

Summary
Because nutrient-sensing nuclear and cytosolic acetylation mediates cellular autophagy, we investigated whether mitochondrial acetylation modulates mitochondrial autophagy (mitophagy). Knockdown of GCN5L1, a component of the mitochondrial acetyltransferase machinery, diminished mitochondrial protein acetylation and augmented mitochondrial enrichment of autophagy mediators. This program was disrupted by SIRT3 knockdown. Chronic GCN5L1 depletion increased mitochondrial turnover and reduced mitochondrial protein content and/or mass. In parallel, mitochondria showed blunted respiration and enhanced ‘stress-resilience’. Genetic disruption of autophagy mediators Atg5 and p62 (also known as SQSTM1), as well as GCN5L1 reconstitution, abolished deacetylation-induced mitochondrial autophagy. Interestingly, this program is independent of the mitophagy E3-ligase Parkin (also known as PARK2). Taken together, these data suggest that deacetylation of mitochondrial proteins initiates mitochondrial autophagy in a canonical autophagy-mediator-dependent program and shows that modulation of this regulatory program has ameliorative mitochondrial homeostatic effects.
Key words: GCN5L1, SIRT3, Mitochondrial autophagy, Acetylation, Parkin
Introduction
Macroautophagy controls turnover of cellular content to optimize homeostasis and recycle intracellular energy reserves. Interestingly, organelle autophagy can function selectively or in concert with macroautophagy to remove damaged organelles. This program is operational in mitochondria, where mitochondrial autophagy (mitophagy) selectively removes organelles during development and in response to stressors (Youle and Narendra, 2011). The contribution of mitophagy to ‘intracellular recycling’ under fasting or starvation conditions has not been clearly delineated in mammals. In yeast, disruption of mitophagy impairs starvation-induced mitochondrial clearance resulting in impaired mitochondrial fidelity (Kurihara et al., 2012).
During nutrient limitation, protein acetylation status confers adaptations to starvation and calorific restriction (Hirschey et al., 2010; Someya et al., 2010). Moreover, nuclear and cytosolic acetyltransferase and deacetylase enzymes regulate macroautophagy (Hamaï and Codogno, 2012; Lee et al., 2008; Yi et al., 2012), suggesting a link between the acetylome, cellular response to starvation and autophagy.
SIRT3 is the major mitochondrial deacetylase, regulating mitochondrial metabolism, redox status and cell death in a nutrient-dependent manner (Webster et al., 2012). Whether the mitochondrial acetylome regulates mitophagy is uncertain, and the ability of experimenters to interrogate this post-translational modification through Sirt3 cDNA transfection is limited because SIRT3 overexpression results in deacetylation of mitochondrial, cytosolic and nuclear proteins (Bao et al., 2010; Iwahara et al., 2012; Sundaresan et al., 2008). Recently, GCN5L1 has been identified as an essential component of the mitochondrial acetyltransferase program, and its genetic depletion selectively diminishes mitochondrial protein acetylation (Scott et al., 2012). We exploited this finding to investigate whether discrete mitochondrial deacetylation functions as a ‘molecular trigger’ to initiate mitochondrial autophagy, and to explore functional consequences of induction of this program.
Results and Discussion
Transient GCN5L1 knockdown promotes mitochondrial enrichment of autophagy mediators in a SIRT3-dependent manner
Investigations of mitophagy utilize composite measurements of the recruitment of autophagy mediators to the mitochondria, ubiquitylation of mitochondrial proteins, assessment of mitochondrial mass and evidence of mitochondrial inclusion into autophagosomes (Klionsky et al., 2012). To test whether manipulation of the mitochondrial acetylome modulates mitophagy, we measured mitochondrial enrichment of autophagy mediators [including the LC3-phosphatidylethanolamine conjugate LC3-II and p62 (also known as SQSTM1)] and mitochondrial protein ubiquitylation in response to siRNA-mediated knockdown (KD) of GCN5L1 or SIRT3. Isolated mitochondria from GCN5L1 KD HepG2 cells showed higher levels of LC3-II, p62 and protein ubiquitylation (Fig. 1A,B). In contrast, the mitochondrial LC3-II, p62 and protein ubiquitylation levels were similar following SIRT3 KD and transfection of scrambled siRNA (Fig. 1A,B). Confocal microcopy confirmed mitochondrial accumulation of these autophagy mediators as there was increased colocalization of GFP-tagged LC3 with dsRed-labeled mitochondria upon GCN5L1 KD (Fig. 1C,D) but not upon SIRT3 KD (supplementary material Fig. S1A). In parallel, p62, ubiquitin and the lysosomal protein Lamp1 showed enhanced localization to mitochondria following GCN5L1 KD (Fig. 1E; supplementary material Fig. S1B,C). Electron micrograph results mirrored these findings with evidence of more autophagic vacuoles and autolysosomes in GCN5L1 KD, which was further enhanced by bafilomycin inhibition of autophagic degradation (supplementary material Fig. S1D).
Fig. 1.
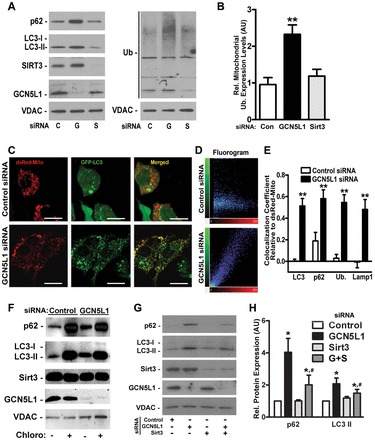
Depletion of GCN5L1 leads to mitochondrial accumulation of autophagy factors. (A) Western blots of isolated mitochondria from control (C), GCN5L1 (G) and SIRT3 (S) siRNA HepG2 cells with antibodies directed against p62, LC3, SIRT3, GCN5L1 and ubiquitylation (Ub) with VDAC as a loading control. (B) Relative mitochondrial protein ubiquitylation levels (representative blot shown in A) in the three groups. (C) Confocal microscopy in control or GCN5L1 siRNA cells showing colocalization of ds-Red-labeled mitochondria with GFP–LC3. Scale bars: 10 µm. (D) 2D fluorograms showing colocalization of ds-Red-Mito and GFP–LC3 as a distribution of pairs of pixel intensities (with greater diagonal alignment correlating to higher colocalization). (E) Quantification of the colocalization coefficient between ds-Red-Mito and LC3, p62, ubiquitin and Lamp1 displayed as Pearson coefficients in the colocalized volume (1, perfect correlation; 0, no correlation; –1, perfect inverse correlation). (F) Mitochondrial accumulation of autophagy mediators in response to the inhibition of lysosomal function by chloroquine. (G,H) Representative western blot analysis of concurrent SIRT3 and GCN5L1 siRNA on p62 and LC3-II mitochondrial accumulation in HepG2 cells. Protein levels are relative to VDAC. Control samples were normalized to 1, with KD levels determined relative to control values. Data are expressed as the mean±s.e.m. (n = 5 replicates per group). *P<0.05 versus scrambled siRNA control, #P<0.05 versus GCN5L1 siRNA levels.
Although not functionally characterized in autophagy, the cytosolic fraction of GCN5L1 (also known as BLOC1S1) has been shown to interact with non-lysosomal proteins involved in the biogenesis of lysosome-related organelles (Starcevic and Dell'Angelica, 2004). In this context we evaluated whether GCN5L1 KD preferentially initiated mitochondrial autophagy and/or affects global autophagy. We measured whole-cell levels of p70 S6K phosphorylation, p62 levels and the ratio of the cytosolic LC3 form LC3-I to LC3-II. Whole-cell levels of these mediators were not altered by GCN5L1 siRNA (supplementary material Fig. S2A), supporting a selective mitochondrial response to GCN5L1 KD. Additionally, we found that autophagy induction was intact, as evident by a similar response to rapamycin in control and GCN5L1 KD cells (supplementary material Fig. S2B). To validate this, we assayed dual RFP–GFP-labeled LC3 fluorescence stability. As GFP is more susceptible to lysosomal degradation, the quantification of RFP-labeled punctae represents successful LC3 delivery to the autolysosome and intact autophagic flux and lysosomal function (Klionsky et al., 2012). Confocal microscopy confirmed similar red punctae formation in control and GCN5L1 KD cells, further supporting the conclusion that autophagic flux is intact (supplementary material Fig. S2C,D). To exclude a ‘block’ in autophagic flux accounting for the increased ‘signature’ of mitochondrial autophagy, chloroquine was administered to disrupt lysosomal acidification. Chloroquine increased isolated mitochondrial autophagy markers in control and GCN5L1 KD cells, indicating intact flux (Fig. 1F). Overall these data suggest that GCN5L1 KD does not disrupt lysosomal biogenesis or function.
Because depletion of GCN5L1 and SIRT3 had opposing effects on mitochondrial protein acetylation, we evaluated whether the GCN5L1 KD effects were attenuated by simultaneous SIRT3 KD. The mitochondrial enrichment of p62 and LC3-II was diminished by concurrent SIRT3 and GCN5L1 KD (Fig. 1G,H). These counter-regulatory effects of the mitochondrial acetylome mirror the regulation of macroautophagy through genetic and pharmacologic modulation of acetylation (Eisenberg et al., 2009; Lee et al., 2008; Morselli et al., 2011; Yi et al., 2012).
Stable GCN5L1 knockdown results in mitochondrial autophagy with a reduction in mitochondrial protein content
In yeast, mitophagy-mediated mitochondrial depletion is discernable after prolonged starvation (Kurihara et al., 2012), consistent with the low intrinsic rate of mitochondrial turnover. Our data mirror this, as despite acute induction of mitophagy by GCN5L1 siRNA, there was no change in mitochondrial mass or protein levels (supplementary material Fig. S3A,B). To assess whether prolonged mitochondrial deacetylation reduces mitochondrial content, we generated stable GCN5L1 KD cells by lentiviral shRNA infection. Chronic GCN5L1 KD resulted in a similar attenuation of mitochondrial protein acetylation, in parallel with enrichment of mitochondrial LC3-II, p62 and protein ubiquitylation (Fig. 2A,B). Here, mitochondrial mass and mitochondrial protein levels were diminished (Fig. 2C–E). In contrast, levels of cytosolic, nuclear and endoplasmic reticulum proteins were not modified by GCN5L1 shRNA KD (supplementary material Fig. S3C,D).
Fig. 2.
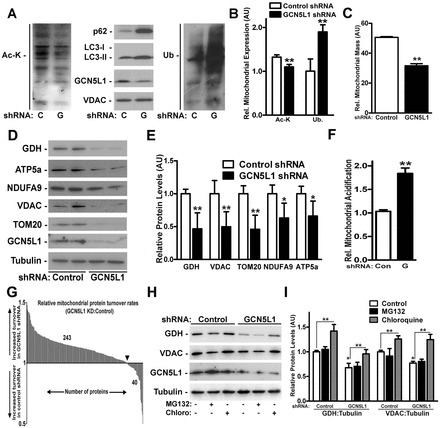
Chronic GCN5L1 KD attenuated mitochondrial mass and protein levels via autophagic degradation. (A) Representative immunoblot of mitochondrial protein acetylation and the accumulation of autophagy mediators in isolated mitochondria following chronic lentiviral GCN5L1 (G) or control (C) shRNA expression in HepG2 cells. (B) Quantification of mitochondrial protein acetylated at lysine residues and ubiquitin (Ub) levels in control and GCN5L1 shRNA HepG2 cells. (C) Relative mitochondrial mass in lentiviral-infected HepG2 cells. (D) Representative immunoblot of whole-cell levels of the mitochondrial proteins glutamate dehydrogenase (GDH), ATP5a, NDUFA9 (complex I), VDAC and TOM20 following expression of GCN5L1 shRNA. (E) Quantification of mitochondrial proteins levels assayed in whole-cell preparations from control and GCN5L1 shRNA cells (F) Quantification of mKeima acidification as a measure of mitophagy in shRNA-infected cells (n = 3) relative to the untreated control (set at 1). (G) Histogram depicting the relative rate of incorporation of the heavy label into mitochondrial proteins in GCN5L1 shRNA cells to that in control shRNA HepG2 cells 48 hours after label introduction. (H) Representative immunoblot of whole-cell levels of mitochondrial proteins following lentiviral GCN5L1 shRNA expression in response to MG132 and chloroquine. (I) Quantification of mitochondrial protein levels relative to tubulin normalized to the control shRNA samples (set at 1). Data are expressed as the mean±s.e.m. with n≥4 experiments per study. *P<0.05 versus shRNA control group, **P<0.01 versus controls.
To determine whether chronic mitochondrial deacetylation increased turnover by lysosomal hydrolysis, we quantified mitochondrial flux through the autophagic degradation pathway. Lysosomal-dependent loss of the mitochondrial-enriched pH-sensitive fluorescent protein mKeima (Katayama et al., 2011) increased with GCN5L1 shRNA, as measured by the fluorescence change upon mKeima delivery to the acidic lysosome (Fig. 2F; supplementary material Fig. S3E). In parallel, stable isotope labeling of amino acids in cell culture (SILAC) showed increased mitochondrial protein turnover in all mitochondrial compartments following chronic GCN5L1 depletion (Fig. 2G; supplementary material Fig. S3F; Table S1).
To further validate that the reduction in mitochondrial protein levels occurred by lysosomal degradation, we compared changes in the levels of glutamate dehydrogenase (GDH) and voltage-dependent anion carrier (VDAC) in response to the proteosome and lysosomal inhibitors, MG132 and chloroquine. In both control and GCN5L1 KD cells, chloroquine increased mitochondrial protein levels above those in vehicle-treated controls, whereas no significant change was seen after MG132 treatment. These data indicate that, under these experimental conditions, mitochondrial protein degradation is mediated by autophagy, rather than the proteosome (Fig. 2H,I).
The canonical autophagy mediators Atg5 and p62, but not Parkin, are required for GCN5L1-KD-mediated mitochondrial autophagy
We next investigated whether canonical autophagy mediators were necessary for GCN5L1 KD mitophagy, using mouse embryonic fibroblast cells (MEFs) deficient in the autophagy mediators Atg5 and p62. Similar to HepG2 cells, GCN5L1 KD in wild-type MEFs attenuated mitochondrial protein lysine acetylation and augmented mitochondrial p62, LC3-II and ubiquitylation (Fig. 3A,B; supplementary material Fig. S4A,B). Atg5 facilitates the lipidation of LC3-I to generate LC3-II, as an essential component of autophagosome assembly, and its absence prevented the accumulation of LC3-II on mitochondria following GCN5L1 KD (Fig. 3A). The adaptor protein p62 links ubiquitylated proteins to LC3-II. Despite the absence of mitochondrial LC3-II in Atg5 KO MEFs, the mitochondrial levels of p62 and ubiquitylation were elevated in the Atg5 KO MEFs, but not further induced in response to GCN5L1 KD (Fig. 3A,B). These data could be consistent with an impaired capacity to perform mitophagy, which is further supported by the finding of increased mitochondrial mass and protein content in Atg5 KO MEFs upon GCN5L1 knockdown (Fig. 3C–E). In p62 KO MEFs, LC3-II does not associate with mitochondria irrespective of GCN5L1 levels (Fig. 3A). In contrast, GCN5L1 KD increased mitochondrial protein ubiquitylation in p62 KO MEFs, indicating that ubiquitylation is upstream of p62 in deacetylation-induced mitophagy (Fig. 3B). The impairment of mitochondrial recycling in these cells is similarly supported by increased mitochondrial protein content and mass (supplementary material Fig. S4C,D) as found by others when canonical autophagy mediators are disrupted (Radoshevich et al., 2010).
Fig. 3.
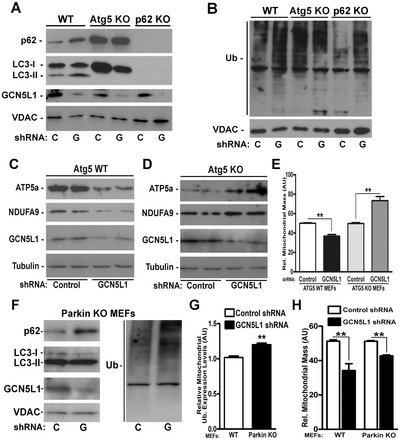
GCN5L1 depletion initiated mitophagy is dependent on Atg5 and p62 but not Parkin. (A,B) Representative mitochondrial protein immunoblots showing accumulation of autophagy mediators p62, LC3 and ubiquitylation in isolated mitochondria from wild-type (WT), Atg5 and p62 KO MEFs following GCN5L1 (G) shRNA and control (C) vector infection. (C,D) Representative whole-cell immunoblots showing mitochondrial proteins ATP5a and NDUFA9 in WT (C) and Atg5 KO (D) MEFs following shRNA infection. (E) Relative mitochondrial mass as measured by mitotracker green fluorescence in WT and Atg5 KO MEFs (n = 7). (F) Representative immunoblot of isolated mitochondria showing mitochondrial enrichment of p62, LC3-II and ubiquitin (Ub) after GCN5L1 shRNA infection in Parkin KO MEFs. (G) Quantification of mitochondrial protein ubiquitylation in Parkin KO MEFs in response to GCN5L1 KD. (H) Relative mitochondrial mass in control versus GCN5L1 shRNA in WT and Parkin KO MEFs (n = 3). Data are expressed as mean±s.e.m., **P<0.01.
In response to robust mitochondrial depolarization, the E3 ubiquitin ligase Parkin functions as an essential mediator of mitophagy (Narendra et al., 2008; Suen et al., 2010). As GCN5L1 KD leads to diminished mitochondrial membrane potential (see below), we investigated whether Parkin is necessary for these GCN5L1 KD effects. We found that Parkin was not required for mitochondrial autophagy induced by GCN5L1 KD, as increased mitochondrial enrichment of p62, LC3-II and ubiquitin was evident in Parkin KO MEFs following GCN5L1 KD (Fig. 3F,G), and mitochondrial mass and proteins levels were reduced by GCN5L1 KD irrespective of Parkin levels (Fig. 3H; supplementary material Fig. S4E,F). Hence, our study supports the hypothesis that mitochondrial protein lysine deacetylation enables lysine residue ubiquitylation as a component of the GCN5L1 KD mitochondrial phenotype. Further studies are required to identify E3 ligase(s) operational in this program.
GCN5L1 depletion increases resistance to mitochondrial stressors
As the dropout of mitochondria is likely to result in less reliance on oxidative phosphorylation for energy, we measured oxygen consumption and glycolysis following GCN5L1 depletion. Consistent with the ‘loss’ of mitochondrial content, GCN5L1 depletion evoked reduced oxygen consumption and a reciprocal increase in glycolysis, as well as a reduction in mitochondrial membrane potential (Fig. 4A–C).
Fig. 4.
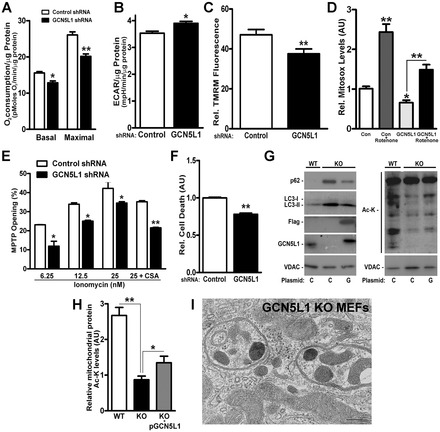
GCN5L1 KD alters mitochondrial function and morphology. (A,B) Basal and maximal [dinitrophenol (100 µM)] oxygen consumption and glycolysis rates in response to chronic GCN5L1 KD in HepG2 cells (n = 3). (C) Cytometric analysis with TMRM to assess relative mitochondrial membrane potential in control and GCN5L1 shRNA HepG2 cells (n = 3). (D) Mitochondrial superoxide generation under basal conditions (control), GCN5L1 shRNA cells and in response to the inhibition of complex I of the electron transfer chain by rotenone (15 µM, 4 hour) relative to control levels (set at 1). (E) Assessment of the relative ionomycin-mediated mitochondrial permeability transition. The addition of cyclosporin A (2 µM, 30 minutes) attenuated this transition. (F) Paraquat-mediated (1.5 mM, 24 hours) cell death in control and GCN5L1 shRNA HepG2 cells. (G) Representative immunoblot of isolated mitochondrial proteins from wild-type (WT) and GCN5L1 KO MEFs to assess reversal of mitochondrial accumulation of autophagy proteins and restoration of acetylation by reconstitution of GCN5L1. (H) Quantification of mitochondrial protein acetylation in KO MEFs in response to transient transfect of GCN5L1. Data are expressed as mean±s.e.m. *P<0.05, **P<0.01 versus respective controls. (I) Representative electron micrograph of a GCN5L1 KO MEFs to visualize mitochondrial inclusions in autophagosome vacuoles and mitochondrial morphology. Scale bar: 200 nm.
Because increased mitophagic flux enhances yeast mitochondrial fidelity (Kurihara et al., 2012) and more efficient mitochondrial function might ameliorate redox stress injury, we explored whether GCN5L1 KD cells are stress resilient. GCN5L1 depletion decreased rotenone-induced reactive oxygen species (ROS) generation, susceptibility to ionomycin-induced mitochondrial permeability transition, and enhanced resistance to paraquat-induced cell death (Fig. 4D–F).
To begin exploring this biology in vivo, GCN5L1-knockout (KO) mice were generated, but these were not viable. However, KO MEFs were harvested and showed the same mitochondrially restricted reduction in protein acetylation (supplementary material Fig. S5A,B) and mitochondrial enrichment of autophagy mediators (Fig. 4G; supplementary material Fig. S5C) as found in the KD studies. In addition, the reconstitution of GCN5L1 in these KO MEFs restored mitochondrial acetylation and reversed the mitochondrial accumulation of p62 and LC3-II (Fig. 4H; supplementary material Fig. S5C). Finally, GCN5L1 KO MEFs showed evidence of large autophagosome vacuoles enriched with mitochondria (Fig. 4I; supplementary material Fig. S6).
Future in vivo studies will be performed as conditional GNC5L1 KO mice are generated, to further explore the functional consequences of mitochondrial deacetylation on mitochondrial autophagy and to identify the mitochondrial targets triggering this program. Preliminary studies to investigate the mechanisms orchestrating starvation-associated mitochondrial quality control are encouraging, in that liver mitochondria from fasted C57BL/6 mice showed reduced GCN5L1 levels, increased SIRT3 levels and mitochondrial accumulation of LC3-II (supplementary material Fig. S5D).
In conclusion, this study finds that the genetic modulation of the mitochondrial acetylome is sufficient to initiate mitochondrial autophagy. We show that chronic GCN5L1 KD resulted in a reduction in mitochondrial content, coupled with an enhancement of mitochondrial stress resilience. This program is dependent on SIRT3 levels, and on the canonical autophagy mediators Atg5 and p62, but is independent of Parkin. The manipulation of this program to enhance mitochondrial autophagy might be a novel approach to further delineate intrinsic pathways operational in nutrient-stress-induced mitophagy, and might ameliorate diseases associated with mitochondrial dysfunction and increased mitochondrial protein acetylation (Kendrick et al., 2011; Wagner et al., 2012). Of note, while our manuscript was under revision, an article showed that SIRT3, under oxidative stress conditions, evoked mitophagy via deacetylation of FoxO3 (Tseng et al., 2013). Further work will be required to investigate whether this exogenous-stress-induced loss of mitochondrial proteins shares features with the mitophagy program induced by discrete mitochondrial protein deacetylation as described here.
Materials and Methods
Cells
KO MEFs were generated from e11.5 embryos or received as gifts (see Acknowledgements). For autophagy, lysosomal and proteosomal functional studies, cholorquine (60 µM), rapamycin (1 µM), bafilomycin (10 nM) or MG132 (7.5 µM) was added to the culture medium for 24 hours. All animal experiments were performed according to approved guidelines.
Protein KD
siRNA transfection was performed using scrambled or Smartpool siRNA targeting SIRT3 and GCN5L1 (Dharmacon) via electroporation. Lentiviral shRNA were generated in HEK293T cells using lentiviral Packaging Mix (Sigma). Viral particle infection to generate stable KD cells occurred under puromycin (2 µg/ml) selection.
Immunofluorescence
Immunofluorescence studies were performed in HepG2 cells transfected with scrambled or GCN5L1 siRNA (Dharmacon), with the co-transfection of plasmids encoding dsRed-mito (Clontech), GFP–ubiquitin (Addgene), GFP–LC3 and RFP, GFP–Lamp1 or p62.
Flow cytometry
Flow cytometry studies were performed to assess the relative mitochondrial membrane potential with TMRM (Invitrogen), mass using Mitotracker Green FM (Invitrogen) and superoxide production following treatment with rotenone (15 µM) for 4 hours by Mitosox (Invitrogen). Induction of autophagy and/or mitophagy was assessed using RFP–GFP–LC3- and mKeima-encoding plasmid constructs, respectively. The ratio of RFP∶GFP signal was determined by geometric mean analysis. For the mKeima studies, samples underwent cytometric analyses while they were excited simultaneously with a 407 nm/605 nm (ex/em) (for neutral pH fluorescence) and at 532 nm/610 nm (ex./em.) (for acidic pH fluorescence). Mitochondrial permeability transition was performed using the MitoProbe Transition Pore Assay Kit (Molecular Probes). Cell death was determined after 24 hours of Parquat treatment and using the Live/Dead Viability/Cytotoxicity Kit (Invitrogen).
SILAC protocol details
Control and GCN5L1 KD HepG2 cells were fed with non-isotopic light medium (6 passages), then chased with isotopic heavy medium (48 hours). Mitochondria were harvested, suspended in a 1% CHAPS buffer, separated by SDS-PAGE, stained in-gel (Coomassie) followed by in-gel trypsin digestion. Peptides were identified by liquid chromatography–tandem mass spectrometry (LC-MS/MS) and proteins with less than two unique peptides were discarded. Mitochondrial-associated proteins were identified [by use of the Gene Ontology (GO) database] and batched by sub-mitochondrial compartment. The heavy∶light ratio was calculated for each protein with the ratio of KD∶control turnover ratios calculated for each unique protein (>1, more KD turnover; <1 = more control turnover).
In vivo mouse experiments
4-month-old C57BL/6 male mice were used for \fasting experiments where mice were either fed ad libitum or fasted for 48 hours. Mice were killed and liver mitochondria isolated by differential centrifugation (Lu et al., 2011). The animal protocol was approved by the NHLBI Animal Care and Use Committee.
Statistics
Immunoblots were analyzed using ImageJ (National Institutes of Health). Data are expressed as the mean±s.e.m. Two-tailed Student t-tests were performed between groups and multiple comparison analysis was performed by ANOVA. P<0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Toren Finkel for the GFP–LC3 plasmid and for Atg5 KO MEFs, Jennifer Lippincott-Schwartz for the GFP–LAMP1 plasmid and Atsushi Miyawaki for the mKeima construct. The Sirt3+/− mice were supplied by Fred Alt and the p62 KO MEFs by Richard Youle.
Footnotes
Author contributions
B.R.W., I.S., K.H., M.V.S. and M.N.S. planned the project; B.R.W., I.S., K.H., J.H.L., Z.L., D.M., Y.C., L.C. and P.S.C. performed the experiments; B.R.W., I.S., Z.L., M.V.S., D.M., Y.C., L.S., P.S.C., M.P.D., J.P.M., C.A.C., M.G. and M.N.S. analyzed data and prepared figures and B.R.W., I.S., M.P.D, J.P.M., C.A.C., M.G. and M.N.S. wrote the paper.
Funding
This work was supported by the National Institute of Heart, Lung, and Blood Institute Division of Intramural Research [grant number HL006047-01 to M.N.S.]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.131300/-/DC1
References
- Bao J., Lu Z., Joseph J. J., Carabenciov D., Dimond C. C., Pang L., Samsel L., McCoy J. P., Jr, Leclerc J., Nguyen P. et al. (2010). Characterization of the murine SIRT3 mitochondrial localization sequence and comparison of mitochondrial enrichment and deacetylase activity of long and short SIRT3 isoforms. J. Cell. Biochem. 110, 238–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg T., Knauer H., Schauer A., Büttner S., Ruckenstuhl C., Carmona-Gutierrez D., Ring J., Schroeder S., Magnes C., Antonacci L. et al. (2009). Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 11, 1305–1314 10.1038/ncb1975 [DOI] [PubMed] [Google Scholar]
- Hamaï A., Codogno P. (2012). New targets for acetylation in autophagy. Sci. Signal. 5, pe29 10.1126/scisignal.2003187 [DOI] [PubMed] [Google Scholar]
- Hirschey M. D., Shimazu T., Goetzman E., Jing E., Schwer B., Lombard D. B., Grueter C. A., Harris C., Biddinger S., Ilkayeva O. R. et al. (2010). SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464, 121–125 10.1038/nature08778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwahara T., Bonasio R., Narendra V., Reinberg D. (2012). SIRT3 functions in the nucleus in the control of stress-related gene expression. Mol. Cell. Biol. 32, 5022–5034 10.1128/MCB.00822-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama H., Kogure T., Mizushima N., Yoshimori T., Miyawaki A. (2011). A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem. Biol. 18, 1042–1052 10.1016/j.chembiol.2011.05.013 [DOI] [PubMed] [Google Scholar]
- Kendrick A. A., Choudhury M., Rahman S. M., McCurdy C. E., Friederich M., Van Hove J. L., Watson P. A., Birdsey N., Bao J., Gius D. et al. (2011). Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem. J. 433, 505–514 10.1042/BJ20100791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D. J., Abdalla F. C., Abeliovich H., Abraham R. T., Acevedo-Arozena A., Adeli K., Agholme L., Agnello M., Agostinis P., Aguirre-Ghiso J. A. et al. (2012). Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8, 445–544 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara Y., Kanki T., Aoki Y., Hirota Y., Saigusa T., Uchiumi T., Kang D. (2012). Mitophagy plays an essential role in reducing mitochondrial production of reactive oxygen species and mutation of mitochondrial DNA by maintaining mitochondrial quantity and quality in yeast. J. Biol. Chem. 287, 3265–3272 10.1074/jbc.M111.280156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I. H., Cao L., Mostoslavsky R., Lombard D. B., Liu J., Bruns N. E., Tsokos M., Alt F. W., Finkel T. (2008). A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 105, 3374–3379 10.1073/pnas.0712145105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z., Bourdi M., Li J. H., Aponte A. M., Chen Y., Lombard D. B., Gucek M., Pohl L. R., Sack M. N. (2011). SIRT3-dependent deacetylation exacerbates acetaminophen hepatotoxicity. EMBO Rep. 12, 840–846 10.1038/embor.2011.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morselli E., Mariño G., Bennetzen M. V., Eisenberg T., Megalou E., Schroeder S., Cabrera S., Bénit P., Rustin P., Criollo A. et al. (2011). Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J. Cell Biol. 192, 615–629 10.1083/jcb.201008167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D., Tanaka A., Suen D. F., Youle R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803 10.1083/jcb.200809125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radoshevich L., Murrow L., Chen N., Fernandez E., Roy S., Fung C., Debnath J. (2010). ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell 142, 590–600 10.1016/j.cell.2010.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott I., Webster B. R., Li J. H., Sack M. N. (2012). Identification of a molecular component of the mitochondrial acetyl transferase program; a novel role for GCN5L1. Biochem. J. 443, 655–661 10.1042/BJ20120118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Someya S., Yu W., Hallows W. C., Xu J., Vann J. M., Leeuwenburgh C., Tanokura M., Denu J. M., Prolla T. A. (2010). Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 143, 802–812 10.1016/j.cell.2010.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starcevic M., Dell'Angelica E. C. (2004). Identification of snapin and three novel proteins (BLOS1, BLOS2, and BLOS3/reduced pigmentation) as subunits of biogenesis of lysosome-related organelles complex-1 (BLOC-1). J. Biol. Chem. 279, 28393–28401 10.1074/jbc.M402513200 [DOI] [PubMed] [Google Scholar]
- Suen D. F., Narendra D. P., Tanaka A., Manfredi G., Youle R. J. (2010). Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc. Natl. Acad. Sci. USA 107, 11835–11840 10.1073/pnas.0914569107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaresan N. R., Samant S. A., Pillai V. B., Rajamohan S. B., Gupta M. P. (2008). SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol. Cell. Biol. 28, 6384–6401 10.1128/MCB.00426-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng A. H., Shieh S. S., Wang D. L. (2013). SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic. Biol. Med. 63, 222–234 10.1016/j.freeradbiomed.2013.05.002 [DOI] [PubMed] [Google Scholar]
- Wagner G. R., Pride P. M., Babbey C. M., Payne R. M. (2012). Friedreich's ataxia reveals a mechanism for coordinate regulation of oxidative metabolism via feedback inhibition of the SIRT3 deacetylase. Hum. Mol. Genet. 21, 2688–2697 10.1093/hmg/dds095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster B. R., Lu Z., Sack M. N., Scott I. (2012). The role of sirtuins in modulating redox stressors. Free Radic. Biol. Med. 52, 281–290 10.1016/j.freeradbiomed.2011.10.484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi C., Ma M., Ran L., Zheng J., Tong J., Zhu J., Ma C., Sun Y., Zhang S., Feng W. et al. (2012). Function and molecular mechanism of acetylation in autophagy regulation. Science 336, 474–477 10.1126/science.1216990 [DOI] [PubMed] [Google Scholar]
- Youle R. J., Narendra D. P. (2011). Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14 10.1038/nrm3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.