

Summary
Transmembrane 4 L six family member 5 (TM4SF5) plays an important role in cell migration, and focal adhesion kinase (FAK) activity is essential for homeostatic and pathological migration of adherent cells. However, it is unclear how TM4SF5 signaling mediates the activation of cellular migration machinery, and how FAK is activated during cell adhesion. Here, we showed that direct and adhesion-dependent binding of TM4SF5 to FAK causes a structural alteration that may release the inhibitory intramolecular interaction in FAK. In turn, this may activate FAK at the cell's leading edge, to promote migration/invasion and in vivo metastasis. TM4SF5-mediated FAK activation occurred during integrin-mediated cell adhesion. TM4SF5 was localized at the leading edge of the cells, together with FAK and actin-organizing molecules, indicating a signaling link between TM4SF5/FAK and actin reorganization machinery. Impaired interactions between TM4SF5 and FAK resulted in an attenuated FAK phosphorylation (the signaling link to actin organization machinery) and the metastatic potential. Our findings demonstrate that TM4SF5 directly binds to and activates FAK in an adhesion-dependent manner, to regulate cell migration and invasion, suggesting that TM4SF5 is a promising target in the treatment of metastatic cancer.
Key words: Cell adhesion, Focal adhesion kinase, Kinase activation, Migration, Tetraspanin
Introduction
Cell migration is critical for development and maintenance of multicellular organisms in addition to the development of pathological conditions, such as cancer metastasis (Yamaguchi and Condeelis, 2007), which involves highly complex processes regulated by coordinated signaling pathways that respond to extracellular matrix (ECM) or soluble factors (Friedl and Wolf, 2009). Cell migration is mediated by the following biological events: (1) leading edges of migrating cells occupying spaces by forming focal adhesions or contacts, (2) rear edges disassembling old focal adhesions, and (3) adhesion-dependent activation of RhoA GTPase regulating cellular actin polymerization and contractility for forward movement (Friedl and Wolf, 2009). As one of the most important signaling molecules activated as a result of cell adhesion and spreading, focal adhesion kinase (FAK) plays a critical role in migration and invasion (Luo and Guan, 2010). FAK overexpression has been detected in diverse primary and metastatic tumor tissues, supporting its pro-tumorigenic and pro-metastatic roles (McLean et al., 2005; Zhao and Guan, 2009). Intensive actin branching and polymerization occur in leading edges of migratory cells, thus forming new focal contacts and adhesions via FAK and RhoA GTPase activation that in turn leads to the activation of downstream actin-organizing effectors, including actin-related protein (Arp2/3), neural Wiskott–Aldrich syndrome protein (N-WASP), and cortactin (Sanz-Moreno and Marshall, 2010).
Cell adhesion causes phosphorylation and activation of focal adhesion molecules that are critically involved in the regulation of morphological changes, migration, and invasion (Danen, 2009). Among these molecules, FAK is a non-receptor Tyr kinase that is autophosphorylated at Tyr397 upon cell adhesion to the ECM. Phosphorylated Tyr397 is a binding site for Src-homology 2-domain containing molecules such as c-Src or phosphoinositide 3-kinase, which phosphorylate other Tyr residues in FAK or phosphatidylinositol, respectively (Schaller, 2010). Although FAK downstream signaling for most adherent cell functions has been intensively investigated, how FAK is activated during cell adhesion remains unknown (Frame et al., 2010).
Membrane-tetraspanning tetraspanins collaborate with integrins for cell adhesion and migration (Berditchevski, 2001). Transmembrane 4 L six family member 5 (TM4SF5) is a membrane glycoprotein with four transmembrane domains whose intracellular loop (ICL; 69R to 91V) and NH2- and COOH-terminal tails are located intracellularly, similar to those of genuine tetraspanins or transmembrane four super-family members (TM4SFs) (Wright et al., 2000). TM4SF5 is highly expressed in diverse clinical cancer tissues, and its overexpression in hepatocytes enhances aberrant proliferation, migration, and invasion (Lee et al., 2011), although it is unknown how cell migration is mechanistically regulated by TM4SF5.
TM4SF5 as a membrane glycoprotein with four transmembrane domains is involved in cell adhesion-related signaling (Lee et al., 2011). TM4SF5 in fibroblasts enhances cell adhesion signaling and FA formation via an increased Tyr925 phosphorylation of FAK on ECM, which are inhibited by serum treatment, suggesting a role of TM4SF5 in regulation of cell-ECM adhesion via coordinative cross-talks between different membrane receptors including integrin α2, TM4SF5 and growth factor receptors (Lee et al., 2006). TM4SF5 in liver epithelial cells results in RhoA inactivation via increased interactions between FAK and RhoGAPs, leading to morphological elongation, EMT, and multilayer growth (Lee et al., 2008). Further interestingly, TM4SF5 in liver epithelial cells in collagen I environment restricts cell spreading and migration via an interaction between TM4SF5 and integrin α2, but an interruption of the interaction recovers spreading and migration (Lee et al., 2009a). TM4SF5 in hepatocarcinoma cells also causes secretion of VEGF depending on activation of FAK/c-Src complex downstream of integrin α5, which can enhance angiogenic activity of neighboring endothelial cells (Choi et al., 2009). Although these previous reports reveal that TM4SF5 collaborates with integrins for different cell functions, they did not reveal how TM4SF5 directly or indirectly activates FAK, a major integrin-mediated signaling molecule important for cell adhesion and migration; normal culture of TM4SF5-positive hepatocarcinoma cells (i.e. even without any adhesion-related stimulation like a reseeding) correlates with an enhanced Tyr577 phosphorylation of FAK higher than that of TM4SF5-null cells (Choi et al., 2009; Lee et al., 2008), which has been the starting observation to pursue this study. Therefore, it remains unknown in the previous studies how direct or indirect is the link between TM4SF5 expression and FAK activation. During our study on TM4SF5-mediated intracellular signaling, we found FAK as a physical intracellular binder of this membrane protein, offering a mechanistic insight into adhesion-dependent FAK activation via a membrane protein.
Here we examined whether TM4SF5 could activate FAK during cell adhesion and how TM4SF5-mediated FAK activation regulates cell migration. We hypothesized that TM4SF5 might directly activate FAK to facilitate cell migration. We found that interaction between FAK and TM4SF5 at the leading edges of migratory cells led to activation of FAK and efficient actin polymerization upon integrin engagement to ECM, resulting in a directional migration and further facilitated invasion and metastasis in vivo. Impaired interactions between both molecules resulted in ineffective FAK activation and migration. Thus our results suggest that TM4SF5 is a membrane protein capable of directly activating FAK during cell adhesion and a promising target in the treatment of cancer metastasis.
Results
Direct interaction between TM4SF5 and FAK
The goal of our study was to understand TM4SF5-mediated intracellular signaling. During our studies on TM4SF5-mediated signaling, TM4SF5 appeared to bind FAK. Because FAK is a non-receptor Tyr kinase, the intracellular regions of TM4SF5 were analyzed for FAK binding using a peptide-pull-down approach. Peptides of the cytosolic NH2- or COOH-terminal domains and the ICL (Fig. 1A) were mixed with whole-cell extracts of SNU449 hepatocytes. Interestingly, FAK bound the ICL; other molecules, such as p130Cas, paxillin, and cortactin, were also found in the complex between the ICL and FAK, although FAK-unrelated molecules like Jab1 and cyclophilin A did not (Fig. 1B). More interestingly, FAK bound to the ICL of TM4SF5, whereas the ICL part of another tetraspan(in) L6 that shows a high sequence identity with TM4SF5 (Wright et al., 2000) did not bind to FAK (Fig. 1C), indicating specific binding between TM4SF5 and FAK. We next determined the interacting regions on TM4SF5 and FAK. Diverse FAK forms were analyzed by pull-down experiments using TM4SF5 peptides. When the peptides were pulled down from extracts prepared from SNU449 cells infected with an adenovirus encoding for either control or diverse FAK forms, the ICL peptide only bound to wild-type (WT) or kinase dead (KD, K454R) FAK, but not to the NH2-terminus (1 to 100 amino acids [aa])-deleted FAK [FAKΔ(1–100)] or to the COOH-terminal region FAK-related non-kinase (FRNK) (Fig. 1D). The ICL peptide also bound to recombinant glutathione S-transferase (GST)-FAK WT, but not to GST-FAKPR1Pr2 [amino acids (aa) 711–877] or to GST-FAKCD (aa 677–1052) (supplementary material Fig. S1A). Further pull-down experiments revealed that both GST-FERM and GST-FAKF1 (GST-fused F1 lobe of the FAK-FERM domain, aa 33–132) bound to the ICL peptide (Fig. 1E). In addition, recombinant GST-TM4SF5, unlike GST alone, bound FAK WT, KD, or FAK-FERM (aa 1–402), but neither to FAKΔ(1–100) nor to FRNK (supplementary material Fig. S1B). These observations suggest that the ICL of TM4SF5 directly binds the F1 lobe of the FAK-FERM domain.
Fig. 1.
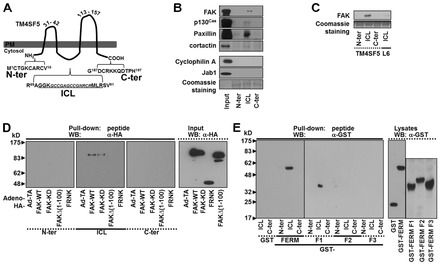
Direct interaction between TM4SF5 and FAK. (A) Structural scheme of TM4SF5. ICL, intracellular loop; C-ter, COOH-terminus; N-ter, NH2-terminus. (B,C) In vitro pull-down using peptides of TM4SF5 cytosolic regions or of the L6 ICL region shows binding between the ICL of TM4SF5, FAK, and others. (D,E) In vitro peptide pull-down using 1 µM peptides for the cytosolic regions of TM4SF5 or L6 and either extracts (500 µg proteins) from SNU449 parental cells infected with various adenovirus for control (Ad-TA) or (HA)3-FAK WT, KD, or N-terminal deletion [Δ(1–100)] (D), or GST-FERM, -F1, -F2, and -F3 domains (E), were performed overnight before immunoblotting. Input control was at 10 µg of proteins. Data shown represent three independent experiments.
TM4SF5-FAK interaction in cells
TM4SF5 is heavily glycosylated and multimerized on the cell surface (supplementary material Fig. S2) (Lee et al., 2006), where it cooperates with integrins to regulate cellular functions (Lee et al., 2011). Since the level of the presumably non-glycosylated form of TM4SF5 (calculated molecular weight; 20.8 kD) at ∼21 kD was unchanged by our homemade rabbit anti-TM4SF5 polyclonal antibody, the naive form was shown throughout in this study, whereas anti-FLAG antibody could show naive and glycosylated TM4SF5 proteins when FLAG-tagged TM4SF5 was exogenously expressed (see below). We thus assessed cells adherent on different ECM components and found that FAK phosphorylations was higher in (stable TM4SF5-expressing) SNU449Tp cells adherent to laminin or fibronectin (FN) than in suspended SNU449Tp or adherent TM4SF5-null SNU449Cp cells (supplementary material Fig. S3A). In addition to SNU449Tp cells, other stable-TM4SF5-expressing clones showed efficient pY397FAK, pY577FAK, and pY118paxillin levels after adhering to FN but not when suspended (supplementary material Fig. S3B), indicating that the binding-mediated effects on FAK and paxillin phosphorylation were not due to cloning artifacts but due to TM4SF5 expression. Given that the cell adhesion process involves dynamic actin remodeling, we examined the interaction between TM4SF5, FAK, and actin-related molecules, such as Arp2 that is shown to bind FAK (Serrels et al., 2007) and cortactin that is a key molecule in actin polymerization (Sanz-Moreno and Marshall, 2010). We observed that ectopic FLAG-TM4SF5 also coimmunoprecipitated FAK and actin polymerization-related molecules such as Arp2 and cortactin, but mock controls did not (Fig. 2A). Endogenous FAK could coimmunoprecipitate endogenous TM4SF5 from Huh7 cells, although it appeared that multimerized TM4SF5 could be more efficiently coimmunoprecipitated than presumably non-glycosylated naive monomer form (Fig. 2B). Anti-FLAG immunoprecipitates from FLAG-TM4SF5 WT cells coimmunoprecipitated FAK and Arp2, whereas immunoprecipitates from FLAG-TM4SF5 ΔICL19 mutant cells, where the ICL deletion was performed by removing 19 aa of the 23 aa in the ICL, did not (Fig. 2C). Additionally, the TM4SF5 ΔICL13 mutant (in which the ICL deletion was performed by removing 13 aa) coimmunoprecipitated FAK, but not Arp2 (Fig. 2C), indicating that the TM4SF5 ΔICL13 mutant might not be able to transduce FAK signaling activity toward actin polymerization. However, the TM4SF5 ΔICL19 mutant could be located at the membrane boundaries (supplementary material Fig. S4A). Further, its level on the plasma membrane was similar to that of TM4SF5 WT on the plasma membranes (supplementary material Fig. S4B).
Fig. 2.
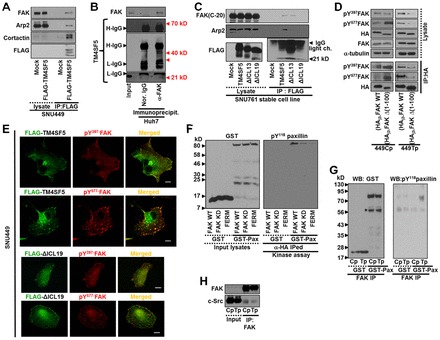
TM4SF5-FAK interaction in cells. (A) SNU449 cells were ectopically transfected with mock or FLAG-tagged TM4SF5 plasmids for 48 h and then harvested for whole-cell extracts, before immunoprecipitation with anti-FLAG antibody and immunoblotting for the indicated molecules. (B) Whole-cell extracts from endogenously TM4SF5-expressing Huh7 cells were immunoprecipitated with anti-FAK antibody, before standard western blots for FAK and TM4SF5. Note that the naïve monomer TM4SF5 at ∼21 kD was bound to FAK, whereas dimerized or multimerized TM4SF5 also appeared to bind efficiently to FAK. H-IgG; heavy IgG, L-IgG; light IgG. (C) Whole-cell lysates from stable SNU761 cells expressing mock, FLAG-TM4SF5 WT, ΔICL13 or ΔICL19 deletion mutants either were immunoprecipitated with anti-FLAG antibody before immunoblotting for the indicated molecules. (D) Whole-cell lysates from SNU449Cp (Cp) or SNU449Tp (Tp) cells infected with adenovirus for (HA)3-FAK WT, or -FAKΔ(1–100) were immunoprecipitated using anti-HA antibody before immunoblotting for the indicated molecules. In parallel, lysates were also immunoblotted. (E) TM4SF5-null SNU449 parental cells were transfected with FLAG-TM4SF5 WT or -TM4SF5 ΔICL19 for 48 h, before indirect immunofluorescence for pY397FAK or pY577FAK using confocal microscopy (top panel) or for pY577FAK, talin, or pY118paxillin using a fluorescence microscopy (bottom panel). Scale bar: 5 µm. (F,G) In vitro FAK assay was performed using recombinant GST-paxillin and either anti-HA, or -myc immunoprecipitates from extracts of SNU449Tp cells infected with different adenovirus for (HA)3-FAK WT, KD, or myc-FAK FERM domain (F), or from extracts of subconfluent SNU449Cp (Cp) or SNU449Tp (Tp) cells (G). (H) Immunoprecipitates using anti-FAK antibody from extracts of subconfluent SNU449Cp (Cp) or SNU449Tp (Tp) cells were immunoblotted for FAK or c-Src. Whole-cell lysates were immunoblotted in parallel. Data shown represent three independent experiments.
When TM4SF5-null SNU449Cp or TM4SF5-stably expressing SNU449Tp cells were infected with an adenovirus coding for FAK WT or FAKΔ(1–100), the phosphorylation of the exogenous FAK forms were found to depend on their capacity to bind TM4SF5, SNU449Cp cells would not be available for the interaction due to insignificant or no TM4SF5 expression, and the total (i.e. exogenous and endogenous) FAK phosphorylation was higher in the FAKΔ(1–100)-infected SNU449Cp cells (Fig. 2D), consistent with the fact that FAKΔ(1–100) is an over-activated form of FAK (Lietha et al., 2007). However, exogenous FAK WT itself showed enhanced Tyr phosphorylation in SNU449Tp cells, whereas exogenous FAKΔ(1–100) showed a markedly reduced phosphorylation, presumably due to the absence of TM4SF5/FAK interaction-mediated FAK phosphorylation (Fig. 2D, IP:HA). Additionally, FLAG-TM4SF5 WT colocalized with pY397FAK or pY577FAK at the membrane boundary and focal adhesions (Fig. 2E, top panel); however, FLAG-TM4SF5 ΔICL19 did not significantly colocalize with the phosphorylated FAK (Fig. 2E, bottom panel). Further, we tested whether FAK in SNU449Tp cells was activated to a greater extent than in SNU449Cp cells via an in vitro kinase assay using exogenous FAK WT or mutant immunoprecipitates and recombinant GST-paxillin. FAK-WT in SNU449Tp cells caused efficient Tyr118 phosphorylation of GST-paxillin, whereas FAK-KD and FAK-FERM could not, although a much reduced Tyr118 phosphorylation was still observed presumably because of the coimmunoprecipitated endogenous FAK or related kinases (Fig. 2F). Moreover, FAK immunoprecipitated from SNU449Tp cell extracts, but not from SNU449Cp cell extracts could efficiently phosphorylate GST-paxillin (Fig. 2G). Because FAK mostly functions as a complex with c-Src (Bolós et al., 2010), c-Src in the FAK immunoprecipitates may phosphorylate the GST-paxillin in our assay. However, the assumption that c-Src is equally coimmunoprecipitated in the immunoprecipitates of either FAK WT or FAK-KD mutant (Wu et al., 2008) and in our study, FAK immunoprecipitates from extracts of both SNU449Cp and SNU449Tp cells showed a similar coimmunoprecipitation of c-Src (Fig. 2H). The c-Src coimmunoprecipitated in the FAK immunoprecipitates thus cannot account for the differential paxillin phosphorylations between the FAK WT and the mutants. Therefore, it is likely that the direct binding between FAK and TM4SF5 can lead to FAK activation during cell adhesion.
TM4SF5 activates FAK upon integrin engagement
Because it is rare to see increased FAK phosphorylation and activation in normally-maintained cultures (that is, subconfluent cells maintained in 10% serum-containing media without any stimulation, such as reseeding to ECM), we next examined via the suppression of endogenous TM4SF5 whether FAK phosphorylation depended on TM4SF5 expression in hepatocytes. Thus, we suppressed endogenous TM4SF5 from HepG2 and Huh7 hepatocytes (Choi et al., 2008) using shRNAs against TM4SF5. Suppression of TM4SF5 resulted in decreased phosphorylation at Tyr397 and Tyr577 of FAK and Tyr118 of paxillin (Fig. 3A), indicating that FAK phosphorylation indeed depends on TM4SF5 expression. To further understand the biological significance of TM4SF5 interaction-mediated FAK activation, we next examined FAK and paxillin phosphorylation in control and TM4SF5-expressing cells either in suspension or reseeded onto FN for different periods of times. Compared with control cells that do not express TM4SF5, such as parental SNU449 (P) or SNU449Cp cells, different TM4SF5-expressing clones (i.e. SNU449Tp and SNU449T16) showed enhanced phosphorylation of FAK and paxillin within a short period after replating (Fig. 3B). When diverse FAK forms were infected into SNU449Tp cells, expression of FAK WT enhanced the phosphorylation of FAK within a short period (i.e. 15 min postreplating) compared with the expression of either control (Ad-TA) or FAK-FERM that does not bind TM4SF5 or does not result in activation, respectively (Fig. 3C). However, the FAK phosphorylation was not significantly differential when diverse FAK forms were transfected into SNU449Cp cells (Fig. 3C). It is thus likely that the direct interaction between TM4SF5 and FAK accelerates FAK activation during the adhesion of cells to the ECM.
Fig. 3.
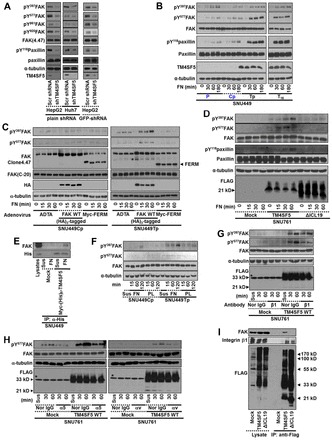
TM4SF5 activates FAK upon integrin engagement. (A) shRNA against control sequence (Con) or TM4SF5 (i.e. plain shRNA or GFP-tagged shRNA against TM4SF5) was transiently transfected to HepG2 or Huh7 cells for 48 h, before harvests of whole cell lysates for standard western blots for the indicated molecules. (B,C) SNU449 cells [SNU449 parental (P), SNU449Cp (Cp, a stable control) cells without TM4SF5 expression, but SNU449Tp (Tp, a stably pooled clone) and SNU449T16 (T16, a single cell derived stable clone) cells, B], or SNU449 cells (SNU449Cp or SNU449Tp cells) infected with adenovirus for control (Ad-TA), (HA)3-FAK WT, or myc-FERM for 24 h (C), were kept in suspension (0) or reseeded on FN for the indicated times (min), before whole-cell lysate preparation to immunoblot for the indicated molecules. (D) Stable SNU761 cells expressing mock, TM4SF5 WT, or ΔICL19 mutant were kept in suspension (0) or reseeded on FN for the indicated times. Whole-cell lysates were prepared and immunoblotted. (E) Myc-(His)6-mock or Myc-(His)6-TM4SF5 were transiently transfected for 48 h and then kept in suspension (Sus) or reseeded on FN-precoated dishes for 2 h, before preparation of whole-cell lysates for immunoprecipitation with anti-(His)6 and standard western blots for the FAK and (His)6-tag. Lysates were analyzed in parallel. (F) Cells were kept in suspension (0) or reseeded on poly-L-lysines (PL) or FN, as explained in the Materials and Methods. (G,H) SNU761 cells stably expressing mock or TM4SF5 WT were preincubated with either normal mouse IgG (Nor-IgG) or neutralizing mouse anti-human integrin β1, α5, αv antibody, before keeping in suspension (Sus) or reseeding on fibronectin for the indicated times (min), as explained in the Materials and Methods. (I) Stable SNU761 cells were harvested and immunoprecipitated with anti-FLAG antibody-precoated agarose beads, before immunoblotting for the indicated molecules in parallel with lysates. * In G and H, bands in Mock (lysate) depict nonspecific bands of anti-FLAG antibody. Data represent three different experiments.
Next, we examined SNU761 hepatocyte cell lines that stably-expressed TM4SF5 WT or the ICL-deleted mutants, ΔICL13 or ΔICL19. When replated on FN, TM4SF5 WT cells showed efficient phosphorylation of FAK and paxillin, whereas ΔICL19 and mock cells either in suspension or in an adhered state showed insignificant phosphorylation of FAK and paxillin (Fig. 3D). Furthermore, the binding between TM4SF5 and FAK depended on cell adhesion, because it did not occur in suspended cells (Fig. 3E). These data indicate that a direct interaction between TM4SF5 membrane protein and FAK occurs during cell adhesion.
We then examined whether TM4SF5-dependent FAK activation might be integrin-dependent. When we maintained cells in suspension or when we replated cells on FN or poly-L-lysine (PL), we clearly observed pY577FAK in SNU449Tp cells on FN at levels higher than those in SNU449Cp cells on FN (Fig. 3F). This difference was not due to the altered expression levels of integrin subtypes on cell surface (supplementary material Fig. S5). Cells on PL were adhered (presumably via charge-mediated adhesion) without spreading. We also compared pY577FAK levels on PL with those on FN to determine the TM4SF5-mediated contribution to FAK phosphorylation. FAK phosphorylation in TM4SF5-expressing SNU449Tp cells on PL was less significant than on FN, but was still higher than those observed in SNU449Tp cells in suspension or TM4SF5-null SNU449Cp cells on FN (Fig. 3F), suggesting that the interaction between TM4SF5 and FAK may have a contribution for FAK activation during cell adhesion. Further, functional blocking of integrin β1 by using its neutralizing antibody resulted in a substantial reduction in pY577FAK levels in SNU761-TM4SF5 WT cells, compared with those of normal mouse IgG-treated SNU761-TM4SF5 WT cells (Fig. 3G). Moreover, functional neutralization of other FN receptors, such as integrin α5 or αv, resulted in a pattern similar to that obtained after integrin β1 neutralization (Fig. 3H), indicating that integrin/FN binding was involved in TM4SF5-mediated FAK activation. However, the antibody neutralization did not result in complete blockings of FAK phosphorylation/activity, probably because the amounts of the neutralizing antibodies were not enough (Fig. 3G,H). Furthermore, TM4SF5 WT bound both FAK and integrin β1, whereas TM4SF5 ΔICL19 did not bind FAK but still bound integrin β1 (Fig. 3I), indicating that the binding interface between TM4SF5 and integrin β1 might be the transmembrane and/or extracellular domains of TM4SF5. Since SNU761-TM4SF5 ΔICL19 cells showed a lower pY397FAK than did SNU761-TM4SF5 WT cells (Fig. 3D), it is thus possible that the interaction between FAK and the ICL of TM4SF5 can be important for a more efficient FAK activation during cell adhesion.
Molecular mechanism of FAK activation through TM4SF5 interaction
To understand the structural aspects of the interaction between the ICL and the (His)6-tagged F1 lobe, standard far-UV circular dichroism (CD) titration experiments were performed. When the ICL peptide was added to the recombinant F1 lobe fragment, there was a profound spectral change in the CD spectra, indicating that the ICL underwent a conformational change upon binding to the (His)6-tagged F1 lobe. The CD spectrum of the ICL alone showed a negative band around 200 nm, without a positive band (presumably due to the unstructured ICL), whereas it adopted a α-helical conformation, probably by becoming more structured in the presence of the F1 lobe, showing a negative band at 210 nm (Fig. 4A, arrow). Further, the CD spectra obtained in trifluoroethanol solution showed peaks with more negative values around 208/220 nm and a less negative peak at 200 nm, as the trifluoroethanol concentration was increased from 0% to 100% (supplementary material Fig. S6). This suggests a possible adaptation of the secondary structure in the F1 lobe by the ICL peptide, although the ICL has many Gly residues that increase the possibility of helix breakage.
Fig. 4.
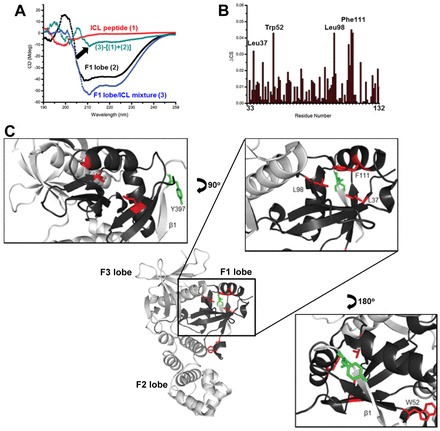
Molecular mechanism of FAK activation by TM4SF5 interaction. (A) CD analysis of the spectral change in the ICL peptide upon binding the F1 lobe. Standard far-UV CD spectra of 48.5 µM ICL peptide (1), 48.5 µM F1 lobe (2), and a mixture of 48.5 µM F1 lobe and 48.5 µM ICL peptide (3). Conformational change in the ICL peptide occurred when titrated into the F1 lobe, which was visualized as (3)−[(1)+(2)]. (B) Chemical shift perturbations of the backbone amide proton (ΔCS) of the F1 lobe after ICL peptide addition. (C) Zoom of the proposed ICL binding region. Ribbon representation of the FAK FERM domain was visualized (PDB ID 2AL6). Pocket region of the F1 lobe consisting of red residues (L37, L98, and F111) that were affected by ICL peptide binding is proposed for the binding region, which is located closely to the C-terminal β-sheet of the F3 lobe and Y397 (green).
Using nuclear magnetic resonance (NMR), we also tried to identify residues in the (His)6-tagged F1 lobe that are critical for binding to the ICL peptide. Upon addition of the peptide, amino acids L37, W52, L98, and F111 underwent large chemical shift changes, which would be in proximity to Tyr397 of FAK (>0.03 ppm, amide proton) (Fig. 4B). A proposed mechanism of interaction between the FAK F1 lobe and the TM4SF5 ICL involves three residues L37, L98, and F111, consisting of the inside pocket region anticipated to contain the key residue that contributes to the recognition of the ICL peptide; this pocket region is spatially close to Y397 located near the β1 sheet (Fig. 4C). As expected, the point mutants L37A or L98A FAK showed enhanced pY397FAK or pY577FAK levels and further activated FAK, leading to greater phosphorylations of Tyr118 of paxillin than that observed by WT FAK (supplementary material Fig. S7). These results indicate that a mutation in the residues that interact with the ICL domain of TM4SF5 may lead to release the inhibitory intramolecular interaction in FAK.
FAK activation through TM4SF5 interaction at the cellular periphery
pEGFP-TM4SF5 WT transiently transfected into TM4SF5-null SNU449 parental cells was localized in the pro-migratory morphological features (supplementary material Fig. S8A–F). We next examined the location of TM4SF5 with regard to the location of pY397FAK and pY577FAK. First, TM4SF5 was observed to localize at the cell periphery and in the intracellular patches (supplementary material Fig. S8G). Only a subpopulation of FLAG-TM4SF5 colocalized with pY397FAK or pY577FAK either along the cell periphery or along the periphery and in intracellular patches, respectively (Fig. 2E). Furthermore, using TM4SF5-null SNU449 or SNU761 hepatocytes transiently transfected with FLAG-TM4SF5, we found that TM4SF5, pY577FAK, talin, and pY118paxillin colocalized to the leading edges of the cells (supplementary material Fig. S9A). Furthermore, pEGFP-TM4SF5-positive cells showed TM4SF5 and actin staining along the cellular periphery (supplementary material Fig. S9B).
Because the interaction between TM4SF5 and FAK led to the formation of complexes containing actin-polymerizing or branching proteins at the leading edges of the cells, it is likely that the TM4SF5/FAK interaction can regulate migration. In the study of chemotactic migration toward 10% fetal bovine serum (FBS), FAK WT-infected SNU449Tp cells showed a dramatic migration, compared with control virus-infected cells (supplementary material Fig. S9C). The pattern of haptotactic migration toward FN was similar to that of chemotaxis (supplementary material Fig. S9D). These observations support that cell migration may be regulated by TM4SF5/FAK interaction-mediated FAK activation at the cell periphery.
TM4SF5 ΔICL19 mutation blocks FAK activation and cell migration
To evaluate the relationship between TM4SF5 and actin reorganization, which is critical for the leading edge formation of migratory cells, we next examined the coimmunoprecipitation of actin polymerization-related molecules in FLAG-TM4SF5 WT or ΔICL19 mutant immunoprecipitates under different signaling contexts, such as suspended or FN-adherent (in the absence of serum) conditions. These physical complexes between FAK and molecules for actin polymerization were formed as cells adhered to FN, although the maximal association of each component in the TM4SF5 immunoprecipitates was different; FAK association gradually increased as cells adhered, pY486cortactin association depended on cell adhesion but declined over time after adhesion, and Arp2 was associated with TM4SF5 both in suspension and in FN-adherent conditions (Fig. 5A). Complex formation did not occur either in the suspended or in the FN-adherent TM4SF5 ΔICL19 cells (Fig. 5A), in which TM4SF5-FAK interaction did not occur (Fig. 2A).
Fig. 5.
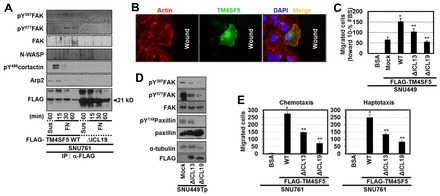
ΔICL19 mutation blocks FAK activation and cell migration. (A) Whole-cell lysates from stable SNU761 cells expressing FLAG-TM4SF5 WT or ΔICL19 deletion mutant in suspension (Sus) or reseeded on FN as described in the Materials and Methods for the indicated times were immunoprecipitated with anti-FLAG antibody before immunoblotting for the indicated molecules. (B) SNU449 cells transfected with pEGFP-TM4SF5 were wounded and incubated for 6 h, before staining with phalloidin. (C) Transwell migration assays (for 12 h) were performed for cells transfected with mock, FLAG-TM4SF5 WT, ΔICL13, or ΔICL19 deletion mutants. Five random images for the migrated cells were calculated and mean ± standard deviation values for graphic presentation. (D) SNU449Tp cells transfected with mock, FLAG-TM4SF5 ΔICL13, or ΔICL19 deletion mutants for 48 h were harvested before standard western blotting for the indicated molecules. (E) Chemotaxis or haptotaxis (for 12 h or 16 h, respectively) of SNU761 cells stably transfected with FLAG-TM4SF5 WT, ΔICL13, or ΔICL19, compared with control migration toward 1% BSA, as explained in the Materials and Methods. *P≤0.05 against mock control; **P≤0.05 against TM4SF5 WT cells. Data represent three independent experiments.
Furthermore, when TM4SF5 location with regard to migratory direction was tested, pEGFP-TM4SF5-positive cells showed TM4SF5 and actin costaining along the cellular periphery toward the free spaces (Fig. 5B). Next, we assessed SNU449 cells transiently transfected with TM4SF5 WT or ICL-deleted mutants for their chemotactic ability. TM4SF5 WT cells migrated more dramatically than did the mock control cells, the TM4SF5 ΔICL13-, or the TM4SF5 ΔICL19-expressing SNU449 cells (Fig. 5C; supplementary material Fig. S10A). We then examined whether the ICL deletion mutants could function as dominant-negatives in TM4SF5-mediated migration. Transfection of TM4SF5 ΔICL13 or of ΔICL19 into SNU449Tp cells decreased FAK and paxillin phosphorylation, although the effects of TM4SF5 ΔICL13 were less prominent than those of ΔICL19 (Fig. 5D). In another hepatic SNU761 cell line, TM4SF5 WT-mediated chemotaxis and haptotaxis were greater than those observed in ICL mutant cells (Fig. 5E; supplementary material Fig. S10B).
TM4SF5 ΔICL19 mutation blocks FAK activity and invasion and metastasis
We next investigated whether TM4SF5/FAK interaction (-mediated FAK activation) affected invasiveness of the cells. TM4SF5 WT-expressing SNU761 cells cultured on fluorophore-conjugated gelatin showed prominent ECM-degrading activities at the cell periphery, whereas control or ICL mutant cells did not (Fig. 6A). We next examined the invasion of cells through matrigel. Compared with control- or FAK mutant-infected SNU449Tp cells, FAK WT-infected cells exhibited dramatically enhanced invasion (Fig. 6B; supplementary material Fig. S10C).
Fig. 6.
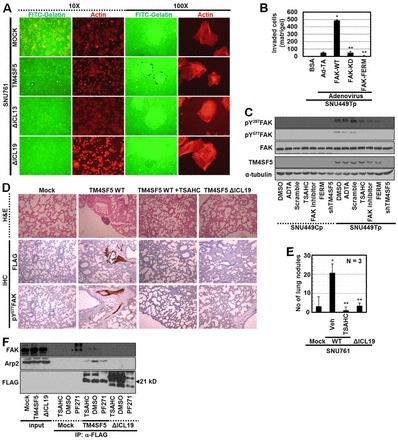
ΔICL19 mutation blocks FAK activity and invasion and metastasis. (A) SNU761 cells stably expressing FLAG-TM4SF5 WT, ΔICL13, or ΔICL19 mutants were cultured on coverglasses precoated with Oregon Green® 488-conjugated gelatin for 24 h. Immunofluorescent microscopic visualization to see degradation of the gelatin and actin were performed. (B) SNU449Tp cells infected with control and diverse adenovirus for FAK forms for 24 h were analyzed for matrigel invasion for 24 h. Invaded cells were stained, imaged and counted for the mean ± standard deviation. (C) Subconfluent cells were treated with diverse reagents or infected with (HA)3-FAK FERM adenovirus or transfected with shRNA against TM4SF5 (shTM4SF5) for 24 h, before harvests and standard western blotting for the indicated molecules. (D) H&E staining or immunohistochemistry of lung tissues using anti-FLAG or pY577FAK antibodies were performed using slices of lung tissue from mice tail-vein-injected with SNU761 cells stably expressing mock, TM4SF5 WT, or ΔICL19 mutant. Mice injected with SNU761-TM4SF5 WT cells were treated with DMSO or TSAHC every other day. (E) Number of nodules on lungs from mice (as in D) to indicate in vivo lung metastasis of TM4SF5 WT (but not ΔICL19 mutant)-expressing SNU761 cells via tail vein injection, depending on TSAHC administration. * P≤0.05 against control virus (Ad-TA; B) or mock (E); **P≤0.05 against FAK WT (B) or TM4SF5 WT (E) cells. (F) Stable SNU761 cells (expressing FLAG-mock, -TM4SF5 WT or -ΔICL19 mutant) were prepared after treatment of DMSO, TSAHC, or FAK inhibitor (PF271) for 24 h. Whole extracts were immunoprecipitated with anti-FLAG antibody, before standard western blotting for the molecules. *Depicts unknown coimmunoprecipitates (at different molecular weights) from FAK inhibitor-treated mock cells. Data represent three independent experiments.
It is important to find the approaches that regulate TM4SF5-mediated FAK activation in order to modulate TM4SF5-mediated migration and invasion capacities. TM4SF5-mediated FAK phosphorylation was tested with or without treatments with different reagents that affect TM4SF5 and/or FAK. The TM4SF5-mediated FAK phosphorylation was unaltered after DMSO treatment, control adenovirus (Ad-TA) infection, or with control shRNA (against a scrambled sequence in TM4SF5) transfection, whereas treatment with an anti-TM4SF5 reagent 4′-(p-toluenesulfonylamido)-4-hydroxychalcone (TSAHC) (supplementary material Fig. S11A,B) or a FAK inhibitor (PF271), myc-FAK-FERM infection, or shTM4SF5 (shRNA against TM4SF5) transfection abolished FAK phosphorylation (Fig. 6C; supplementary material Fig. S11C). Additionally, control compounds lacking the functional group (4′-toluenesulfonylamido group) of TSAHC (i.e. 4′-amino-4-hydroxychalcone and 4′,4-dihydroxychalcone) did not exert any effects on FAK phosphorylation (Lee et al., 2009b).
Increased migration and invasion in cells, in which TM4SF5 can bind FAK, may cause metastasis in vivo. When TM4SF5 WT- or mutant-SNU761 cells were injected into mouse tail veins, immunohistochemistry of the lung tissue showed metastasized-cell masses in the lungs of mice injected with TM4SF5 WT cells, but not with either mock or TM4SF5 ΔICL19 cells (Fig. 6D). TM4SF5 WT cells metastasized efficiently to the lung in vivo, whereas TM4SF5 ΔICL19 cells did not (Fig. 6E; supplementary material Fig. S11D). These observations suggest that the TM4SF5/FAK interaction-mediated FAK activation can enhance migration and invasion of cells, causing lung metastasis in vivo. In addition, coimmunoprecipitation of FAK and Arp2 was observed in the FLAG-TM4SF5 immunoprecipitates from TM4SF5 WT (but not ΔICL19)-expressing cells, which was inhibited by either TSAHC or FAK inhibitor treatment (Fig. 6F). Therefore, TM4SF5 or FAK inhibitors as well as non-binding mutants could block TM4SF5/FAK interaction-mediated FAK phosphorylation. TSAHC was further tested for its ability to block TM4SF5-mediated in vivo lung metastasis. The number of metastatic nodules after the injection of TM4SF5-expressing cells treated with TSAHC dramatically decreased to a level observed in ΔICL19 cell-injected mice (Fig. 6E; supplementary material Fig. S11D). TSAHC treatment caused an insignificant staining of FLAG-TM4SF5-expressing cell masses and Tyr577 FAK phosphorylation in lung tissues (Fig. 6D). These observations suggest that TM4SF5/FAK interaction-mediated phosphorylation (and thus activation) significantly contributes to TM4SF5-mediated metastatic potential.
Discussion
In the present study, we show that TM4SF5 is a membrane protein that directly activates FAK during cell adhesion; the ICL of TM4SF5 directly binds to the F1 lobe of the FAK FERM domain, which appears to release the inhibitory intramolecular interaction in FAK, leading to autophosphorylation (pY397FAK) and activation (pY577FAK). This interaction may cause activation of FAK and actin-polymerizing machinery in the leading edges of migratory cells, and may enhance the metastatic potential of cells presumably during TM4SF5-positive tumor development and progression (Fig. 7).
Fig. 7.
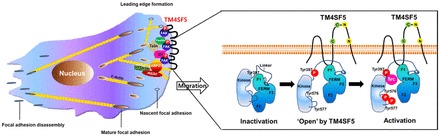
Potential model. This model depicts TM4SF5/FAK interaction-mediated FAK activation at the leading edges of cells, to direct cell migration.
Although integrins are cell adhesion receptors and integrin engagement to the ECM causes the activation and Tyr phosphorylation of diverse signaling or adaptor molecules in FAs, including FAK (Cox et al., 2006), the mechanism by which cell adhesion causes FAK Tyr397 autophosphorylation is not clearly understood (Frame et al., 2010). Recently, it was shown that an intramolecular interaction between the FERM domain (F2 lobe) and the FAK kinase domain (C-lobe centered on Phe596) buries the Tyr397 residue in its inactive form, but upon cell adhesion, this inhibitory interaction is interrupted allowing Tyr397 to be released and trans autophosphorylated (Lietha et al., 2007). In this inactive form of FAK, the Tyr397 autophosphorylation site in the linker between the FERM and the kinase domains is sequestered from the active site by an interaction with the FERM F1 lobe (Frame et al., 2010; Lietha et al., 2007). Thus, the FAK FERM domain is suggested to mediate FAK inhibition at multiple levels including (1) blocking the active site, (2) blocking autophosphorylation, and (3) impeding c-Src recruitment to phosphorylated-Tyr397 (Frame et al., 2010). It was reported that the FAK FERM domain binds phosphatidylinositol-4,5-bisphosphate [PtdIns(4,5)P2] (Cai et al., 2008), suggesting a PtdIns(4,5)P2-mediated conformational change in the FERM domain for FAK activation. Moreover, the FERM domain also binds the c-Met receptor, which mediates FAK phosphorylation, leading to hepatocyte growth factor-enhanced invasion (Chen and Chen, 2006). FAK Tyr194 phosphorylation by c-Met appears to interfere with the inhibitory intramolecular interaction leading to its activation (Chen et al., 2011). The FAK FERM domain also binds EGF receptor, promoting migration via mediation of the intermediary protein SRC3Δ4 after EGF treatment (Long et al., 2010). These previous studies support growth factor (but not adhesion) -mediated FAK activation though interactions of the FERM domain and growth factor receptors. While release of the FERM domain from the kinase domain must occur for Tyr397 to be autophosphorylated, it is currently unknown what causes this release during cell adhesion (Frame et al., 2010). In the present study, we showed that the F1 lobe of the FAK FERM domain was able to bind to the ICL of the TM4SF5 membrane protein and that FAK phosphorylation and activity were enhanced and accelerated after cell adhesion. TM4SF5 is a member of the tetraspan(in) family and is known to collaborate with integrins during cell adhesion and migration (Berditchevski, 2001).
As a membrane glycoprotein with four transmembrane domains, TM4SF5 is involved in cell adhesion-related signaling (Lee et al., 2011). Our previous reports reveal that TM4SF5 collaborates with integrins for different cell functions as explained in the Introduction section, but we could just observe that TM4SF5 expression in hepatocarcinoma cells correlated with enhanced pY577FAK even in normally-cultured conditions without replating (Choi et al., 2009; Lee et al., 2008). However, it was not clear in the previous studies how directly or indirectly the link between TM4SF5 expression and FAK activation occurs. Meanwhile, this current study further reveals that a direct interaction between TM4SF5 and FAK can cause release of the inhibitory intermolecular interaction in FAK leading to its activation, presumably along the leading peripheries of a migratory cell with enhanced FAK/N-WASP/Arp2/cortactin-mediated actin reorganizations.
An NMR study suggested that the binding between the F1 lobe and the ICL induced secondary structural elements of the ICL peptide inside the F1 lobe groove or inside the pocket consisting of L37, L98, and F111 residues. Insertion of the ICL into the pocket of the F1 lobe might reorient the C-terminal β-sheet (β1) and the Tyr397 residue from inside to outside, making its autophosphorylation easier for subsequent c-Src recruitment and Tyr577 phosphorylation (Fig. 7). Although the CD and NMR titration experiments do not provide precise information on the interaction sites, CD spectral changes in the ICL and chemical shift perturbation data of the F1-ICL peptide complex suggest either the reorientation or release of Tyr397 from an intramolecular inhibitory interaction between the FAK FERM and the kinase domain. The K38A mutation in the FAK FERM F1 lobe activates FAK and promotes cell motility (Cohen and Guan, 2005), as well as disrupts Arp2/3 binding for actin polymerization (Serrels et al., 2007). It is thus worthwhile to note that the FERM F1 lobe pocket, which is important for interaction with TM4SF5, includes the L37 residue next to the K38 that is shown to be important for the regulation of FAK activity. Indeed, the L37A or L98A mutations in FAK resulted in dramatically enhanced Tyr397, Tyr577 FAK, and Tyr118 paxillin phosphorylations (supplementary material Fig. S7). Therefore, the observations in our study show that TM4SF5 directly binds to the FAK FERM domain, so that FAK can be released from its inactive form during cell adhesion.
TM4SF5/FAK interaction-mediated FAK activation depends on cell adhesion under certain circumstances, although it is currently unknown how TM4SF5 achieves cell adhesion or what TM4SF5 ligands. We previously reported cross-talk between TM4SF5 and integrins; TM4SF5 interacts extracellularly with integrin α2 (Lee et al., 2010) and intracellularly with integrin α5 (Choi et al., 2009). During cell adhesion, TM4SF5 may collaborate with integrins to activate FAK, presumably via a complex formation that includes TM4SF5, FAK, and integrins. It cannot be ruled out that TM4SF5 might bind and activate FAK at unique locations (i.e. tetraspanin-enriched microdomain or TERM) or integrin-clustered focal adhesions during cell adhesion. TM4SF5-expressing cells replated on PL or pretreated with integrin β1, α5, or αv-neutralizing antibody showed lower FAK phosphorylation levels than those in cells on FN or cells pretreated with normal IgG, indicating a contribution of integrin-mediated FAK phosphorylation. Stable TM4SF5-expressing or control cells showed very similar integrin subunit expression levels on the cell surface, indicating that the differential FAK phosphorylation/activity might not be due to different integrin expression level itself. Meanwhile, we observed in this study that TM4SF5 WT bound both FAK and integrin β1, whereas TM4SF5 ΔICL19 did not bind FAK but still bound integrin β1 (Fig. 3I), and that TM4SF5 ΔICL19-expressing cells showed a lower pY397FAK than did TM4SF5 WT-expressing cells (Fig. 3D, Fig. 5D), it is thus possible that the interaction between FAK and the ICL of TM4SF5 can be important for a more efficient FAK activation under certain circumstances, for example before or during adhesion-mediated integrin clustering/activation.
Our study has used a number of different liver epithelial cells to see the general effects mediated by TM4SF5 expression. TM4SF5 is localized in the pro-migratory morphological features, including the leading edges of cells. Tetraspanins with a membrane topology similar to that of TM4SF5 are suggested to be localized in TERM (Yáñez-Mó et al., 2009), where they form protein-protein complexes in a homophilic or heterophilic manner with other tetraspanins, integrins, or growth factor receptors (Berditchevski, 2001; Stipp et al., 2003). TM4SF5 is shown to regulate not only the phosphorylation of (the present study) but also the localization of FAK and paxillin (Lee et al., 2006), so that TM4SF5 may play a role in the regulation of the dynamic turnover of focal adhesions. We also found that TM4SF5 colocalized with FAK, paxillin, and talin. Although it is not clear whether this colocalization occurs at TERMs, focal adhesions, or both, we observed that a subpopulation of TM4SF5 colocalized with the focal adhesion molecules but the rest of the population did not (Fig. 2E; supplementary material Fig. S9A). Therefore, it is likely that TM4SF5 may be localized both in TERMs and FAs. Furthermore, TM4SF5 further colocalized with phosphorylated FAK and paxillin, Arp2, N-WASP, and Tyr486-phosphorylated cortactin along the leading edges of cells. The interaction between TM4SF5 and Arp2 interestingly also occurs in suspended cells, where FAK is usually unphosphorylated, as previously shown in a study reporting that Arp2 interacts with unphosphorylated FAK (Serrels et al., 2007). However, the interaction between TM4SF5, FAK, and Arp2 was obvious during cell adhesion. It is thus likely that TM4SF5/FAK interaction-mediated FAK phosphorylation/activation can activate actin polymerization and branching machinery, including N-WASP, Arp2, and cortactin, at the leading edges of migratory cells. Furthermore, it was previously reported that TM4SF5 expression in hepatocytes causes RhoA inactivation and concomitant Rac1 activation (Lee et al., 2008), which can also presumably occur at the leading edges of cells (Vicente-Manzanares et al., 2009).
TM4SF5/FAK interaction-mediated FAK activation leads to enhanced migration, invasion, and lung metastasis in vivo. TM4SF5 is highly expressed in many tumor types (Lee et al., 2011) and enhances the speed of cell migration (Lee et al., 2010). Evidences suggest that FAK plays a critical function in new focal contact formation and maturation at the leading cell edges during migration (Schlaepfer and Mitra, 2004). In addition, FAK causes RhoA GTPases activation (Mitra et al., 2005) and is overexpressed in invasive and metastatic tumors (Zhao and Guan, 2009). Therefore, TM4SF5 persistently directs migration in a manner dependent on its interaction with FAK at the leading edges of cells, presumably during the development of metastatic tumors.
With regard to cancer metastasis, our findings suggest that it may be beneficial to identify approaches that can modulate TM4SF5/FAK interaction-mediated FAK activity. Suppression of TM4SF5 or treatment with an anti-TM4SF5 reagent (TSAHC) or a FAK inhibitor attenuated TM4SF5/FAK interaction-mediated FAK phosphorylation and activation. TSAHC was originally screened as an α-glycosidase inhibitor (Seo et al., 2005) and appears to affect the structural integrity or N-glycosylation (at N138/N155) of the extracellular loop 2 (EC2) within TM4SF5, which appears to be important for multilayer growth and migration (Lee et al., 2009b). TSAHC or FAK inhibitor treatment attenuated the interaction between TM4SF5 and FAK, indicating that both the structural aspects of the extracellular region of TM4SF5 and the activity of intracellular FAK are able to modulate the interaction between TM4SF5 and FAK. It is thereby likely that the linkage between TM4SF5 and FAK allows cells to bi-directionally communicate with extracellular cues during migration. In addition, mutations in which the interacting region of either TM4SF5 or FAK is deleted caused insignificant FAK activation, migration, invasion and in vivo lung metastasis, supporting the biological significance of the TM4SF5/FAK interaction in the regulation of cell migration. Moreover, growth rates between cells expressing TM4SF5 WT and deletion mutants did not show any difference (unpublished data), indicating that TM4SF5/FAK interaction-mediated activation may not be important for tumor growth, but may be crucial for metastasis.
Taken together, our results show that the interaction between TM4SF5 and FAK causes FAK activation in a cell adhesion-dependent manner, recapitulating a cell adhesion-dependent mechanism in which a membrane protein activates FAK at the leading edges of migratory cells. Impairing this interaction may be effective to prohibit the migration of diverse TM4SF5-positive tumor cells, especially during metastases.
Materials and Methods
Cell culture
Control (SNU449, SNU761 parental, SNU449Cp, and SNU761-mock), TM4SF5 WT (SNU449Tp and SNU761-TM4SF5 WT), or mutant (SNU761-TM4SF5 ΔICL13, or ΔICL19) -expressing human hepatocellular carcinoma cells have been described previously (Lee et al., 2008) or prepared by G418 (A.G. Scientifics, San Diego, CA) selection following transfection of FLAG-mock, -TM4SF5 WT, -TM4SF5 ΔICL13, or -TM4SF5 ΔICL19 plasmids into TM4SF5-null SNU761 hepatocytes. Stable cells were maintained in RPMI-1640 (WelGene, Daegu, Korea) containing 10% FBS, G418 (250 µg/ml) and antibiotics (Invitrogen, Grand Island, NY). Endogenously TM4SF5-expressing HepG2 and Huh7 cells were maintained in Dulbecco's modified Eagle medium, high glucose (DMEM-H, WelGene) containing 10% FBS and antibiotics (Invitrogen).
Peptides and cDNA mutagenesis
The TM4SF5 cDNA sequence (NM-003963, 197 aa) was conjugated to pFLAG, and deletion mutations in the ICL were constructed to produce 69RA-90SV (ΔICL19 with a deletion of 71GGKGCCGAGCCGNRCRMLR89) or 69RAGGK-87MLRSV (ΔICL13 with a deletion of 74GCCGAGCCGNRCR86) (Fig. 2A). For in vitro peptide pull-down, TM4SF5 peptides 1MCTGKCARCV10 (N-terminally cytosolic), 69RAGGKGCCGAGCCGNRCRMLRSV91 (ICL), 187GDCRKKQDTPH197 (C-terminally cytosolic), or L6 (M90657, 202 aa) peptide 72QDDCCGCCGHENCGKRCAMLS92 (ICL) were commercially synthesized to contain N-terminally-linked biotin without modifications and then purified (90%, Peptron Inc., Santa Clara, CA). Adenovirus encoding (HA)3-FAK WT, different mutants (kinase dead K454R, KD; FAKΔ(1–100) with a deletion from aa 1 to 100; FAK-related non-kinase, FRNK), or myc-FAK FERM (aa 1–402) have been previously explained (Lim et al., 2008). GST-FERM F1 (aa 33–132), F2 (aa 128–252), or F3 (aa 253–362) lobe constructs have been previously described (Lim et al., 2008). Similarly, GST-FAKNT (aa 1–423), GST-FAKPR1PR2 (aa 711–877), and GST-FAKCD (aa 677–1052) have also been previously described (Oh et al., 2009). pEGFP-TM4SF5, myc-(His)6-TM4SF5, FLAG-TM4SF5, (HA)3-FAK point mutation (Y397F, Y577F, L37A, or L98A), and GST-paxillin constructs were engineered and confirmed by direct sequence analyses.
Extract preparation and western blots
Subconfluent cells in media containing 10% FBS were harvested, or cells infected or transiently transfected with adenovirus or plasmids, respectively, for 24 or 48 h were either harvested for whole-cell lysates or analyzed by indirect immunofluorescence. In some cases, cells were transiently transfected with shRNA against control scrambled sequence or TM4SF5 [shTM4SF5 (Lee et al., 2006) or GFP-shTM4SF5 (Origene, Rockville, MD)] for 48 h; treated with DMSO or pharmacological inhibitors, including TSAHC (20 μM) against TM4SF5 (Lee et al., 2009b) or PF271 (1.0 µM) against FAK (Pasapera et al., 2010) for 24 h; or pretreated with normal mouse IgG or neutralizing anti-integrin β1, α5, or αv antibody (Invitrogen, clone P4C10, P1D6, LM609, respectively, 20 µg/ml) for 30 min in the middle of rocking prior to replating. For replating, cells were trypsinized and washed with serum-free media containing 1% BSA twice before rocking (60 rpm) at 37°C for 1 h and then either kept in suspension or replated on PL (Sigma, St. Louis, MO) or FN (10 µg/ml)-precoated culture dishes for the indicated times. The cells were then harvested using a lysis buffer containing 1% Brij58, 0.1% sodium dodecyl sulfate (SDS), 150 mM NaCl, 20 mM HEPES, pH 7.4, 2 mM MgCl2, 2 mM CaCl2, and protease inhibitors, or processed for immunostaining. Primary antibodies used are included in supplementary material Table S1.
In vitro pull-down
In vitro pull-down experiments using biotin-conjugated peptides of the cytosolic TM4SF5 region, or using GST-fused TM4SF5 and extracts from cells infected or transfected with different FAK WT or mutants were performed using streptavidin agarose beads or immunoprecipitates using protein A/G beads (Upstate Biotech., Billerica, MA), as previously described (Oh et al., 2009) or as following. pEBG-FERM, F1, F2, or F3 (mammalian GST fusion protein expression vector) were transfected to HEK293 cells for 48 h. The cytosolic peptides of TM4SF5 (1 µM) was incubated with the whole extracts of HEK293 cells using the Brij58-containing lysis buffer (see above) overnight at 4°C. The proteins bound to the beads were washed with ice-cold lysis buffer (3 times) and phosphate buffered saline (3 times), prior to sample preparation with 2× SDS-PAGE sample buffer. The proteins were then subjected to standard western blots. The GST-FAK fragments bound to Glutathione-Sepharose 4 Fast Flow (Amersham Bioscience, Piscataway, NJ) were assumed to be functionally folded because these have successfully been used for in vitro binding to a tyrosine kinase Fer (Oh et al., 2009).
In vitro kinase assay
In vitro FAK assay was performed using anti-HA or -myc immunoprecipitates prepared from whole extracts of cells infected with adenovirus for (HA)3-FAK WT, KD mutant, or myc-FAK FERM, or using anti-FAK immunoprecipitates from SNU449Cp or SNU449Tp cells for Tyr118 phosphorylation of recombinant GST or GST-paxillin, as previously described (Kim et al., 2008).
Coimmunoprecipitation
Whole-cell extracts were immunoprecipitated with anti-HA, or anti-FLAG antibody-precoated agarose beads (Sigma) overnight, prior to immunoblotting for the indicated molecules. Alternatively, cell extracts were incubated with anti-(His)6 or FAK antibody overnight prior to protein A/G bead precipitation for 2 h. Immunoprecipitated proteins were boiled in 2× SDS-PAGE sample buffer before subjecting them to standard western blots.
Indirect immunofluorescence
Confocal images were acquired on a microscope (LSM 510, Zeiss) using a 40×/1.4 NA (oil) Plan-neofluar or 63×/1.4 NA Apochromat objective lens (Zeiss). Green fluorescent protein (GFP, or fluorescein isothiocyanate FITC) and tetramethylrhodamine isothiocyanate (TRITC) were excited using the 488 nm laser line of an Ar ion laser and the 543 nm laser line of a He-Ne laser (CVI Melles Griot, Carlsbad, CA), respectively. All confocal image processing was performed using the Zeiss LSM 5 image examiner software. Epifluorescent images were acquired on a microscope (BX51TR, Olympus) using 40×/0.75 or 100×/1.3 (oil) NA U plan semi Apochromat objective lens (Olympus). The filters for GFP or yellow fluorescent protein (YFP) were XF105-2 or XF115-2, respectively. A JVC KY-F75U CCD digital camera with IEEE 1394 interface was used. The TOMORO image analyzer software (Techsan Community Co., Korea) was used for image analysis. For quantification of fluorescence intensities, nonsaturated images were captured with a full-open pinhole. For multichannel imaging, each fluorescent dye was imaged sequentially in the frame-interlace mode to eliminate cross-talk between the channels. Cells were transiently transfected with pEGFP-TM4SF5, FLAG-TM4SF5 WT, or -TM4SF5 ΔICL19 for 48 h, prior to visualization or staining, by using phalloidin for detection of actin or an antibody against pY397FAK, pY577FAK, pY118paxillin, or talin. In some cases, cells were transiently transfected with pEGFP-TM4SF5, and a wound was made in the middle of the confluent cell layer, which was washed twice prior to actin staining. GFP-positive cells along the wound edges were then analyzed for actin and TM4SF5 staining.
Cell surface protein analysis
Localization of TM4SF5 WT and ΔICL19 mutant on plasma membranes was analyzed using Pierce® Cell Surface Isolation Kit (Pierce, Rockford, IL) by following the manufacturer's protocol. Subconfluent SNU761-mock, -TM4SF5 WT, or TM4SF5 ΔICL19 mutant cells were biotinylated for 30 min at 4°C with ensuring even coverage of the cells with the labeling solution of the kit, prior to preparation of biotinylated surface protein samples. Immunoblotting of the samples were performed in parallel with lysates, using anti-FLAG antibody.
Transwell migration or invasion assay
Stable SNU449 or SNU761 cells were analyzed for migration or invasion using transwell chambers with 8-µm pores (Corning, Corning, NY), as described previously (Lee et al., 2010). In some cases, SNU449Tp cells were transiently transfected with FLAG TM4SF5 WT or deletion mutants (ΔICL19 or ΔICL13) for 48 h prior to the migration assay. The migration assay was performed separately either for haptotaxis via coating the membrane with FN (10 µg/ml) or for chemotaxis using 10% FBS.
Invasive protrusion analysis
Invasive protrusions of cells on Oregon Green® 488-conjugated-gelatin (Invitrogen) were analyzed as described in a previous report (Lee et al., 2010).
In vivo lung metastasis analysis
SNU761 cells stably expressing TM4SF5 WT or ΔICL19 (5×106 cells/100 µl RPMI-1640) were injected into the lateral tail vein of female BALB/c-n/n mice (n = 3, Orient. Co. Ltd). In some cases, 50 mg/kg TSAHC (in 40% DMSO) was intraperitoneally injected every other day. All animal procedures were performed according to the procedures approved in the in the Seoul National University Laboratory Animal Maintenance Manual and Institutional Review Board agreement. All mice were sacrificed 6 weeks after cell injection, and their lungs were weighed and harvested. Metastatic colonies were counted macroscopically on the lung surface after staining with Bouin's solution (Sigma). Formalin-fixed lungs were stained with hematoxylin and eosin or processed for immunohistochemistry using anti-FLAG M2 (Sigma) or -pY577FAK antibody, before visualization at 100×. Alternatively, the lung pieces were frozen in liquid N2 until extract preparation, prior to standard western blot analysis.
Protein sample preparation for NMR study
The amplified DNA fragment of the F1 lobe (aa 33–132) was cloned into the expression vector pET-21a (Novagen, Darmstadt, Germany). The resulting construct [(His)6-tagged F1 lobe] contains eight nonnative residues at the C-terminus (LEHHHHHH) that facilitate protein purification. The 13C/15N-labeled protein was overexpressed in Escherichia coli strain BL21 (DE3) cells using modified M9 medium supplemented with 50 mg/L ampicillin at 37°C. Cells were transferred into a M9 medium containing 1 g/L 13C-glucose (Cambridge Isotope Laboratories, Inc., Andover, MA) and 1 g/L 15NH4Cl (Cambridge Isotope Laboratories, Inc.) and grown to an OD600 of 0.5. Isopropyl β-D-1-thiogalactopyranoside was added to a final concentration of 0.5 mM and cells were cultured for 4 h at 37°C. Harvested cells were lysed by sonication in 20 mM Tris buffer (pH 7.9) containing 0.5 M NaCl, 1 mM phenylmethylsulfonyl fluoride (PMSF), and protease inhibitor cocktail (Roche, St. Nutley, NJ). After centrifugation, inclusion bodies were solubilized using 8 M urea buffer. The lysate was applied to a Ni2+ affinity column (Chelating Sepharose Fast Flow resin, Pharmacia) and eluted by addition of 200 mM imidazole at room temperature. NMR samples were concentrated to ∼1.5 mM using Amicon Ultra concentrator (5000 MWCO; Millipore, Billerica, MA) in pH 6.0, 20 mM MES, 50 mM NaCl, 1 mM DTT, 0.1 mM PMSF, and 200 mM perdeutrated SDS. The identity and purity of the protein were analyzed by FPLC, SDS-PAGE and NMR. The NMR sample was an ∼1.5 mM 15N- and 15N/13C-labeled protein prepared in a 90% H2O/10% D2O buffer solution.
Peptide preparation for structural study
A 23-residue peptide corresponding to the ICL region (Arg69-Val91; NH2-RAGGKGCCGAGCCGNRCRMLRSV-COOH) of TM4SF5 was commercially synthesized and purified to 96.9% purity (AnyGen, Gwangju, Korea).
CD titration experiments
Standard far-UV CD spectra of the purified (His)6-tagged F1 lobe, ICL and F1-ICL complex were recorded using a 2 mm path length cuvette in a J-715 spectropolarimeter (Jasco, Easton, MD) equipped with a peltier temperature control system (Model PTC-348WI). Spectra were acquired after the addition of increasing amounts of ICL peptide up to a molar ratio of 3∶1 ICL peptide:/F1 lobe. Final records were averaged over three scans collected at a speed of 10 nm/min over a 190–250 nm interval, subtracting the blank.
NMR spectroscopy
All NMR spectra were acquired at 308 K on a 900 MHz Bruker AVANCE spectrometer using a cryoprobe. The ratio of the (His)6-tagged-F1 protein versus the ICL peptide was 1∶3 (11 mg/ml for 1 mM: 3 mM) for the experiments. The backbone assignments were performed manually from the following series of experiments: 3D transverse relaxation optimized spectroscopy (TROSY)-type HNCO, HN(CA)CO, HNCA, HN(CO)CA, HNCACB, and HN(CO)CACB spectra. Therefore, to avoid peak broadening effects by increased molecular weights caused due to the molecular micelles of SDS, which was used for the refolding of (His)6-tagged F1 proteins, we performed TROSY that is useful for high molecular samples rather than HSQC pulse sequence. Chemical shifts were referenced to sodium 2,2-dimethyl-2-sila-pentane-5-sulfonate (DSS) externally. The NMRPipe/NMRDraw (Delaglio et al., 1995) and NMRView (Johnson, 2004) programs were used for data processing and spectral analysis, respectively. The unlabeled ICL peptides were titrated into the F1 lobe. The chemical shift changes were monitored via 1H-15N TROSY and the difference in the amide proton chemical shift was detected by comparing the trHNCA spectra. The perturbation of the HN chemical shift along the aa sequence was quantified.
Statistical methods
Student's t-tests were performed for comparisons of mean values to determine significance. P≤0.05 was considered significant.
Supplementary Material
Acknowledgments
O.J. performed the experiments. S.B.J., D.H.K., H.K.C., and B.J.L. helped with structural studies. S.C., H.J.K., Y.J.C., S.A.L., T.K.K., H.K., M.K., M.S.L., S.Y.P., J.R., D.J. and H.J.K. helped with animal studies and experimental tool preparations. K.H.P. helped with TSAHC drug synthesis. S.T.L. and D.D.S. helped with constructs and virus and discussed for experimental designs. J.W.L. designed experiments, analyzed data, and wrote the manuscript. All authors discussed the data. The authors have no conflicts of interest.
Footnotes
Funding
This work was supported by the Korea Healthcare Technology R&D Project, MHWFA, Republic of Korea [grant number A092006 to B.-J.L.];, a grant from High Field NMR Research Program of Korea Basic Science Institute (to B.-J. L.); the National Research Foundation (NRF) by the MEST of Korea for Tumor Microenvironment Global Core Research Center (GCRC) grant, [grant number 2012-0004891]; senior researchers program (Leap research) [grant number 2012-0005606]; and Global Frontier Project grant [grant number NRF-M1AXA002-2011-0028411], and a grant of the Korean Health Technology R&D Project [grant number A100727], MHWFA, Republic of Korea to J.W.L.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.100586/-/DC1
References
- Berditchevski F. (2001). Complexes of tetraspanins with integrins: more than meets the eye. J. Cell Sci. 114, 4143–4151 [DOI] [PubMed] [Google Scholar]
- Bolós V., Gasent J. M., López–Tarruella S., Grande E. (2010). The dual kinase complex FAK-Src as a promising therapeutic target in cancer. Onco. Targets Ther. 3, 83–97 10.2147/OTT.S6909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X., Lietha D., Ceccarelli D. F., Karginov A. V., Rajfur Z., Jacobson K., Hahn K. M., Eck M. J., Schaller M. D. (2008). Spatial and temporal regulation of focal adhesion kinase activity in living cells. Mol. Cell. Biol. 28, 201–214 10.1128/MCB.01324-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S. Y., Chen H. C. (2006). Direct interaction of focal adhesion kinase (FAK) with Met is required for FAK to promote hepatocyte growth factor-induced cell invasion. Mol. Cell. Biol. 26, 5155–5167 10.1128/MCB.02186-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T. H., Chan P. C., Chen C. L., Chen H. C. (2011). Phosphorylation of focal adhesion kinase on tyrosine 194 by Met leads to its activation through relief of autoinhibition. Oncogene 30, 153–166 10.1038/onc.2010.398 [DOI] [PubMed] [Google Scholar]
- Choi S., Oh S-R., Lee S-A., Lee S-Y., Ahn K., Lee H-K., Lee J. W. (2008). Regulation of TM4SF5-mediated tumorigenesis through induction of cell detachment and death by tiarellic acid. Biochim. Biophys. Acta 1783, 1632–1641 10.1016/j.bbamcr.2008.04.008 [DOI] [PubMed] [Google Scholar]
- Choi S., Lee S. A., Kwak T. K., Kim H. J., Lee M. J., Ye S. K., Kim S. H., Kim S., Lee J. W. (2009). Cooperation between integrin α5 and tetraspan TM4SF5 regulates VEGF-mediated angiogenic activity. Blood 113, 1845–1855 10.1182/blood-2008-05-160671 [DOI] [PubMed] [Google Scholar]
- Cohen L. A., Guan J. L. (2005). Residues within the first subdomain of the FERM-like domain in focal adhesion kinase are important in its regulation. J. Biol. Chem. 280, 8197–8207 10.1074/jbc.M412021200 [DOI] [PubMed] [Google Scholar]
- Cox B. D., Natarajan M., Stettner M. R., Gladson C. L. (2006). New concepts regarding focal adhesion kinase promotion of cell migration and proliferation. J. Cell. Biochem. 99, 35–52 10.1002/jcb.20956 [DOI] [PubMed] [Google Scholar]
- Danen E. H. (2009). Integrin proteomes reveal a new guide for cell motility. Sci. Signal. 2, pe58 10.1126/scisignal.289pe58 [DOI] [PubMed] [Google Scholar]
- Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995). NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 10.1007/BF00197809 [DOI] [PubMed] [Google Scholar]
- Frame M. C., Patel H., Serrels B., Lietha D., Eck M. J. (2010). The FERM domain: organizing the structure and function of FAK. Nat. Rev. Mol. Cell Biol. 11, 802–814 10.1038/nrm2996 [DOI] [PubMed] [Google Scholar]
- Friedl P., Wolf K. (2009). Proteolytic interstitial cell migration: a five-step process. Cancer Metastasis Rev. 28, 129–135 10.1007/s10555-008-9174-3 [DOI] [PubMed] [Google Scholar]
- Johnson B. A. (2004). Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 278, 313–352 [DOI] [PubMed] [Google Scholar]
- Kim Y. B., Choi S., Choi M. C., Oh M. A., Lee S. A., Cho M., Mizuno K., Kim S. H., Lee J. W. (2008). Cell adhesion-dependent cofilin serine 3 phosphorylation by the integrin-linked kinase.c-Src complex. J. Biol. Chem. 283, 10089–10096 10.1074/jbc.M708300200 [DOI] [PubMed] [Google Scholar]
- Lee S. Y., Kim Y. T., Lee M. S., Kim Y. B., Chung E., Kim S., Lee J. W. (2006). Focal adhesion and actin organization by a cross-talk of TM4SF5 with integrin α2 are regulated by serum treatment. Exp. Cell Res. 312, 2983–2999 10.1016/j.yexcr.2006.06.001 [DOI] [PubMed] [Google Scholar]
- Lee S. A., Lee S. Y., Cho I. H., Oh M. A., Kang E. S., Kim Y. B., Seo W. D., Choi S., Nam J. O., Tamamori–Adachi M.et al. (2008). Tetraspanin TM4SF5 mediates loss of contact inhibition through epithelial-mesenchymal transition in human hepatocarcinoma. J. Clin. Invest. 118, 1354–1366 10.1172/JCI33768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. A., Kim Y. M., Kwak T. K., Kim H. J., Kim S., Ko W., Kim S. H., Park K. H., Kim H. J., Cho M.et al. (2009a). The extracellular loop 2 of TM4SF5 inhibits integrin α2 on hepatocytes under collagen type I environment. Carcinogenesis 30, 1872–1879 10.1093/carcin/bgp234 [DOI] [PubMed] [Google Scholar]
- Lee S. A., Ryu H. W., Kim Y. M., Choi S., Lee M. J., Kwak T. K., Kim H. J., Cho M., Park K. H., Lee J. W. (2009b). Blockade of four-transmembrane L6 family member 5 (TM4SF5)-mediated tumorigenicity in hepatocytes by a synthetic chalcone derivative. Hepatology 49, 1316–1325 10.1002/hep.22777 [DOI] [PubMed] [Google Scholar]
- Lee S. A., Kim T. Y., Kwak T. K., Kim H., Kim S., Lee H. J., Kim S. H., Park K. H., Kim H. J., Cho M.et al. (2010). Transmembrane 4 L six family member 5 (TM4SF5) enhances migration and invasion of hepatocytes for effective metastasis. J. Cell. Biochem. 111, 59–66 10.1002/jcb.22662 [DOI] [PubMed] [Google Scholar]
- Lee S. A., Park K. H., Lee J. W. (2011). Modulation of signaling between TM4SF5 and integrins in tumor microenvironment. Front. Biosci. 16, 1752–1758 10.2741/3818 [DOI] [PubMed] [Google Scholar]
- Lietha D., Cai X., Ceccarelli D. F., Li Y., Schaller M. D., Eck M. J. (2007). Structural basis for the autoinhibition of focal adhesion kinase. Cell 129, 1177–1187 10.1016/j.cell.2007.05.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S-T., Chen X. L., Lim Y., Hanson D. A., Vo T-T., Howerton K., Larocque N., Fisher S. J., Schlaepfer D. D., Ilic D. (2008). Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol. Cell 29, 9–22 10.1016/j.molcel.2007.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long W., Yi P., Amazit L., LaMarca H. L., Ashcroft F., Kumar R., Mancini M. A., Tsai S. Y., Tsai M. J., O'Malley B. W. (2010). SRC-3δ4 mediates the interaction of EGFR with FAK to promote cell migration. Mol. Cell 37, 321–332 10.1016/j.molcel.2010.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M., Guan J. L. (2010). Focal adhesion kinase: a prominent determinant in breast cancer initiation, progression and metastasis. Cancer Lett. 289, 127–139 10.1016/j.canlet.2009.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean G. W., Carragher N. O., Avizienyte E., Evans J., Brunton V. G., Frame M. C. (2005). The role of focal-adhesion kinase in cancer – a new therapeutic opportunity. Nat. Rev. Cancer 5, 505–515 10.1038/nrc1647 [DOI] [PubMed] [Google Scholar]
- Mitra S. K., Hanson D. A., Schlaepfer D. D. (2005). Focal adhesion kinase: in command and control of cell motility. Nat. Rev. Mol. Cell Biol. 6, 56–68 10.1038/nrm1549 [DOI] [PubMed] [Google Scholar]
- Oh M-A., Choi S., Lee M. J., Choi M-C., Lee S-A., Ko W., Cance W. G., Oh E-S., Buday L., Kim S-H.et al. (2009). Specific tyrosine phosphorylation of focal adhesion kinase mediated by Fer tyrosine kinase in suspended hepatocytes. Biochim. Biophys. Acta 1793, 781–791 10.1016/j.bbamcr.2009.01.015 [DOI] [PubMed] [Google Scholar]
- Pasapera A. M., Schneider I. C., Rericha E., Schlaepfer D. D., Waterman C. M. (2010). Myosin II activity regulates vinculin recruitment to focal adhesions through FAK-mediated paxillin phosphorylation. J. Cell Biol. 188, 877–890 10.1083/jcb.200906012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz–Moreno V., Marshall C. J. (2010). The plasticity of cytoskeletal dynamics underlying neoplastic cell migration. Curr. Opin. Cell Biol. 22, 690–696 10.1016/j.ceb.2010.08.020 [DOI] [PubMed] [Google Scholar]
- Schaller M. D. (2010). Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. J. Cell Sci. 123, 1007–1013 10.1242/jcs.045112 [DOI] [PubMed] [Google Scholar]
- Schlaepfer D. D., Mitra S. K. (2004). Multiple connections link FAK to cell motility and invasion. Curr. Opin. Genet. Dev. 14, 92–101 10.1016/j.gde.2003.12.002 [DOI] [PubMed] [Google Scholar]
- Seo W. D., Kim J. H., Kang J. E., Ryu H. W., Curtis–Long M. J., Lee H. S., Yang M. S., Park K. H. (2005). Sulfonamide chalcone as a new class of α-glucosidase inhibitors. Bioorg. Med. Chem. Lett. 15, 5514–5516 10.1016/j.bmcl.2005.08.087 [DOI] [PubMed] [Google Scholar]
- Serrels B., Serrels A., Brunton V. G., Holt M., McLean G. W., Gray C. H., Jones G. E., Frame M. C. (2007). Focal adhesion kinase controls actin assembly via a FERM-mediated interaction with the Arp2/3 complex. Nat. Cell Biol. 9, 1046–1056 10.1038/ncb1626 [DOI] [PubMed] [Google Scholar]
- Stipp C. S., Kolesnikova T. V., Hemler M. E. (2003). Functional domains in tetraspanin proteins. Trends Biochem. Sci. 28, 106–112 10.1016/S0968-0004(02)00014-2 [DOI] [PubMed] [Google Scholar]
- Vicente–Manzanares M., Choi C. K., Horwitz A. R. (2009). Integrins in cell migration – the actin connection. J. Cell Sci. 122, 199–206 10.1242/jcs.018564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright M. D., Ni J., Rudy G. B. (2000). The L6 membrane proteins – a new four-transmembrane superfamily. Protein Sci. 9, 1594–1600 10.1110/ps.9.8.1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L., Bernard–Trifilo J. A., Lim Y., Lim S. T., Mitra S. K., Uryu S., Chen M., Pallen C. J., Cheung N. K., Mikolon D.et al. (2008). Distinct FAK-Src activation events promote α5β1 and α4β1 integrin-stimulated neuroblastoma cell motility. Oncogene 27, 1439–1448 10.1038/sj.onc.1210770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H., Condeelis J. (2007). Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim. Biophys. Acta 1773, 642–652 10.1016/j.bbamcr.2006.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yáñez–Mó M., Barreiro O., Gordon–Alonso M., Sala–Valdés M., Sánchez–Madrid F. (2009). Tetraspanin-enriched microdomains: a functional unit in cell plasma membranes. Trends Cell Biol. 19, 434–446 10.1016/j.tcb.2009.06.004 [DOI] [PubMed] [Google Scholar]
- Zhao J., Guan J. L. (2009). Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 28, 35–49 10.1007/s10555-008-9165-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.