

Summary
Soluble guanylyl cyclase (sGC) is the principal receptor for nitric oxide (NO) and crucial for the control of various physiological functions. The β1 subunit of sGC is obligatory for the biological stability and activity of the sGC heterodimer. MicroRNAs (miRNAs) are important regulators of gene expression and exert great influences on diverse biological activities. The aim of the present study was to determine whether or not the expression of sGCβ1 is specifically regulated by miRNAs. We report that miR-34c-5p directly targets sGCβ1 under hypoxia. Bioinformatics analysis of the sGCβ1 3′-untranslated region (3′-UTR) revealed a putative binding site for miR-34b-5p and miR-34c-5p, but only miR-34c-5p inhibited luciferase activity through interaction with sGCβ1 3′-UTR in HEK293T cells. Site-directed mutagenesis of the putative miR-34c-5p binding site abolished the negative regulation of luciferase expression. Overexpression of miR-34c-5p repressed the expression of sGCβ1 in stable cell lines, which was reversed by miR-34c-5p-specific sponge. Inoculation of mouse lung tissues in vitro with lentivirus bearing miR-34c-5p significantly decreased both the expression of sGCβ1 and NO-stimulated sGC activity, which was also rescued by miR-34c-5p-specific sponge. Furthermore, we identified the putative Sp1-binding site in the promoter region of miR-34c-5p. Luciferase reporter constructs revealed that Sp1 directly binds to the wild-type promoter of miR-34c-5p, which was confirmed by chromatin immunoprecipitation. In summary, these findings reveal that miR-34c-5p directly regulates sGCβ1 expression, and they identify the key transcription factor Sp1 that governs miR-34c-5p expression during hypoxia.
Key words: MicroRNA, miR-34c-5p, Soluble guanylyl cyclase, Hypoxia
Introduction
The nitric oxide (NO)-soluble guanylyl cyclase (sGC)–cGMP pathway has been implicated in a variety of biological processes, such as vasodilatation, inhibition of platelet aggregation and leukocyte adhesion, neuromodulation and neurotransmission (Sharina et al., 2003; Sharina et al., 2011). To mediate these effects, endogenous and exogenous NO binds to the ferrous iron of sGC heme prosthetic group, triggering a conformational change of sGC followed by the activation of the sGC enzyme. sGC in turn converts GTP to the second messenger cGMP, which exerts physiological effects by the activation of cGMP-dependent protein kinase, cGMP-regulated phosphodiesterases and cGMP-gated ion channels (Francis et al., 2010).
sGC is gaining attention as a therapeutic target in cardiopulmonary disease, with several sGC agonists currently in clinical development (Stasch et al., 2011). sGC functions as an obligatory heterodimer, consisting of a large α-subunit and a smaller haem-binding β-subunit. In mammals, two isoforms of each subunit are currently known to exist, termed α1,2 and β1,2. To date, β2-containing heterodimer has not been identified in native tissues, only α1β1 and α2β1 heterodimers have been found, at the protein level (Sharina et al., 2003). The β1 subunit contains an evolutionarily conserved N-terminal heme-binding domain, which is crucial for the stimulatory effect of NO (Evgenov et al., 2006). The deletion of the β1 subunit in mice greatly affects neonatal survival, blood pressure and neuronal phenotypes (Friebe and Koesling, 2009).
MicroRNAs (miRNAs) represent an evolutionarily conserved family of small non-coding RNAs, which post-transcriptionally regulate gene expression, predominantly through sequence-specific interactions with the 3′-untranslated region (3′-UTR) of target mRNAs (Valastyan et al., 2009; Smith-Vikos and Slack, 2012). miRNAs modulate diverse biological processes, including cell proliferation, differentiation and apoptosis (Kasinski and Slack, 2011). Increasing evidence indicates that aberrant miRNAs expression is involved in many diseases such as cardiovascular disease (Abdellatif, 2012), cancer (Deng et al., 2011; Schramedei et al., 2011) and autoimmune disease (Du et al., 2009; Ma et al., 2011). Hence, miRNAs are emerging as promising target for therapeutics.
Despite the critical role of the β1 subunit in the NO signaling pathway, our understanding of underlying mechanisms that govern regulation of the β1 subunit remains very limited. The pleiotropic nature of gene regulation by miRNAs prompted us to hypothesize that certain miRNAs might exert a suppressive effect on the expression of β1 subunit. The present work was to determine the role of miRNAs in regulating β1 subunit expression and sGC activity.
Results
Hypoxia decreases sGCβ1 expression in mouse lung
To investigate the effects of hypoxia on sGCβ1 expression, mice were exposed to 8% O2 for 72 hours or 120 hours. Western blot analysis showed that sGCβ1 expression in the crude homogenates obtained from mouse lungs exposed to 72 hours and 120 hours hypoxia was significantly decreased compared to normoxic lungs (Fig. 1A). After 120 hours hypoxic exposure, sGCβ1 expression was reduced by 52% compared with the normoxic control (P<0.01). In the contrast, the levels of sGCβ1 from mouse kidney and heart were not changed (Fig. 1A).
Fig. 1.
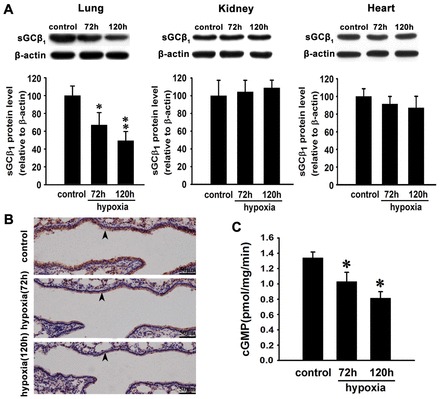
Hypoxia decreases sGCβ1 subunit expression and NO-stimulated sGC activity in mouse lung. (A) Western blot analysis and quantification of sGCβ1 subunit expression in lungs, kidneys and hearts of mice exposed to hypoxia for 72 hours or 120 hours. Expression levels are normalized to β-actin and expressed as the means ± s.e.m. (n = 4; *P<0.05, **P<0.01 versus control). (B) Immunohistochemical microscopy to assess sGCβ1 subunit expression in lungs of mice exposed to normoxia and hypoxia for 72 hours or 120 hours. Arrowheads indicate the sGCβ1-positive signals in the vascular wall. (C) NO-stimulated sGC activity in mouse lungs exposed to normoxia and hypoxia. There was a significant decrease of cGMP levels in mouse lungs after 72 hours and 120 hours of hypoxia treatment compared with normoxia-treated animals. Data are expressed as means ± s.e.m. (n = 4; *P<0.05 versus control).
To further confirm the effect of hypoxia on sGCβ1, we performed immunohistochemistry using paraffin-embedded lung tissues. The results showed a substantial decrease of sGCβ1 expression in the vascular wall of hypoxia-treated mouse lung compared with normoxic control (Fig. 1B).
Hypoxia decreases NO-stimulated sGC activity
To determine whether or not sGC activity was affected by hypoxia, we measured the cGMP levels in response to DETA NONOate in the crude homogenate supernatant. DETA NONOate is a stable NO donor and releases NO in a spontaneous manner with a half-life of 20 hours at 37°C (Zheng et al., 2011). A significant decrease in sGC activity occurred in mouse lung exposed to hypoxia at 72 hours and 120 hours compared with those of normoxic controls (Fig. 1C; P<0.05).
The sGCβ1 subunit is a functional target of miR-34c-5p
To examine whether certain miRNAs are involved in regulating sGCβ1 during hypoxia, we employed an online public platform, Starbase (Yang et al., 2011) (http://starbase.sysu.edu.cn/index.php), which contains five prediction software algorithms (TargetScan, PicTar, PITA, miRanda and RNA22), to identify potential miRNAs that target the sGCβ1 subunit. miRNAs that could be identified by at least two prediction algorithms were chosen as the candidate miRNAs and miR-34b-5p and miR-34c-5p were selected. Quantitative real-time PCR (qRT-PCR) was performed to determine the expression of miR-34b-5p and miR-34c-5p during normoxia and hypoxia. The results showed that miR-34b-5p was upregulated by 2- and 4.75-fold, and miR-34c-5p by 2.5- and 8.75-fold after 72 hours and 120 hours hypoxic exposure, respectively (Fig. 2A).
Fig. 2.
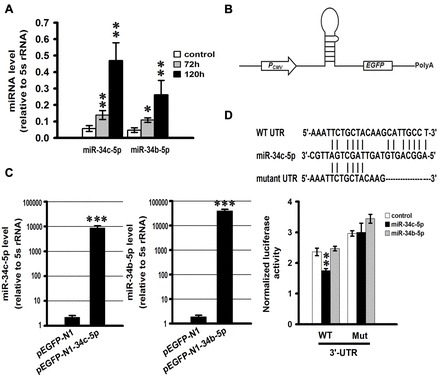
sGCβ1 subunit is a direct target of miR-34c-5p. (A) The expression of miR-34b-5p and miR-34c-5p was increased in mouse lungs treated with hypoxia. miR-34b-5p and miR-34c-5p expression was determined by quantitative real-time PCR (qRT-PCR) in the mouse lungs from the normoxic and hypoxic groups at 72 hours and 120 hours. The levels of miRNAs are relative to the control 5 s rRNA. Data are expressed as means ± s.e.m. (n = 3; *P<0.05, **P<0.01 versus control). (B) Schematic representation of the expression vector for miR-34b-5p and miR-34c-5p. (C) In vitro validation of miR-34b-5p and miR-34c-5p overexpression after transfection of HEK293T cells with pEGFP-N1-34b-5p and pEGFP-N1-34c-5p. Data are expressed as means ± s.e.m. (n = 3; ***P<0.001). (D) Wild-type putative miR-34c-5p binding site in sGCβ1 3′-UTR and its mutant form (top). Luciferase activity measured in HEK293T cells co-transfected with pEGFP-N1-34b-5p, pEGFP-N1-34c-5p, or empty plasmid and a luciferase reporter plasmid carrying either wild-type or mutant form of sGCβ1 3′-UTR (bottom). Firefly luciferase activity is normalized to Renilla luciferase expression levels. Data are expressed as means ± s.e.m. (n = 3; **P<0.01).
To determine whether miR-34b-5p and miR-34c-5p directly target the 3′-UTR of sGCβ1 mRNA, reporter plasmids were constructed by inserting wild-type and mutant 3′-UTR of sGCβ1 mRNA bearing miR-34b-5p and miR-34c-5p putative binding sites into pGL3-control vector immediately downstream of the luciferase coding sequence. miR-34b-5p and miR-34c-5p overexpression plasmids were also prepared by cloning pre-miR-34b-5p and pre-miR-34c-5p into pEGFP-N1 (Fig. 2B). The ability of plasmids to overexpress miR-34b-5p and miR-34c-5p was verified in HEK293T cells by qRT-PCR (Fig. 2C).
We co-transfected reporter plasmids with the overexpression vectors of miR-34b-5p or miR-34c-5p into HEK293T cells to perform luciferase reporter assays. miR-34c-5p significantly inhibited the luciferase activity of a reporter bearing the wild-type sGCβ1 3′-UTR (Fig. 2D) but not that of a reporter with a mutated sGCβ1 3′-UTR stripped of the miR-34c-5p conserved seed-match sequence. In contrast, miR-34b-5p did not inhibit the luciferase activity of the reporter containing the wild-type sGCβ1 3′-UTR (Fig. 2D).
Generation of a miR-34c-5p-specific sponge
miRNA sponge is a competitive inhibitors of miRNA developed by Ebert et al. which contains many tandem binding sites complementary to a mature miRNA of interest (Ebert et al., 2007). We constructed a miR-34c-5p-specific sponge by inserting tandemly arrayed miR-34c-5p binding sites into pEGFP-C1 immediately downstream of an EGFP coding sequence finally containing two, four or six binding sites (Fig. 3A). Binding sites for miR-34c-5p were perfectly complementary in the seed region with a bulge at positions 9–12 to prevent cleavage by Argonaute 2. For the lentiviral sponge, NheI- and BamHI-digested fragments were subcloned into pCDH-CMV-MCS-EF1-copGFP.
Fig. 3.
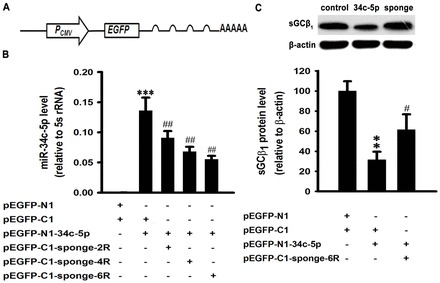
Overexpression of miR-34c-5p negatively regulates sGCβ1 levels in COS-7-sGCβ1 stable cell lines. (A) Schematic representation of the miR-34c-5p-specific sponge construct. (B) Effects of miR-34c-sponge on expression of miR-34c-5p. The miR-34c-5p levels were measured by qRT-PCR in HEK293T cells at 48 hours after transfection with pEGFP-N1-34c-5p and miR-34c-sponge containing two to six tandem bulged (at positions 9–12) miR-34c-5p binding sites. Data are expressed as means ± s.e.m. (n = 3; ***P<0.001 versus empty vector group; ##P<0.01 versus miR-34c-5p overexpression group). (C) Western blot analysis of sGCβ1 expression in COS-7-sGCβ1 treated with pEGFP-N1-miR-34c-5p or miR-34c-5p sponge or empty vector. Expression levels are normalized to β-actin and expressed as the means ± s.e.m. (n = 3; **P<0.01 versus empty vector group, #P<0.05 versus miR-34c-5p overexpression group). R, one repeat of the miR-34c-5p binding site.
To validate the efficacy and specificity of the miRNA sponge, we transfected HEK293T cells with the miR-34c-5p expression vector and the miR-34c-5p-specific sponge containing two, four or six binding sites. All of the designs were able to partially silence miR-34c-5p, and the sponge bearing six binding sites showed stronger blockage than others (Fig. 3B).
Ectopic expression of miR-34c-5p decreases sGCβ1 protein levels
To assess the direct reaction between miR-34c-5p and sGCβ1, stable cell lines expressing sGCβ1 with its 3′-UTR were set up by lentiviral infection of COS-7 cells, which did not contain endogenous sGCβ1. Firstly, we cloned sGCβ1 with its 3′-UTR into the pEGFP-C1 vector to produce an EGFP-fused protein. Then, NheI and EcoRI-digested fragments were inserted into pCDH-CMV-MCS-EF1-Puro vector to package the lentivirus. We infected the COS-7 cells with lentivirus and established a cell line stably expressing sGCβ1 with its 3′-UTR by puromycin selection, which we designated COS-7-sGCβ1. When COS-7-sGCβ1 was transfected with the miR-34c-5p expression vector, western blots indicated that the level of sGCβ1 was decreased by about 69% (P<0.01), which was attenuated by the miR-34c-5p-specific sponge (Fig. 3C).
Lentivirus-mediated overexpression of miR-34c-5p downregulates sGCβ1 expression and decreases sGC activity in mouse lung in vitro
To provide definitive proof of the inhibitory effect of miR-34c-5p on sGCβ1, mouse lung tissue blocks were inoculated with miR-34c-5p-expressing lentivirus in vitro. miR-34c-5p significantly downregulated sGCβ1 expression (Fig. 4A,B). When infected together with the miR-34c-5p-specific sponge, the inhibition was reversed (Fig. 4A,B). Correspondingly, overexpression of miR-34c-5p markedly decreased the activity of sGC compared with the control, and the miR-34c-5p-specific sponge attenuated the negative effect of miR-34c-5p on the activity of sGC (Fig. 4C).
Fig. 4.
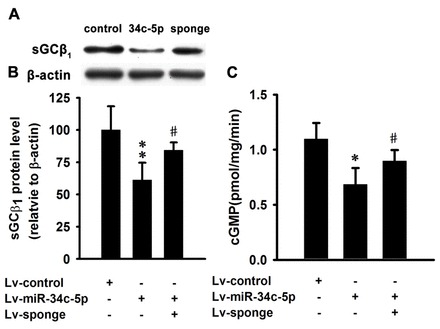
Lentivirus-mediated overexpression of miR-34c-5p downregulates sGCβ1 expression and decreases sGC activity in mouse lung in vitro. (A) Western blot analysis of sGCβ1 subunit expression in lungs inoculated with lentivirus expressing miR-34c-5p or miR-34c-5p-sponge or control lentivirus. (B) Quantification of the sGCβ1 subunit expression in the western blots. Expression levels are normalized to β-actin and expressed as the means ± s.e.m. (n = 3; **P<0.01 versus Lv-control group, #P<0.05 versus Lv-miR-34c-5p group). (C) Overexpression of miR-34c-5p markedly decreased the activity of sGC in mouse lung in vitro, which was attenuated by miR-34c-sponge. Data are expressed as means ± s.e.m. (n = 3; *P<0.05 versus Lv-control group, #P<0.05 versus Lv-miR-34c-5p group).
miR-34c-5p represses the expression of sGCβ1 in mouse pulmonary smooth muscle cells during hypoxia
miRNAs regulate their targets by repressing the translational efficiency and/or decreasing mRNA levels. We treated mouse pulmonary smooth muscle cells with 1% O2 for 24 hours. Hypoxia significantly decreased the level of sGCβ1 mRNA in the cells. The effect was prevented by silencing the miR-34c-5p with sponge (Fig. 5A). Hypoxia also significantly decreased the protein level of sGCβ1 and the negative effect was attenuated by miR-34c-5p-specific sponge (Fig. 5B).
Fig. 5.
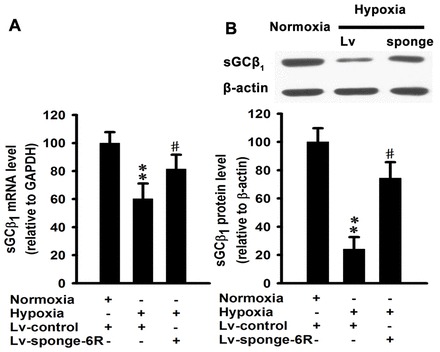
miR-34c-5p mediates hypoxia-induced downregulation of mRNA and protein levels of sGCβ1 in mouse pulmonary smooth muscle cells. (A) Cells were exposed to 1% O2 under control conditions or treated with miR-34c-specific sponge for 24 hours. mRNA expression of sGCβ1 was determined by qRT-PCR. The levels of mRNAs were relative to the control GAPDH. Data are expressed as means ± s.e.m. (n = 4; **P<0.01 versus normoxia, #P<0.05 versus hypoxia). (B) Western blot analysis of sGCβ1 subunit expression in mouse pulmonary smooth muscle cells exposed to 1% O2 under control conditions or treated with miR-34c-specific sponge for 24 hours. Expression levels are normalized to β-actin and expressed as means ± s.e.m. (n = 4; **P<0.01 versus normoxia, #P<0.05 versus hypoxia).
Sp1 mediates hypoxia-induced upregulation of miR-34c-5p in mouse
To determine how the miRNA expression is regulated at the transcriptional level, we analyzed the promoter region of miR-34c-5p using bioinformatics and observed that it contains a binding site for the transcription factor Sp1, which is a redox-sensitive transcription factor. qRT-PCR showed that the level of Sp1 mRNA was elevated after exposed to hypoxia for 72 hours and 120 hours (Fig. 6A). A dual luciferase reporter assay was conducted to analyze whether Sp1 is involved in the regulation of miR-34c-5p. The results showed that Sp1 bound to the wild-type promoter and promoted the upregulation of luciferase. However, a mutation introduced into the Sp1-binding site attenuated the upregulation of luciferase induced by Sp1 (Fig. 6B,C), indicating that Sp1 can positively regulate miR-34c-5p expression during hypoxia.
Fig. 6.
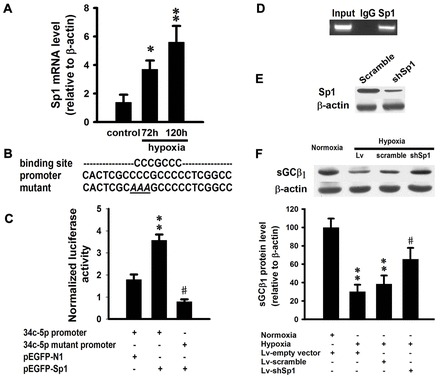
Sp1 transcriptionally regulates miR-34c-5p expression during hypoxia. (A) mRNA expression of Sp1 in mouse lungs exposed to hypoxia for 72 hours or 120 hours. Data are expressed as means ± s.e.m (n = 3; *P<0.05, **P<0.01 versus control). (B) Sp1-binding site and Sp1 expression vector. Data are expressed as means ± s.e.m. (n = 3; **P<0.05 versus empty vector group, #P<0.001 versus wild-type promoter group). (D) ChIP analysis of the miR-34c-5p promoter using pulmonary smooth muscle cells. PCR was performed to detect miR-34c-5p promoter containing a putative binding site for Sp1. (E) Western blot to confirm knockdown of Sp1 protein induced by Sp1 shRNA. (F) Western blot analysis of sGCβ1 expression in pulmonary smooth muscle cells treated with 1% O2 or Sp1 shRNA. Expression levels are normalized to β-actin and expressed as the means ± s.e.m. (n = 4; **P<0.01 versus normoxia, #P<0.05 versus Lv-scramble group).
To further demonstrate the regulatory role of Sp1 in miR-34c-5p expression, we performed a chromatin immunoprecipitation (ChIP) assay using mouse pulmonary smooth muscle cells infected with lentivirus bearing Sp1. Immunoprecipitation of chromatin-bound DNA using an antibody to Sp1 and IgG was followed by PCR that amplified the 240 bp region spanning the length of the Sp1-binding site. As shown in the ChIP assay, anti-Sp1 antibody was capable of immunoprecipitating the miR-34c-5p promoter fragment containing the Sp1 site, whereas immunoprecipitation using anti-IgG failed (Fig. 6D).
To confirm the regulatory role of Sp1 in hypoxia-induced downregulation of sGCβ1, we designed the lentivirus-mediated shRNA for Sp1 and infected mouse pulmonary smooth muscle cells. Western blotting showed that shRNA could efficiently interfere with the expression of Sp1 (Fig. 6E). Then cells were exposed to 1% O2 with or without shRNA for Sp1. Hypoxia significantly decreased the expression of sGCβ1 (Fig. 6F). When coupled with Sp1 silencing by shRNA, the negative effect of hypoxia on sGCβ1 was relieved (Fig. 6F).
Hif-1α has no influence on the expression of miR-34c-5p in mouse pulmonary smooth muscle cells during hypoxia
Hif-1α is the principal regulator of hypoxia-inducible gene expression (Semenza, 2012). To investigate its role in the regulation of miR-34c-5p, we designed the lentivirus-mediated shRNA for Hif-1α and infected the mouse pulmonary smooth muscle cells during hypoxia. As shown, shRNA efficiently interfered with the expression of Hif-1α during hypoxia (Fig. 7A). Then we examined the regulatory effect of Hif-1α on miR-34c-5p by using an Hif-1α-specific shRNA during hypoxia in the mouse pulmonary smooth muscle cells. The results show that Hif-1α has no effect on the regulation of miR-34c-5p expression (Fig. 7B).
Fig. 7.
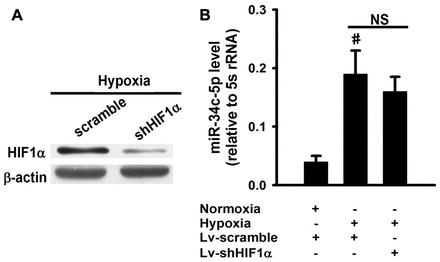
Hif-1α has no effect on the expression of miR-34c-5p during hypoxia in pulmonary smooth muscle cells. (A) Western blot to confirm knockdown of Hif-1α protein induced by lentivirus shRNA for Hif-1α during hypoxia. (B) Analysis of the expression of miR-34c-5p in pulmonary smooth muscle cells treated with 1% O2 or shRNA for Hif-1α. The levels of miRNAs were relative to the control 5 s rRNA. Data are expressed as means ± s.e.m. (n = 3; #P<0.01 versus normoxia). NS, non-significant.
Discussion
sGC is the key enzyme mediating the biological actions of NO. Abnormal expression of sGC has been implicated in the pathogenesis of various cardiovascular diseases, including pulmonary hypertension, atherosclerosis and tolerance to organic nitrates (Hussain et al., 1999; Oppermann et al., 2008). Melichar et al. observed that both subunits and specific activity of sGC are significantly reduced in the aortas of chronically hypercholesterolemic rabbits (Melichar et al., 2004). Reduced sGC expression also correlates with senescence (Klöss et al., 2000) and hypertension induced by high salt intake (Kagota et al., 2001). Moreover, inflammatory cytokines, such as lipopolysaccharide (Pedraza et al., 2003) and reactive oxygen species (Gerassimou et al., 2007), could also regulate the expression of sGC. However, upregulation of the β1 subunit level has been linked to altered vasodilator response in aortic rings from rats after myocardial infraction (Bauersachs et al., 1999). The β1 subunit is obligatory for the activity of sGC. Therefore, regulation of β1 subunit expression emerges as an important factor in the regulation of sGC function under physiological and pathophysiological conditions. The present work investigated the role of miRNAs in the expression of sGCβ1 subunit.
microRNAs are potent mediators in a wide range of disease and are emerging as new drug targets (Thum, 2012). The aberrant expression of miRNAs is associated with various cardiovascular diseases, such as myocardial infarction, cardiac hypertrophy and heart failure (Li et al., 2010; Thum, 2012). Recently, miRNAs have gained more attention in the regulation of pulmonary vascular system. Caruso et al. examined global miRNA profiles during the development of pulmonary artery hypertension caused by chronic hypoxia and monocrotaline in rats (Caruso et al., 2010). Courboulin et al. reported that miR-204 was downregulated in both human and rodent pulmonary arterial hypertension (Courboulin et al., 2011). The delivery of synthetic miR-204 to the lungs of animals with pulmonary arterial hypertension significantly reduced the severity of the disease. Pullamsetti et al. took advantage of antagonists of miR-17 to improve heart and lung function in experimental pulmonary hypertension related to lung vascular and right ventricular remodeling (Pullamsetti et al., 2012). By using a network-based bioinformatic approach coupled with confirmatory in vivo experiments Parikh et al. provided convincing evidence for the central role of miR-21 in the pathogenesis of pulmonary hypertension (Parikh et al., 2012). Guo et al. found that miRNA-328 was an important protecting factor in pulmonary artery constriction and remodeling by targeting insulin growth factor 1 receptor and l-type calcium channel-α1C (Guo et al., 2012). All these findings emphasize the crucial role of miRNAs in pulmonary system.
Here we report that both the expression of sGCβ1 and the activity of sGC were significantly decreased in the lungs of mice after exposure to hypoxia. In contrast, sGCβ1 expression in the kidney and heart of the mouse was not affected by hypoxia. It suggests that hypoxia-induced downregulation of sGC is specific for the lungs, and such an effect may in part contribute to hypoxic pulmonary vasoconstriction (Gao and Raj, 2010; Sylvester et al., 2012). It is of interest to note that in our in vivo study the protein levels of sGCβ1 in the lungs were reduced after the mice were exposed to hypoxia of 8% O2 for 72 hours. Under in vitro conditions, treatment with 1% O2 for 24 hours significantly decreased sGCβ1 protein levels of mouse pulmonary smooth muscle cells. It appears that the length of hypoxic exposure required for sGC downreguation decreases when the severity of hypoxia increases. These results indicate that under in vivo conditions chronic hypoxia may inhibit sGC activity of the lungs through the suppression of the expression of sGCβ1.
Bioinformatic and functional analysis revealed that the sGCβ1 subunit is a direct target of miR-34c-5p. miR-34c-5p belongs to the miR-34 family, which consists of miR-34a, miR-34b and miR-34c. The physiological and pathophysiological importance of this family in diseases, especially cancer, is becoming increasingly apparent. The miR-34 family may act as a tumor suppressor by inducing apoptosis and cell-cycle arrest (He et al., 2007; Hermeking, 2010), and miR-34a in particular, plays an essential role in restraining somatic reprogramming by repression of pluripotency genes, including Nanog, Sox2 and Mycn (Choi et al., 2011). miR-34c is implicated in the late steps of spermatogenesis (Bouhallier et al., 2010) and sperm-borne miR-34c is important for the first cell division by modulation of Bcl-2 expression (Liu et al., 2012b). Recently, the miR-34 family has been shown to be involved in the nervous system. miR-34c could act as a repressor of stress-induced anxiety by targeting stress-related corticotrophin releasing factor receptor type 1 (CRFR1) and play a physiological function in regulating the central stress response (Haramati et al., 2011). By targeting silent information regulator 1 (SIRT1), miR-34a regulates mouse neural stem cell differentiation (Aranha et al., 2011). miR-34c levels are elevated in the hippocampus of Alzheimer's disease patients and corresponding mouse models, and attenuation of miR-34c rescues learning ability in these mouse models (Zovoilis et al., 2011). More recently miR-34 has been found to be involved in aging and neurodegeneration in Drosophila (Liu et al., 2012a). Additionally, miR-34c induced by bone morphogenetic protein 2 (BMP2) regulates Notch signaling during bone development by directly targeting Notch1, Notch2 and Jag1. It highlights a possible mechanism modulating the proliferative effect of Notch in committed osteoblast progenitors, which may be important in the pathogenesis of osteosarcomas (Bae et al., 2012).
Among the miR-34 family members, miR-34b and miR-34c share a same intergenic overlapping transcript and are regulated by a common promoter (He et al., 2007; Wong et al., 2011). In the present study we found that miR-34b-5p was also markedly upregulated during hypoxia. However, functional analysis demonstrated that miR-34b-5p was unable to transcriptionally regulate sGCβ1 although they differ just by two nucleotides. These results suggest that sGCβ1 is a specific direct target of miR-34c-5p but not of miR-34b-5p. Similarly, Cardinaud et al. showed that a one-base shift significantly changes the list of targets of miR-34b-5p, which should differ to some extent from the list of targets for miR-34c and therefore T-cell leukemia/lymphoma 1 (TCL1A) is targeted by miR-34b-5p but not by miR-34c (Cardinaud et al., 2009). Cannell et al. found that miR-34c could inhibit the luciferase activity of wild-type c-Myc 3′-UTR, whereas overexpression of miR-34b resulted in the repression the luciferase activity but this failed to reach significance (Cannell et al., 2010). In contrast, Bae et al. reported that miR-34b and miR-34c share the same putative target sites of Notch1 and decrease Notch1 expression during osteogenesis of C2C12 cells (Bae et al., 2012).
Several observations in our study supported that miR-34c-5p directly regulates the expression of sGC. First, we set up a cell line stably expressing sGCβ1 plus a 3′-UTR, by infection of COS-7. The COS-7 cell line, derived from the kidney of the African green monkey, does not contain endogenous sGCβ1 (Sayed et al., 2008). Western blot analysis indicated that overexpression of miR-34c-5p repressed the expression of sGCβ1, which was reversed by miR-34c-5p-specific sponge. Second, both the expression of sGCβ1 and sGC activity were also inhibited by lentivirus-mediated overexpression of miR-34c-5p in mouse lung. This effect was relieved by miR-34c-5p-specific sponge. These data suggests that miR-34c-5p specifically represses the expression of sGCβ1. In mammals, miRNAs can downregulate gene expression using two distinct post-transcriptional mechanisms, including decreased translational efficiency (Selbach et al., 2008) and the destabilization of mRNAs that are associated with mRNA deadenylation (Guo et al., 2010). In our work, silencing miR-34c-5p with sponge attenuated hypoxia-induced downregulation of mRNA and protein expression of sGCβ1, which implies miR-34c-5p-induced mRNA decay partially contributes to sGCβ1 downregulation during hypoxia.
miRNA expression can be regulated at both the transcriptional and posttranscriptional levels (Yu et al., 2012). A few studies have focused on the regulation of miR-34c. He et al. and Corney et al. reported that p53 can directly regulate miR-34c (He et al., 2007; Corney et al., 2007). Cannell et al. demonstrated that miR-34c could be epigenetically silenced by the p38 MAPK/MK2 pathway (Cannell et al., 2010). More recently, Yu et al. identified that Sp1 could target the miR-34c promoter in tumor-initiating cells (Yu et al., 2012). Hypoxia may induce the expression of a number of miRNAs, which are divided into hypoxia-inducible factor 1-dependent and -independent miRNAs (Loscalzo, 2010). By using shRNA to knockdown Hif-1α during hypoxia in mouse pulmonary smooth muscle cells, we identified that hypoxia could regulate miR-34c-5p expression in a Hif-1α-independent manner. In order to better understand the regulation of miR-34c-5p during hypoxia, we analyzed the promoter region of miR-34c-5p and observed that it contains a canonical binding site for the transcription factor Sp1, which is an hypoxia-sensitive transcription factor. Our data show that the level of Sp1 mRNA was significantly upregulated during hypoxia. To corroborate the importance of Sp1 in the regulation of miR-34c-5p promoter activity, we conducted a luciferase assay to confirm the interaction of Sp1 with the miR-34c-5p promoter. This was identical to the previous work performed in tumor-initiating cells by Yu et al. (Yu et al., 2012). Additional chromatin immunoprecipitation using the region of the promoter of miR-34c-5p carrying the Sp1-binding site demonstrated that Sp1 was capable of promoter interaction. To confirm the role of Sp1 in hypoxia-induced repression of sGCβ1, we successfully established the shRNA for Sp1. Knockdown of Sp1 attenuated hypoxia-induced downregulation of sGCβ1 during hypoxia in mouse pulmonary smooth muscle cells. Taken together, our data suggest the regulatory effect of Sp1 in hypoxia-induced upregulation of miR-34c-5p.
In summary, our present study, provides the first evidence that sGCβ1 can be regulated by a miRNA, specifically miR-34c-5p, during hypoxia and Sp1 is responsible for hypoxia-induced regulation of miR-34c-5p (a schematic overview of the signaling pathway revealed by our experiments can be found in Fig. 8). These findings provide a new understanding of the regulation of sGCβ1 expression. Decreased expression and activity of sGC has been documented under certain pathophysiological conditions and in a number of diseases such as hypoxic pulmonary vasoconstriction and pulmonary hypertension (Friebe and Koesling, 2009; Sylvester et al., 2012). Our present findings suggest that an increased expression of miR-34c-5p following hypoxic exposure could be involved in the development of hypoxic pulmonary hypertension.
Fig. 8.
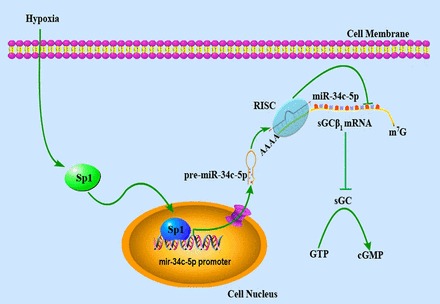
Summary diagram of the hypoxia–Sp1–miR-34c-5p signaling that regulates sGCβ1 subunit expression. Hypoxia upregulates Sp1 expression. Sp1 in turn transcriptionally promotes miR-34c-5p expression by direct interaction with the promoter of miR-34c-5p in the nucleus. After transport into the cytosol, miR-34c-5p exerts a translational blockade of sGCβ1 subunit by binding in the 3′-UTR and then decreases sGC activity. sGC, soluble guanylyl cyclase; RISC, RNA-induced silencing complex.
Materials and Methods
Animal experiments
Female CD-1 mice (23∼27 g) were exposed to hypoxia as previously described (Li et al., 1999; Kirsch et al., 2008). Briefly, the mice were randomly divided into normoxic and hypoxic groups and placed in a normobaric chamber and exposed to hypoxia (8% O2) or normoxia for 72 hours or 120 hours. Animals were housed at 20∼24°C and on a 12-hour:12-hour light-dark cycle with free access to food and water. The protocols and animal use were approved by the Animal Care and Use Review Committees of Peking University Health Science Center. The study conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996).
Culture of primary mouse pulmonary smooth muscle cells and treatment
Cultures of primary mouse pulmonary smooth muscle cells (MPSMCs) were prepared from explants of endothelium- and adventitia-stripped pulmonary arteries of CD-1 mice as described with modification (Filippov et al., 1997). Cells were maintained in DMEM containing 20% FBS (Gibco, USA), 100 units/ml penicillin and 100 µg/ml streptomycin, and incubated at 37°C with 5% CO2. The purity of the MPSMCs was confirmed by immunoreactivity for α-actin (BioGenex) and greater than 98% of the subcultured MPSMCs had positive staining. All the following experiments involving MPSMCs were performed with passage number ≤5.
Hypoxic cell cultures were prepared using an hypoxia workstation at 1% O2, 5% CO2, 37°C. For RNA interference, 20 hours after lentivirus infection, the cells were cultured under normoxic or hypoxic conditions for 24 hours before harvest.
Quantitative real-time PCR
Total RNA was isolated with TRIzol regent (Invitrogen, USA) according to the manufacturer's instructions. To measure the expression level of mRNAs, cDNA was synthesized using a First Strand cDNA Synthesis Kit for RT-PCR (Toyobo, Japan). Mature miRNA quantification was as previously described (Shi and Chiang, 2005; Carlsbecker et al., 2010). 1 µg of total RNA including small RNAs was polyadenylated with ATP by poly(A) polymerase (New England Biolabs, USA) at 37°C for 1 hour using in a 10-µl reaction mixture. cDNA from polyadenylated RNA was reverse-transcribed with ReverTra Ace-α-Transcriptase (Toyobo, Japan) and 0.5 µg poly(T) adapter. qRT-PCR reactions were performed on an ABI Prism 7100 (Applied Biosystems) for 15 seconds at 95°C, 5 seconds at 95°C and 31 seconds at 60°C for 40 cycles. mRNA and miRNA quantification data were normalized to GAPDH and 5S rRNA, respectively. Relative expressions of mature miRNA and mRNA were determined by a comparative method (2−ΔΔCT).
Overexpression and inhibition of miR-34b-5p and miR-34c-5p
miR-34b/c-5p and ∼100 bp of flanking sequence was amplified from mouse genomic DNA (Du et al., 2009) and cloned into pEGFP-N1 (Clontech). Primer sequences: pre-miR-34b-5p: 5′-GAATTCAGCCTGAGGCACCTCTCGCT-3′ and 5′-GGATCCGGTTACTTGCACTTAGACCT-3′; pre-miR-34c-5p: 5′-GAATTCCACCAAGGCAGCGACTAGAGTCAACC-3′ and 5′-GGATCCCCCAAACCACTAATAGTATGGTAAGAA-3′. A PCR fragment was inserted between the CMV promoter and the EGFP coding sequence in the pEGFP-N1 vector using EcoRI and BamHI sites (Haramati et al., 2011). The resulting recombinant plasmids were designated pEGFP-N1-34b-5p and pEGFP-N1-34c-5p, respectively. The constructs were sequence verified.
The miR-34c-5p sponge, designated pEGFP-C1-sponge, was constructed by annealing, purifying and cloning oligonucleotides containing two to six tandem bulged (at positions 9–12) miR-34c-5p binding sites with 4-nt spacers into 3′-UTR of pEGFP-C1 vector (Ebert et al., 2007), into which the stop codon TAA was introduced downstream of the EGPF coding sequence.
For lentivirus-mediated miR-34c-5p overexpression, the miR-34c-5p fragment from pEGFP-N1-34c-5p digested with EcoRI and BamHI was cloned into lentiviral vector pCDH-CMV-MCS-EF1-copGFP (System Bioscience, USA) downstream of the CMV promoter, and is referred to as Lv-miR-34c-5p. For lentiviral miR-34c-5p sponge, the NheI–BamHI fragment of pEGFP-34c-sponge was also cloned into pCDH-CMV-MCS-EF1-copGFP, and is referred to as Lv-sponge. Virus was produced and target cells were infected according to the users' manual (System Bioscience, USA).
Construction of the luciferase reporter of sGCβ1 3′-UTR
To generate luciferase reporter vectors containing a putative miR-34b/c-5p binding site, sense and antisense strands of the oligonucleotides of the sGCβ1 3′-UTR were synthesized, annealed and subcloned into the pGL3-control vector (Promega, USA) in a single XbaI site immediately downstream of the stop codon of the luciferase gene (Qin et al., 2010). A mutated form of sGCβ1 3′-UTR was used to remove all complementarity to nucleotides 1–7 of the miR-34c-5p seed region (Haramati et al., 2011). Cloning orientation was verified by sequencing.
Preparation of Sp1 and sGCβ1 plus 3′-UTR constructs
The Sp1 coding sequence was amplified from cDNA using primers: 5′-CTCGAGATGAGCGACCAAGATCACT-3′ and 5′-GGTACCGCGAAACCATTGCCACTGATA-3′. The fragment was cloned into pEGFP-N1 using XhoI and KpnI digestion, and designated pEGFP-Sp1.
Lentiviral sGCβ1 plus 3′-UTR expression vector was constructed using cDNA as template: forward primer, 5′-CTCGAGTATACGGTTTCGTGAACCATG-3′ and the reverse primer, 5′-GAATTCACAGCAGTGTCCTAGCTATG-3′. The amplified fragments were inserted into pEGFP-C1 by XhoI and EcoRI digestion. The resulting plasmid was digested with NheI and EcoRI and ligated into pCDH-CMV-MCS-EF1-Puro (System Bioscience, USA), and designated Lv-sGCβ1. Virus was produced and target cells were infected according to the users' manual (System Bioscience, USA).
Generation of stable cell lines expressing sGCβ1 plus 3′-UTR
To generate sGCβ1 plus 3′-UTR stable cell lines, COS-7 cells were infected with Lv-sGCβ1 virus or control Lv-EGFP which expresses only EGFP. Cells were grown in the presence of 1 mg/ml puromycin to select the stably infected clones. Single clones were selected to generate monoclonal cell lines, termed COS-7-sGCβ1.
Western blot analysis
Western blot was performed as described with modifications (Wang et al., 2010b). Mouse lung tissues or cultured cells were homogenized in RIPA buffer containing 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Nonidet P40, 0.5% sodium deoxycholate, 0.1% SDS, 5 mM EGTA, 2 mM EDTA, 10 mM NaF and protease inhibitors. After centrifugation at 12,000 rpm for 20 minutes at 4°C, the supernatant was collected and protein concentration measured using the Bradford assay. Aliquots of the extract containing about 30 µg of protein were subjected to reducing 10% SDS-PAGE and transferred to PVDF membranes for 45 minutes on ice. The membranes were blocked in 5% non-fat milk containing 20 mM Tris-HCl, pH 7.6, 137 mM NaCl, 0.05% Tween 20 (TBST) for 1 hour at room temperature, and then incubated with rabbit anti-guanylate cyclase β1 polyclonal antibody (1∶5000, Cayman), rabbit anti-Sp1 polyclonal antibody (1∶500, Millipore), mouse anti-Hif-1α monoclonal antibody (1∶300, Abcam) and mouse anti-β-actin monoclonal antibody (1∶5000, Merck) overnight at 4°C. After washing with TBST three times, the membranes were incubated in horseradish-peroxidase-conjugated secondary antibody for 1 hour at room temperature. Immunoreactive proteins were detected using the chemiluminescence detection method (SuperSignal West Pico Chemiluminescent Substrate; Thermo Fisher Scientific Inc., Rockford, IL, USA), and quantified by transmittance densitometry using volume integration with Gel-Pro software. To ensure even loading of the samples, the protein levels in each specimen were normalized to β-actin.
Immunohistochemistry
Immunohistochemistry was performed as described before (Wang et al., 2010b). Sections were dewaxed for 30 minutes at room temperature in xylene and rehydrated to water through an ethanol series. The slices were washed with 0.01 M PBS before incubating in 3% H2O2 for 10 minutes to quench the endogenous peroxidase activity. They were then placed in 10 mM citrate buffer (pH 6.0) and microwaved for 10 minutes at 750 W for antigen retrieval. After blocking with 0.5% BSA in PBS for 1 hour, the slices were incubated overnight at 4°C with the rabbit anti-sGCβ1 polyclonal antibody (1:500, Cayman) and then washed three times with PBS for 5 minutes. HRP-conjugated anti-rabbit secondary antibody (Zymed Laboratories, San Francisco, CA, USA) was used at a dilution of 1∶200 for a 60-minute incubation at room temperature. The slides were then washed three times with PBS for 5 minutes and the staining reaction was carried out using 3,3′-diaminobenzidine tetrahydrochloride (DAB) as the chromogen. The sections were re-stained with Hematoxylin for 1 minute, and put in water for 2 minutes. They were then differentiated with 0.3% acid alcohol for two quick dips, put in water for 2 minutes, dehydrated in ethanol, cleared in xylene and a coverslip placed on top with neutral balsam. The slices were photographed with an Olympus DP70 microscope.
Construction of mouse miR-34c-5p promoter and its mutant
Mouse genomic DNA was prepared with EasyPure Genomic DNA Kit (TransGen Biotech, China). miR-34c-5p promoter was PCR amplified from mouse genomic DNA. The forward primer was 5′-GGTACCTCTCAAGAATCTGTCTCTC-3′. The reverse primer was 5′-AAGCTTCAGCACCGCACTACAATCA-3′. The promoter fragment was cloned into the pGL3-basic vector (Promega, USA). The introduction of mutations in the putative Sp1-binding site was performed with the Fast Mutagenesis System site-directed mutagenesis kit (TransGen Biotech, China) using the wild-type vector as a template. The mutated primer: 5′-TCCCCCACTCGCAAAGCCCCCT-3′ (sense), 5′-TTTGCGAGTGGGGGAGGGGTGG-3′ (antisense). The construct was sequenced to check that only the desired mutations had been introduced (Wang et al., 2010a).
Transduction of mouse lung explants in vitro
The procedure was performed as described previously (McNeilly et al., 2007; Chan et al., 2009; Grivel and Margolis, 2009). Briefly, after rinsing three times with PBS, mice lungs were cut into ∼2×2×1 mm pieces. Four pieces of lung were subsequently placed into each well of 24-well tissue plates containing culture medium (F-12 nutrient mixture) and incubated at 37°C with 5% CO2 in a humidified atmosphere. After 6 hours, explants were inoculated with Lv-34c-5p and/or Lv-34c-sponge along with empty lentivirus as a control, for 12 hours. Cultures were then washed three times in PBS followed by 48-hour incubation in fresh medium prior to analysis.
sGC activity
sGC activity was assayed as described previously (Li et al., 1999; Sayed et al., 2007). Lung tissue was homogenized in buffer containing 50 mM Hepes (pH 8.0), 1 mM EDTA, 150 mM NaCl and protease inhibitors. Extracts were centrifuged at 12,000 rpm for 20 minutes at 4°C. Supernatants (40 µg) were incubated for 10 minutes at 37°C in a reaction mixture containing 50 mM Hepes (pH 8.0), 500 µM GTP, 1 mM DTT, and 5 mM MgCl2, 1 mM 3-isobutyl-1-methylxanthine in the presence or absence of NO donor DETA NONOate (10−5 M). The reaction was terminated by the addition of HCl to a final concentration of 0.1 M. cGMP was measured using a commercially available cGMP enzyme-linked immunoassay according to the manufacturer's instructions (Monoclonal Anti-cGMP Antibody Based Direct cGMP ELISA Kit, NewEast Biosciences). sGC enzyme activity is expressed as picomoles of cGMP produced per minute per milligram of lung protein.
Lentivirus-based RNAi
Vectors used for RNAi were constructed by inserting a synthesized 59-mer oligonucleotide containing a specific sequence for Sp1 and Hif-1α into pSicoR vector. The target sequence for Sp1 was: 5′-GAGACATAAACGTACACAT-3′; and for Hif-1α: 5′-GCTGACCAGTTACGATTGT-3′. The oligos were resuspended in annealing buffer and heated to 95°C for 5 minutes and then cooled to room temperature to generate double-stranded DNA. The double-stranded oligos were clone into lentivirus vector pSicoR. Lentivirus particles were produced by transient transfection of HEK293T cells according to standard protocols.
Luciferase reporter assay
Luciferase activity assay was performed using the Dual-Luciferase reporter assay system (Promega) according to the manufacturer's instructions. For sGCβ1 3′-UTR luciferase assay, HEK293T cells were co-transfected with wild-type or mutant reporter constructs and miRNA expression vector (pEGFP-N1-34b-5p or pEGFP-N1-34c-5p) with empty vector as control. pRL-TK (Promega) was co-transfected as a normalization control. Cells were lysed 48 hours after transfection, and firefly luciferase and Renilla luciferase activities were determined with a Dual-Luciferase Reporter System (Promega).
For miR-34c-5p promoter luciferase assay, HEK293T cells were co-transfected with wild-type and mutant reporter constructs along with pEGFP-Sp1 expression vector or an empty vector. Luciferase activity was determined as above.
ChIP assay
Mouse pulmonary smooth muscle cells (MPSMCs) were infected with lentivirus bearing Sp1. ChIP assays were performed according to the manufacturer's instructions (EZ-ChIPTM; Millipore). Briefly, cells were fixed with 1% formaldehyde and harvested. Input DNA was collected after pre-clearing lysate with protein G agarose beads prior to immunoprecipitation with anti-Sp1 antibody (Millipore) or with a non-relevant IgG. A 240 bp region of the miR-34c-5p promoter was amplified by PCR using specific primers (5′-CTCAAGAATCTGTCTCTCCATCTC-3′; 5′-AACAACTTCTTCACGGCTTCCAGA-3′).
Statistical analysis
Statistical analysis was performed using SPSS version 12.0 (SPSS, Chicago, IL, USA). Data are expressed as means ± s.e.m. The difference between means was determined by one-way ANOVA followed by a Student–Newman–Keuls test for multiple comparisons. A probability value of P<0.05 was taken to be statistically significant.
Acknowledgments
The authors thank Haibing Wang (State Key Laboratory of Reproductive Biology, Institute of Zoology, Chinese Academy of Sciences) for kindly providing the normobaric hypoxic chamber.
Footnotes
Funding
This work was supported by the National Natural Science Foundation of China [grant numbers 81270341 to Y.G., 30900511 to H.Q., 81001433 to D.D.]; and the National Heart, Lung, and Blood Institute USA, [grant numbers HL059435 and HL075187 to J.U.R.]. Deposited in PMC for release after 12 months.
References
- Abdellatif M. (2012). Differential expression of microRNAs in different disease states. Circ. Res. 110, 638–650 10.1161/CIRCRESAHA.111.247437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranha M. M., Santos D. M., Solá S., Steer C. J., Rodrigues C. M. (2011). miR-34a regulates mouse neural stem cell differentiation. PLoS ONE 6, e21396 10.1371/journal.pone.0021396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae Y., Yang T., Zeng H. C., Campeau P. M., Chen Y., Bertin T., Dawson B. C., Munivez E., Tao J., Lee B. H. (2012). miRNA-34c regulates Notch signaling during bone development. Hum. Mol. Genet. 21, 2991–3000 10.1093/hmg/dds129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauersachs J., Bouloumié A., Fraccarollo D., Hu K., Busse R., Ertl G. (1999). Endothelial dysfunction in chronic myocardial infarction despite increased vascular endothelial nitric oxide synthase and soluble guanylate cyclase expression: role of enhanced vascular superoxide production. Circulation 100, 292–298 10.1161/01.CIR.100.3.292 [DOI] [PubMed] [Google Scholar]
- Bouhallier F., Allioli N., Lavial F., Chalmel F., Perrard M. H., Durand P., Samarut J., Pain B., Rouault J. P. (2010). Role of miR-34c microRNA in the late steps of spermatogenesis. RNA 16, 720–731 10.1261/rna.1963810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannell I. G., Kong Y. W., Johnston S. J., Chen M. L., Collins H. M., Dobbyn H. C., Elia A., Kress T. R., Dickens M., Clemens M. J.et al. (2010). p38 MAPK/MK2-mediated induction of miR-34c following DNA damage prevents Myc-dependent DNA replication. Proc. Natl. Acad. Sci. USA 107, 5375–5380 10.1073/pnas.0910015107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinaud B., Moreilhon C., Marcet B., Robbe–Sermesant K., LeBrigand K., Mari B., Eclache V., Cymbalista F., Raynaud S., Barbry P. (2009). miR-34b/miR-34c: a regulator of TCL1 expression in 11q- chronic lymphocytic leukaemia? Leukemia 23, 2174–2177 10.1038/leu.2009.125 [DOI] [PubMed] [Google Scholar]
- Carlsbecker A., Lee J. Y., Roberts C. J., Dettmer J., Lehesranta S., Zhou J., Lindgren O., Moreno–Risueno M. A., Vatén A., Thitamadee S.et al. (2010). Cell signalling by microRNA165/6 directs gene dose-dependent root cell fate. Nature 465, 316–321 10.1038/nature08977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruso P., MacLean M. R., Khanin R., McClure J., Soon E., Southgate M., MacDonald R. A., Greig J. A., Robertson K. E., Masson R.et al. (2010). Dynamic changes in lung microRNA profiles during the development of pulmonary hypertension due to chronic hypoxia and monocrotaline. Arterioscler. Thromb. Vasc. Biol. 30, 716–723 10.1161/ATVBAHA.109.202028 [DOI] [PubMed] [Google Scholar]
- Chan R. W., Chan M. C., Wong A. C., Karamanska R., Dell A., Haslam S. M., Sihoe A. D., Chui W. H., Triana–Baltzer G., Li Q.et al. (2009). DAS181 inhibits H5N1 influenza virus infection of human lung tissues. Antimicrob. Agents Chemother. 53, 3935–3941 10.1128/AAC.00389-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y. J., Lin C. P., Ho J. J., He X., Okada N., Bu P., Zhong Y., Kim S. Y., Bennett M. J., Chen C.et al. (2011). miR-34 miRNAs provide a barrier for somatic cell reprogramming. Nat. Cell Biol. 13, 1353–1360 10.1038/ncb2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corney D. C., Flesken–Nikitin A., Godwin A. K., Wang W., Nikitin A. Y. (2007). MicroRNA-34b and MicroRNA-34c are targets of p53 and cooperate in control of cell proliferation and adhesion-independent growth. Cancer Res. 67, 8433–8438 10.1158/0008-5472.CAN-07-1585 [DOI] [PubMed] [Google Scholar]
- Courboulin A., Paulin R., Giguère N. J., Saksouk N., Perreault T., Meloche J., Paquet E. R., Biardel S., Provencher S., Côté J.et al. (2011). Role for miR-204 in human pulmonary arterial hypertension. J. Exp. Med. 208, 535–548 10.1084/jem.20101812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng M., Tang H., Zhou Y., Zhou M., Xiong W., Zheng Y., Ye Q., Zeng X., Liao Q., Guo X.et al. (2011). miR-216b suppresses tumor growth and invasion by targeting KRAS in nasopharyngeal carcinoma. J. Cell Sci. 124, 2997–3005 10.1242/jcs.085050 [DOI] [PubMed] [Google Scholar]
- Du C., Liu C., Kang J., Zhao G., Ye Z., Huang S., Li Z., Wu Z., Pei G. (2009). MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat. Immunol. 10, 1252–1259 10.1038/ni.1798 [DOI] [PubMed] [Google Scholar]
- Ebert M. S., Neilson J. R., Sharp P. A. (2007). MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 4, 721–726 10.1038/nmeth1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evgenov O. V., Pacher P., Schmidt P. M., Haskó G., Schmidt H. H., Stasch J. P. (2006). NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nat. Rev. Drug Discov. 5, 755–768 10.1038/nrd2038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippov G., Bloch D. B., Bloch K. D. (1997). Nitric oxide decreases stability of mRNAs encoding soluble guanylate cyclase subunits in rat pulmonary artery smooth muscle cells. J. Clin. Invest. 100, 942–948 10.1172/JCI119610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis S. H., Busch J. L., Corbin J. D., Sibley D. (2010). cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 62, 525–563 10.1124/pr.110.002907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friebe A., Koesling D. (2009). The function of NO-sensitive guanylyl cyclase: what we can learn from genetic mouse models. Nitric Oxide 21, 149–156 10.1016/j.niox.2009.07.004 [DOI] [PubMed] [Google Scholar]
- Gao Y., Raj J. U. (2010). Regulation of the pulmonary circulation in the fetus and newborn. Physiol. Rev. 90, 1291–1335 10.1152/physrev.00032.2009 [DOI] [PubMed] [Google Scholar]
- Gerassimou C., Kotanidou A., Zhou Z., Simoes D. C., Roussos C., Papapetropoulos A. (2007). Regulation of the expression of soluble guanylyl cyclase by reactive oxygen species. Br. J. Pharmacol. 150, 1084–1091 10.1038/sj.bjp.0707179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivel J. C., Margolis L. (2009). Use of human tissue explants to study human infectious agents. Nat. Protoc. 4, 256–269 10.1038/nprot.2008.245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H., Ingolia N. T., Weissman J. S., Bartel D. P. (2010). Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466, 835–840 10.1038/nature09267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L., Qiu Z., Wei L., Yu X., Gao X., Jiang S., Tian H., Jiang C., Zhu D. (2012). The microRNA-328 regulates hypoxic pulmonary hypertension by targeting at insulin growth factor 1 receptor and L-type calcium channel-α1C. Hypertension 59, 1006–1013 10.1161/HYPERTENSIONAHA.111.185413 [DOI] [PubMed] [Google Scholar]
- Haramati S., Navon I., Issler O., Ezra–Nevo G., Gil S., Zwang R., Hornstein E., Chen A. (2011). MicroRNA as repressors of stress-induced anxiety: the case of amygdalar miR-34. J. Neurosci. 31, 14191–14203 10.1523/JNEUROSCI.1673-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L., He X., Lim L. P., de Stanchina E., Xuan Z., Liang Y., Xue W., Zender L., Magnus J., Ridzon D.et al. (2007). A microRNA component of the p53 tumour suppressor network. Nature 447, 1130–1134 10.1038/nature05939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermeking H. (2010). The miR-34 family in cancer and apoptosis. Cell Death Differ. 17, 193–199 10.1038/cdd.2009.56 [DOI] [PubMed] [Google Scholar]
- Hussain M. B., Hobbs A. J., MacAllister R. J. (1999). Autoregulation of nitric oxide-soluble guanylate cyclase-cyclic GMP signalling in mouse thoracic aorta. Br. J. Pharmacol. 128, 1082–1088 10.1038/sj.bjp.0702874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagota S., Tamashiro A., Yamaguchi Y., Sugiura R., Kuno T., Nakamura K., Kunitomo M. (2001). Downregulation of vascular soluble guanylate cyclase induced by high salt intake in spontaneously hypertensive rats. Br. J. Pharmacol. 134, 737–744 10.1038/sj.bjp.0704300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasinski A. L., Slack F. J. (2011). Epigenetics and genetics. MicroRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy. Nat. Rev. Cancer 11, 849–864 10.1038/nrc3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch M., Kemp–Harper B., Weissmann N., Grimminger F., Schmidt H. H. (2008). Sildenafil in hypoxic pulmonary hypertension potentiates a compensatory up-regulation of NO-cGMP signaling. FASEB J. 22, 30–40 10.1096/fj.06-7526com [DOI] [PubMed] [Google Scholar]
- Klöss S., Bouloumié A., Mülsch A. (2000). Aging and chronic hypertension decrease expression of rat aortic soluble guanylyl cyclase. Hypertension 35, 43–47 10.1161/01.HYP.35.1.43 [DOI] [PubMed] [Google Scholar]
- Li D., Zhou N., Johns R. A. (1999). Soluble guanylate cyclase gene expression and localization in rat lung after exposure to hypoxia. Am. J. Physiol. 277, L841–L847 [DOI] [PubMed] [Google Scholar]
- Li Q., Song X. W., Zou J., Wang G. K., Kremneva E., Li X. Q., Zhu N., Sun T., Lappalainen P., Yuan W. J.et al. (2010). Attenuation of microRNA-1 derepresses the cytoskeleton regulatory protein twinfilin-1 to provoke cardiac hypertrophy. J. Cell Sci. 123, 2444–2452 10.1242/jcs.067165 [DOI] [PubMed] [Google Scholar]
- Liu N., Landreh M., Cao K., Abe M., Hendriks G. J., Kennerdell J. R., Zhu Y., Wang L. S., Bonini N. M. (2012a). The microRNA miR-34 modulates ageing and neurodegeneration in Drosophila. Nature 482, 519–523 10.1038/nature10810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W. M., Pang R. T., Chiu P. C., Wong B. P., Lao K., Lee K. F., Yeung W. S. (2012b). Sperm-borne microRNA-34c is required for the first cleavage division in mouse. Proc. Natl. Acad. Sci. USA 109, 490–494 10.1073/pnas.1110368109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loscalzo J. (2010). The cellular response to hypoxia: tuning the system with microRNAs. J. Clin. Invest. 120, 3815–3817 10.1172/JCI45105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma F., Xu S., Liu X., Zhang Q., Xu X., Liu M., Hua M., Li N., Yao H., Cao X. (2011). The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon-γ. Nat. Immunol. 12, 861–869 10.1038/ni.2073 [DOI] [PubMed] [Google Scholar]
- McNeilly T. N., Tennant P., Luján L., Pérez M., Harkiss G. D. (2007). Differential infection efficiencies of peripheral lung and tracheal tissues in sheep infected with Visna/maedi virus via the respiratory tract. J. Gen. Virol. 88, 670–679 10.1099/vir.0.82434-0 [DOI] [PubMed] [Google Scholar]
- Melichar V. O., Behr–Roussel D., Zabel U., Uttenthal L. O., Rodrigo J., Rupin A., Verbeuren T. J., Kumar, H S., Schmidt H. H. (2004). Reduced cGMP signaling associated with neointimal proliferation and vascular dysfunction in late-stage atherosclerosis. Proc. Natl. Acad. Sci. USA 101, 16671–16676 10.1073/pnas.0405509101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppermann M., Dao V. T., Suvorava T., Bas M., Kojda G. (2008). Effect of oral organic nitrates on expression and activity of vascular soluble guanylyl cyclase. Br. J. Pharmacol. 155, 335–342 10.1038/bjp.2008.269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh V. N., Jin R. C., Rabello S., Gulbahce N., White K., Hale A., Cottrill K. A., Shaik R. S., Waxman A. B., Zhang Y. Y.et al. (2012). MicroRNA-21 integrates pathogenic signaling to control pulmonary hypertension: results of a network bioinformatics approach. Circulation 125, 1520–1532 10.1161/CIRCULATIONAHA.111.060269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedraza C. E., Baltrons M. A., Heneka M. T., García A. (2003). Interleukin-1 beta and lipopolysaccharide decrease soluble guanylyl cyclase in brain cells: NO-independent destabilization of protein and NO-dependent decrease of mRNA. J. Neuroimmunol. 144, 80–90 10.1016/j.jneuroim.2003.08.034 [DOI] [PubMed] [Google Scholar]
- Pullamsetti S. S., Doebele C., Fischer A., Savai R., Kojonazarov B., Dahal B. K., Ghofrani H. A., Weissmann N., Grimminger F., Bonauer A.et al. (2012). Inhibition of microRNA-17 improves lung and heart function in experimental pulmonary hypertension. Am. J. Respir. Crit. Care Med. 185, 409–419 10.1164/rccm.201106-1093OC [DOI] [PubMed] [Google Scholar]
- Qin X., Wang X., Wang Y., Tang Z., Cui Q., Xi J., Li Y. S., Chien S., Wang N. (2010). MicroRNA-19a mediates the suppressive effect of laminar flow on cyclin D1 expression in human umbilical vein endothelial cells. Proc. Natl. Acad. Sci. USA 107, 3240–3244 10.1073/pnas.0914882107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayed N., Baskaran P., Ma X., van den Akker F., Beuve A. (2007). Desensitization of soluble guanylyl cyclase, the NO receptor, by S-nitrosylation. Proc. Natl. Acad. Sci. USA 104, 12312–12317 10.1073/pnas.0703944104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayed N., Kim D. D., Fioramonti X., Iwahashi T., Durán W. N., Beuve A. (2008). Nitroglycerin-induced S-nitrosylation and desensitization of soluble guanylyl cyclase contribute to nitrate tolerance. Circ. Res. 103, 606–614 10.1161/CIRCRESAHA.108.175133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramedei K., Mörbt N., Pfeifer G., Läuter J., Rosolowski M., Tomm J. M., von Bergen M., Horn F., Brocke–Heidrich K. (2011). MicroRNA-21 targets tumor suppressor genes ANP32A and SMARCA4. Oncogene 30, 2975–2985 10.1038/onc.2011.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbach M., Schwanhäusser B., Thierfelder N., Fang Z., Khanin R., Rajewsky N. (2008). Widespread changes in protein synthesis induced by microRNAs. Nature 455, 58–63 10.1038/nature07228 [DOI] [PubMed] [Google Scholar]
- Semenza G. L. (2012). Hypoxia-inducible factors in physiology and medicine. Cell 148, 399–408 10.1016/j.cell.2012.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharina I. G., Martin E., Thomas A., Uray K. L., Murad F. (2003). CCAAT-binding factor regulates expression of the beta1 subunit of soluble guanylyl cyclase gene in the BE2 human neuroblastoma cell line. Proc. Natl. Acad. Sci. USA 100, 11523–11528 10.1073/pnas.1934338100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharina I. G., Cote G. J., Martin E., Doursout M. F., Murad F. (2011). RNA splicing in regulation of nitric oxide receptor soluble guanylyl cyclase. Nitric Oxide 25, 265–274 10.1016/j.niox.2011.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi R., Chiang V. L. (2005). Facile means for quantifying microRNA expression by real-time PCR. Biotechniques 39, 519–525 10.2144/000112010 [DOI] [PubMed] [Google Scholar]
- Smith–Vikos T., Slack F. J. (2012). MicroRNAs and their roles in aging. J. Cell Sci. 125, 7–17 10.1242/jcs.099200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasch J. P., Pacher P., Evgenov O. V. (2011). Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation 123, 2263–2273 10.1161/CIRCULATIONAHA.110.981738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylvester J. T., Shimoda L. A., Aaronson P. I., Ward J. P. (2012). Hypoxic pulmonary vasoconstriction. Physiol. Rev. 92, 367–520 10.1152/physrev.00041.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thum T. (2012). MicroRNA therapeutics in cardiovascular medicine. EMBO Mol. Med 4, 3–14 10.1002/emmm.201100191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valastyan S., Reinhardt F., Benaich N., Calogrias D., Szász A. M., Wang Z. C., Brock J. E., Richardson A. L., Weinberg R. A. (2009). A pleiotropically acting microRNA, miR-31, inhibits breast cancer metastasis. Cell 137, 1032–1046 10.1016/j.cell.2009.03.047 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wang K., Long B., Zhou J., Li P. F. (2010a). miR-9 and NFATc3 regulate myocardin in cardiac hypertrophy. J. Biol. Chem. 285, 11903–11912 10.1074/jbc.M109.098004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S. M., Fu L. J., Duan X. L., Crooks D. R., Yu P., Qian Z. M., Di X. J., Li J., Rouault T. A., Chang Y. Z. (2010b). Role of hepcidin in murine brain iron metabolism. Cell. Mol. Life Sci. 67, 123–133 10.1007/s00018-009-0167-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong K. Y., Yu L., Chim C. S. (2011). DNA methylation of tumor suppressor miRNA genes: a lesson from the miR-34 family. Epigenomics 3, 83–92 10.2217/epi.10.74 [DOI] [PubMed] [Google Scholar]
- Yang J. H., Li J. H., Shao P., Zhou H., Chen Y. Q., Qu L. H. (2011). starBase: a database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res. 39 Database issue, D202–D209 10.1093/nar/gkq1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F., Jiao Y., Zhu Y., Wang Y., Zhu J., Cui X., Liu Y., He Y., Park E. Y., Zhang H.et al. (2012). MicroRNA 34c gene down-regulation via DNA methylation promotes self-renewal and epithelial-mesenchymal transition in breast tumor-initiating cells. J. Biol. Chem. 287, 465–473 10.1074/jbc.M111.280768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X., Ying L., Liu J., Dou D., He Q., Leung S. W., Man R. Y., Vanhoutte P. M., Gao Y. (2011). Role of sulfhydryl-dependent dimerization of soluble guanylyl cyclase in relaxation of porcine coronary artery to nitric oxide. Cardiovasc. Res. 90, 565–572 10.1093/cvr/cvr016 [DOI] [PubMed] [Google Scholar]
- Zovoilis A., Agbemenyah H. Y., Agis–Balboa R. C., Stilling R. M., Edbauer D., Rao P., Farinelli L., Delalle I., Schmitt A., Falkai P.et al. (2011). microRNA-34c is a novel target to treat dementias. EMBO J. 30, 4299–4308 10.1038/emboj.2011.327 [DOI] [PMC free article] [PubMed] [Google Scholar]