

Glioma, the most common central nervous system cancer in adults, has poor prognosis. We identify a new glioma risk SNP, rs1920116 (near TERC), that reached genome-wide significance (Pcombined=8.3×10−9) in a meta-analysis of high-grade glioma GWAS and replication data (1644 cases, 7736 controls). This region has previously been associated with mean leukocyte telomere length (LTL). We therefore examined the relationship between LTL and both this new risk locus and other previously established glioma risk loci using data from a recent GWAS of LTL (N=37684)1. Glioma risk alleles near TERC and TERT were strongly associated with longer LTL (P=5.5×10−20 and 4.4×10−19, respectively). In contrast, glioma risk alleles near RTEL1 were inconsistently associated with LTL and suggested the presence of distinct causal alleles. No other glioma risk loci were associated with LTL. The identification of glioma risk alleles near TERC and TERT which also associate with telomere length implicates telomerase in gliomagenesis.
Glioblastoma, the most common and aggressive form of glioma, has a median survival time of just 15 months2. Both inherited and acquired genetic variation influence gliomagenesis. Previous glioma GWAS have discovered seven risk loci, including two near genes involved in telomere dynamics (TERT and RTEL1)3-7. Acquired mutations in the TERT promoter increase TERT expression and are observed in ~75% of glioblastomas8-10.
To identify novel glioma risk loci, we imputed thirty 500kb regions with at least one SNP having a P-value <1.0×10−4 in our previous high-grade glioma GWAS (online methods)6. The discovery phase meta-analysis combined the new imputation and existing genotyping array data from The University of California, San Francisco Adult Glioma Study (AGS)6,11 with new data from The Cancer Genome Atlas (TCGA)12 and the Wellcome Trust Case-Control Consortium (WTCCC)13 (1013 cases and 6595 controls). The replication phase used criteria defined in the original high-grade glioma GWAS, evaluating SNPs with P<1.0×10−6 in an additional 631 glioblastoma cases and 1141 controls from The Mayo Clinic and UCSF. All individuals were of European ancestry. Supplementary Figure 1 and Supplementary Table 1 summarize the study design and study populations.
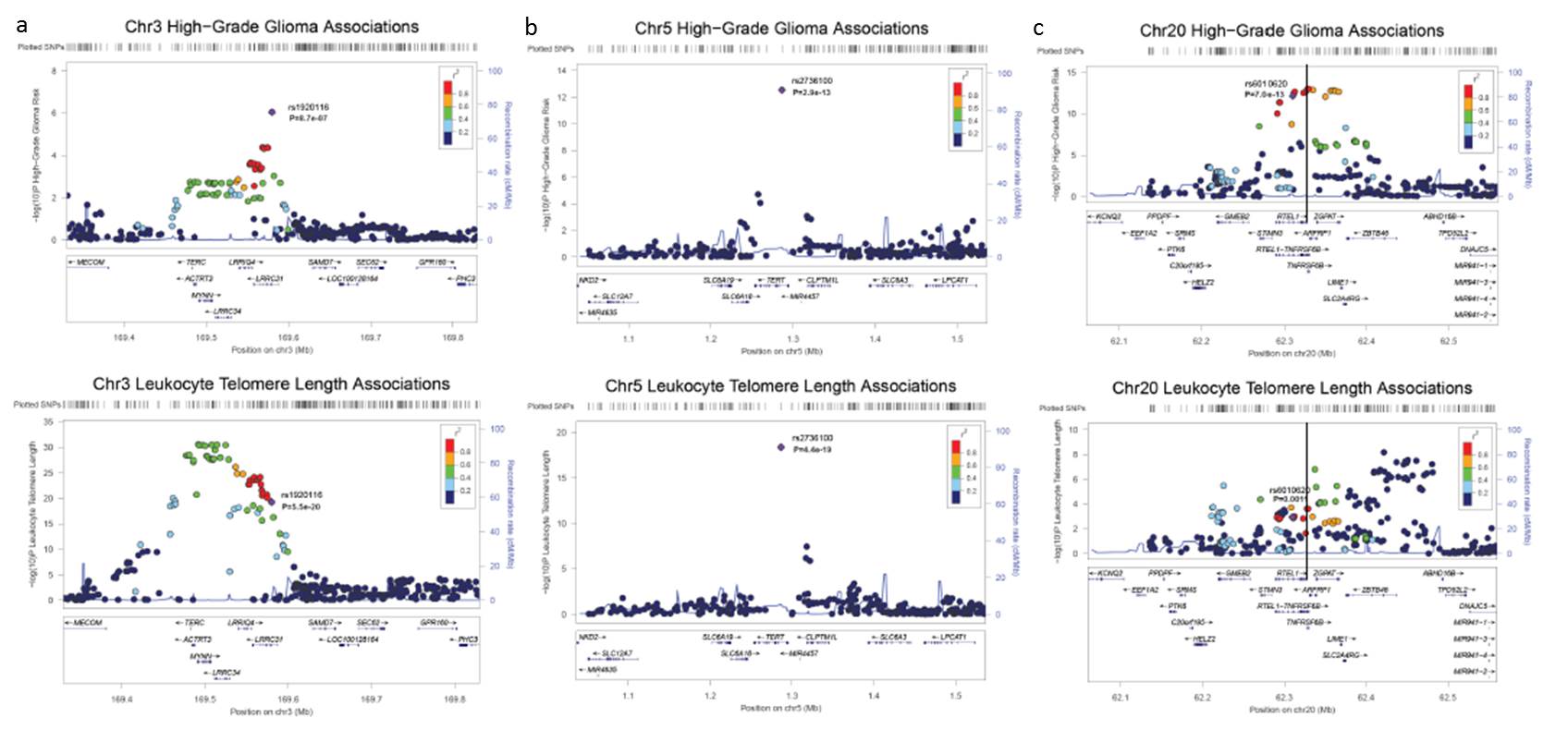
From the discovery phase, only rs1920116 on chromosome 3q26.2 was associated with glioma risk at P<1.0×10−6 (P=8.7×10−7). The association at rs1920116 was also significant in the replication dataset (P=3.4×10−3). The combined P-value for all 1644 cases and 7736 controls in the discovery and replication data (8.3×10−9) achieved genome-wide statistical significance (Table 1). Interestingly, the association peak for this novel glioma risk locus encompasses the telomerase RNA component gene, TERC (Figure 1a).
Table 1.
Associations of high-grade glioma risk with SNPs near TERC, TERT, and RTEL1 in the UCSF and Mayo Clinic Adult Glioma Studies, The Cancer Genome Atlas (TCGA) and The Wellcome Trust Consortium.
| SNP | Chr. | Position | Risk Allele | Gene | Study | Number cases / controls |
RAF cases / controls |
OR (95% CI) | P |
|---|---|---|---|---|---|---|---|---|---|
| rs1920116 | 3 | 169579971 | G | TERC | Discovery GWAS | 1013 / 6595 | 0.77 / 0.72 | 1.31 (1.18-1.46) | 8.7×10−7 |
| Replication | 631 / 1141 | 0.76 / 0.71 | 1.27 (1.09-1.51) | 3.4×10−3 | |||||
| Combined Total | 1644 / 7736 | 0.77 / 0.72 | 1.30 (1.19-1.42) | 8.3×10−9 | |||||
| rs2736100 | 5 | 1286516 | C | TERT | Discovery GWAS | 1013 / 6595 | 0.60 / 0.51 | 1.43 (1.30-1.57) | 2.9×10−13 |
| Replication | 605 / 1141 | 0.57 / 0.49 | 1.29 (1.12-1.49) | 5.8×10−4 | |||||
| Combined Total | 1618 / 7736 | 0.59 / 0.51 | 1.39 (1.28-1.50) | 1.4×10−15 | |||||
| rs6010620 | 20 | 62309839 | G | RTEL1 | Discovery GWAS | 1013 / 6595 | 0.84 / 0.76 | 1.53 (1.36-1.72) | 7.0×10−13 |
| Replication | 631 / 1141 | 0.83 / 0.75 | 1.64 (1.37-1.97) | 1.4×10−7 | |||||
| Combined Total | 1644 / 7736 | 0.84 / 0.76 | 1.56 (1.42-1.72) | 4.7×10−19 |
RAF: risk allele frequency
Figure 1. SNP association plots for high-grade glioma risk and mean leukocyte telomere length at 3q26.2 (TERC), 5p15.33 (TERT), and 20q13.33 (RTEL1).

Telomere associations (grey squares) are from the telomere length genome-wide association meta-analysis (N=37684). Glioma associations (red circles) are from the genome-wide discovery meta-analysis, combining data from the UCSF Adult Glioma Study, The Cancer Genome Atlas, and the Wellcome Trust Case-Control Consortium (N=1013 cases and 6595 controls). Red diamonds are from the combined glioma discovery and UCSF and Mayo Clinic replication analyses (1644 cases, 7736 controls).
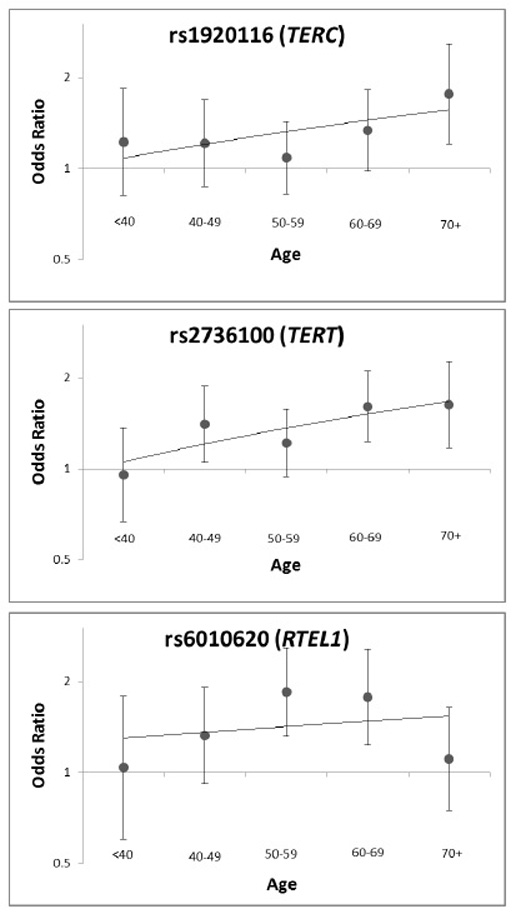
Table 1 and Figures 1b-1c highlight findings for the other telomere-related glioma risk loci, TERT and RTEL1, in these datasets. Interestingly, glioma risk alleles near TERC, TERT and RTEL1 have similar effect sizes and are the major allele in all three regions. SNPs in TERC, TERT and RTEL1 were independently associated with high-grade glioma risk; modeling the effect of one SNP while controlling for the presence of the others did not attenuate associations. SNPxSNP interaction tests did not reveal any effect modification (Pmin=0.11). Each additional risk allele in TERC, TERT and RTEL1 (range 0-6) contributed to glioma risk in a monotonic increasing fashion (Supplementary Figure 2). We previously observed that the effect of TERT and RTEL1 SNPs on glioma risk increases with age14. We observe modest evidence of this effect at rs1920116 near TERC (P=0.098) (Supplementary Figure 3).
To determine if glioma risk loci influence telomere length in an independent dataset, we examined SNP data from a recent GWAS of mean LTL conducted in 37,684 individuals of European descent1. LTL association data were available for lead glioma risk SNPs near TERC, TERT, EGFR, CCDC26, CDKN2B, PHLDB1, and RTEL13,4,6. The top glioma risk alleles near TERC and TERT were strongly associated with longer LTL (P=5.5×10−20 and 4.4×10−19, respectively) (Table 1). In contrast, the glioma risk allele at rs6010620 in RTEL1 was modestly associated with shorter LTL (P=1.1×10−3) (Table 1). Other known glioma risk loci were not associated with LTL (Supplementary Table 2). The lack of association with LTL at these other loci supports the hypothesis that multiple mechanisms contribute to glioma risk, of which telomere dynamics may be one15. The presence of genes causally implicated in carcinogenesis at glioma risk loci not associated with LTL supports this (e.g. EGFR, CDKN2B).
Next we comprehensively compared the direction and statistical significance of the associations in the TERC, TERT and RTEL1 regions with both high-grade glioma risk and mean LTL. For all SNPs, the glioma risk allele was set as the reference allele. We then evaluated whether the glioma risk allele was associated with longer or shorter LTL. Alleles in the TERC and TERT regions were consistently associated with both increased glioma risk and longer LTL (Figures 2a and 2b). In contrast, some glioma risk alleles near RTEL1 were associated with longer LTL and others with shorter LTL (Figure 2c). The LTL associations near RTEL1 have substantially more modest p-values than those observed near the telomerase components TERC and TERT.
Figure 2. SNP association plots indicating the –log10P for association with high-grade glioma risk (y-axis) and mean leukocyte telomere length (x-axis).

Colors correspond to whether the glioma risk allele is associated with longer (blue) or shorter (orange) leukocyte telomere length.
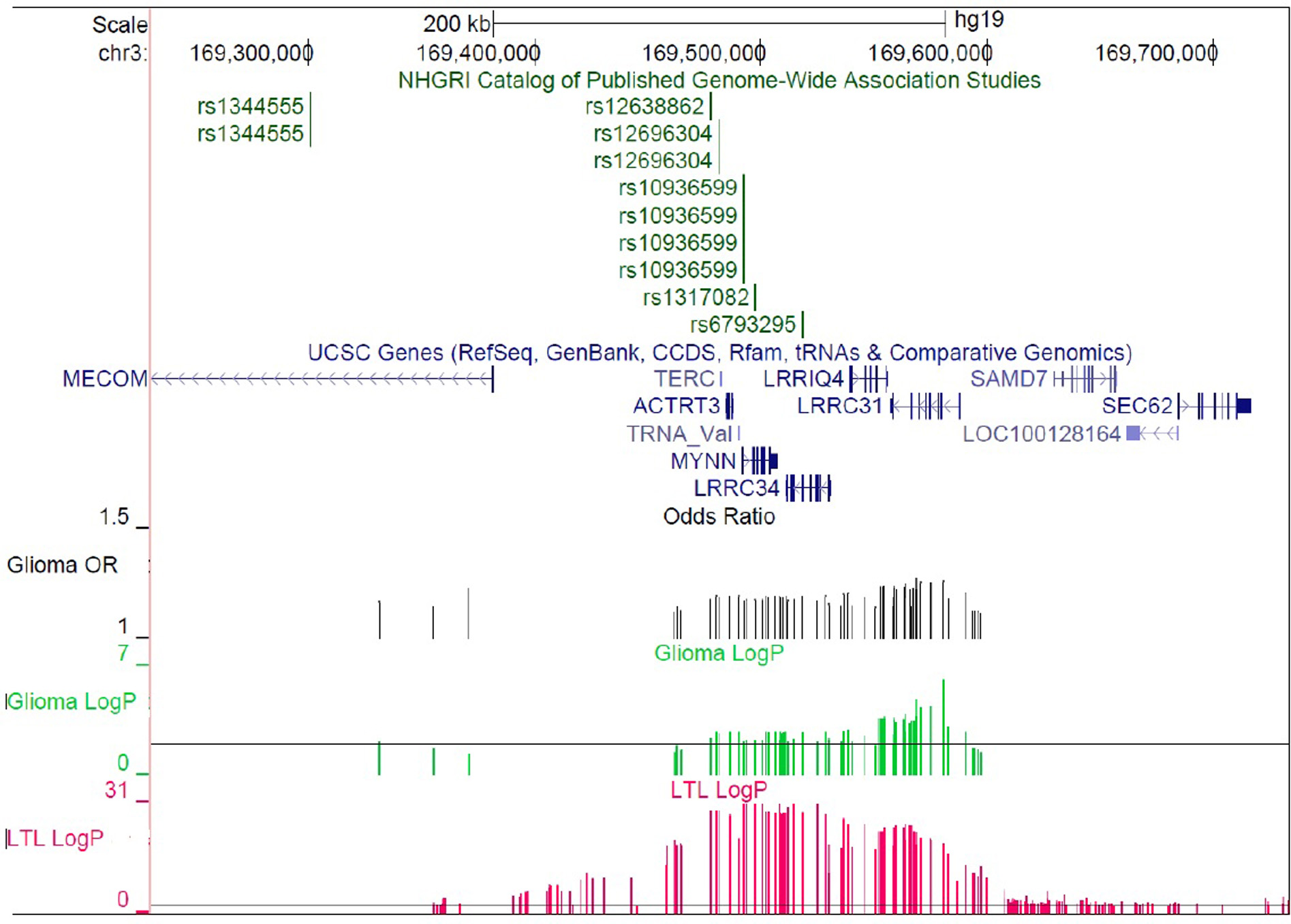
The glioma and LTL association peaks on 3q26.2, containing TERC and five additional genes, span a ~200kb region with low recombination rates and strong linkage disequilibrium (LD) (Supplementary Figure 4a). Although the association peak for LTL is located ~85kb centromeric to the glioma association peak, every 3q26.2 SNP which was associated with glioma at P<0.01 (N=54 SNPs) was also associated with increased LTL at P<1.0×10−5. Similarly, of 67 SNPs on 3q26.2 associated with increased LTL at P<1.0×10−10, 65 (97%) were associated with increased glioma risk at P<0.05. This included the top LTL association, rs10936599 (PGlioma=7.6×10−3; OR=1.17). Although distinct causal alleles may underlie the LTL and glioma association signals near TERC, such alleles likely reside on the same extended haplotype.
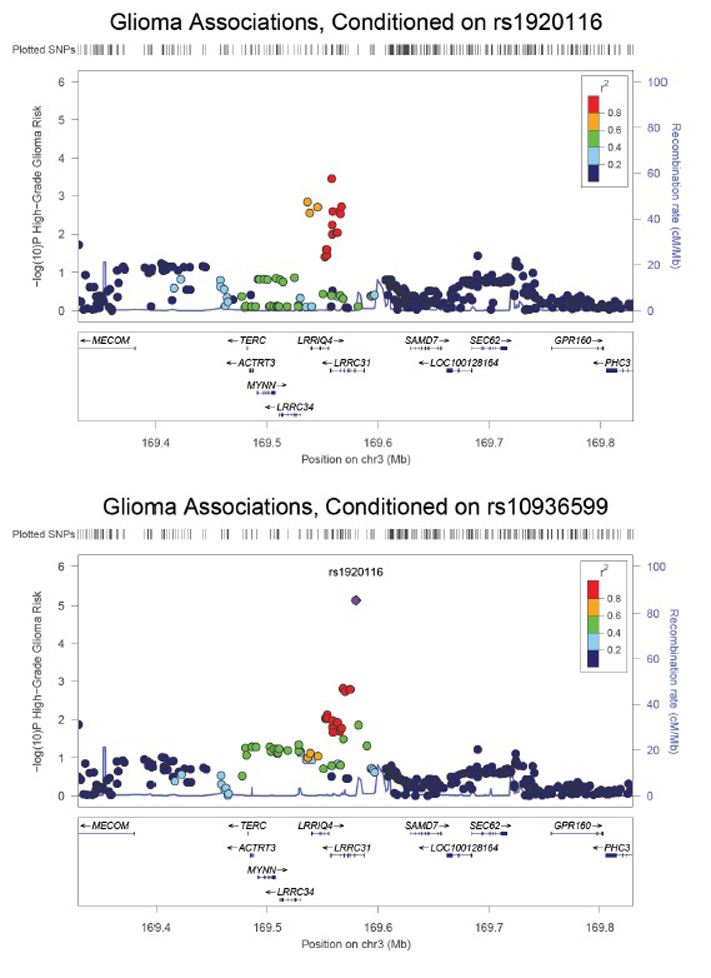
The odds ratios for high-grade glioma risk appear relatively uniform throughout the 3q26.2 region (Supplementary Figure 5), similar to observations of this region’s relationship to colorectal cancer16. Conditional analyses suggest that multiple 3q26.2 SNPs may influence glioma risk, as controlling for either the lead glioma SNP (rs1920116) or the lead LTL SNP (rs10936599) did not fully attenuate the association between glioma risk and nearby alleles (Supplementary Figure 6). Differences in the statistical significance of the LTL and glioma risk associations may also relate to differences in sample size and analytic technique (i.e. linear regression for LTL vs. logistic regression for glioma), or to the tissue type under study (i.e. leukocytes for LTL vs. astrocytes for glioma). Variants regulating TERC expression, and possible downstream effects on telomere length, may differ across tissues due to differential transcription factor expression or other tissue-divergent regulatory mechanisms. A full analysis of the potential functional consequences of 3q26.2 SNPs appears in Supplementary Table 3 and indicates that rs1920116 may have long-range effects on gene expression.
In the TERT region on 5p15.33, with high recombination rates and low LD (Supplementary Figure 4b), rs2736100 is the most statistically significant association for both glioma risk and LTL. A previous study found multiple independent TERT SNPs that influenced LTL and breast cancer risk17. Our data suggest that, in the case of glioma, TERT variants are simultaneously associated with increased glioma risk and longer LTL.
The glioma and LTL association peaks near RTEL1 display substantially less overlap than those near TERC and TERT. Indeed, the RTEL1 SNPs most significantly associated with glioma risk are not in LD with the top LTL SNPs, suggesting that different causal alleles influence the two phenotypes (Supplementary Figure 4c). This is particularly intriguing because the RTEL1 protein interacts with proliferating cell nuclear antigen (PCNA), a processivity factor for DNA polymerase18. The RTEL1-PCNA interaction is essential for replication fork stability and suppression of telomere fragility, but in the absence of this interaction, RTEL1 can still disassemble telomere loops and inhibit telomere shortening18. Thus, genetic variation that limits the RTEL1-PCNA interaction might influence glioma risk without directly influencing telomere length. The PCNA interaction motif (PIP box) is encoded by the 34th exon of RTEL1, near our most significant glioma associations.
Because telomere maintenance is a universal requirement of oncogenic progression and telomere length displays substantial interindividual variability19,20, telomere length is a promising epidemiologic risk factor for cancer. Mean telomere length decreases with age (declining 20–40 bp per year) and has been proposed as a biomarker of health23. Although shorter telomere length is causally linked to increased coronary artery disease1,24, both longer and shorter telomere length have been associated with increased cancer risk25-27.
Although data connecting telomere length in astrocytes with LTL is lacking, telomere length in individuals is highly correlated across tissues28,29. Recent research indicates that environmental insults alter telomere length and that the magnitude of these changes is linked to telomere length at baseline30. This correlation suggests that stress-induced telomere extension occurs through the same pathway as canonical telomere extension. Therefore, heritable variation may influence telomere dynamics in both normal and pre-malignant cells.
Another SNP near TERC is significantly associated with colorectal cancer risk (lead SNP: rs10936599)31. The colorectal cancer risk allele is also associated with increased glioma risk (OR=1.17, P=7.6×10−3) and longer telomeres (P=2.5×10−31) in our data. Idiopathic pulmonary fibrosis risk is significantly associated with SNPs near TERC (rs6793295) and TERT (rs2736100)32. For both SNPs, the pulmonary fibrosis risk allele is associated with significantly shorter telomere length (P=1.1×10−28 and 4.4×10−19, respectively) and with decreased glioma risk (P=2.1×10−3 and 2.9×10−13) in our data. These observations suggest that both longer and shorter telomere length may be pathogenic, depending on the disease under consideration.
We identify a novel high-grade glioma susceptibility locus near TERC, and demonstrate that the glioma risk alleles near TERC and TERT may also influence telomere length. Top glioma risk SNPs near TERC, TERT and RTEL1 all have risk allele frequencies greater than 50% among our control sample. Considering that increased telomere length protects against cardiovascular disease33, but may concurrently increase risk of some cancers, these alleles may be simultaneously influenced by both positive and negative selective pressures. This balancing selection underscores the complexity of telomere-based pathways of disease susceptibility.
METHODS
Ethics statement
Glioma studies were approved by the University of California, San Francisco Committee on Human Research and The Mayo Clinic Office for Human Research Protection. Informed consent was obtained from all study participants. The genome-wide meta-analysis of mean leukocyte telomere length (LTL) obtained approval by local ethics committees as previously outlined1.
Study design
The overall study design is summarized in Supplementary Figure 1. The study had four stages. Stage 1 and Stage 2 were part of a combined discovery phase, while Stage 3 involved replication and Stage 4 examined function of top-loci in an independent dataset with LTL measurements.
Stage 1 was a genome-wide association study of high-grade glioma cases (N=690) and controls (N=3992), results of which were first published in 20096. This GWAS analyzed UCSF Adult Glioma Study cases (N=620) and controls (N=602), TCGA cases genotyped on Illumina arrays (N=70), and Illumina iControls (N=3390). Results of this pooled analysis were used to generate a list of regions which had not undergone attempted replication in the 2009 publication (p > 1.0×10−6) but which showed suggestive associations (p<1×10−4). This yielded thirty independent loci containing at least 1 SNP with a p-value between 1.0×10−4 and 1.0×10−6 for additional study. Imputation to 1000 Genomes was performed for 500kb regions, centered on the most statistically significant SNP from each of these thirty regions (Supplementary Table 4). Eigenstrat-adjusted case-control associations were calculated for genotyped and imputed SNPs.
Stage 2 analyzed Affymetrix 6.0 SNP array data from new TCGA glioblastoma cases (N=323) and Wellcome Trust Case-Control Consortium controls (N=2603). Imputation to 1000 Genomes was performed for the thirty 500kb regions identified in Stage 1, and case-control comparisons were made for genotyped and imputed SNPs. Eigenstrat-adjusted case-control association results from Stage 1 and Stage 2 were combined using meta-analysis to identify SNPs meeting the original a priori threshold for attempted replication (p<1.0×10−6).
Stage 3 attempted replication of the single SNP from Stage 2 that had a p-value less than 1.0×10−6 (rs1920116 near TERC, p=8.7×10−7). Targeted genotyping of this SNP using either TaqMan or Sequenom assays was performed in an additional 328 Mayo Clinic glioblastoma cases, 303 UCSF glioblastoma cases, 766 Mayo Clinic controls and 375 UCSF controls. SNP data for these samples were also available for previously established high-grade glioma risk loci in TERT and RTEL1 which emerged from Stage 1 and were first published in 20096.
Stage 4 combined data from a GWAS meta-analysis of mean LTL (N=37,684), published in 20131, with our high-grade glioma association data to investigate the direction and statistical significance of associations between glioma risk alleles and mean LTL.
Study population
Characteristics of cases and controls from UCSF, The Mayo Clinic, Illumina, The TCGA and the Wellcome Trust Case-Control Consortium are summarized in Supplementary Table 1. All individuals were of European ancestry and were older than 18 years of age. UCSF cases and controls were taken from the San Francisco Bay Area Adult Glioma Study (AGS). A total of 923 high-grade glioma cases and 977 controls from the UCSF AGS were used in this study (Stages 1 and 3). A total of 328 glioblastoma cases and 766 controls from the Mayo Clinic were used in this study (Stage 3). A total of 37,684 individuals of European descent, aged >18 years, were included in the genome-wide analyses of mean LTL (Stage 4).
Subject recruitment
All UCSF AGS cases were adults (>18 years of age) with newly diagnosed histologically confirmed glioma (International Classification of Disease for Oncology, morphology codes 9380-9481). Population-based cases from six San Francisco Bay Area counties were ascertained using the Cancer Prevention Institute of California’s early case ascertainment system from May 1997 to August 1999 (Series 2), from November 2001 to September 2005 (Series 3), and from September 2006 to September 2009 (Series 4). Clinic-based cases diagnosed between 2002-2006 (Series 3), 2006-2010 (Series 4), and 2009-2012 (Series 5) of the same histologies were recruited from the UCSF Neuro-oncology Clinic, regardless of place of residence. From 1991-2010, population-based controls from the same residential area as the population-based cases were identified using random digit dialing and were frequency matched to population-based cases on age, gender and ethnicity. After 2010, all controls were selected from the UCSF general medicine phlebotomy clinic. Clinic-based controls were matched to clinic-based glioma cases on age, gender and ethnicity. Tumor specimens and pathology reports were reviewed by UCSF neuropathologists. Consenting participants provided blood, buccal and/or saliva specimens and information during an in-person or telephone interview.
The Mayo Clinic cases include individuals with newly diagnosed glioblastoma between 2005 and 2012. Cases were identified within 24 hours of diagnosis, except for those who had their initial diagnosis elsewhere, followed by verification at the Mayo Clinic. Pathologic diagnosis was confirmed by review of the primary surgical material for all cases by two Mayo Clinic neuropathologists. The control group consisted of consented individuals who had a general medical exam at the Mayo Clinic. Matching variables were sex, date of birth (within 2.5 years), self-identified race (Hispanic white, non-Hispanic white, American Indian, African American, Asian, Pacific Islander, Other) and residence. Geographic region of residence was matched in three zones based on the distance to the Mayo Clinic Rochester: Olmsted County; the rest of Minnesota, Wisconsin, Iowa, North Dakota and South Dakota; and the rest of the United States and Canada. Individuals under age 18 and those with a history of a brain tumor were excluded.
A total of 37,684 individuals from 15 cohorts were used in the GWAS meta-analysis of mean LTL. Samples were collected and analyzed by the ENCODE Telomere Consortium, comprised of European and Australian collaborating institutions. These institutions are distinct from those collecting the glioma case-control samples. Details of the ENCODE Telomere Consortium cohorts and subject recruitment procedures can be found in Supplementary Table 1 of Codd et al., 20131.
Measurement of telomere length
Telomere measurements were performed on blood-derived DNA. Assays were conducted at one of four centralized laboratories to ensure data harmonization. Mean LTL was measured using a quantitative PCR–based technique in all samples34,35. This method expresses telomere length as a ratio (T/S) of telomere repeat length (T) to copy number of a single copy gene (S), in each sample. To standardize across plates, either a calibrator sample or a standard curve was used for quantification. LTL measurements were made in five separate laboratories. Details for the methods used are provided elsewhere1. The majority of the samples (67% of the total) were run in a single laboratory with mean inter-run coefficients of variation for LTL measurements in individual cohorts ranging between 2.7% and 3.9%. Ranges in T/S ratios were found to vary between cohorts measured in different laboratories, largely owing to differences in the calibrator or standard DNA used. Standardized LTL in each cohort was generated using a Z-transformation approach.
Sample preparation and genotyping
Stage 1
For UCSF AGS discovery samples used in Stage 1 of the study, DNA was isolated from whole blood using Gentra Puregene DNA isolation kit (Qiagen) and quantified using Picogreen reagent (Invitrogen). Genotyping was conducted by deCODE Genetics. Samples were randomized before plating on specimen plates provided by deCODE Genetics. The genotyping assay panel used was Illumina’s HumanCNV370-Duo BeadChip. In addition to randomization of samples and the quality control measures provided by deCODE Genetics, we included two duplicate samples per plate and one CEPH24 trio (parents and child) per plate. A total of 51 duplicate samples were plated with average concordance >99%. DNA was re-extracted for any samples with call rates <98% and genotyped again. Samples with call rates <98% were excluded from analysis (N=1), as were samples with mismatched reported and genotyped sex (N=1). Although all subjects were of self-reported European-ancestry, one subject was removed because of not clustering with those of European ancestry in Eigenstrat analyses. The assay panel contained a total of 353,202 SNP probes, of which 326,506 biallelic SNPs had call rates >98% and Hardy-Weinberg equilibrium p-values >0.0001 (among controls), and were included in analyses.
We downloaded HumanHap550 platform genotyping data from blood specimen DNA for 72 glioblastoma cases from The Cancer Genome Atlas (TCGA) for use in Stage 1. Although 72 were identified as white, our analyses showed that one had non-European ancestry and another appeared to duplicate an AGS case, leaving 70 TCGA cases in Phase 1 analyses.
We assembled an independent control genotype dataset of 3,390 non-redundant European-ancestry controls from the Illumina iControlDB. The subjects are anonymous, with information only on age, sex and ethnicity. We checked for evidence of non-European ancestry and sample duplicates or related subjects (IBS > 1.6) among AGS samples, TCGA cases and Illumina iControls by performing multidimensional scaling (MDS) analysis on 20 bootstrap samples of 1,000 random autosomal biallelic SNPs. Following these quality assessment measures, we obtained a total of 3,390 European-ancestry Illumina iControls from three different panels with up to 306,154 autosomal SNPs overlapping the HumanHap370duo panel used for the AGS subjects. The iControls samples were genotyped on the Illumina HumanHap300 (n = 319 subjects), HumanHap550v1 (n = 1,519) and HumanHap550v3 (n = 1,552).
Stage 2
Genome-wide SNP data for 323 TCGA glioblastoma patients, not included in Stage 1 analyses, were downloaded from TCGA for use in Stage 2 analyses12. These data were generated following publication of the original UCSF AGS genome-wide association study (2009)6 and do not overlap with samples included in those analyses. Genotyping was performed using blood-derived DNA specimens, assayed on the Affymetrix 6.0 genotyping array. Control genotype data for 2603 European-ancestry control samples genotyped on the Affymetrix 6.0 were downloaded from the Wellcome Trust Case-Control Consortium13. We excluded subjects showing evidence of non-European ancestry, as well as duplicate samples and related subjects (IBS > 1.6) among the TCGA cases and WTCCC. We also used genome-wide SNP data to ensure there was no overlap between TCGA cases included in Stage 2 and those UCSF and TCGA cases included in Stage 1 analyses. SNPs with call rates <0.98 or HWE p-value <0.001 (in controls) were excluded to remove poorly genotyped SNPs.
Stage 3
DNAs for cases and controls were isolated from whole blood or saliva samples. UCSF samples were whole genome amplified as previously described36. UCSF and Mayo samples were genotyped using a TaqMan assay from Applied Biosystems (ABI: C_26010927_10). Cases and controls were randomized on 96-well plates, containing HapMap trios and 5 duplicate samples per plate. All trio genotypes displayed Mendelian consistency and duplicates showed genotype concordance. Genotypes for a small subset of UCSF samples were generated on a custom Sequnom panel (n=25 cases). All cluster plots were visually inspected. To exclude poorly genotyped SNPs, any SNP with a Hardy-Weinberg Equilibrium (HWE) P-value < 0.001 in controls, stratified by site, was removed from further analyses. The TERT and RTEL1 genotypes for samples in Stage 3 were generated on an Illumina GoldenGate custom genotyping array, as previously described11. No SNP failed HWE checks in Stage 3.
Stage 4
All discovery cohorts for the genome-wide meta-analysis of leukocyte telomere length had genotype information generated on a standard genotyping platform from Illumina or Affymetrix, and include imputed genotypes based on HapMapII CEU reference data. Detailed information about individual genotyping platforms is provided elsewhere1. All study-specific files underwent extensive quality control procedures before meta-analysis. All files were checked for completeness. Allele frequencies were checked for compliance with HapMap. In addition to study-specific quality control filters, SNP results of a study were included in the meta-analysis only if the imputation quality score was >0.5 and if the minor allele frequency was >1%. Only SNPs that were available in >50% of the total sample size over all studies were analyzed, resulting in a total of 2,362,330 SNPs in the meta-analysis.
Statistical analyses
Stage 1
Genome-wide SNP data for high-grade glioma cases and controls were analyzed using logistic regression, adjusted for the first two ancestry-informative principal components, using R and Eigenstrat. Single SNP association results were computed assuming an allelic additive model for 0, 1 or 2 copies of the minor allele (equivalent to a Cochran-Armitage test for trend).
Imputation was performed for 500kb regions centered on thirty top SNPs using the Impute2 v2.1.2 software and its standard Markov chain Monte Carlo algorithm and default settings for targeted imputation37. All 1,000 Genomes Phase I interim release haplotypes were provided as the imputation reference panel38. SNPs with imputation quality (info) scores less than 0.80 or posterior probabilities less than 0.90 were excluded to remove poorly imputed SNPs. Any SNP with a minor allele frequency <1% in case subjects was excluded from association tests. Association statistics for imputed and directly genotyped SNPs were calculated using logistic regression in SNPTESTv2, using an allelic additive model39. A missing-data likelihood score-test was applied to the imputed variants to produce standard errors which account for the additional uncertainty inherent in the analysis of imputed genotypes. The effect of individual SNPs on glioma risk was calculated while adjusting for the first 2 principal components from Eigenstrat40.
Stage 2
As in Stage 1, thirty 500kb regions were imputed using the Impute2 v2.1.2 software and the 1,000 Genomes Phase I reference panel. SNPs with imputation quality (info) scores less than 0.80 or posterior probabilities less than 0.90 were excluded to remove poorly imputed SNPs. Association statistics for imputed and directly genotyped SNPs were calculated using logistic regression in SNPTEST v2, using an allelic additive model. SNP association statistics were adjusted for the first two principal components, generated by Eigenstrat. SNP association statistics from Stage 1 and Stage 2 analyses were combined using the program META41. As a measure for between study heterogeneity, I2 was calculated42. For SNPs with I2 ≤ 40%, fixed-effects models were applied; random-effects models were applied for SNPs with I2 > 40%.
Stage 3
Any SNP with a p-value in the Stage 2 meta-analysis of thirty regions surpassing our a priori significance threshold of 1×10−6 was selected for replication. For the single SNP that met this threshold, replication was attempted in 328 glioblastoma cases and 766 controls from The Mayo Clinic and 303 cases and 375 controls from UCSF. UCSF cases included in Stage 3 were not included in the original Stage 1 GWAS due to date of recruitment or insufficient DNA quantity. Case-control associations were evaluated using logistic regression, assuming an allelic additive model. Associations were calculated separately for each site (Mayo and UCSF) and then combined using fixed effects meta-analysis with the META software41. A summary meta-analysis of all high-grade glioma cases and controls was calculated by combining results from Stage 1, Stage 2 and the two replication series from Stage 3 using fixed effects meta-analysis with the META software. SNPs showing association with high-grade glioma risk with P < 5.0×10−8, corresponding to a Bonferroni correction of one million independent tests, were considered to be have achieved genome-wide statistical significance by current convention43.
Stage 4
In the GWAS of LTL, SNP array data from each of fifteen cohorts underwent genome-wide SNP imputation using HapMapII CEU as the reference, as previously described1. Within each cohort, SNP associations with LTL were analyzed by linear regression assuming additive effects, with adjustment for age, sex and study-specific covariates where appropriate (e.g. smoking). This ensured that all regression analyses were done uniformly, but permitted studies to adjust for additional relevant site-specific covariates (e.g. ancestry-related principal components). Standard errors of each study were corrected using study-specific lambda estimates for the genomic control procedure. Meta-analysis of all individual study associations was conducted using inverse variance weighting in STATA. As a measure of between study heterogeneity, I2 was calculated42. For SNPs with I2 ≤ 40%, fixed-effects models were applied; random-effects models were applied for SNPs with I2 > 40%. The overall inflation factor lambda of the meta-analyzed results was 1.007, and results were further corrected for this using the genomic control procedure.
The glioma and LTL SNP datasets were harmonized by mapping all positions to Build37/hg19 and setting the allele which increased glioma risk as the reference allele in association tests of both traits. As a result, all odds ratios for associations between SNPs and high-grade glioma are >1.0. Similarly, associations between SNPs and LTL are calculated with the glioma risk allele as the reference. A positive Beta value from the linear regression of mean LTL indicates that the glioma risk allele is associated with increased telomere length. Likewise, a negative Beta value from the linear regression of mean LTL indicates that the glioma risk allele is associated with decreased telomere length. Glioma risk loci showing association with LTL at a p-value <6.25×10−3 (i.e. 0.05/8 loci) were considered to be statistically significant.
Testing the independence of glioma risk loci and for the presence of interaction
We tested for evidence of gene-gene interaction for top hit SNPs in the telomere-associated genes TERC, TERT and RTEL1. The associations of rs1920116, rs2736100 and rs6010620 with high-grade glioma risk were modeled jointly to assess whether the observed associations were independent or possibly synergistic in nature. First, all three SNPs were included in a single logistic regression model to determine if controlling for glioma risk variants in related genes attenuated any of the SNP associations. Inclusion of an allelic additive interaction term in the model (rs1920116*rs2736100, rs1920116*rs6010620, rs2736100*rs6010620) was performed to test for the presence of any significant effect modification (p<0.05). Combining the risk allele dosage into a single ordinal variable representing the total number of risk SNPs in a telomere-associated gene (range 0-6) was done to determine if the alleles contributed to glioma risk in a monotonic increasing fashion (Supplementary Figure 2). To test whether age modified the effect of these SNPs on glioma risk, an allelic additive interaction term was included in the model (e.g. rs1920116*age). Additionally, odds ratios for glioma were calculated in case-control analyses stratified into 10-year intervals of age, as previously described (Supplementary Figure 3)14.
Genomic annotation of SNPs
Fifty-four SNPs on 3q26.2 that had p-values <0.01 in the glioma discovery dataset were annotated for their potential to regulate nearby genes. We used HaploReg44 and RegulomeDB45 to examine whether any of these SNPs are located in putative promoters, enhancers, DNase hypersensitivity sites or transcription factor binding sites. Sequence conservation was assessed using Genomic Evolutionary Rate Profiling (GERP)46. Associations between SNP genotype and mRNA expression levels were assessed with Spearman’s rho, analyzed using the Stranger, et al. (2012) dataset47 and tools available in Genevar 3.3.048. Analyses were restricted to Caucasian lymphoblastoid samples, and a p-value threshold of 0.05 was used.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 2.
Association of SNPs near TERC, TERT, and RTEL1 with mean leukocyte telomere length in the UK genome-wide association meta-analysis.
| SNP | Chr. | Position | Alleleb | Gene | Number | AF | Beta (SE)b | P |
|---|---|---|---|---|---|---|---|---|
| rs1920116 | 3 | 169579971 | G | TERC | 37,489 | 0.71 | 0.0733 (0.0080) | 5.5×10−20 |
| rs2736100 | 5 | 1286516 | C | TERT | 25,842 | 0.49 | 0.0783 (0.0087) | 4.4×10−19 |
| rs6010620 | 20 | 62309839 | G | RTEL1 | 37,607 | 0.77 | −0.0278 (0.0085) | 1.1×10−3 |
Allele listed is the risk allele for high-grade glioma.
Beta estimates are from regressing mean leukocyte telomere length on each additional copy of the glioma risk allele. Negative Beta values indicate that the glioma risk allele is associated with shorter leukocyte telomere length, while positive Beta values indicate that the glioma risk allele is associated with longer leukocyte telomere length.
AF: allele frequency
ACKNOWLEDGMENTS
Work at University of California, San Francisco was supported by the National Institutes of Health (grant numbers R25CA112355, R01CA52689, P50CA097257, R01CA126831 and R01CA139020), as well as the National Brain Tumor Foundation, the UCSF Lewis Chair in Brain Tumor Research, the UCSF Robert Magnin Newman chair in Neuro-Oncology, and by donations from families and friends of John Berardi, Helen Glaser, Elvera Olsen, Raymond E. Cooper, and William Martinusen. Work at the Mayo Clinic was supported by the National Institutes of Health (grant numbers P50CA108961 and P30 CA15083), the National Institute of Neurological Disorders and Stroke (grant number RC1NS068222Z), the Bernie and Edith Waterman Foundation, and the Ting Tsung and Wei Fong Chao Family Foundation. Work at University of Leicester was undertaken under the framework of European Union Framework 7 ENGAGE Project (HEALTH-F4-2007-201413). VC and NJS are supported by the British Heart Foundation.
This project was supported by the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health, through UCSF-CTSI Grant Number UL1 RR024131. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
The collection of cancer incidence data used in this study was supported by the California Department of Public Health as part of the statewide cancer reporting program mandated by California Health and Safety Code Section 103885; the National Cancer Institute’s Surveillance, Epidemiology and End Results Program under contract HHSN261201000140C awarded to the Cancer Prevention Institute of California, contract HHSN261201000035C awarded to the University of Southern California, and contract HHSN261201000034C awarded to the Public Health Institute; and the Centers for Disease Control and Prevention’s National Program of Cancer Registries, under agreement # U58DP003862-01 awarded to the California Department of Public Health. The ideas and opinions expressed herein are those of the author(s) and endorsement by the State of California Department of Public Health, the National Cancer Institute, and the Centers for Disease Control and Prevention or their Contractors and Subcontractors is not intended nor should be inferred.
The results published here are in part based upon data generated by The Cancer Genome Atlas managed by the NCI and NHGRI. Information about TCGA can be found at http://cancergenome.nih.gov. This study makes use of data generated by the Wellcome Trust Case-Control Consortium. A full list of the investigators who contributed to the generation of the data is available from www.wtccc.org.uk. Funding for the project was provided by the Wellcome Trust under award 076113 and 085475.
Footnotes
CONFLICT OF INTEREST STATEMENT: The authors do not have any conflicts of interest, financial or otherwise.
AUTHOR CONTRIBUTIONS: K.M.W., M.R.W. and J.K.W. led the study at UCSF, R.B.J. led the study at The Mayo Clinic, and N.J.S led the study at the University of Leicester. K.M.W., V.C., R.B.J., M.R.W., M.P., and T.R. contributed to manuscript preparation. Study coordination was the responsibility of T.K. at Mayo Clinic and T.R. and L.S.M. at UCSF. K.M.W. and V.C. co-directed and conducted biostatistics and bioinformatic analyses with additional support from P.A.D., J.E.E.-P., M.L.K., A.M., P.M.B., T.R., H.S., A.R.P., I.V.S., P.vdH. and the ENGAGE Consortium Telomere Group. Laboratory work was performed by T.K., under the direction of R.B.J. at Mayo Clinic and by H.M.H., S.Z., and B.C. under the direction of J.K.W. and J.L.W. at UCSF. Pathology support was provided by T.T. Subject enrollment or clinical record review was performed or facilitated by M.D.P., S.M.C., M.S.B., B.P.O., and D.H.L. and P.vdH.
REFERENCES
- 1.Codd V, et al. Identification of seven loci affecting mean telomere length and their association with disease. Nat Genet. 2013;45:422–7. 427e1–2. doi: 10.1038/ng.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stupp R, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–66. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 3.Sanson M, et al. Chromosome 7p11. EGFR) variation influences glioma risk. Hum Mol Genet. 2011;20:2897–904. doi: 10.1093/hmg/ddr192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shete S, et al. Genome-wide association study identifies five susceptibility loci for glioma. Nat Genet. 2009;41:899–904. doi: 10.1038/ng.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stacey SN, et al. A germline variant in the TP53 polyadenylation signal confers cancer susceptibility. Nat Genet. 2011;43:1098–103. doi: 10.1038/ng.926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wrensch M, et al. Variants in the CDKN2B and RTEL1 regions are associated with high-grade glioma susceptibility. Nat Genet. 2009;41:905–8. doi: 10.1038/ng.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jenkins RB, et al. A low-frequency variant at 8q24.21 is strongly associated with risk of oligodendroglial tumors and astrocytomas with IDH1 or IDH2 mutation. Nat Genet. 2012;44:1122–5. doi: 10.1038/ng.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Killela PJ, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013;110:6021–6. doi: 10.1073/pnas.1303607110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horn S, et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339:959–61. doi: 10.1126/science.1230062. [DOI] [PubMed] [Google Scholar]
- 10.Huang FW, et al. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339:957–9. doi: 10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walsh KM, et al. Analysis of 60 reported glioma risk SNPs replicates published GWAS findings but fails to replicate associations from published candidate-gene studies. Genet Epidemiol. 2013;37:222–8. doi: 10.1002/gepi.21707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walsh KM, et al. Genetic variants in telomerase-related genes are associated with an older age at diagnosis in glioma patients: evidence for distinct pathways of gliomagenesis. Neuro Oncol. 2013;15:1041–7. doi: 10.1093/neuonc/not051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Melin BS, Nordfjall K, Andersson U, Roos G. hTERT cancer risk genotypes are associated with telomere length. Genet Epidemiol. 2012;36:368–72. doi: 10.1002/gepi.21630. [DOI] [PubMed] [Google Scholar]
- 16.Jones AM, et al. TERC polymorphisms are associated both with susceptibility to colorectal cancer and with longer telomeres. Gut. 2012;61:248–54. doi: 10.1136/gut.2011.239772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bojesen SE, et al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat Genet. 2013;45:371–84. 384e1–2. doi: 10.1038/ng.2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vannier JB, et al. RTEL1 is a replisome-associated helicase that promotes telomere and genome-wide replication. Science. 2013;342:239–42. doi: 10.1126/science.1241779. [DOI] [PubMed] [Google Scholar]
- 19.Chang S, Khoo CM, Naylor ML, Maser RS, DePinho RA. Telomere-based crisis: functional differences between telomerase activation and ALT in tumor progression. Genes Dev. 2003;17:88–100. doi: 10.1101/gad.1029903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vasa-Nicotera M, et al. Mapping of a major locus that determines telomere length in humans. Am J Hum Genet. 2005;76:147–51. doi: 10.1086/426734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fitzpatrick AL, et al. Leukocyte telomere length and cardiovascular disease in the cardiovascular health study. Am J Epidemiol. 2007;165:14–21. doi: 10.1093/aje/kwj346. [DOI] [PubMed] [Google Scholar]
- 22.Codd V, et al. Common variants near TERC are associated with mean telomere length. Nat Genet. 2010;42:197–9. doi: 10.1038/ng.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Denham J, et al. Longer leukocyte telomeres are associated with ultra-endurance exercise independent of cardiovascular risk factors. PLoS One. 2013;8:e69377. doi: 10.1371/journal.pone.0069377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brouilette SW, et al. Telomere length, risk of coronary heart disease, and statin treatment in the West of Scotland Primary Prevention Study: a nested case-control study. Lancet. 2007;369:107–14. doi: 10.1016/S0140-6736(07)60071-3. [DOI] [PubMed] [Google Scholar]
- 25.Ma H, et al. Shortened telomere length is associated with increased risk of cancer: a meta-analysis. PLoS One. 2011;6:e20466. doi: 10.1371/journal.pone.0020466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wentzensen IM, Mirabello L, Pfeiffer RM, Savage SA. The association of telomere length and cancer: a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2011;20:1238–50. doi: 10.1158/1055-9965.EPI-11-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hou L, Zhang X, Gawron AJ, Liu J. Surrogate tissue telomere length and cancer risk: shorter or longer? Cancer Lett. 2012;319:130–5. doi: 10.1016/j.canlet.2012.01.028. [DOI] [PubMed] [Google Scholar]
- 28.Wilson WR, et al. Blood leucocyte telomere DNA content predicts vascular telomere DNA content in humans with and without vascular disease. Eur Heart J. 2008;29:2689–94. doi: 10.1093/eurheartj/ehn386. [DOI] [PubMed] [Google Scholar]
- 29.Okuda K, et al. Telomere length in the newborn. Pediatr Res. 2002;52:377–81. doi: 10.1203/00006450-200209000-00012. [DOI] [PubMed] [Google Scholar]
- 30.Romano GH, et al. Environmental stresses disrupt telomere length homeostasis. PLoS Genet. 2013;9:e1003721. doi: 10.1371/journal.pgen.1003721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Houlston RS, et al. Meta-analysis of three genome-wide association studies identifies susceptibility loci for colorectal cancer at 1q41, 3q26.2, 12q13.13 and 20q13.33. Nat Genet. 2010;42:973–7. doi: 10.1038/ng.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fingerlin TE, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. 2013;45:613–20. doi: 10.1038/ng.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Samani NJ, van der Harst P. Biological ageing and cardiovascular disease. Heart. 2008;94:537–9. doi: 10.1136/hrt.2007.136010. [DOI] [PubMed] [Google Scholar]
- 34.Cawthon RM. Telomere length measurement by a novel monochrome multiplex quantitative PCR method. Nucleic Acids Res. 2009;37:e21. doi: 10.1093/nar/gkn1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cawthon RM. Telomere measurement by quantitative PCR. Nucleic Acids Res. 2002;30:e47. doi: 10.1093/nar/30.10.e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hansen HM, Wiemels JL, Wrensch M, Wiencke JK. DNA quantification of whole genome amplified samples for genotyping on a multiplexed bead array platform. Cancer Epidemiol Biomarkers Prev. 2007;16:1686–90. doi: 10.1158/1055-9965.EPI-06-1024. [DOI] [PubMed] [Google Scholar]
- 37.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abecasis GR, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marchini J, Howie B. Genotype imputation for genome-wide association studies. Nat Rev Genet. 2010;11:499–511. doi: 10.1038/nrg2796. [DOI] [PubMed] [Google Scholar]
- 40.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 41.Liu JZ, et al. Meta-analysis and imputation refines the association of 15q25 with smoking quantity. Nat Genet. 2010;42:436–40. doi: 10.1038/ng.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ. 2003;327:557–60. doi: 10.1136/bmj.327.7414.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pe’er I, Yelensky R, Altshuler D, Daly MJ. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet Epidemiol. 2008;32:381–5. doi: 10.1002/gepi.20303. [DOI] [PubMed] [Google Scholar]
- 44.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40:D930–4. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boyle AP, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–7. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cooper GM, et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–13. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stranger BE, et al. Patterns of cis regulatory variation in diverse human populations. PLoS Genet. 2012;8:e1002639. doi: 10.1371/journal.pgen.1002639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang TP, et al. Genevar: a database and Java application for the analysis and visualization of SNP-gene associations in eQTL studies. Bioinformatics. 2010;26:2474–6. doi: 10.1093/bioinformatics/btq452. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.