cell per segment. In stage 13 embryos, bilaterally symmetrical expression in one or two cells per VC segment is visible (Fig. 2A,B). Given that all VC neuroblasts (NBs) are present by late stage 11 (Doe, 1992), CORL-positive cells are unlikely to be NBs but instead are ganglion mother cells (GMCs) or mature cells derived from early NB divisions. During stages 14-15 CORL in the VC expands to groups of three cells per segment with larger groups in the posterior. These are located peripherally (towards the surface) roughly four to five cell diameters from the midline (Fig. 2C-E). At stage 16, one instance of new CORL VC expression is seen in a single pair of cells at the midline within abdominal segment 3 (Fig. 2G).
Fig. 2.
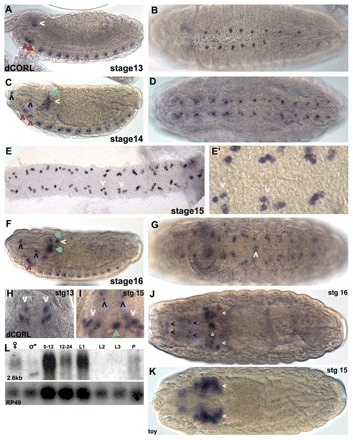
Drosophila CORL embryonic expression is restricted to CNS cells. (A) Lateral stage 13: in the brain, CORL is seen within the ventral region of the tritocerebrum (right red arrowhead) and the deutocerebrum (left red arrowhead), as well as in a central region of the ocular protocerebrum (white arrowhead). (B) Ventral 13: in the VC, one or two CORL cells per segment are detected. (C) Lateral 14: in the brain, more CORL-positive cells are present within the ventral region of the tritocerebrum (right red arrowhead), the deutocerebrum (left red arrowhead) and the ocular protocerebrum (white arrowhead). A single CORL cell dorsal to the large group of cells in the ocular protocerebrum has appeared (green arrowhead). Single cells at the dorsal/anterior tip (black arrowhead) and the ventral/medial region of the pharynx (blue arrowhead) are now visible. (D) Ventral 14: in the VC, there are groups of two or three CORL cells per segment. (E,E′) Ventral 15: in a filleted VC, sets of three CORL cells per segment (white arrowheads) are present, although groups with four or five cells are present in posterior segments. (F) Lateral 16: in the brain, CORL cells in the ocular protocerebrum now form a star-shaped pattern with the largest group in the center (filled green arrowhead) and four smaller groups located dorsal, dorsal/posterior (white arrowhead), posterior and ventral to the central group. In the anterior/dorsal (black arrowhead) and ventral/medial regions of the pharynx (blue arrowhead), as well as in the ventral region of the trito- and the deutocerebrum, CORL has expanded and the latter two have merged into a continuous row of cells (red arrowhead). The dorsal-most CORL cell has migrated to just below the dorsal surface (green arrowhead). (G) Ventral 16: in the VC, CORL persists in three to five cells per segment and new expression is observed in a single pair of cells adjacent to the midline in A3 (white arrowhead). (H-K) Dorsal views of the brain with anterior upwards in H,I and towards the left in J,K. Arrow colors indicating CORL brain expression are consistent between lateral and dorsal views. (H) Stage 13: CORL is seen in two groups of ocular protocerebral cells (white arrowheads). (I) Stage 15: in the brain, the number of CORL cells in the ocular protocerebral (white arrowheads) and medial regions (green arrowhead) increases and expression in two pairs of cells anterior to the brain is apparent (blue arrowheads). (J) Stage 16: the number of CORL cells in the ocular protocerebral region appears stable, the medial CORL cells now appear associated with the esophagus (filled green arrowhead) and a new set of paired cells at the anterior are detected (black arrowheads). (K) Stage 15: toy is visible in MB neurons in the ocular protocerebrum (white arrowheads). (L) CORL hybridized to mRNA from adult females, adult males, 0- to 12- and 12- to 24-hour-old embryos, first, second and third instar larvae, and pupae. A single transcript of 2.6 kb that corresponds in size to the CORL 5′RACE extended cDNA is detected in all lanes except L2 and L3. The blot was stripped and rehybridized with Rp49 (ubiquitously expressed) showing that that variation in CORL expression intensity is probably due to variation in RNA loading.
In the developing brain, at stage 13 CORL first appears in one or two cells within the ventral-most part of the trito- and deutocerebrum, and in small groups of cells in lateral regions of the ocular protocerebrum (Fig. 2A, lateral; 2H, dorsal). During stages 14-16, the number of CORL cells significantly expands in the ocular protocerebrum (Fig. 2C,F, lateral; 2H-J, dorsal). The CORL cells are present in roughly the same region of the ocular protocerebrum as those expressing the adjacent gene twin of eyeless (toy; Fig. 2J,K). Interestingly, at this stage toy is present in MB neurons and toy mutants die with visible MB defects (Furukubo-Tokunaga et al., 2009). The MB is the site of many cognitive functions and receives input from a variety of sources (Truman et al., 1994; Davis, 1996).
During stages 13-15 CORL transcripts become detectable anterior to the brain in pairs of cells that flank the pharynx (Fig. 2C, lateral; 2H,I, dorsal). By stage 16, CORL cells in the ocular protocerebrum form a five-pointed star pattern with a large cluster in the center and smaller groups located at the tips (Fig. 2F, lateral; 2J, dorsal). A thorough analysis of CORL expression in the embryo will be reported elsewhere. Northerns revealed a single CORL transcript of 2.6 kb present at all stages except second and third larval instars (Fig. 2L). Overall, these studies suggest that CORL expression is restricted to specific subsets of CNS cells.
CORL mutant adults display MB defects
CORL RNA is ubiquitously visible in the CNS of third instar larvae at a low level with several areas of strong expression. In the brain, CORL is prominent in roughly circular groups of cells in the dorsal/anterior region of the brain where the MB is located (Fig. 3A). CORL is also notable in subesophageal cells in the dorsal/posterior region of the brain. In the VC, three to five CORL cells per segment are evident but their identity is unknown. No expression is seen in the optic lobe of the brain, in imaginal disks or in any other larval tissue.
Fig. 3.
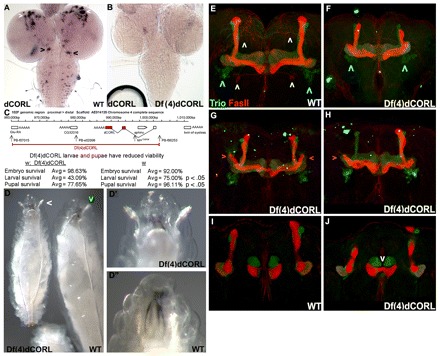
CORL mutants display MB defects. (A,B) Third instar larval CNS in dorsal view. (A) Wild type shows CORL expression in groups of brain cells in the anterior/dorsal (white arrowheads) and subesophageal regions (posterior; black arrowheads). Additional CORL cells are scattered between these groups, present in the VC and there is low-level ubiquitous expression. (B) In Df(4)dCORL, CORL expression, including low level ubiquitous expression, is absent. (C) Df(4)dCORL deletion map and lethal phase analysis. The region precisely deleted via FLP-FRT-mediated intrachromosomal recombination in Df(4)dCORL is shown. The locations of the two FRT-bearing Piggy-bac insertions employed to generate Df(4)dCORL, a Piggy-bac insertion in CG32016 and a splicing mutation of sphinx are shown. Stage of lethality data reveals significant larval and pupal lethality for Df(4)dCORL homozygotes. (D) Age-matched sibling larvae are shown to scale. Left: Df(4)dCORL in dorsal view is roughly half the size of its heterozygous sibling and displays prematurely everted anterior spiracles (white arrowhead). Right: phenotypically wild-type sibling [Df(4)dCORL/In(4)CiD] in ventral/lateral view, the anterior spiracle of which does not protrude (green arrowhead). (D′,D″) High-magnification dorsal views of the contrasting topology of the anterior spiracles (images not to scale). (E,F) Stacked confocal images of the adult MB (view is anterior with dorsal towards the top) stained for the membrane-associated protein Trio (green: α′, β′ and γ lobes in adults) and the transmembrane protein Fas2 (red: α, β and γ lobes). Defects in Df(4)dCORL MB are lobes that are shortened or mis-shapen and a β neuron that crosses the midline. Outside the MB, Trio-positive neurons ventrolateral to the MB are normal (blue arrowheads), Fas2-positive neurons ventral to the MB are missing (yellow arrowheads) and Fas2- or Trio-positive neurons of unknown origin between the dorsal lobes of the MB are missing (white arrowheads). (G,H) Df(4)dCORL MBs show bilateral or unilateral extensions of the γ-lobe (pink arrowheads), indicating a defect in neuronal pruning that is a hallmark of EcR-B1 loss in the MB. (I,J) Single confocal slice of the adult MB (same view as above). The majority of the β-lobe is fused across the midline (white arrowhead) in Df(4)dCORL.
CORL is located in chromosome region 102D on the difficult to study chromosome 4. Nevertheless, we generated a small deletion removing CORL [Df(4)dCORL; Fig. 3B,C] using the FLP-FRT method of Parks et al. (Parks et al., 2004). FLP-induced recombination between two syntenic FRT sites precisely deletes the intervening region and leaves behind a functional FRT. We confirmed the endpoints of Df(4)dCORL via two-sided PCR and verified that the CORL locus is absent by genomic Southern blotting (supplementary material Fig. S2A,B). In addition to CORL, Glu-RA (metabotropic glutamate receptor), sphinx (non-coding RNA) and CG32016 are removed by Df(4)dCORL. toy is adjacent to CORL on the distal side (Furukubo-Tokunaga et al., 2009) but is not affected by Df(4)dCORL (Fig. 3C).
The stage of lethality studies revealed that 57% of Df(4)dCORL homozygotes die as larvae and an additional 22% die as pupae with 21% surviving to adulthood. This is significantly more lethality at both stages than in the parental strains. We conclude that the lethality of homozygous individuals is due to the loss of CORL because, as described in detail below, tissue-specific expression of UAS.dCORL rescues Df(4)dCORL mutant phenotypes in that tissue (Fig. 4G). Consistent with this conclusion, we noted that the parental FRT strains are fully viable, indicating an absence of secondary lethals and that adults homozygous for deletions of Glu-RA (Bogdanik et al., 2004), sphinx (Dai et al., 2008) or Pbac{RB}e02096 in CG32016 are fully viable. Thus, we consider all Df(4)dCORL pre-adult phenotypes to result from the loss of dCORL.
Fig. 4.
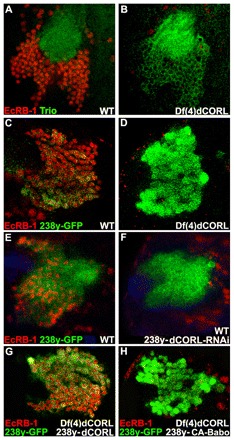
EcR-B1 MB expression is absent in Df(4)dCORL larval brains. (A,B) Ventral/posterior slice of MB cells near the calyx stained for Trio (green; membranes of all MB larval neurons but not MB neuroblasts or GMC) and EcR-B1 (red). The calyx is composed of bundled MB dendrites (green circle). In wild type, Trio and EcR-B1 (nuclear and thus not present in the calyx) are co-expressed in MB neurons. In Df(4)dCORL, EcR-B1 is absent from MB neurons (with one exception) but present in nearby non-MB neurons. (C,D) Anterior/dorsal slice of MB cells with 238y.Gal4 driving GFP (green; cytoplasmic in all MB neurons) and stained for EcR-B1 (red). In wild type, 238y-GFP and EcR-B1 are co-expressed in MB neurons but EcR-B1 is absent in Df(4)dCORL MB neurons. (E,F) Anterior/dorsal slice of wild-type MB with 238y.Gal4 driving GFP or GFP+dCORL-RNAi stained for Tll (blue; MB neuroblasts and GMC but not MB neurons), GFP (green) and EcR-B1 (red). CORL-RNAi eliminates EcR-B1 from MB neurons but not from cells outside the MB. (G) In Df(4)dCORL MB, expression of UAS.dCORL by 238y.Gal4 at 18°C rescues EcR-B1 (red) expression cell-autonomously (GFP; green). (H) Anterior/dorsal slice of Df(4)dCORL MB cells with 238y.Gal4 driving CA-Babo+GFP (green) that are stained for EcR-B1 (red). Note that loss of CORL prevents CA-Babo from activating EcR-B1.
Df(4)dCORL mutants display a complex phenotype. Df(4)dCORL larvae exhibit poor growth, developmental delay, wander away from food precociously and prematurely evert their anterior spiracles (Fig. 3D). Df(4)dCORL adults have greatly reduced lifespan and poor female fertility. Given the presence of dCORL transcripts in the anterior/dorsal region of the brain where the MB is located and the role of the MB in a variety of behaviors, we examined the MB in Df(4)dCORL adults. These MBs display numerous structural defects (Fig. 3E-J). Among the defects are: shortened, misshapen and missing dorsal lobes, β neurons that cross the midline and γ-lobe pruning defects that can reflect the loss of EcR-B1 expression (Zheng et al., 2003).
CORL is required for Smad2 activation of EcR-B1 larval MB expression
We then investigated the role of CORL in larval MBs. Here, circulating Myoglianin (Myo) signals via the type I receptor Baboon (Babo) and Smad2 to activate expression of EcR-B1 (Zheng et al., 2003; Zheng et al., 2006; Awasaki et al., 2011). We examined EcR-B1 MB expression in both Df(4)dCORL and CORL-RNAi flip-out clone genotypes. The presence of a Smad2-binding hydrophobic region suggested the hypothesis that CORL may cooperate with Smad2 upstream of EcR-B1. A prediction of this hypothesis was that loss of CORL would lead to loss of EcR-B1 in the MB.
An alternative hypothesis we tested first is that CORL is a general MB factor rather than one specific for Smad2-dependent events. We examined three Smad2-independent MB markers in Df(4)dCORL larvae: Dachshund (Dac; nuclear in neurons with functions in α and β lobe maturation) (Martini et al., 2000), Tailless (Tll; nuclear in NBs and GMCs with functions in proliferation) (Kurusu et al., 2009) and Prospero (Pros; nuclear in GMCs then cortical in subset of neurons with functions in differentiation) (Doe et al., 1991). Expression of these proteins in wild-type and Df(4)dCORL larval MB was indistinguishable (supplementary material Fig. S2C-L), as were the levels of mitotic marker phospho-Histone3 (not shown). There is no evidence that CORL is a general regulator of transcription in the MB. A second alternative hypothesis we examined was that CORL is required for expression of Activin (Act). This seemed reasonable because Act has significant expression in a pair of subesophageal neurons whose axons trace a path similar to the distribution of CORL cells in the larval brain. We found that Act expression is unaltered in Df(4)dCORL (supplementary material Fig. S4M-O).
By contrast, experiments with the MB markers Trio (all MB neurons in larvae; Fig. 4A,B) or 238y.Gal4 (all MB neurons; Fig. 4C,D) in Df(4)dCORL larvae revealed that EcR-B1 was absent in the MB of most individuals. EcR-B1 in non-brain neural tissues was unaffected in Df(4)dCORL larvae (e.g. ring gland; supplementary material Fig. S2K,L). EcR-B1 expression in the VC was greatly reduced (supplementary material Fig. S2M,O) in Df(4)dCORL larvae, suggesting that CORL plays roles in the CNS outside the MB. An analysis of those functions will be reported elsewhere.
The loss of EcR-B1 expression in the MB of Df(4)dCORL larvae is phenocopied by 238y.Gal4 driving dCORL-RNAi in wild-type larvae (Fig. 4E,F). Rescue experiments employing 238y.Gal4 driving UAS.dCORL in Df(4)dCORL larvae significantly restored EcR-B1 expression only in 238y.Gal4 expressing cells (compare Fig. 4G,D). The rescue experiment formally demonstrates that it is the deletion of CORL that leads to the loss of EcR-B1 in Df(4)dCORL larvae. The data are consistent with the hypothesis that CORL cooperates with Smad2 upstream of EcR-B1.
We then examined flip-out clones of CORL-RNAi, Smad2-RNAi and Medea-RNAi. If these studies phenocopy the loss of EcR-B1 MB expression in Df(4)dCORL larvae (which is rescued by UAS.dCORL) it would further support our hypothesis that CORL facilitates Smad2 signaling upstream of EcR-B1. We include Medea owing to its established role as a partner for Smad2, although a role in EcR-B1 activation has not yet been formally shown. In the ventral/posterior region near the calyx, EcR-B1 is prominent in the cell bodies of MB neurons. EcR-B1 at this location is unaffected by co-expression of GFP and CORL-scrambled-RNAi in flip-out control clones (Fig. 5A). With CORL-RNAi, EcR-B1 expression is lost in a large MB clone but not in single cell MB clones or in clones outside the MB (Fig. 5B). Large clones containing multiple cells are caused by flip-out in an MB neuroblast. In all neurons derived from the flipped-out NB, CORL-RNAi would be present prior to Smad2 signaling upstream of EcR-B1 activation. In these cells, CORL-RNAi prevents Smad2 activation of EcR-B1. In cells adjacent to large clones, EcR-B1 is not affected, indicting that knockdown of CORL cell-autonomously eliminates EcR-B1. Alternatively, single cell clones reflect a flip-out in a differentiated MB neuron. In these flipped-out neurons, EcR-B1 was already activated by Smad2 signaling or by either of two non-Smad pathways upstream of EcR-B1 [Rho-GTPases (Ng, 2008); Ftz-F1 (Boulanger et al., 2011)]. In differentiated MB neurons, knockdown of CORL does not affect EcR-B1, indicating that dCORL does not participate in EcR-B1 maintenance.
Fig. 5.
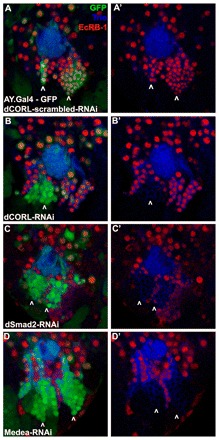
Drosophila CORL-, Smad2- and Medea-RNAi eliminate EcR-B1 in posterior/ventral MB clones. Single confocal slices of flip-out clones in the ventral/posterior region of the MB of wild-type larval brains stained for Trio (blue), EcR-B1 (red) and GFP (green). Left column is three color and the right column is two color of Trio (blue) and EcR-B1 (red). (A,A′) Control expressing CORL-scrambled-RNAi has one medium and one large clone inside the domain of EcR-B1 (arrowheads). EcR-B1 is not affected by CORL-scrambled-RNAi. (B,B′) CORL-RNAi has one large (arrowhead) and several single cell clones inside the domain of EcR-B1. In the large clone, originating in a MB neuroblast, EcR-B1 is absent. In single cell clones originating in differentiated neurons, either inside or outside the MB, EcR-B1 is unaffected. (C,C′) Smad2-RNAi has two large clones (arrowheads) and several single cell clones inside the EcR-B1 domain. Smad2-RNAi phenocopies CORL-RNAi with EcR-B1 absent in large clones but unaffected in single cell clones. (D,D′) Medea-RNAi has two large clones (arrowheads) and several single cell clones inside the domain of EcR-B1. Medea-RNAi phenocopies CORL-RNAi and Smad2-RNAi with EcR-B1 absent in large clones but unaffected in single cell clones.
The phenotype of CORL-RNAi clones mimics exactly the phenotype seen in Smad2-RNAi (Fig. 5C) and Medea-RNAi clones (Fig. 5D). In these three RNAi genotypes, EcR-B1 expression is lost in large clones generated by flip-out in an MB neuroblast but not in single cell clones where the flip-out occurred in a differentiated MB neuron or in clones outside the MB. To further improve our confidence in the data, we conducted a second set of flip-out studies in the anterior/dorsal region of the MB adjacent to the NB/GMC cluster. Data from these anterior/dorsal studies perfectly mimicked our analysis of the ventral/posterior region (supplementary material Fig. S3A-D). Both sets of flip-out clone data further support the hypothesis that CORL contributes to Smad2 signaling upstream of EcR-B1 activation in the larval MB.
In a rigorous test of this hypothesis, we examined whether loss of CORL could prevent a constitutively active form of Babo (CA-Babo; Brummel et al., 1999) from stimulating EcR-B1 expression in the MB of Df(4)dCORL larvae. The hypothesis predicts that EcR-B1 will be absent in this experiment, which was observed when employing 238y.Gal4 to drive CA-Babo (compare Fig. 4H with 4G). This epistasis data strongly demonstrates a requirement for CORL in Babo-Smad2 signaling upstream of EcR-B1 activation in larval MB.
Mouse Corl1 strongly binds Smad3 and binding is enhanced by TGFβ stimulation
To determine whether the requirement for CORL in EcR-B1 activation was based on physical interactions between CORL and Smad2, we analyzed mouse Corl1 in biochemical studies. We examined the ability of mouse Corl1 to bind every Smad protein and the influence of receptor activation on mouse Corl1-Smad binding. First, we found that mouse Corl1 interacted strongly with mouse Smad3 (a homolog of Drosophila Smad2) and weakly with mouse Smad8 (homolog of Mad; Fig. 6A). Second, we demonstrated that mouse Corl1-Smad3 complex formation is strongly increased by TGFβ receptor activation. Comparison with the well-known TGFβ repressor Ski (which binds mouse Smad2/3 and mouse Smad4) revealed that Ski interacts with mouse Smad3 more strongly than mouse Corl1 (Fig. 6B). The extraordinary sequence conservation of CORL proteins and the biochemical data for mouse Corl1 strongly suggest that the requirement for CORL in Smad2 signaling is based upon CORL-Smad2 physical interactions.
Fig. 6.
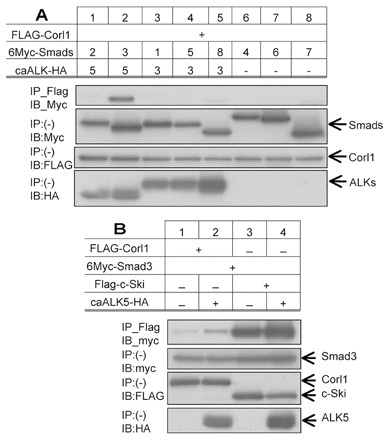
Mouse Corl1 binding to Smad3 is enhanced by TGFβ/Activin signaling. (A) The interaction of FLAG-tagged mouse Corl1 and Ski with Myc-tagged mouse Smads was examined by immunoprecipitation followed by immunoblotting in 293T cells. Constitutively active ALK5 and ALK3 (CA-ALK5 and CA-ALK3) tagged with HA were used to activate TGFβ/Activin and BMP signal transduction, respectively. Top panel shows that mouse Corl1 strongly binds Smad3 when stimulated by activated receptor. The lower three panels indicate expression levels for each protein in the experiment. (B) Examination of the ability of CA-ALK5 (TGFβ/Activin receptor) to stimulate interaction of Smad3 with mouse Corl1 and Ski shows activated receptor induces an increase in Smad3 binding by both proteins and that Ski has greater affinity for Smad3 than mouse Corl1.
Ectopic expression of CORL antagonizes Babo and Tkv signaling
Given that Sno/Ski proteins have both negative and positive effects on TGFβ signaling, we examined CORL flip-out clones in the MB. In both anterior/dorsal (Fig. 7A) and ventral/posterior clones (Fig. 7B), overexpression of CORL led to cell-autonomous loss of EcR-B1 expression. Consistent with these results, luciferase assays of TGFβ/Activin signaling showed that mouse Corl1 and Ski can repress TGFβ/Activin responses in a dose-dependent manner (supplementary material Fig. S4). This led us to wonder whether CORL overexpression functions as a general Smad inhibitor. We drove CORL in wing disks, where it is not normally expressed, and found that this led to vein truncations that phenocopy expression of Sno and dominant-negative Mad (protein encoded by the Mad1 allele; Fig. 7C-F). Thus, ectopic CORL can inhibit Dpp-Tkv-Mad signaling that is required for vein formation. To rigorously test this hypothesis, we examined the ability of ectopic CORL to block CA-Tkv (Haerry et al., 1998) from stimulating vein overgrowth (Fig. 7G). Co-expression of CORL and CA-Tkv significantly, though not completely, suppressed the CA-Tkv phenotype (Fig. 7H), demonstrating that ectopic CORL can non-specifically antagonize Smad signaling.
Fig. 7.
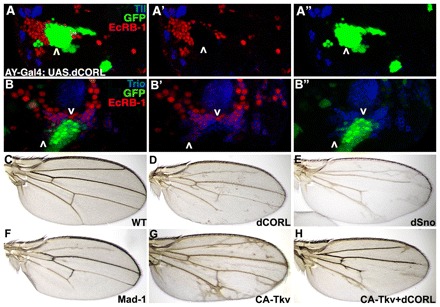
Drosophila CORL overexpression blocks EcR-B1 and generates vein truncations. (A-A″) Single confocal slice of UAS.dCORL flip-out clones in the anterior/dorsal region of the MB stained for Tll (blue), EcR-B1 (red) and GFP (green). EcR-B1 is lost in the clone (above the arrowhead), whereas Tll is unaffected. Images are three color (A), two color (EcR-B1+Tll; A′) and two color (Tll+GFP; A″). (B-B″) Single confocal slice of UAS.dCORL flip-out clones in the ventral/posterior region of the MB stained for Trio (blue), EcR-B1 (red) and GFP (green). EcR-B1 is lost in the clone (below the downward arrowhead) but present in cells adjacent to the clone (above the upward arrowhead). Trio is unaffected at either location. Images are three color (B), two color (EcR-B1 and Trio; B′) and two color (Trio and GFP; B″). (C) Wild-type wing. (D) T80;dCORL wing with truncations of L2, L5 and the posterior crossvein. (E) T80;dSno wing with a more severe phenotype, including truncated L3, L4 and loss of the anterior crossvein. (F) T80;Mad1 wing similar to CORL. (G) T80;CA-Tkv wing with numerous ectopic veins. (H) T80;CA-Tkv+dCORL wing with few ectopic veins and truncations of L2, L4 and L5, indicating CORL can suppress the CA-Tkv vein overgrowth phenotype.
DISCUSSION
CORL function in TGFβ signaling and neural development
Although the core elements of the TGFβ signal transduction pathway (ligands, receptors and Smads) and their various subfamily specificities have been known for over a decade, efforts to uncover new mechanisms influencing this potent cell fate/cell cycle regulator continue unabated. Previously unknown mechanisms of truly general application are occasionally uncovered (e.g. mono- and deubiquitylation) (Dupont et al., 2012) but the majority of interest and effort in the fields of developmental biology and oncology focus on the identification of new factors that influence TGFβ-dependent cell fate or cell cycle decisions in a single cell type. Analysis of these new pathway components will provide information on as yet unknown developmental processes and suggest potential targets for therapeutics.
Here, we have moved this effort forwards significantly by demonstrating that CORL is a new player in TGFβ signal transduction: as a facilitator of Smad2 signaling in loss-of-function studies; and as an antagonist of Smad2 and Mad signaling when ectopically expressed. This finding has wide implications because CORL belongs to a highly conserved gene family that is closely related to the family of Sno/Ski proteins, which also appear to have a dual role in regulating TGFβ signaling. There are clear similarities between CORL and Sno data from fly loss- and gain-of-function phenotypes. Loss-of-function studies showed that, in both Df(4)dCORL and CORL-RNAi genotypes, the loss of CORL led to the loss of Babo-Smad2-dependent EcR-B1 expression in the larval MB. In SnoEx4B/Snosh1402 genotypes, the loss of Sno led to reduced expression of cell cycle markers such as phospho-histone 3 in the larval optic lobe (Takaesu et al., 2006). Gain-of-function studies showed that overexpression of CORL or Sno in wing disks antagonized Mad signaling, leading to vein defects.
This functional correspondence does not extend to CORL and Sno expression during development, suggesting that although they may have similar effects on TGFβ signaling, they do so at different times and in different tissues. CORL is probably expressed as a single transcript that encodes one protein and is restricted to the embryonic and larval CNS where it is present at high levels in a small number of cells. Sno is expressed as multiple transcripts that become multiple proteins very broadly in both embryos and larvae. Sno is expressed in many tissues that do not express CORL, such as the embryonic epidermis and larval imaginal disks (Takaesu et al., 2006). In the embryonic CNS, CORL expression precedes Sno by two developmental stages (12 versus 14) but given its ubiquity in this tissue, Sno is almost certainly present in cells that express CORL beginning at stage 14, although this has not been formally shown. Within the third instar larval CNS, the only cells with significant Sno expression are in the optic lobe (Quijano et al., 2010) where CORL is not expressed.
Biochemical differences between CORL and Sno proteins dictate that they accomplish their similar effects on TGFβ signaling via distinct mechanisms. At the amino acid level, the key distinction is likely to be the absence in CORL of the Smad4-binding TCHW motif in Sno (supplementary material Fig. S1C). Only the threonine residue is present in mammalian CORL proteins (Fig. 1C). By contrast, all Sno/Ski and CORL family members contain a hydrophobic region upstream of the Sno homology domain that strongly binds Smad2/3. Thus, the ability of CORL family members to bind preferentially to the TGFβ/Activin transducer Smad3 (mouse CORL1 explicitly and Drosophila CORL by analogy, based on sequence conservation exceeding that of the cross-functional proteins Dpp and BMP4) is distinct from the ability of Sno to bind Smad2, Mad and Medea.
The ability of Sno to bind multiple Smads allows it to function as a ‘pathway switch’ by shunting Medea between pathways – reducing Dpp/Mad while stimulating Activin/Smad2. Positive and negative actions on TGFβ signaling are inherent in the mechanism of action of Sno. The effect of CORL on Smad2 signaling cannot be explained in that way. Our model proposes instead that the dual activity of CORL is based on its function as a dose-sensitive co-factor for Smad2, as suggested by the mouse Corl1 luciferase assays. In this model, loss or gain of CORL in an otherwise wild-type individual leads to aberrant Smad2 signaling and loss of EcR-B1, either through the loss of a required co-factor or by the sequestration of Smad2 into nonfunctional complexes when overwhelmed by excess CORL. CORL-mediated inhibition of Dpp-Tkv-Mad signaling when ectopically expressed may arise from its ability to weakly recognize Mad, as suggested by the weak binding of mouse Corl1 to Smad8 (Fig. 6A) and its sequestration into non-functional complexes.
This model explains the cell-type specificity of EcR-B1 activation in MB neurons by circulating Myo (Awasaki et al., 2011) via ubiquitous babo and Smad2 (Zheng et al., 2003; Brummel et al., 1999): MB neurons are the only cells exposed to Myo that have sufficient CORL function to facilitate Smad2 transcriptional activity. However, the model requires two caveats. First, CORL loss of function dose effects are only penetrant if the loss exceeds 50% as heterozygous Df(4)dCORL flies appear wild type. Second, CORL is not a universal Smad2 co-factor as Smad2 regulates Ecdysone biosynthesis in the ring gland without CORL (Gibbens et al., 2011).
Implications for mammalian CORL in TGFβ signaling, development and disease
Significant levels of conservation for both neural-specific expression and amino acid sequence for CORL proteins suggest that the function of CORL in TGFβ/Activin signaling (facilitation of Smad2 activity) and CORL developmental roles (tissue-specific activation of gene expression) will be conserved in vertebrates. This hypothesis is supported by a recent paper describing a loss-of-function phenotype for mouse Corl2 in mice (Wang et al., 2011). In addition to gross cerebellar defects, homozygous mouse Corl2 mutant mice lack sonic hedgehog expression in Purkinje cells. The authors view this result through the lens of overexpression studies showing that mouse Corl2 represses BMP signaling in cell culture to generate a two-step explanation for the phenotype: BMP repression is necessary for sonic hedgehog activation. Viewed through the lens of the CORL loss-of-function MB phenotype, a one-step explanation is obtained: mouse Corl2 facilitates TGFβ/Activin signaling upstream of sonic hedgehog activation.
In adult mammals, the presence of human and mouse CORL proteins in Purkinje cells implies a role in motor control, the primary physiological function of the cerebellum. A large number of ataxias are caused by disruption of normal Purkinje cell function (Orr, 2010). A study connecting mutations in the chromosomal region containing FUSSEL15 (human CORL1/SKOR1) to the familial movement disorder restless leg syndrome (Kemlink et al., 2009) suggests the possibility that loss of human CORL1 is a factor in the genesis and/or progression of a subset of ataxias. The possibility that Df(4)dCORL adults may be a new model for ataxia is under investigation.
In summary, loss-of-function studies demonstrated a formal requirement for CORL in Babo-Smad2 signaling, while gain-of-function studies showed that ectopic expression of CORL antagonizes Smad signaling nonspecifically. Our model for the dual activity of CORL proposes that it functions as a tissue-specific, dose-sensitive co-factor for Smad2. The conservation of neural expression and strong sequence homology for all CORL proteins suggests that they are a new family of co-factors for Smads.
Supplementary Material
Acknowledgments
We thank Yuto Kamiya, Charlotte Konikoff, Tomohiro Ogami, Barrett Pfeiffer, Alice Schmid and Robert Wisotzkey for technical assistance. We appreciate insightful discussions and/or support from Kevin Cook, Manyuan Long, Mike O’Connor and Tetsuya Tabata. Reagents were provided by Children’s Hospital Oakland, Developmental Studies Hybridoma Bank, Drosophila Genomics Resource Center, Exelixis Collection, Indiana Stock Center, Manyuan Long, Mike O’Connor, Yuichi Ono, John Reinitz and Tetsuya Tabata.
Footnotes
Funding
The Miyazono lab is supported by Global Center of Excellence Program (Integrative Life Science Based on the Study of Biosignaling Mechanisms) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and the Newfeld lab by Science Foundation Arizona.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.079442/-/DC1
References
- Arndt S., Poser I., Schubert T., Moser M., Bosserhoff A.-K. (2005). Cloning and functional characterization of a new Ski homolog, Fussel-18, specifically expressed in neuronal tissues. Lab. Invest. 85, 1330–1341 [DOI] [PubMed] [Google Scholar]
- Arndt S., Poser I., Moser M., Bosserhoff A. K. (2007). Fussel-15, a novel Ski/Sno homolog protein, antagonizes BMP signaling. Mol. Cell. Neurosci. 34, 603–611 [DOI] [PubMed] [Google Scholar]
- Aso Y., Grübel K., Busch S., Friedrich A. B., Siwanowicz I., Tanimoto H. (2009). The mushroom body of adult Drosophila characterized by GAL4 drivers. J. Neurogenet. 23, 156–172 [DOI] [PubMed] [Google Scholar]
- Awasaki T., Saito M., Sone M., Suzuki E., Sakai R., Ito K., Hama C. (2000). The Drosophila trio plays an essential role in patterning of axons by regulating their directional extension. Neuron 26, 119–131 [DOI] [PubMed] [Google Scholar]
- Awasaki T., Huang Y., O’Connor M. B., Lee T. (2011). Glia instruct developmental neuronal remodeling through TGF-β signaling. Nat. Neurosci. 14, 821–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanik L., Mohrmann R., Ramaekers A., Bockaert J., Grau Y., Broadie K., Parmentier M. (2004). The drosophila metabotropic glutamate receptor DmGluRA regulates activity-dependent synaptic facilitation and fine synaptic morphology. J. Neurosci. 24, 9105–9116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulanger A., Clouet-Redt C., Farge M., Flandre A., Guignard T., Fernando C., Juge F., Dura J. M. (2011). ftz-f1 and Hr39 opposing roles on EcR expression during Drosophila mushroom body neuron remodeling. Nat. Neurosci. 14, 37–44 [DOI] [PubMed] [Google Scholar]
- Broadie K. S., Bate M. (1993). Development of the embryonic neuromuscular synapse of Drosophila melanogaster. J. Neurosci. 13, 144–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummel T., Abdollah S., Haerry T. E., Shimell M. J., Merriam J., Raftery L., Wrana J. L., O’Connor M. B. (1999). The Drosophila Activin receptor Baboon signals through dSmad2 and controls cell proliferation but not patterning during larval development. Genes Dev. 13, 98–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Graca L., Zimmerman K., Mitchell M., Kozhan-Gorodetska M., Sekiewicz K., Morales Y., Patterson G. (2003). Daf-5 is a Ski homolog that functions in a neuronal TGF-β pathway to regulate C. elegans dauer development. Development 131, 435–446 [DOI] [PubMed] [Google Scholar]
- Dai H., Chen Y., Chen S., Mao Q., Kennedy D., Landback P., Eyre-Walker A., Du W., Long M. (2008). The evolution of courtship behaviors through the origination of a new gene in Drosophila. Proc. Natl. Acad. Sci. USA 105, 7478–7483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis R. L. (1996). Physiology and biochemistry of Drosophila learning mutants. Physiol. Rev. 76, 299–317 [DOI] [PubMed] [Google Scholar]
- Doe C. Q. (1992). Molecular markers for identified neuroblasts and ganglion mother cells in the Drosophila central nervous system. Development 116, 855–863 [DOI] [PubMed] [Google Scholar]
- Doe C. Q., Chu-LaGraff Q., Wright D. M., Scott M. P. (1991). The prospero gene specifies cell fates in the drosophila central nervous system. Cell 65, 451–464 [DOI] [PubMed] [Google Scholar]
- Dupont S., Inui M., Newfeld S. J. (2012). Regulation of TGF-β signal transduction by mono- and deubiquitylation of Smads. FEBS Lett. 586, 1913–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukubo-Tokunaga K., Adachi Y., Kurusu M., Walldorf U. (2009). Brain patterning defects caused by mutations of the twin of eyeless gene in Drosophila melanogaster. Fly (Austin) 3, 263–269 [DOI] [PubMed] [Google Scholar]
- Gibbens Y. Y., Warren J. T., Gilbert L. I., O’Connor M. B. (2011). Neuroendocrine regulation of Drosophila metamorphosis requires TGFbeta/Activin signaling. Development 138, 2693–2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haerry T. E., Khalsa O., O’Connor M. B., Wharton K. A. (1998). Synergistic signaling by two BMP ligands through the SAX and TKV receptors controls wing growth and patterning in Drosophila. Development 125, 3977–3987 [DOI] [PubMed] [Google Scholar]
- Jahchan N. S., Luo K. (2010). SnoN in mammalian development, function and diseases. Curr. Opin. Pharmacol. 10, 670–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata M., Inoue H., Hanyu A., Imamura T., Miyazono K. (1998). Smad proteins exist as monomers in vivo and undergo homo- and hetero-oligomerization upon activation by serine/threonine kinase receptors. EMBO J. 17, 4056–4065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemlink D., Polo O., Frauscher B., Gschliesser V., Högl B., Poewe W., Vodicka P., Vavrova J., Sonka K., Nevsimalova S., et al. (2009). Replication of restless legs syndrome loci in three European populations. J. Med. Genet. 46, 315–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konikoff C. E., Wisotzkey R. G., Stinchfield M. J., Newfeld S. J. (2010). Distinct molecular evolutionary mechanisms underlie the functional diversification of the Wnt and TGFbeta signaling pathways. J. Mol. Evol. 70, 303–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosman D., Small S., Reinitz J. (1998). Rapid preparation of a panel of polyclonal antibodies to Drosophila segmentation proteins. Dev. Genes Evol. 208, 290–294 [DOI] [PubMed] [Google Scholar]
- Kronhamn J., Frei E., Daube M., Jiao R., Shi Y., Noll M., Rasmuson-Lestander A. (2002). Headless flies produced by mutations in the paralogous Pax6 genes eyeless and twin of eyeless. Development 129, 1015–1026 [DOI] [PubMed] [Google Scholar]
- Kurusu M., Maruyama Y., Adachi Y., Okabe M., Suzuki E., Furukubo-Tokunaga K. (2009). A conserved nuclear receptor, Tailless, is required for efficient proliferation and prolonged maintenance of mushroom body progenitors in the Drosophila brain. Dev. Biol. 326, 224–236 [DOI] [PubMed] [Google Scholar]
- Marquez R. M., Singer M. A., Takaesu N. T., Waldrip W. R., Kraytsberg Y., Newfeld S. J. (2001). Transgenic analysis of the Smad family of TGF-beta signal transducers in Drosophila melanogaster suggests new roles and new interactions between family members. Genetics 157, 1639–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini S. R., Roman G., Meuser S., Mardon G., Davis R. L. (2000). The retinal determination gene, dachshund, is required for mushroom body cell differentiation. Development 127, 2663–2672 [DOI] [PubMed] [Google Scholar]
- Minaki Y., Nakatani T., Mizuhara E., Inoue T., Ono Y. (2008). Identification of a novel transcriptional corepressor, Corl2, as a cerebellar Purkinje cell-selective marker. Gene Exp. Pat. 8, 418–423 [DOI] [PubMed] [Google Scholar]
- Miyata T., Ono Y., Okamoto M., Masaoka M., Sakakibara A., Kawaguchi A., Hashimoto M., Ogawa M. (2010). Migration, early axonogenesis, and Reelin-dependent layer-forming behavior of early/posterior-born Purkinje cells in the developing mouse lateral cerebellum. Neural Dev. 5, 23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuhara E., Nakatani T., Minaki Y., Sakamoto Y., Ono Y. (2005). Corl1, a novel neuronal lineage-specific transcriptional corepressor for the homeodomain transcription factor Lbx1. J. Biol. Chem. 280, 3645–3655 [DOI] [PubMed] [Google Scholar]
- Newfeld S. J., Gelbart W. M. (1995). Identification of two Drosophila TGF-β family members in the grasshopper Schistocerca americana. J. Mol. Evol. 41, 155–160 [DOI] [PubMed] [Google Scholar]
- Ng J. (2008). TGF-β signals regulate axonal development through distinct Smad-independent mechanisms. Development 135, 4025–4035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr H. T. (2010). Nuclear ataxias. Cold Spring Harb. Perspect. Biol. 2, a000786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks A. L., Cook K. R., Belvin M., Dompe N. A., Fawcett R., Huppert K., Tan L. R., Winter C. G., Bogart K. P., Deal J. E., et al. (2004). Systematic generation of high-resolution deletion coverage of the Drosophila melanogaster genome. Nat. Genet. 36, 288–292 [DOI] [PubMed] [Google Scholar]
- Quijano J. C., Stinchfield M. J., Zerlanko B., Gibbens Y. Y., Takaesu N. T., Hyman-Walsh C., Wotton D., Newfeld S. J. (2010). The Sno oncogene antagonizes Wingless signaling during wing development in Drosophila. PLoS ONE 5, e11619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santelli E., Leone M., Li C., Fukushima T., Preece N., Olson A., Ely K., Reed J., et al. (2005). Structural analysis of Siah1-Siah-interacting protein interactions and insights into the assembly of an E3 ligase multiprotein complex. J. Biol. Chem. 280, 34278–34287 [DOI] [PubMed] [Google Scholar]
- Sarker K., Wilson S., Bonni S. (2005). SnoN is a cell type-specific mediator of transforming growth factor-beta responses. J. Biol. Chem. 280, 13037–13046 [DOI] [PubMed] [Google Scholar]
- Sarker K. P., Kataoka H., Chan A., Netherton S. J., Pot I., Huynh M. A., Feng X., Bonni A., Riabowol K., Bonni S. (2008). ING2 as a novel mediator of transforming growth factor-beta-dependent responses in epithelial cells. J. Biol. Chem. 283, 13269–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu K., Sato M., Tabata T. (2011). The Wnt5/planar cell polarity pathway regulates axonal development of the drosophila mushroom body neuron. J. Neurosci. 31, 4944–4954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl G., Basler K. (1993). Organizing activity of wingless protein in Drosophila. Cell 72, 527–540 [DOI] [PubMed] [Google Scholar]
- Takaesu N. T., Herbig E., Zhitomersky D., O’Connor M. B., Newfeld S. J. (2005). DNA-binding domain mutations in SMAD genes yield dominant-negative proteins or a neomorphic protein that can activate WG target genes in Drosophila. Development 132, 4883–4894 [DOI] [PubMed] [Google Scholar]
- Takaesu N. T., Hyman-Walsh C., Ye Y., Wisotzkey R. G., Stinchfield M. J., O’connor M. B., Wotton D., Newfeld S. J. (2006). dSno facilitates baboon signaling in the Drosophila brain by switching the affinity of Medea away from Mad and toward dSmad2. Genetics 174, 1299–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truman J., Talbot W., Fahrbach S., Hogness D. (1994). Ecdysone receptor expression in the CNS correlates with stage-specific responses to ecdysteroids during Drosophila and Manduca development. Development 120, 219–234 [DOI] [PubMed] [Google Scholar]
- Wang B., Harrison W., Overbeek P. A., Zheng H. (2011). Transposon mutagenesis with coat color genotyping identifies an essential role for Skor2 in sonic hedgehog signaling and cerebellum development. Development 138, 4487–4497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X., Wang J., Haerry T. E., Wu A. Y., Martin J., O’Connor M. B., Lee C. H., Lee T. (2003). TGF-β signaling activates steroid hormone receptor expression during neuronal remodeling in the Drosophila brain. Cell 112, 303–315 [DOI] [PubMed] [Google Scholar]
- Zheng X., Zugates C., Lu Z., Shi L., Bai J., Lee T. (2006). Baboon/dSmad2 TGF-β signaling is required during late larval stage for development of adult-specific neurons. EMBO J. 25, 615–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.