

Summary
The endoplasmic reticulum (ER) forms a network of sheets and tubules that extends throughout the cell. Proteins required to maintain this complex structure include the reticulons, reticulon-like proteins, and dynamin-like GTPases called atlastins in mammals and Sey1p in Saccharomyces cerevisiae. Yeast cells missing these proteins have abnormal ER structure, particularly defects in the formation of ER tubules, but grow about as well as wild-type cells. We screened for mutations that cause cells that have defects in maintaining ER tubules to grow poorly. Among the genes we found were members of the ER mitochondria encounter structure (ERMES) complex that tethers the ER and mitochondria. Close contacts between the ER and mitochondria are thought to be sites where lipids are moved from the ER to mitochondria, a process that is required for mitochondrial membrane biogenesis. We show that ER to mitochondria phospholipid transfer slows significantly in cells missing both ER-shaping proteins and the ERMES complex. These cells also have altered steady-state levels of phospholipids. We found that the defect in ER to mitochondria phospholipid transfer in a strain missing ER-shaping proteins and a component of the ERMES complex was corrected by expression of a protein that artificially tethers the ER and mitochondria. Our findings indicate that ER-shaping proteins play a role in maintaining functional contacts between the ER and mitochondria and suggest that the shape of the ER at ER–mitochondria contact sites affects lipid exchange between these organelles.
Key words: Endoplasmic reticulum, Mitochondria, Lipid trafficking, Membrane contact sites, Reticulons, ERMES complex, Sey1p
Introduction
The endoplasmic reticulum (ER) forms an elaborate, dynamic network of sheets and tubules that extends throughout the cytoplasm. The complex structure of the ER is determined by a number of factors (Shibata et al., 2009). One is the tethering of the ER to the cytoskeleton. In mammalian cells, microtubules play a role in ER biogenesis (Terasaki et al., 1986; Waterman-Storer and Salmon, 1998). Some new ER tubules are pulled out from the ER by extending microtubules. ER tubules have also been observed to slide along microtubules. Actin filaments appear to play a similar role in plants and yeasts (Kachar and Reese, 1988; Prinz et al., 2000). A number of proteins that maintain the structure of the ER have also been identified. The reticulons are a large family of highly abundant ER shaping proteins that are ubiquitously expressed in all eukaryotic cells. The yeast S. cerevisiae expresses two reticulons, Rtn1p and Rtn2p, and the reticulon-like protein Yop1p. A mutant missing all three proteins has dramatic changes in ER structure including a significant decrease in the number of ER tubules (Voeltz et al., 2006; West et al., 2011). It has also been shown that the reticulon Rtn1p and Yop1p can directly tubulate membranes in vitro (Hu et al., 2008). Another family of proteins that play a role in maintaining ER shape are called atlastins; these are dynamin-like GTPases proteins that mediate ER–ER fusion (Hu et al., 2009; Orso et al., 2009). Knock down of atlastins cause the formation of long unbranched ER tubules in mammalian cells. Sey1p is the functional ortholog of the atlastins in yeast. Cells missing Sey1p and either Rtn1p or Yop1p have a reduced number of ER tubules, although cells missing only Rtn1p or Yop1p do not, indicating that Sey1p plays a role in maintaining ER tubules (Hu et al., 2009). Like atlastins, Sey1p also mediates homotypic ER fusion (Anwar et al., 2012).
The ER has been found to make close contact with other organelles (Toulmay and Prinz, 2011; Elbaz and Schuldiner, 2011). At these regions, often called membrane contact sites (MCSs), the ER membrane and a second organelle are closely apposed, typically within about 20 nm of one another. MCSs are thought to be zones where signals and small molecules such as lipids and calcium are exchanged between organelles. Close contacts between the ER and mitochondria have been proposed to play a critical role in mitochondria membrane biogenesis, which requires the import of phospholipids into mitochondria. At ER–mitochondria MCSs, phospholipids are exchanged between these organelles by unknown mechanisms (Voelker, 2009; Prinz, 2010). The transport of phosphatidylserine (PS) from the ER to mitochondria has been most heavily studied. PS is synthesized in the ER and can be converted to phosphatidylethanolamine (PE) by the enzyme PE decarboxylase (Psd), which resides in the mitochondrial inner membrane. Therefore the conversion of PS to PE can be used to estimate the transport of PS from the ER to mitochondria. Work in mammalian cells first suggested that ER to mitochondria PS transport occurs at regions of close contact between these organelles (Voelker, 1985; Vance, 1990; Voelker, 1989). MCSs were also found to play a role in PS transport to mitochondria in yeast, even though PS biosynthesis differs between mammals and yeast (Simbeni et al., 1991).
How ER–mitochondrial contacts are maintained is not well understood. A number of proteins have been proposed to maintain these contacts in mammalian cells (Toulmay and Prinz, 2011; Elbaz and Schuldiner, 2011). Whether similar complexes exist in yeast is not known. In S. cerevisiae, the ER mitochondria encounter structure (ERMES) complex plays a role in maintaining functional contacts between these organelles (Kornmann et al., 2009). This complex contains four proteins: Mmm1p in the ER membrane, Mdm34p and Mdm10p in the mitochondrial outer membrane (OMM), and Mdm12p, a soluble protein. All four proteins are needed for complex formation; in cells missing any one of the ERMES proteins, the remaining proteins fail to localize to MCSs. Interestingly, green fluorescent protein (GFP) fusions to ERMES components localize to about 1–10 punctae per cell, suggesting that either there are merely a few ER–mitochondria junctions per cell, or that the ERMES complex is only at some of these junctions. Consistent with a role for ERMES proteins in maintaining ER–mitochondrial contacts, phospholipids exchange between the ER and mitochondria was found to slow in cells missing any one of the ERMES proteins (Kornmann et al., 2009). A role for the ERMES complex in mitochondrial lipid homeostasis is also suggested by the genetic interaction of genes encoding ERMES proteins and those required for cardiolipin (CL) biosynthesis (Kornmann et al., 2011), which occurs in mitochondria. However, the role of ERMES in lipid exchange between the ER and mitochondria remains unclear; a recent study found little or no decrease in the transfer of phosphatidylserine (PS) in cells missing ERMES proteins (Nguyen et al., 2012).
The goal of this study was to better understand the functions of ER-tubulating proteins. Surprisingly, S. cerevisiae mutants missing these proteins grow about as well as wild-type cells. A strain lacking Rtn1p, Rtn2p, and Yop1p has only a slight growth defect and no defect in vesicular trafficking from the ER was detected (Voeltz et al., 2006). More recently, we constructed a strain lacking these proteins and Sey1p and found that it also grew about as well as wild-type cells (C. V., unpublished observation). To better understand the role of ER morphology in cell physiology, we screened for mutations that cause cells missing ER-shaping proteins to grow poorly. We found that cells missing ERMES proteins and ER-shaping proteins have severe growth defects, a decreased rate of phospholipid transfer from the ER to mitochondria, and an altered mitochondrial phospholipids composition. These results suggest an unexpected role of ER-tubulating proteins in maintaining functional ER–mitochondria contact sites.
Results
In order to investigate the functions of reticulon and reticulon-like proteins, we screened for mutations that cause cells missing these proteins to grow poorly. S. cerevisiae has two reticulons, Rtn1p, Rtn2p, and one reticulon-like protein, Yop1p, and cells missing all three are viable but have abnormal ER morphology (Voeltz et al., 2006). Cells missing only Rtn1p and Yop1p also have defects in ER morphology, probably because Rtn2p is less abundant than the other two proteins (Voeltz et al., 2006). Therefore we used a strain missing Rtn1p and Yop1p for synthetic genetic array analysis (SGA). This technique entails the systematic creation of triple mutants missing RTN1, YOP1, and one of the ∼5000 nonessential genes in S. cerevisiae. The ability of the resulting strains to grow on synthetic complete (SC) medium was determined and the results are shown in supplementary material Table S1. Among the strains that had substantial growth defects were those lacking MDM34 and MDM12, which encode two of the four components of the ERMES complex.
To confirm this finding, we constructed strains lacking Rtn1p, Yop1p, and either Mdm10p, Mdm12p, Mdm34p, or Mmm1p. All four strains had substantial growth defects compared to wild-type cells, those missing Rtn1p and Yop1p, or strains lacking one of the ERMES components (Fig. 1). Therefore there are strong genetic interactions between RTN1, YOP1, and the genes encoding all four members of the ERMES complex. It should also be noted that when all of the strains missing any one member of the ERMES complex, Rtn1p, and Yop1p were plated on solid medium, rapidly growing colonies were often seen along with more slowly growing colonies (Fig. 1). The cells in these colonies probably contained suppressing mutations.
Fig. 1.
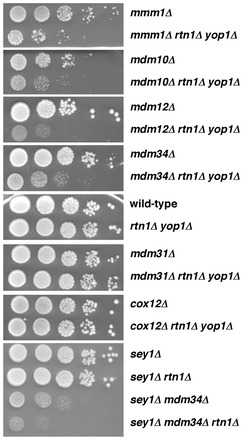
Cells missing ER-tubulating proteins and ERMES proteins grow poorly. Cells with the indicated genotypes were grown to stationary growth phase in YPD, serial 10-fold dilutions were spotted on YPD plates, and the plates were incubated at 30°C for 3 days. Representative examples of at least three independent experiments are shown.
Because cells missing proteins in the ERMES complex have abnormally shaped mitochondria (Burgess et al., 1994; Sogo and Yaffe, 1994; Berger et al., 1997; Kornmann et al., 2009), we wanted to determine if deletion of other genes that affect mitochondrial shape also cause cells missing Rtn1p and Yop1p to grow poorly. Cells lacking Mdm31p, an inner mitochondrial membrane (IMM) protein of unknown function, have abnormal mitochondrial shape (Dimmer et al., 2005). We found that a strain missing Mdm31p, Rtn1p, and Yop1 grew about as well as cells missing only Mdm31p (Fig. 1), suggesting that the growth defects of cells missing ERMES components, Rtn1p, and Yop1p was not caused by the abnormal mitochondrial morphology in these cells. We also did not find genetic interactions between deletion of YOP1 and RTN1 and genes encoding proteins needed for oxidative phosphorylation. Cox12p is subunit VIb of cytochrome c oxidase and cells lacking this protein have decreased respiratory growth (LaMarche et al., 1992). We found that cox12Δ rtn1Δ yop1Δ cells grew as well as cells missing only Cox12p (Fig. 1).
These findings suggest that cells with defects in maintaining ER morphology grow poorly when they also lack one of the proteins in the ERMES complex. We wondered if other strains with abnormal ER shape also grow poorly when they lack one of the proteins in the ERMES complex. Cells missing Sey1p and either Yop1p or Rtn1p have defects in ER shape that are similar to those of cells missing Rtn1p and Yop1p (Hu et al., 2009). We found that sey1Δ rtn1Δ mdm34Δ cells grew much more slowly than sey1Δ rtn1Δ cells (Fig. 1C), indicating that strains lacking proteins required to maintain ER shape grow poorly when they are also missing one of the members of the ERMES complex. Surprisingly, we found that sey1Δ mdm34Δ cells grew more slowly than sey1Δ cells or mdm34Δ cells (Fig. 1A,C). This was unexpected because sey1Δ cells have a relatively normal ER morphology (Hu et al., 2009). It is possible that Sey1p indirectly affects mitochondrial function by a mechanism that is independent of its role in maintaining ER shape.
Because ER shape is abnormal in rtn1Δ yop1Δ cells, we wondered if ERMES protein localization was altered in these cells. It has previously been shown that ERMES proteins localize to about 1–10 punctae per cell and that in cells missing one of the ERMES proteins the localization of remaining members changes dramatically: Mmm1–GFP is found all over the ER, Mdm12–GFP is in the cytosol, and Mdm10–GFP and Mdm34–GFP are all over the surface of mitochondria (Kornmann et al., 2009). If ERMES complex assembly were compromised in rtn1Δ yop1Δ cells, ERMES proteins would have a significantly different localization in rtn1Δ yop1Δ cells than they do in wild-type cells. We found that localization of Mdmd10-GFP, Mdm34–GFP, Mdm12–GFP, and Mmm1–GFP was similar when they were expressed in wild-type or rtn1Δ yop1Δ cells (Fig. 2). These findings suggest that ERMES complex formation is not altered in cells with abnormal ER shape.
Fig. 2.
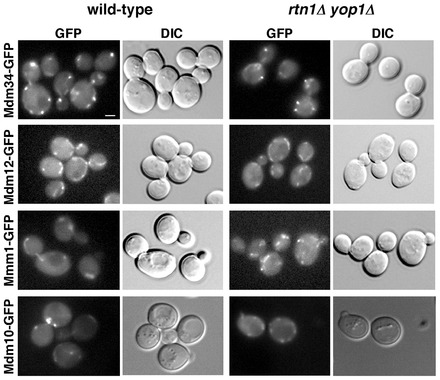
Localization of ERMES complex proteins is not altered in rtn1Δ yop1Δ cells. Wild-type or rtn1Δ yop1Δ cells expressing GFP fusions to the indicated ERMES proteins were visualized live. Fluorescent images (GFP) and differential interference contrast (DIC) images of the cells are shown. The fusions were expressed from the chromosome under the endogenous promoter except for Mdm10–GFP, which was expressed from a plasmid under the MDM10 promoter. Scale bar: 1 µm.
ER to mitochondria PS transfer decreases in cells missing ER-shaping proteins and Mdm34p
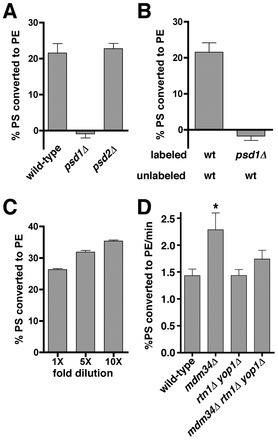
Cells lacking ER-shaping proteins and proteins in the ERMES complex may grow poorly because they have a defect in lipid exchange between the ER and mitochondria, a process that is required for mitochondrial membrane biogenesis and lipid metabolism. To determine if phospholipid transfer from the ER to mitochondria slows in these strains, we measured the amount of newly synthesized PS that is converted to PE. PS is synthesized in the ER (Zinser et al., 1991) and can be decarboxylated to PE by Psd1p, which is in the IMM (Trotter et al., 1993; Clancey et al., 1993), or Psd2p, which is in the Golgi complex or vacuole (Trotter et al., 1995). Therefore in cells missing Psd2p, the conversion of newly synthesized PS to PE indicates that the PS has been transferred from the ER to mitochondria. To determine the amount of PS that is transferred from the ER to mitochondria, we labeled various strains lacking Psd2p with [3-3H]serine. We have previously shown that, using the labeling conditions described in Materials and Methods, cells produce PS and PE at a linear rate and that little or none of the radiolabeled PE is converted to PC (Raychaudhuri and Prinz, 2008). The strains were labeled with [3H]serine for 30 minutes and the percentage of [3H]PS converted to PE was calculated. For psd2Δ cells, this was about 40% (Fig. 3A). In psd1Δ psd2Δ cells ∼20% of the [3H]PS synthesized was converted to PE even though this strain cannot decarboxylate PS (Trotter et al., 1995). The most likely explanation for this is that some of the radiolabel from [3H]serine can be incorporated into PE by mechanisms other than decarboxylation of PS. Serine is used in the biosynthesis of sphingosine and degradation of the sphingosine derivative dihydrosphingosine-1-phosphate by Dpl1p yields ethanolamine phosphate (Henry et al., 2012). When cells are labeled with [3H]serine, Dpl1p can produce [3H]ethanolamine phosphate, which in turn can be used to make [3H]PE. This pathway probably accounts for most of the [3H]PE made in psd1Δ psd2Δ cells since we have found that when psd1Δ psd2Δ dpl1Δ cells are labeled with [3H]serine almost no [3H]PE is generated (Raychaudhuri and Prinz, 2008). Thus, the percentage of [3H]PS converted to PE in psd1Δ psd2Δ cells is the lowest that can be achieved when PS decarboxylation is ablated. If [3H]PS transport to mitochondria in psd2Δ cells were completely blocked, the percentage of [3H]PS converted to PE would be the same as that found in psd1Δ psd2Δ cells.
Fig. 3.
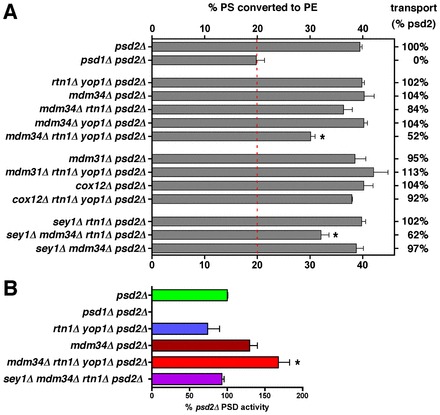
Cells missing ER-shaping proteins and the ERMES complex have defects in PS transfer from the ER to mitochondria. (A) Cells with the indicated genotypes were grown in SC without ethanolamine (except psd1Δ psd2Δ cells) and labeled with [3H]serine for 30 minutes. The percentage of [3H]PS converted to [3H]PE was determined (mean ± s.d., n = 3–6 independent experiments). The dashed red line indicates the amount of conversion that occurred in psd1Δ psd2Δ cells. The percentage transport was calculated taking the amount of conversion in psd1Δ psd2Δ cells as 0% and the amount in psd2Δ cells as 100%. (B) The Psd activity of the indicated strains. Psd activity was normalized to that of psd2Δ cells (mean ± s.d., n = 3–8 independent experiments). *Statistical difference from psd2Δ (P<0.05, two-tailed t-test) in both panels.
We wanted to determine the percentage of [3H]PS that is converted to PE in cells missing Rtn1p, Yop1p, and one of the ERMES proteins. In mdm34Δ rtn1Δ yop1Δ psd2Δ cells, we found that ∼30% percent of [3H]PS was converted to [3H]PE, suggesting that PS transfer from the ER to mitochondria is reduced about 50% relative to the amount in psd2Δ cells (Fig. 3A). We were unable to label cells missing ER-shaping proteins and components of the ERMES complex other than Mdm34p because these strains readily acquired fast growing suppressors that made it impossible to grow the large cultures required for the [3H]serine labeling experiments. However, because the ERMES complex fails to assemble in cells lacking Mdm34p (Kornmann et al., 2009), it seems likely that ERMES complex function is ablated in cells missing Mdm34p. We found that cells missing Psd2p and either Mdm34p or Rtn1p and Yop1p did not have a decrease in the amount of [3H]PS that is converted to PE (Fig. 3A), suggesting that PS transfer from the ER to mitochondria is not altered in cells missing only the ERMES complex or the ER-shaping proteins Rtn1p and Yop1p. A previous study has found that ER structure is normal in cells missing either Rtn1p or Yop1p but abnormal in cells missing both (Voeltz et al., 2006). Therefore we measured the percentage of PS that was converted to PE in cells missing Psd2p, Mdm34p and either Rtn1p or Yop1p and found that it was not reduced (Fig. 3A). This finding suggests that PS transfer from mitochondria to the ER is normal in missing Mdm34p and either Rtn1p or Yop1p because ER structure is normal in these cells. We also determined if other mutations that affect ER shape reduce PS transport when they are combined with a deletion of MDM34. It has been shown that ER structure is abnormal in cells missing Sey1p and Rtn1p (Hu et al., 2009). We measured the percentage of [3H]PS converted to PE in sey1Δ mdm34Δ rtn1Δ psd2Δ cells and found that PS transfer to mitochondria was reduced in this strain while it was not reduced in cells missing Psd2p and either Sey1p and Mdm34p or Sey1p and Rtn1p (Fig. 3A). It should be noted that the total amount of [3H]PS produced per OD600 of cells labeled varied substantially between the strains used in Fig. 3A (supplementary material Fig. S1A). The reasons for this are not known but there was no correlation between the amount of [3H]PS produced and the percentage converted to PE. Therefore differences in the amount of [3H]PS converted to PE were not caused by differences in the amount of [3H]PS synthesized during [3H]serine labeling. We also wanted to rule out that strains that converted a reduced amount of [3H]PS to PE had a decrease in the amount of Psd1p in mitochondria. We measured Psd activity in a number of the strains shown in Fig. 3A and found that they all had at least as much Psd activity as psd2Δ cells, if not more (Fig. 3B). Psd1p also localized to mitochondria normally in strains in which the percentage of [3H]PS converted to PE decreased. Psd1–GFP was expressed in psd2Δ, mdm34Δ rtn1Δ yop1Δ psd2Δ, and sey1Δ rtn1Δ mdm34Δ psd2Δ cells and was found to localize to mitochondria in all three strains (Fig. 4A). We confirmed that Psd1–GFP is functional by showing that it allowed a psd1Δ psd2Δ strain to grow without ethanolamine supplementation (Fig. 4B). Taken together, these findings suggest that cells that lack Mdm34p (and therefore the ERMES complex) and have abnormal ER shape have defects in PS transfer from the ER to mitochondria.
Fig. 4.
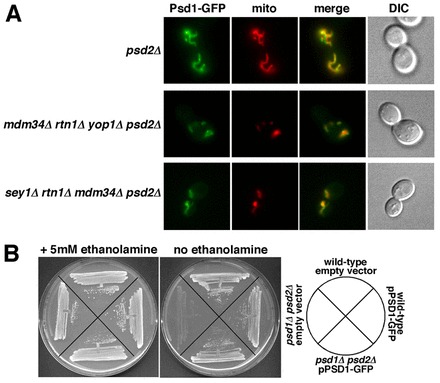
The localization of Psd1–GFP is not altered in cells lacking ER-shaping proteins and Mdm34p. (A) Cells with the indicated genotypes, expressing Psd1–GFP and Mito-Red were visualized live. (B) The Psd1–GFP fusion is functional. Strains with the indicated genotypes were plated on SC medium with or without ethanolamine. Representative examples of three independent experiments are shown.
Defects in mitochondrial shape and oxidative phosphorylation do not affect ER to mitochondria PS transfer
Because cells missing Mdm34p have abnormally shaped mitochondria, we wondered if mitochondrial shape affects ER to mitochondria PS transfer. To test this, we labeled mdm31Δ rtn1Δ yop1Δ psd2Δ cells with [3H]serine and measured the percentage of [3H]PS converted to PE. This strain converted about as much [3H]PS to PE as psd2Δ cells (Fig. 3A), suggesting that abnormal mitochondrial shape does not affect PS transfer from the ER to mitochondria. Even though cells missing Mdm31p have abnormally shaped mitochondria, they are able to grow on non-fermentable carbon sources (Dimmer et al., 2005), indicating that they are able to carry out oxidative phosphorylation. Consistent with this, we found that mdm31Δ rtn1Δ yop1Δ psd2Δ cells were able on YPG, a medium that contains the nonfermentable carbon source glycerol (supplementary material Fig. S1A). Since cells missing any one of the ERMES proteins do not have functional mitochondria and cannot grow on YPG (Dimmer et al., 2002; Kornmann et al., 2009), we wondered if other mutants with nonfunctional mitochondria had defects in PS transfer from the ER to mitochondria. Cells lacking Cox12p do not have functional mitochondria and cannot grow on YPG (supplementary material Fig. S2A) (Dimmer et al., 2002). When we labeled cox12Δ psd2Δ and cox12Δ rtn1Δ yop1Δ psd2Δ strains with [3H]serine, we found they converted about the same percentage of [3H]PS to PE as psd2Δ cells (Fig. 3A), indicating that ablation of oxidative phosphorylation does not altered PS transfer from the ER to mitochondria. Taken together, these results suggest that defects in mitochondrial morphology and oxidative phosphorylation do not affect PS transfer from ER to mitochondria.
Mitochondria proliferate when yeast is grown in YPG or other media with nonfermentable carbon sources. The proliferation of mitochondria probably requires increased lipid transport from ER to mitochondria for membrane biogenesis. Although cells missing Mdm34p or other ERMES proteins do not grow on YPG (Dimmer et al., 2002; Kornmann et al., 2009), we found that cells lacking ER-shaping proteins grow on YPG (supplementary material Fig. S1A). Therefore we asked if PS transfer from the ER to mitochondria was altered when cells are cultured in YPG. When strains lacking ER-shaping proteins or Mdm31 were labeled with [3H]serine in YPG, we found that they converted about the same percentage of [3H]PS to PE as psd2Δ cells (supplementary material Fig. S2B). Therefore cells with defect in maintaining ER shape or mitochondrial shape can transfer PS from the ER to mitochondria normally whether they are grown in media with fermentable or nonfermentable carbon sources.
Steady-state phospholipid levels are altered in cells missing ER-shaping proteins and Mdm34p
Because PS transfer from the ER to mitochondria is reduced in cells missing ER-shaping proteins and the ERMES protein Mdm34p, we wondered if steady-state levels of lipids were altered in these strains. We determined the relative amounts of the four major phospholipids in yeast: PS, PE, phosphatidylcholine (PC), and phosphotidylinositol (PI). We found that cells with defects in ER to mitochondria PS transfer also had changes in steady state levels of lipids; they had reduced levels of PE and increased amounts of PS and PI (Fig. 5A). These cells also had an altered distribution of phospholipids in mitochondria. We purified mitochondria from strains labeled to steady-state with [3H]acetate and determined the relative abundance of the major glycerophospholipids in cells: PC, PE, PS, PI, phosphatidic acid (PA), and cardiolipin (CL). The strains with defects in PS transfer from the ER to mitochondria, mdm34Δ rtn1Δ yop1Δ psd2Δ and sey1Δ rtn1Δ mdm34Δ psd2Δ cells, had decreased levels of PS and PE in mitochondria, consistent with a defect in ER to mitochondria PS transfer (Fig. 5B). The PS results are particularly notable since the total amount of PS in these strains is elevated compared to wild-type cells (Fig. 5A). Therefore, PS accumulates in the membranes of strains lacking Mdm34p and ER-shaping proteins but PS is not elevated in mitochondria, consistent with the idea that ER to mitochondria PS transfer is reduced in these strains.
Fig. 5.
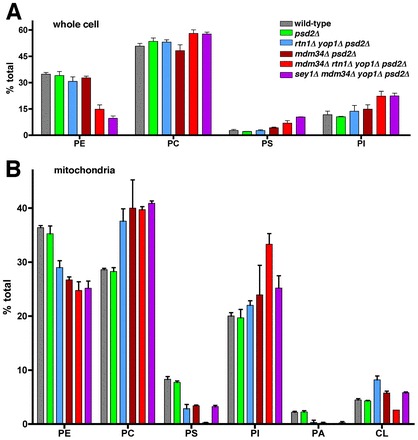
The steady-state distribution of phospholipids in cells lacking Mdm34p and ER-shaping proteins is altered. (A) Relative amounts of the four major phospholipids in cells (mean ± s.d., n = 3 independent experiments). Cells were grown in SC medium without ethanolamine. (B) Relative amount of phospholipids in mitochondria (mean ± s.d., n = 3 independent experiments). The strains used in B are the same as in A. Cells were grown for at least three generations in SC without ethanolamine and containing [3H]acetate and the total amount of phospholipids in purified mitochondria were determined.
In vitro ER to mitochondria PS transfer is not decreased in cells missing ER-shaping proteins and ERMES proteins
Because cells missing ER-shaping proteins and Mdm34p have reduced PS transfer from the ER to mitochondria, we wondered if a similar defect would be seen when transfer occurs in vitro. To study PS transfer in vitro, we used a method adapted from Daum and co-workers (Achleitner et al., 1999), which used crude mitochondria to measure PS transfer. Crude mitochondria were isolated by lysing spheroplasts with a dounce and centrifuging the resulting lysate at ∼10,000 g, which pellets mitochondria together with associated ER. PS transfer was then measured with a two-step reaction: first, radiolabled PS was synthesized by PS synthase in ER membranes (Zinser et al., 1991) and, second, PS transfer was assessed by determining the rate at which the newly synthesized PS was converted to PE, a reaction that occurs in the IMM (Trotter et al., 1993; Clancey et al., 1993). In the first step, [3H]serine was added to crude mitochondria in a buffer that contains Mn2+, which is needed by PS synthase (Nikawa and Yamashita, 1981) but inhibits Psd1p (Lamping et al., 1991). After a 20-minute incubation, the Mn2+ was removed by chelation and PS decarboxylation began. Using this assay, we found that PS to PE conversion was linear for 15 minutes (not shown), as previously described (Achleitner et al., 1999). Psd1p is responsible for all the PS decarboxylation that occurs in this assay even though the membranes were prepared from cells that contain both Psd1p and Psd2p. We found that no PS to PE conversion occurred in membranes isolated from cells lacking Psd1p (Fig. 6A), indicating that Psd2p is either not present in the crude mitochondria membranes used in the assay or is not active. Therefore the PS to PE conversion that occurs in vitro is only the result of PS transfer to mitochondria, where Psd1p resides.
Fig. 6.
ER-shaping proteins and Mdm34p are not required for efficient PS transfer from the ER to mitochondria in vitro. (A) Crude mitochondria were incubated with [3H]serine and Mn2+. After 20 minutes at 30°C, an excess of unlabeled serine and EDTA was added; chelation of Mn2+ by EDTA inhibits PS synthase and allows Psd1p to function. The samples were incubated at 30°C for either 0 or 15 minutes. The percentage of [3H]PS converted to [3H]PE after 15 minutes minus the amount at zero minutes was calculated. (B) Crude mitochondria from wild-type or psd1Δ cells were incubated with [3H]serine and Mn2+ (‘labeled’ membrane). After 20 minutes, an excess of unlabeled serine and EDTA were added together with unlabeled crude mitochondria from wild-type cells or psd1Δ cells (‘unlabeled’ membranes). (C) Crude mitochondria were labeled with [3H]serine. After 20 minutes the samples were diluted 1×, 5×, or 10×, and an excess of unlabeled serine and EDTA were added to the reaction. The samples were incubated for 15 minutes and the percentage of [3H]PS converted to [3H]PE was determined. (D) Crude mitochondria were labeled as in A and samples were taken at 0, 5, 10, and 15 minutes. These were used to calculate the rate of [3H]PS conversion to [3H]PE. All panels show mean ± s.d., n = 3 independent experiments. *Statistical difference from wild type (P<0.05, two-tailed t-test).
In this assay, PS transfer probably occurs between ER membranes and mitochondria that remain associated during mitochondria isolation. We found that if we labeled membranes from psd1Δ cells and then mixed them with membranes from wild-type cells, none of the [3H]PS in the membranes from psd1Δ cells was converted to PE (Fig. 6B). This suggests that the ER-derived membranes where [3H]PS occurred could not associate with wild-type mitochondria in vitro in such a way that PS transfer was possible. This also suggests that the [3H]PE produced in the assay is probably not the result of mitochondrial lysis. Consistent with the hypothesis that ER and mitochondrial membranes do not dissociate in our assay, we found that the amount of PS to PE conversion in our in vitro assay was not decreased by dilution (Fig. 6C). Together, these findings suggest that ER-derived membranes and mitochondria remain associated during PS synthesis, transfer to mitochondria, and conversion to PE.
We determined the rate of PS to PE conversion with crude mitochondria derived from wild-type cells and those missing ER-shaping proteins and the ERMES component Mdm34p. Surprisingly, we found that the rate of PS transfer did not decrease or even slightly increased with mitochondria derived from the mutant strains (Fig. 6D). This result could not be explained by differences in the total amount of [3H]serine synthesized in these experiments; for reasons that are not known crude mitochondria derived from rtn1Δ yop1Δ cells were able to produce more [3H]PS than those from the other strains used, but these differences do not correlate with the rate of [3H]PS transferred measured (supplementary material Fig. S1B). Therefore, even though PS transfer from ER to mitochondria slows in cells missing Mdm34p, Rtn1p, and Yop1p in vivo, no decrease in the rate of PS transport was found in vitro.
An artificial ER–mitochondria tether restores PS transfer in cells missing ER-shaping proteins and ERMES proteins
The decreased PS transfer from the ER to mitochondria in mdm34Δ rtn1Δ yop1Δ psd2Δ cells suggests that ER–mitochondria junctions are abnormal in this strain. We wondered if increasing the number of ER–mitochondria junctions in this strain would increase the amount of PS transferred from ER to mitochondria. To test this, we expressed a fusion protein that artificially tethers ER and mitochondria in mdm34Δ rtn1Δ yop1Δ psd2Δ cells and determined the conversion of [3H]PS to PE. The ChiMERA protein contains GFP fused to sequences that insert into the ER and mitochondria and when this fusion is expressed in cells it tethers these organelles (Kornmann et al., 2009). We expressed ChiMERA in both mdm34Δ rtn1Δ yop1Δ psd2Δ and psd2Δ cells and found that PS to PE conversion increased in both strains (Fig. 7A), indicating that an increase in ER–mitochondrial tethering can compensate for the decrease in PS transfer found in mdm34Δ rtn1Δ yop1Δ psd2Δ cells. Interestingly, ChiMERA increased the percent of PS converted PE in both psd2Δ and mdm34Δ rtn1Δ yop1Δ psd2Δ (Fig. 7A), suggesting that increasing the number of contact sites between the ER and mitochondria increases PS transfer between these organelles even in cells that have normal ER–mitochondria junctions. To confirm that ChiMERA alters lipid metabolism in both strains, we measured the steady-state levels of phospholipids in psd2Δ and mdm34Δ rtn1Δ yop1Δ psd2Δ cells either containing or lacking ChiMERA. Remarkably, ChiMERA increased the amount of PE and decreased the amount of PI in both strains (Fig. 7B). Thus, the increased amount of PS transfer from ER to mitochondria in cells expressing ChiMERA resulted in an increased steady-state level of PE. Since CDP-diacylglycerol is a precursor for both PI and PS (Henry et al., 2012), it may be that increased PE production from PS in cells expressing ChiMERA depletes the pools of CDP-diacylglycerol available for PI biosynthesis. It was not possible to determine phospholipids levels were altered in the mitochondria from cells expressing ChiMERA since we were unable to isolate purified mitochondria from these cells (not shown), perhaps because they have a substantially increased amount of ER-derived membranes associated with them. Surprisingly, even though ChiMERA restored ER to mitochondria PS transfer in mdm34Δ rtn1Δ yop1Δ psd2Δ cells, it did not correct their growth defect (Fig. 6C). The growth defect in this strain is probably at least partially caused by its propensity to lose mitochondrial DNA. It has previously been shown that cells missing some ERMES proteins rapidly lose mitochondrial DNA (Hobbs et al., 2001; Dimmer et al., 2002). We confirmed that the mdm34Δ rtn1Δ yop1Δ psd2Δ strain we used for these studies lacked mitochondrial DNA (not shown). This may explain why ChiMERA failed to cause this strain to grow as rapidly as psd2Δ cells expressing ChiMERA. Attempts to make a mdm34Δ rtn1Δ yop1Δ psd2Δ strain that retained mitochondrial DNA were not successful.
Fig. 7.
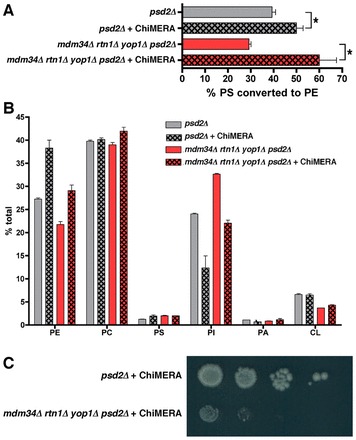
An artificial ER–mitochondria tether restores PS transfer from ER to mitochondria in mdm34Δ rtn1Δ yop1Δ psd2Δ cells. (A) Cells with the indicated genotypes and containing the plasmid that expresses the ER–mitochondria tethering ChiMERA protein were labeled with [3H]serine as in Fig. 3A (mean ± s.d., n = 3 independent experiments). *P<0.05, two-tailed t-test. (B) Strains were grown in SC supplemented with [3H]acetate for at least three generations. Lipids were extracted and quantified (mean ± s.d., n = 3 independent experiments). (C) Cells were grown to mid-logarithmic growth phase in SC medium, serial 10-fold dilutions were spotted on SC plates, and the plates incubated at 30°C for 4 days. A representative example of three independent experiments is shown.
Discussion
Regions of close contact between the ER and mitochondria are thought to be zones where lipids are exchanged between these organelles. In this study we found that PS transfer from the ER to mitochondria slows in cells missing Mdm34p, one of the proteins in the ERMES complex, and either Rtn1p and Yop1 or Rtn1p and Sey1p. Because the ERMES complex fails to assemble in cells lacking Mdm34p (Kornmann et al., 2009), it seems likely that ER to mitochondria PS transfer is reduced in cells missing any of the ERMES proteins and ER-shaping proteins, a conclusion that is supported by our discovery that all four genes encoding ERMES proteins interact genetically with RTN1 and YOP1. Thus, our findings suggest that both the ERMES complex and proteins required to maintain tubular ER are needed for optimal lipid exchange between the ER and mitochondria. Our discovery that increasing ER–mitochondria junctions with an artificial tether restores PS transfer from the ER to mitochondria in rtn1Δ yop1Δ mdm34Δ cells also supports this conclusion. Surprisingly, although ER to mitochondria PS transfer slows in rtn1Δ yop1Δ mdm34Δ cells in vivo, we found that it did not slow in vitro. It may be that the structure of ER–mitochondria junctions maintained by ER-shaping proteins and the ERMES complex in vivo is not retained in vitro. Alternatively, proteins that are not present in the in vitro reactions may regulate lipid transfer in cells.
What role ER-shaping proteins may play in maintaining the structure of the ER at ER–mitochondria junctions remains to be determined. It seems unlikely that the number of junctions between the ER and mitochondria is significantly reduced in cells missing ER-shaping proteins since in vitro PS transport occurs normally in crude mitochondria derived from mdm34Δ rtn1Δ yop1Δ psd2Δ cells. Rather, ER-shaping proteins are probably needed to maintain ER structure at ER–mitochondria junctions in order to allow optimal lipid exchange in vivo. It is also possible that deletion of the reticulons and Sey1p indirectly affects lipid metabolism and that these proteins do not play a direct role in structuring ER–mitochondria junctions. However, we found that steady-state levels of the major phospholipids in rtn1Δ yop1Δ cells were very similar to those in wild-type cells.
What role ER–mitochondria tethering plays in lipid exchange between these organelles remains an important question. Interestingly, we found that artificially tethering the ER and mitochondria increases PS transport not only in cells with defects in this process but in wild-type cells as well. This increase in transfer is significant enough that it elevates the steady-state levels of PE in cells. These findings suggest that the extent of tethering between the ER and mitochondria is an important determinant of the efficiency of lipid exchange between these organelles. Regulation of the extent of tethering between the ER and mitochondria and the structure of regions of contact may be an important part of how cells regulate lipid exchange between these organelles.
Previous work indicated that phospholipid exchange between the ER and mitochondria slows in cells missing the ERMES complex (Kornmann et al., 2009). In contrast, we found that PS transfer from the ER to mitochondria did not slow significantly in cells missing Mdm34p both in vitro and in vivo. Our finding is consistent with a recent study that found little or no difference in the rate of PS transfer from the ER to mitochondria in cells missing the components of the ERMES complex (Nguyen et al., 2012). This difference may be explained by the assays used to assess lipid exchange between the ER and mitochondria. The work by Kornmann and co-workers measured the rate of PS conversion to PC, a process that requires that PS be moved from the ER to mitochondria, converted to PE, transferred back to the ER, and converted to PC. It may be that the movement of PE from mitochondria to ER is more inhibited than PS transfer from the ER to mitochondria in cells lacking the ERMES complex. How the ERMES complex and proteins needed to maintain tubular ER facilitate lipid exchange between the ER and mitochondria is an important question for future studies.
Materials and Methods
Strains, plasmids, and growth media
Strains and plasmids used in this study are listed in the supplementary material Table S2. Cells were grown in three media: YPD (1% yeast extract, 2% peptone, 2% glucose), YPG (1% yeast extract, 2% peptone, 3% glycerol), and SC (2% glucose, 0.67% yeast nitrogen base without amino acids, and amino acid dropout mix from BIO101). Where indicated, ethanolamine was added to 5 mM.
SGA analysis
SGA Analysis was performed using media and techniques as previously described (Tong and Boone, 2006). An rtn1Δ yop1Δ strain was mated to the haploid deletion mutant array at a density of 1536 spots per plate using a Singer RoToR HDA robot. The resulting diploids were copied in triplicate onto enriched sporulation medium and incubated at 25°C for 7 days. MATa haploid cells were generated by germination on SC-His/Arg/Lys +Can/Thia. Triple mutants were selected by two rounds of incubation on SC-His/Arg/Lys/Ura +Can/Thia/G418/NAT medium. A control set of single mutants was generated by two rounds of incubation on SC-His/Arg/Lys +Can/Thia/G418.
Synthetic sick phenotypes were identified by imaging plates using a flatbed scanner and comparing the growth of each spot on the triple mutant plate to the corresponding spot on the control plate. This was achieved using Colony software (Tong and Boone, 2006) to normalize spot sizes and correct for edge effects (the tendency for colonies at the edges of the array to grow faster). For each position in the array, the ratio of the normalized pixel area of the triple mutant to that of the single mutant was calculated. ‘Hits’ were defined as those positions in which this ratio was below 0.8 for all three replicates and the corresponding P-value from a one-tailed t-test below 0.05.
Lipid extraction and analysis
Lipids were extracted as described (Parks et al., 1985). All lipid analysis was performed on cells grown in SC. If whole cells were used for extraction they were first lysed in a Mini-BeadBeater-8 (BioSpec). Quantification of total glycerophospholipids for Fig. 5A and Psd assays were performed as described (Raychaudhuri and Prinz, 2008). Analysis of total glycerophospholipids for Fig. 7B and of purified mitochondria was preformed on cells labeled for at least three generations with [3H]acetate (American Radiolabeled Chemicals). Cultures (50 ml) were grown in SC with 25 µCi of [3H]acetate to an OD600 of 0.4–0.6. The medium did not contain ethanolamine. For analysis of whole cells, cells were lysed in a Mini-BeadBeater-8. Purified mitochondria were obtained as described (Nunnari et al., 2002). Lipids were extracted and separated by thin layer chromatography (TLC) as described (Vaden et al., 2005). TLC plates were scanned on a RITA Star Thin Layer Analyzer (Raytest).
Mitochondrial extracts and in vitro [3H]serine labeling
Cells were grown in YPD medium to an optical density of ∼0.3 at 600 nm, washed once with water, and resuspended in 1 ml 0.1 M Tris-SO4 (pH 9.4) containing 10 mM DTT. After incubation at 30°C for 10 min, the cells were washed once with spheroplast buffer (1.2 M sorbitol, 20 mM Tris pH 7.4) and resuspended in 1.5 ml of the same buffer containing 1 mg/ml zymolyase 20T (Seikagaku Biobusiness, Japan). After incubation for 60 min at 30°C, cells were pelleted (5 min, 500 g) and washed twice with spheroplast buffer. Cells were resuspended in ice-cold lysis buffer (0.6 M mannitol, 2 0 mM Tris pH 7.4, 1 mM EDTA, 1 mM PMSF and protease inhibitors (Roche) and lysed with a dounce using the B-pestle. The extract was spun twice (5 min, 3000 g) to remove unlysed cells and debris. The supernatant was spun at 9600 g for 10 min and the pellet containing crude mitochondria was resuspended in lysis buffer using a dounce (B-pestle).
The method of labeling crude mitochondria with [3H]serine was adapted from (Achleitner et al., 1999). 1–2 µg of crude mitochondria in 1 ml of lysis buffer were heated to 30°C and 0.6 µM MnCl2 and 10 µCi of L-[3-3H]serine (American Radiolabeled Chemicals) were added. After 20 minutes, 0.5 mM serine and 5 mM EDTA were added. Samples of 200 µl were taken after 0, 5, 10, and 15 minutes and added to 6 ml of chloroform–methanol (1∶2). Lipids were extracted and PS, PE, and PC were separated by HPLC (Wang et al., 2003) and counted by liquid scintillation counting.
In vivo labeling with [3H]serine
Cells were labeled with L-[3-3H]serine (American Radiolabeled Chemicals) as described (Raychaudhuri and Prinz, 2008) with the following modifications. Cells were grown in SC without ethanolamine or in YPG. About 10 OD600 units of cells in logarithmic growth phase (OD600 = 0.5–0.65) were pelleted and resuspended in 25 ml of fresh prewarmed medium and grown at 30°C. After about 10 minutes, 10 µg/ml Myriocin (Sigma-Aldrich; stock = 500 µg/ml in methanol) was added to the medium, the cells were grown for 30 minutes, and 10 µg/ml Cerulenin (Sigma-Aldrich; stock = 5 mg/ml in DMSO) was added to the medium. About 5 minutes later, 50 µCi of [3H]serine was added to the medium and the cells were grown for an additional 30 minutes, when cells were grown in SC, or 60 minutes when cells were grown in YPG. The culture was then added to an equal volume of ice-cold water and it was washed once with ice-cold water. Lipids were extracted, separated by HPLC, and extracted as described in the previous section.
Fluorescence microscopy
Cells were imaged live at room temperature by using an Olympus BX61 microscope, a UplanApo 100×/1.35 NA lens, a QImaging Retiga EX camera, and IVision software (version v 4.0.5).
Psd assay
Psd assay were performed as previously described (Raychaudhuri and Prinz, 2008) except that the concentration of the substrate, 1-oleoyl-2-(12-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl)-sn-glycero-3-phosphoserine (Avanti Polar Lipids), was 500 µM.
Supplementary Material
Acknowledgments
We thank B. Kornmann and J. Nunnari for plasmids and O. Cohen-Fix and K. Anwar for critical reading of the manuscript.
Footnotes
Funding
This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases. B.P.Y and C.J.L were supported by CIHR, NSERC, CFI, MSFHR and the Tula Foundation. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.105635/-/DC1
References
- Achleitner G., Gaigg B., Krasser A., Kainersdorfer E., Kohlwein S. D., Perktold A., Zellnig G., Daum G. (1999). Association between the endoplasmic reticulum and mitochondria of yeast facilitates interorganelle transport of phospholipids through membrane contact. Eur. J. Biochem. 264, 545–553 10.1046/j.1432-1327.1999.00658.x [DOI] [PubMed] [Google Scholar]
- Anwar K., Klemm R. W., Condon A., Severin K. N., Zhang M., Ghirlando R., Hu J., Rapoport T. A., Prinz W. A. (2012). The dynamin-like GTPase Sey1p mediates homotypic ER fusion in S. cerevisiae. J. Cell Biol. 197, 209–217 10.1083/jcb.201111115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger K. H., Sogo L. F., Yaffe M. P. (1997). Mdm12p, a component required for mitochondrial inheritance that is conserved between budding and fission yeast. J. Cell Biol. 136, 545–553 10.1083/jcb.136.3.545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess S. M., Delannoy M., Jensen R. E. (1994). MMM1 encodes a mitochondrial outer membrane protein essential for establishing and maintaining the structure of yeast mitochondria. J. Cell Biol. 126, 1375–1391 10.1083/jcb.126.6.1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancey C. J., Chang S. C., Dowhan W. (1993). Cloning of a gene (PSD1) encoding phosphatidylserine decarboxylase from Saccharomyces cerevisiae by complementation of an Escherichia coli mutant. J. Biol. Chem. 268, 24580–24590 [PubMed] [Google Scholar]
- Dimmer K. S., Fritz S., Fuchs F., Messerschmitt M., Weinbach N., Neupert W., Westermann B. (2002). Genetic basis of mitochondrial function and morphology in Saccharomyces cerevisiae. Mol. Biol. Cell 13, 847–853 10.1091/mbc.01-12-0588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmer K. S., Jakobs S., Vogel F., Altmann K., Westermann B. (2005). Mdm31 and Mdm32 are inner membrane proteins required for maintenance of mitochondrial shape and stability of mitochondrial DNA nucleoids in yeast. J. Cell Biol. 168, 103–115 10.1083/jcb.200410030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaz Y., Schuldiner M. (2011). Staying in touch: the molecular era of organelle contact sites. Trends Biochem. Sci. 36, 616–623 10.1016/j.tibs.2011.08.004 [DOI] [PubMed] [Google Scholar]
- Henry S. A., Kohlwein S. D., Carman G. M. (2012). Metabolism and regulation of glycerolipids in the yeast Saccharomyces cerevisiae. Genetics 190, 317–349 10.1534/genetics.111.130286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs A. E., Srinivasan M., McCaffery J. M., Jensen R. E. (2001). Mmm1p, a mitochondrial outer membrane protein, is connected to mitochondrial DNA (mtDNA) nucleoids and required for mtDNA stability. J. Cell Biol. 152, 401–410 10.1083/jcb.152.2.401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J., Shibata Y., Voss C., Shemesh T., Li Z., Coughlin M., Kozlov M. M., Rapoport T. A., Prinz W. A. (2008). Membrane proteins of the endoplasmic reticulum induce high-curvature tubules. Science 319, 1247–1250 10.1126/science.1153634 [DOI] [PubMed] [Google Scholar]
- Hu J., Shibata Y., Zhu P. P., Voss C., Rismanchi N., Prinz W. A., Rapoport T. A., Blackstone C. (2009). A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell 138, 549–561 10.1016/j.cell.2009.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachar B., Reese T. S. (1988). The mechanism of cytoplasmic streaming in characean algal cells: sliding of endoplasmic reticulum along actin filaments. J. Cell Biol. 106, 1545–1552 10.1083/jcb.106.5.1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornmann B., Currie E., Collins S. R., Schuldiner M., Nunnari J., Weissman J. S., Walter P. (2009). An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 325, 477–481 10.1126/science.1175088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornmann B., Osman C., Walter P. (2011). The conserved GTPase Gem1 regulates endoplasmic reticulum-mitochondria connections. Proc. Natl. Acad. Sci. USA 108, 14151–14156 10.1073/pnas.1111314108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMarche A. E., Abate M. I., Chan S. H., Trumpower B. L. (1992). Isolation and characterization of COX12, the nuclear gene for a previously unrecognized subunit of Saccharomyces cerevisiae cytochrome c oxidase. J. Biol. Chem. 267, 22473–22480 [PubMed] [Google Scholar]
- Lamping E., Kohlwein S. D., Henry S. A., Paltauf F. (1991). Coordinate regulation of phosphatidylserine decarboxylase in Saccharomyces cerevisiae. J. Bacteriol. 173, 6432–6437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T. T., Lewandowska A., Choi J. Y., Markgraf D. F., Junker M., Bilgin M., Ejsing C. S., Voelker D. R., Rapoport T. A., Shaw J. M. (2012). Gem1 and ERMES do not directly affect phosphatidylserine transport from ER to mitochondria or mitochondrial inheritance. Traffic 13, 880–890 10.1111/j.1600-0854.2012.01352.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikawa J. I., Yamashita S. (1981). Characterization of phosphatidylserine synthase from Saccharomyces cerevisiae and a mutant defective in the enzyme. Biochim. Biophys. Acta 665, 420–426 10.1016/0005-2760(81)90254-X [DOI] [PubMed] [Google Scholar]
- Nunnari J., Wong E. D., Meeusen S., Wagner J. A. (2002). Studying the behavior of mitochondria. Methods Enzymol. 351, 381–393 10.1016/S0076-6879(02)51859-0 [DOI] [PubMed] [Google Scholar]
- Orso G., Pendin D., Liu S., Tosetto J., Moss T. J., Faust J. E., Micaroni M., Egorova A., Martinuzzi A., McNew J. A.et al. (2009). Homotypic fusion of ER membranes requires the dynamin-like GTPase atlastin. Nature 460, 978–983 10.1038/nature08280 [DOI] [PubMed] [Google Scholar]
- Parks L. W., Bottema C. D., Rodriguez R. J., Lewis T. A. (1985). Yeast sterols: yeast mutants as tools for the study of sterol metabolism. Methods Enzymol. 111, 333–346 10.1016/S0076-6879(85)11020-7 [DOI] [PubMed] [Google Scholar]
- Prinz W. A. (2010). Lipid trafficking sans vesicles: where, why, how? Cell 143, 870–874 10.1016/j.cell.2010.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz W. A., Grzyb L., Veenhuis M., Kahana J. A., Silver P. A., Rapoport T. A. (2000). Mutants affecting the structure of the cortical endoplasmic reticulum in Saccharomyces cerevisiae. J. Cell Biol. 150, 461–474 10.1083/jcb.150.3.461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychaudhuri S., Prinz W. A. (2008). Nonvesicular phospholipid transfer between peroxisomes and the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 105, 15785–15790 10.1073/pnas.0808321105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata Y., Hu J., Kozlov M. M., Rapoport T. A. (2009). Mechanisms shaping the membranes of cellular organelles. Annu. Rev. Cell Dev. Biol. 25, 329–354 10.1146/annurev.cellbio.042308.113324 [DOI] [PubMed] [Google Scholar]
- Simbeni R., Pon L., Zinser E., Paltauf F., Daum G. (1991). Mitochondrial membrane contact sites of yeast. Characterization of lipid components and possible involvement in intramitochondrial translocation of phospholipids. J. Biol. Chem. 266, 10047–10049 [PubMed] [Google Scholar]
- Sogo L. F., Yaffe M. P. (1994). Regulation of mitochondrial morphology and inheritance by Mdm10p, a protein of the mitochondrial outer membrane. J. Cell Biol. 126, 1361–1373 10.1083/jcb.126.6.1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasaki M., Chen L. B., Fujiwara K. (1986). Microtubules and the endoplasmic reticulum are highly interdependent structures. J. Cell Biol. 103, 1557–1568 10.1083/jcb.103.4.1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong A. H., Boone C. (2006). Synthetic genetic array analysis in Saccharomyces cerevisiae. Methods Mol. Biol. 313, 171–192 [DOI] [PubMed] [Google Scholar]
- Toulmay A., Prinz W. A. (2011). Lipid transfer and signaling at organelle contact sites: the tip of the iceberg. Curr. Opin. Cell Biol. 23, 458–463 10.1016/j.ceb.2011.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotter P. J., Pedretti J., Voelker D. R. (1993). Phosphatidylserine decarboxylase from Saccharomyces cerevisiae. Isolation of mutants, cloning of the gene, and creation of a null allele. J. Biol. Chem. 268, 21416–21424 [PubMed] [Google Scholar]
- Trotter P. J., Pedretti J., Yates R., Voelker D. R. (1995). Phosphatidylserine decarboxylase 2 of Saccharomyces cerevisiáe. Cloning and mapping of the gene, heterologous expression, and creation of the null allele. J. Biol. Chem. 270, 6071–6080 [DOI] [PubMed] [Google Scholar]
- Vaden D. L., Gohil V. M., Gu Z., Greenberg M. L. (2005). Separation of yeast phospholipids using one-dimensional thin-layer chromatography. Anal. Biochem. 338, 162–164 10.1016/j.ab.2004.11.020 [DOI] [PubMed] [Google Scholar]
- Vance J. E. (1990). Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 265, 7248–7256 [PubMed] [Google Scholar]
- Voelker D. R. (1985). Disruption of phosphatidylserine translocation to the mitochondria in baby hamster kidney cells. J. Biol. Chem. 260, 14671–14676 [PubMed] [Google Scholar]
- Voelker D. R. (1989). Reconstitution of phosphatidylserine import into rat liver mitochondria. J. Biol. Chem. 264, 8019–8025 [PubMed] [Google Scholar]
- Voelker D. R. (2009). Genetic and biochemical analysis of non-vesicular lipid traffic. Annu. Rev. Biochem. 78, 827–856 10.1146/annurev.biochem.78.081307.112144 [DOI] [PubMed] [Google Scholar]
- Voeltz G. K., Prinz W. A., Shibata Y., Rist J. M., Rapoport T. A. (2006). A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell 124, 573–586 10.1016/j.cell.2005.11.047 [DOI] [PubMed] [Google Scholar]
- Wang F., Zhao Y., Wang P. (2003). Separation and determination of phospholipids in biological samples by high-performance liquid chromatography. J. Chromatogr. Sci. 41, 142–144 [DOI] [PubMed] [Google Scholar]
- Waterman–Storer C. M., Salmon E. D. (1998). Endoplasmic reticulum membrane tubules are distributed by microtubules in living cells using three distinct mechanisms. Curr. Biol. 8, 798–807 10.1016/S0960-9822(98)70321-5 [DOI] [PubMed] [Google Scholar]
- West M., Zurek N., Hoenger A., Voeltz G. K. (2011). A 3D analysis of yeast ER structure reveals how ER domains are organized by membrane curvature. J. Cell Biol. 193, 333–346 10.1083/jcb.201011039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinser E., Sperka–Gottlieb C. D., Fasch E. V., Kohlwein S. D., Paltauf F., Daum G. (1991). Phospholipid synthesis and lipid composition of subcellular membranes in the unicellular eukaryote Saccharomyces cerevisiae. J. Bacteriol. 173, 2026–2034 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.