

Abstract
Chronic stress may be a risk factor for developing Alzheimer’s disease (AD), but most studies of the effects of stress in models of AD utilize acute adverse stressors of questionable clinical relevance. The goal of this work was to determine how chronic psychosocial stress affects behavioral and pathological outcomes in an animal model of AD, and to elucidate underlying mechanisms. A triple-transgenic mouse model of AD (3xTgAD mice) and nontransgenic control mice were used to test for an affect of chronic mild social stress on blood glucose, plasma glucocorticoids, plasma insulin, anxiety and hippocampal Aβ, ptau and BDNF levels. Despite the fact that both control and 3xTgAD mice experienced rises in corticosterone during episodes of mild social stress, at the end of the 6 week stress period 3xTgAD mice displayed increased anxiety, elevated levels of Aβ oligomers and intraneuronal Aβ, and decreased BDNF levels, whereas control mice did not. Findings suggest 3xTgAD mice are more vulnerable than control mice to chronic psychosocial stress, and that such chronic stress exacerbates Aβ accumulation and impairs neurotrophic signaling.
Keywords: Alzheimer’s disease, amyloid oligomers, psychosocial stress, corticosterone, hippocampus, BDNF
1. Introduction
Approximately 4.5 million Americans are currently living with a diagnosis of Alzheimer’s disease (AD), the most common form of dementia and cognitive decline (Hebert, 2003). There are some cases of familial AD and known genetic predispositions to the disease; however, the vast majority of AD cases are sporadic and likely due to a variety of environmental factors (Mattson, 2004). Among circumstances that may increase the risk of developing AD, head trauma (van den Heuvel et al., 2007), a sedentary lifestyle (Liang et al., 2010), and chronic stress (Simard et al., 2009) have emerged from both clinical and experimental studies. The occurrence of a major stressful event may lower the age of onset of familial AD (Mejia et al., 2003). People vary greatly in their response to stressful events, and those most vulnerable to adverse outcomes exhibit a trait that can be described as negative affectivity, neuroticism or distress proneness (Watson and Clark, 1984; Lockenhoff et al., 2009; Weiss et al., 2009). Patients with a high level of distress proneness are 2.7 times more likely to develop AD than those not prone to distress, and this trait is also associated with a more rapid progression of the disease (Wilson et al., 2006, 2007). Animal studies show that adverse stress not only exacerbates impairments in learning but also causes an earlier onset of neurological symptoms in several different rodent models of AD (Srivareerat et al., 2009; Catania et al., 2009; Jeong et al., 2006; Dong et al., 2004). Yet despite the clinical and experimental evidence demonstrating a role for stress in the onset and progression of AD, few studies to date have evaluated the effects of chronic stress on behavioral and biochemical outcomes in animal models of AD; instead more severe acute highly adverse stressors in lieu of long-term clinically relevant ones.
Social stress is an unavoidable aspect of complex societies and can have a series of adverse affects of an organism (Sapolsky, 2005; Mooring et al., 2006; Wolf et al., 2009). Research on primates has shown that socially hierarchical societies cause numerous deleterious health effects on individuals as a result of social stressors in both low and high-ranking individuals, including hormonal, inflammatory, reproductive and cardiovascular effects (Sapolsky, 2005). Stress induces activation of the hypothalamic-pituitary-adrenal (HPA) axis which leads to increased release of steroid hormones known as glucocorticoids from the adrenal cortex (cortisol in humans, corticosterone in rodents) (Nelson, 1972; Munck and Guyre, 1986). Circulating glucocorticoids, while necessary for normal life function, can exert a host of adverse effects on the central nervous system (CNS) when in excess. These effects include dendritic atrophy, impaired neurogenesis, impaired synaptic plasticity and cell death (McEwen, 2001, 2007; Baran et al., 2005; Stranahan et al., 2008). In addition to these structural and physiological effects, social stressors, or exogenous corticosterone, causes impairments in spatial memory, contextual memory and object recognition as well as heightened anxiety (Krugers et al., 1997; Li et al., 2008; Mitra and Sapolsky, 2008). Increases in anxiety from stress are more pronounced in aged rodents compared to young, which could indicate an age-related vulnerability to stress (Shoji and Mizoguchi, 2010). It is likely that this is due to an age-related alteration in the HPA axis; indeed, levels of circulating glucocorticoids increase with age in both clinical and experimental studies (Landfield et al., 1978; Brett et al., 1983; Vancauter et al., 1996; Kudielka et al., 2004). Yet despite the high prevalence of social stressors in modern society and evidence for increasing susceptibility to the effects of stress with age, most studies of social stressors utilize acute adverse stressors that lack clinical relevance and few quantify outcomes related to anxiety behaviors.
In addition to age, disease state may also influence vulnerability to stress, particularly in the case of neurodegenerative diseases which often cause damage to regions of the brain involved in regulating the HPA axis and the stress response. The hippocampus, a region of the brain considered critical for learning and memory, is prone to damage in AD including cell loss, dendritic atrophy and impaired synaptic function, potentially due to accumulations in the protein Aβ (Mattson, 2004; Cequeira et al., 2007). In addition, the hippocampus directly regulates the HPA axis, as demonstrated by studies that show that hippocampal CA3 lesions cause increases in corticosterone levels (Herman et al., 1989; Jacobson and Sapolsky, 1991; Roozendaal et al., 2001). It is possible that in AD, an increase in Aβ accumulation in the hippocampus causes disinhibition of the HPA axis which then leads to an increase in basal glucocorticoid levels (Kulstad et al., 2005; Breyhan et al., 2009; Nuntagij et al., 2009). Further, increases in glucocorticoid levels may promote accelerated Aβ production and a decrease in Aβ degradation in AD (Green et al., 2006). This implies that early progression of AD pathology could be leading to the initiation of a powerful positive-feedback loop that exacerbates AD pathology via dis-inhibition of the HPA axis, production of glucocorticoids and increased production and aggregation of Aβ. Indeed, elevated basal levels of corticosterone are further increased in AD animal models with the application of adverse stress implying an elevated stress response in AD which may lead to exacerbated AD pathology and, potentially, a worsening of cognitive deficits (Green et al., 2006; Jeong et al., 2006; Dong et al., 2008; Lee et al., 2009). Circulating glucocorticoids are elevated in both clinical and experimental studies of AD, indicating a dysregulation of the HPA-axis and the potential for a particular susceptibility to stress in AD (Davis et al., 1986; Umegaki et al., 2000; Rasmuson et al., 2001; Csernansky et al., 2006). Yet this scenario has not established experimentally.
The goal of this study was to utilize a clinically relevant model of long-term social stress to determine whether pre-symptomatic AD mice display greater vulnerability to stress than wild-type controls including increases in corticosterone, changes in anxiety behaviors and worsening of AD pathology. Results presented here begin to outline susceptibility to stress for AD that may be related to decreases in levels of the neuroprotective protein brain-derived neurotrophic factor (BDNF).
2. Materials and Methods
2.1 Mice
Animal care and experimental procedures followed National Institutes of Health guidelines and were approved by the National Institute on Aging Animal Care and Use Committee. Twelve month-old male triple transgenic AD (3xTgAD) mice (n=25) harboring PS1M146V, APPSwe, and tauP301L transgenes (Oddo, et al., 2003) that had been backcrossed to C57BL/6 mice for seven generations were group housed (4–5 per cage) and maintained under a 12 hr light and dark cycle. Age-matched male C57BL/6 wild type (WT) mice were maintained in the same animal room and were used as controls (n=27).
2.2 Stress Protocol
Mice (n=18 WT; n=16 3xTgAD) underwent six weeks of chronic mild social stress. Briefly, the cage composition of group-housed mice was changed for 6 hours per day for 2–3 days per week at random followed by return to their home cage (Alzoubi et al., 2009; Gerges et al., 2001). Cage compositions were randomized in order to maintain intensity of social stress. Cage composition was restricted to same-strain mice only. Control mice remained in their group-housed cages for the duration of the study with no change in cage compostion.
2.3 Blood Draw
Blood samples were drawn from the tail either 2 hours after the beginning of stress, the end of the stress period (6 hours), or the following morning after an overnight fast. For each stress session for which blood samples were drawn, only one of the above time points was probed to reduce any effect of the blood draw on the levels of the outcomes being probed (ie, if a blood sample was drawn at the 2 hour time point it was not drawn again at 6 hours). Briefly, scissors were cleaned with ethanol and the tip of tail (< 5 mm of tissue) was cut off. Blood was gathered using a heparinized micro-hematocrit capillary tube (Fisher Scientific; Pittsburgh, PA) and clotisol (Agri-med; St Louis, MO) was used to stop the bleeding. Samples were used to quantify either corticosterone, glucose or insulin. For quantification of corticosterone and glucose two sets of data gathered from blood draws done during different weeks of the study were averaged together (2 hour timepoint: weeks 3 and 4; 6 hour timepoint: weeks 2 and 4; morning fasting timepoint: weeks 4 and 5). For insulin data one blood draw for each timepoint was used (2 hour timepoint: week 3; 6 hour timepoint: week 4; morning fasting timepoint: week 5).
2.4 Quantification of Corticosterone Glucose and Insulin Levels
Whole-blood glucose concentration was quantified at the time of blood draw using a Freestyle Glucose meter (Abbott Laboratories; Abbott Park, IL). Blood samples were then centrifuged at 12,500 rpm for 12 minutes at 4° and plasma supernatant was removed and stored at −80°. Plasma corticosterone was quantified using an RIA kit (MP Biomedicals; Solon, OH) according to manufacturer’s instructions. Insulin was quantified using a commercially-available ELISA kit (Crystal Chem Inc.; Downers Grove, IL). Baseline, morning, non-fasting levels of corticosterone (n=12 WT; n=8 3xTgAD), glucose and insulin (n=4 WT; n=8 3xTgAD), were established in a separate pilot study and are presented for reference to current results.
2.5 Behavior Testing
At the end of the 6-week stress period mice were tested for anxiety behavior using an open field testing paradigm (MEDOFA-MS system; Med Associates). Briefly, mice were placed in the center of an open field, and exploration was assessed for 5 min. Cages were routinely cleaned with ethanol following each session. The dimensions of the arena were 40 cm × 40 cm, of which the peripheral 10 cm were considered as the residual zone and the central 30 cm were considered as zone 1.
2.6 Tissue Collection
Within one week of behavior testing, all mice were deeply anesthetized and decapitated for tissue harvest. The brain was exposed and separated into right and left hemispheres. The left hippocampus was dissected, immediately frozen on dry ice and stored at −80° for western blot analysis of Aβ, phosphorylated tau (ptau), total tau and ELISA quantification of BDNF. The right hemisphere was post-fixed in a 4% paraformaldehyde solution for 20 minutes for immunohistochemical analysis of Aβ and ptau. Brain tissue was then transferred to 30% sucrose/PBS and stored for 3 days at 4°C. Samples were freeze-mounted with OCT medium (Triangle Biomedical Sciences; Durham, NC) for coronal cryosectioning.
2.7 Immunohistochemistry
Brain tissue was assayed for Alzheimer’s pathology using antibodies to Aβ and phosphorylated tau (ptau). Immunohistochemical analyses were performed on brain tissue from a subset of 3xTgAD mice (n=4 control; n=12 stress).
Six to eight brain sections (40 μm) from each mouse were mounted on superfrost slides and prepared for immunohistochemical staining. A polyclonal antibody raised against Aβ (Cell Signaling Cat no. 2454; Danvers, MA) was used that detects several forms of Aβ including Aβ-40 and Aβ-42 oligomers. Phosphorylated tau (ptau) was detected using a monoclonal antibody (Pierce Endogen Cat no. MN1040; Rockford, IL) shown to label neurofibrillary tangles (Spillatini et al., 1996). Slides were incubated with boiling citric acid, quenched in 0.3% peroxide solution and blocked with PBS with bovine serum albumin and tween. Primary antibodies (1:200) were applied overnight followed by treatment with a biotinylated goat anti-rabbit immunoglobulin G. Sections were developed using 3,3-diaminobenzidine (Vector Labs; Burlingame, CA) dehydrated in an ethanol series, and coverslipped using Permount (Fisher Chemical; Fairlawn, NJ). Previous studies were performed to determine optimal antibody dilutions. A negative control with no primary antibody was always included for verification of specificity of immunohistochemical techniques.
Both Aβ and ptau staining in the hippocampus were analyzed using quantitative densitometry. Hippocampi were photographed at 100× magnification using a digital camera and stereomicroscope system and Axiovision software (Zeiss; Thornwood, NY). Pictures were cropped to include the region of interest (900 × 300 pixels). Pictures were then inverted and analyzed for pixel intensity. Values were averaged and compared.
2.8 Immunoblot analysis
A subset of samples were analyzed by immunoblotting for Aβ and ptau to quantify AD pathology in the hippocampus. Hippocampi (n=5 control/WT, n=5 control/AD, n=10 stress/WT, n=8 stress/AD) were homogenized in RIPA buffer with protease inhibitor on ice. Protein concentrations were determined using a Bradford assay. Immunoblots were performed using 50 μg of total protein extract separated on 6–12% SDS-PAGE gels and then transferred electrophoretically to polyvinylidene difluoride (PVDF) (for p-tau and total tau) or nitrocellulose (for Aβ and actin) membranes. Membranes were blocked with 5% milk in TBS-t, washed in TBS-t and incubated overnight at 4° in a primary antibody (1:1000), either Aβ 6E10 (Santa Cruz Biotechnology; Santa Cruz, CA), phf-tau AT 180 (Pierce Endogen; Rockford, IL), total tau T46 (Invitrogen; Carlsbad, CA) or actin (Sigma-Aldrich; St. Louis, MO). Membranes were then incubated in a secondary antibody solution for 30 minutes (1:5000, then in the presence of horseradish peroxidase-conjugated anti-mouse IgG) for 30 minutes. The optical density of immunoreactive bands was detected and quantified by chemiluminescence using the ECL system.
2.9 BDNF ELISA
A commercially available ELISA kit (R&D Systems, blank) was used to quantify BDNF levels in the same hippocampi used for western blot according to manufacturer’s instructions. Results were analyzed using total protein data from the Bradford assay and expressed as pg BDNF per mg total protein for each sample.
2.10 Statistics
A two-way ANOVA was used to test for an effect of strain (WT, 3xTgAD) or stress (control, stress) on whole blood glucose, blood plasma corticosterone, insulin and distance traveled in the open field. Quantitative densitometric analysis of Aβ and ptau staining were analyzed by t-test to probe for a difference between control and stress for AD mice. BDNF ELISA results were analyzed using a one-way ANOVA followed by pairwise comparisons using post-hoc Bonferroni tests. Significance was defined as p<0.05.
3. Results
3.1 3xTgAD mice exhibit perturbed endocrine and metabolic responses to stress
Stress activates the HPA axis resulting in increases in circulating glucocorticoids. We therefore quantified plasma corticosterone in blood samples taken at different time points after the beginning of the stress period to determine how 3xTgAD and wild type (WT) control mice respond to mild social stress. Basal blood plasma corticosterone in 3xTgAD mice was three times higher than that of WT mice at the 2 hour time point. This increase was significant and was preserved during the 6 hour time point as well, and a two-way ANOVA established a significant effect of strain at both 2 hours and 6 hours after the beginning of the stress period (p<0.004 ; Fig 1A). Two hours of mild social stress caused a significant increase in plasma corticosterone levels in both WT and 3xTgAD mice compared to controls (p=0.02). After 6 hours of mild stress, corticosterone returned to baseline levels in WT mice (Fig 1A). However, 3xTgAD mice continued to display elevated corticosterone compared to non-stressed controls (Fig 1A). Interestingly, after an overnight fast, non-stressed mice displayed a marked increase in corticosterone levels, representing seven-fold and two-fold increases over basal (nonfasting) levels for WT and 3xTgAD mice respectively (p<0.001). Mice that experienced mild social stress did not display elevations in plasma corticosterone after overnight fasting (Fig 1A).
Figure 1. 3xTgAD mice exhibit elevated basal plasma corticosterone levels and prolonged responses to social stress compared to WT control mice.

Blood plasma corticosterone (A), whole-blood glucose (B) and blood plasma insulin (C) levels at 2 hours, 6 hours and the morning after a 6 hour period of social stress in WT and 3xTgAD mice (AD). Measurements taken in the morning were done after an overnight fast. Basal corticosterone was increased in AD mice compared to WT mice (A) and although both strains displayed a rise in corticosterone in stressed mice compared to non-stressed controls, this increase was shorter in WT mice, which displayed normal corticosterone levels by 6 hours after the onset of stress compared to AD mice which still displayed elevated circulating corticosterone at this time point. Interestingly, after an overnight fast, corticosterone levels were dramatically increased in mice that had not previously been exposed to social stress, but not in mice that experienced stress (A). This phenomenon was noted in both strains. Basal glucose levels were lower in AD mice compared to WT mice (B) and at 2 hours after the beginning of stress, both strains experienced a non-significant rise in glucose levels. By 6 hours, both strains displayed a significant (p< 0.001) decrease in glucose levels, but by the morning after a 6 hour stress session, glucose was again elevated in stressed mice compared to non-stressed controls, an increase which was significant for both strains (p< 0.03; B). Similar to glucose levels, insulin levels were also decreased at 6 hours after the beginning of stress, however this was non-significant and, in contrast to glucose patterns, came after a small drop in insulin at the 2 hour time point (C).
Whole blood glucose levels were lower in 3xTgAD mice compared to WT mice at all time points tested, and a significant effect of strain was noted each time point (p<0.01 ; Fig 1B). Mild social stress caused a small, non-significant, increase in glucose levels for both 3xTgAD and WT mice at 2 hours (Fig 1B). However, at 6 hours after the onset of mild stress, both 3xTgAD and WT mice experienced a drop in glucose compared to non-stressed mice, and a significant effect of stress was noted (p=0.01; Fig 1B). By the morning after social stress, and after an overnight fast, both WT and 3xTgAD mice displayed a significant rise in corticosterone levels in the mild stress group compared to non-stressed group (p=0.03; Fig 1B).
As the results of plasma glucose measurements suggested a metabolic aberrancy in 3xTgAD mice, we measured plasma insulin levels to better elucidate the metabolic profile of 3xTgAD mice. Similar to glucose levels, insulin levels were decreased at 6 hours after the beginning of social stress, with a greater decrease in 3xTgAD mice compared to WT mice (Fig 1C). Morning fasting insulin levels were low in all mice and not significantly different for stressed mice versus non-stressed mice of either genotype.
3.2 Chronic mild social stress causes an increase in anxiety behaviors in AD mice, but not in WT mice
The open field is used to test for anxiety-like behavior by measuring the distance traveled in different regions of an open field. Results are analyzed by region where zone 1 is the area in the center of the field and the residual is the area around zone 1 near the walls of the apparatus. More distance traveled in zone 1 is considered less anxious behavior and in general, less movement corresponds to higher levels of anxiety as well. For both regions of the open field, the distance traveled by 3xTgAD mice was less than the distance traveled by WT mice. A significant effect of strain was measured for both regions and for the total distance traveled (p<0.04) (Fig 2). Both strains displayed a decrease in distance traveled in the open field for stressed mice compared to non-stressed mice indicating an increase in anxiety due to chronic stress (Fig 2). However, the magnitude of the decrease in distance traveled was significantly greater for 3xTgAD mice compared to WT mice (p<0.03).
Figure 2. 3xTgAD mice exhibit an anxiety phenotype that is exacerbated by mild chronic social stress.
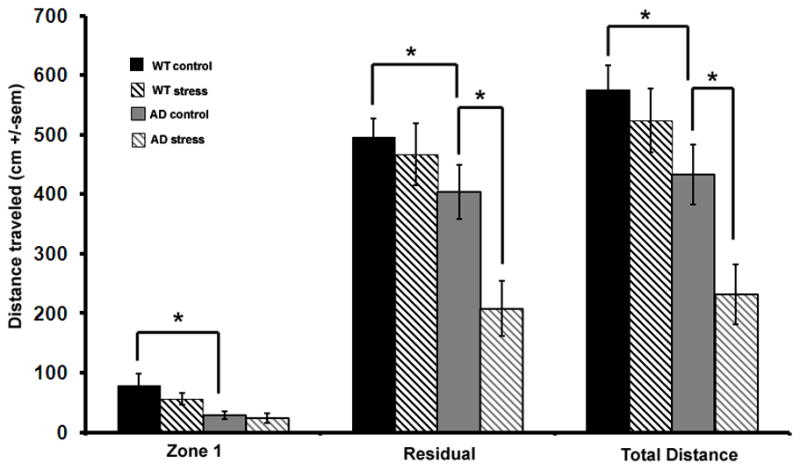
The distance traveled in the open field, a test of anxiety. Less distance traveled corresponds to greater anxiety. Results are expressed as distance traveled in zone 1 (the center area), the residual (area nearest the perimeter of the apparatus) and total distance traveled for wild type and AD mice. Basal anxiety was higher in AD mice compared to WT mice. After 6 weeks of social stress, AD mice displayed heightened anxiety compared to non-stressed controls whereas WT mice did not.
3.3 3xTgAD mice display an increase in intraneuronal Aβ but no change in ptau levels in response to chronic social stress
AD is characterized by accumulations of Aβ and hyperphosphorylation of tau. Further, activation of the HPA axis and an increase in circulating corticosterone can cause alterations in both Aβ and tau in the brains of rodents (Stein-Behrens et al., 1994; Kang et al., 2007; Sayer et al., 2008; Sotiropoulos et al., 2008). We therefore quantified Aβ (Fig 3A–3D) and ptau (Fig 3E–3H) levels in the hippocampus using immunoblotting, and also used immunohistochemistry to visualize the cellular localization of staining in the hippocampus (Fig 3). The immunoblots revealed the presence of Aβ oligomers in samples from 3xTgAD mice, with the amounts of Aβ oligomers being greater in the mice that experienced social stress compared to non-stressed controls (Fig 3D). Quantification of immunoblot results confirmed this trend. As such, immunohistochemical analysis of the localization of Aβ staining focused on brain tissue sections from 3xTgAD mice only. Aβ immunoreactivity was concentrated in neurons in the CA1 region of the hippocampus (Fig 3A & 3B). Mice that experienced social stress displayed an increase in intraneuronal Aβ compared to non-stressed mice. This was confirmed by quantitative densitometry which showed a significant increase in pixel intensity for stressed compared to non-stressed 3xTgAD mice (Fig 3C; p=0.049).
Figure 3. Mild social stress enhances intraneuronal Aβ immunoreactivity, but not tau hyperphosphorylation in hippocampal neurons of 3xTgAD mice.
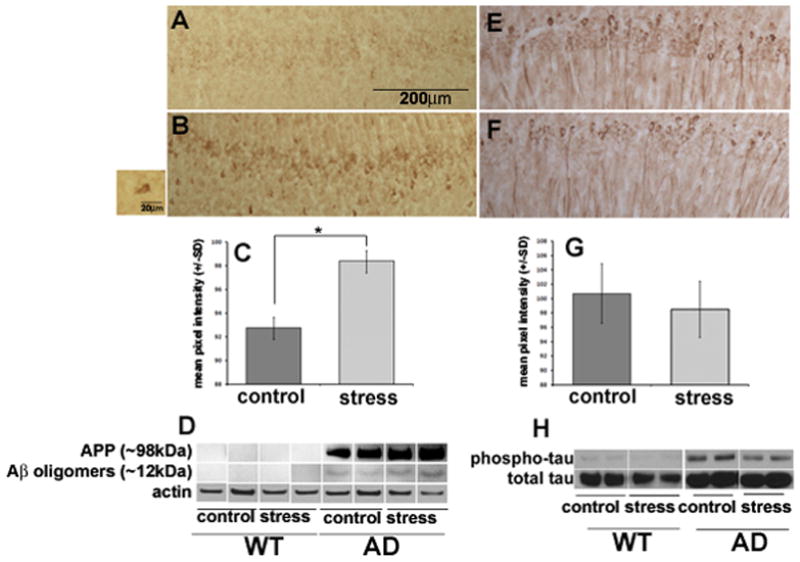
Results for Aβ (A–D) and ptau (E–H) immunostaining, densitometry and western blotting. Images show an increase in Aβ in the CA1 region in AD mice after social stress (B) compared to non-stressed controls (A). Insert box shows intraneuronal Aβ reactivity at a higher magnification. Qualitative results are confirmed by densitometry quantifying mean pixel intensity (C) showing a significant (p=0.049) increase in pixel intensity for stressed samples compared to control. Western blot results confirm staining (D) and show no Aβ aggregates in the ~70kDa range for wild-type mice, either control or stressed. Immunohistochemistry for ptau shows no differences in expression between stressed and unstressed AD mice (E&F), which is confirmed by densitometry (G) and western blot (H).
Phosphorylated tau immunoreactivity was present in CA1 neurons from all 3xTgAD mice, with the intensity of the ptau immunoreactivity being similar in CA1 neurons of 3xTgAD mice that had been subjected to social stress compared to non-stressed 3xTgAD mice (Fig. 3E, F). Immunoblots demonstrated that ptau levels were much greater in the hippocampus of 3xTgAD mice compared to WT mice and that in 3xTgAD mice social stress did not have a significant effect on ptau levels (Fig. 3H). Pixel intensity for immunohistochemical results for stressed versus non-stressed AD mice confirmed trends showing no difference between the two groups (Fig 3G).
3.4 Hippocampal BDNF levels are decreased by social stress in 3xTgAD mice, but not in control mice
At the end of the six-week stress period hippocampal BDNF levels were evaluated to determine whether this neurotrophin was regulated by chronic mild stress. Interestingly, hippocampal BDNF levels were elevated in non-stressed 3xTgAD mice compared to non-stressed WT mice (p=0.03). Mild stress slightly increased BDNF levels in control mice compared to unstressed WT mice, whereas BDNF levels were significantly decreased in 3xTgAD mice after chronic mild stress compared to non-stressed 3xTgAD mice (p=0.01) (Fig 4).
Figure 4. Mild chronic social stress results in a decrease in levels of BDNF in the hippocampus of 3xTgAD mice.
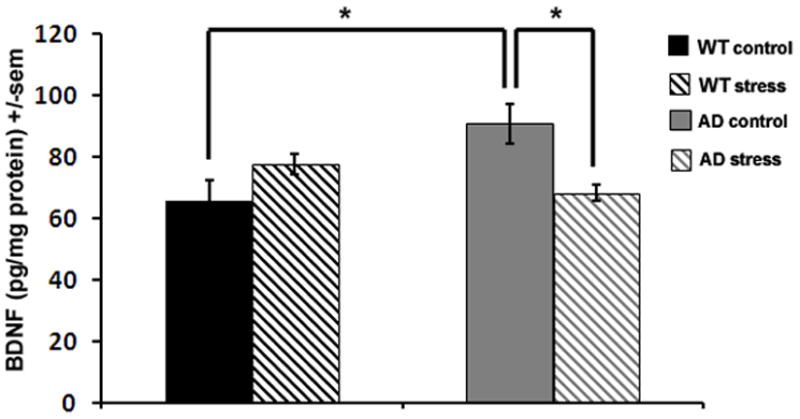
Hippocampal BDNF levels as measured by ELISA. AD mice displayed elevated hippocampal BDNF levels compared to WT mice, that were reduced in stressed 3xTgAD mice compared to non-stressed controls.
4. Discussion
The goal of the current study was to determine whether 3xTgAD mice display vulnerability to chronic stress and whether chronic stress exacerbates AD pathology and behavioral outcomes. Results indicate that during an individual 6 hour stress period 3xTgAD mice experienced a sustained rise in plasma corticosterone levels, whereas WT mice exhibited a more transient elevation of plasma corticosterone levels. Glucose levels were reduced in 3xTgAD mice compared to WT mice regardless of whether or not they were subjected to stress. The 3xTgAD mice exhibited an anxiety phenotype which was exacerbated by chronic social stress. Social stress also enhanced the accumulation of intraneuronal Aβ immunoreactivity in the CA1 region of the hippocampus. Collectively, these results indicate that a level of intermittent social stress that may be considered mild for normal mice has pronounced effects on AD mice. This not only implies a specific susceptibility to stress in AD, but also introduces a novel tool for the study of how chronic stress may modulate the appearance of symptoms and the progression of AD experimentally.
Social stress caused an increase in anxiety behaviors for AD mice that was not present in nontransgenic mice. The heightened anxiety in the 3xTgAD mice compared to WT mice was associated with a longer-lasting rise in corticosterone levels for each stress session. At 2 hours after the beginning of social stress both strains displayed elevated corticosterone, but by 6 hours corticosterone returned to baseline levels for WT mice, whereas 3xTgAD mice still displayed elevated circulating corticosterone levels. It is noteworthy that the stressor used in this study was continuous over the 6 hour period, potentially indicating a lack of habituation in the 3xTgAD mice rather than an impaired recovery. Nonetheless, over the course of the six week study, mice were exposed to social stress 2–3 times per week, meaning that the cumulative exposure to elevated corticosterone levels was likely much greater for stressed 3xTgAD mice compared to both stressed control mice and non-stressed 3xTgAD mice. Previous studies have found strong correlations between stress, glucocorticoids and anxiety (McEwen, 2000; de Kloet et al., 1999). Drugs that block glucocorticoid synthesis or signaling also reduce anxiety behaviors, implying a direct role for glucocorticoids in mediating stress-induced anxiety behaviors (Pereira et al., 2010; Myers and Greenwood-Van Meerveld, 2007). Similar to our findings, Sterniczuk et al. (2010) found that female 3xTgAD females exhibit a higher level of fear and anxiety than wild-type controls.
In addition to a direct effect from glucocorticoids, the effects of Aβ on the excitability of hippocampal neurons might play a role in the anxiety of 3xTgAD mice (Kapogiannis and Mattson, 2011), given the known roles of the hippocampus in anxiety and depression (Deltheil et al., 2008). Our results indicate an increase in Aβ aggregates in the hippocampus of stressed AD mice compared to WT mice (Fig. 3) which could imply an increase in other regions of the brain as well. In addition to accumulation in hippocampal CA1 neurons, Aβ levels are elevated in neurons in the amygdala of 3xTgAD mice, which may contribute to the elevated level of anxiety in these mice (Espana et al., 2010). Further, 3 hour restraint stress or the application of a corticotrophin-releasing-factor receptor agonist directly to the amygdala both induced increased anxiety and intracellular APP and Aβ implying that AD pathology in the amygdala can be influenced by stress or activation of the HPA axis (Ray et al., 2011). Finally, recent clinical work shows that while neuroticism is associated with an increased risk for developing AD, the specific aspects of neuroticism that are most closely related to this phenomenon are anxiety and vulnerability to stress (Wilson et al., 2006; 2011).
Glucocorticoids exert their biological effects via two receptors, the high affinity mineralocorticoid receptor (MR), predominantly expressed in the hippocampus, and the glucocorticoid receptor (GR), expressed in particularly high amounts in CA1 neurons in the hippocampus (Reul and de Kloet, 1985; de Kloet et al., 1999; Nishi et al., 2007; Patel et al., 2008; Romeo et al., 2008). Several studies in both primates and rodents show that in non-AD subjects, exposure to stressors such as restraint stress or social isolation causes a decrease in expression of both protein and mRNA for GR in the hippocampus (Arabadzisz et al., 2010; Romeo et al., 2008; Chen et al., 2008). It is possible that chronic stress causes the opposite effect on GR expression in AD. As evidence, Dong et al. showed that social isolation, widely considered an adverse stressor, caused an increase in GR expression in the hippocampus in a transgenic model of AD (Dong et al. 2008). It is hypothesized that differential regulation of the GR contributes to the different anxiety behaviors following this mild stressor. It is also possible that AD causes a loss of GR-positive neurons which contributes to aberrant responses to increased corticosterone compared to WT controls. Indeed, the CA1 region, site of GR expression, is an area of considerable cell loss in AD (Mueller 2010; Akram 2008).
Our findings suggest that 3xTgAD mice have altered energy metabolism when subjected to social stress. In general, long-term elevation of glucocorticoid levels can result in hyperglycemia by increasing insulin resistance and gluconeogenesis, consistent with elevation of glucose levels at the 2 hour time point during a social stress session. However, glucose levels were significantly decreased by 6 hours after the beginning of stress, in both 3xTgAD and WT mice (Fig. 1B). This is likely due to an overall decrease in energy as the mice respond to social stress by first mobilizing, then depleting, energy. Increased levels of glucocorticoids are generally linked with metabolic shifts including changes in eating behavior, food preferences and body weight (Harris et al., 1998; Moles et al., 2006; van Leeuwen et al., 1997). Interestingly, the two strains displayed similar patterns of glucose alterations in response to stress, despite differences in corticosterone patterns. The latter results are consistent with a primary alteration in the brain of the 3xTgAD mice that results in heightened anxiety and hyperactivation of the HPA axis response to social stress.
Elevations in glucocorticoids are associated with memory impairments and basal cortisol levels are higher in AD patients than non-diseased controls implying a role for dysregulation of the HPA axis in AD (Davis et al., 1986; Umegaki et al., 2000; McEwen, 2001 Rasmuson et al., 2001; Csernansky et al., 2006). Our results show an increase in basal corticosterone in a rodent model of AD, which is in agreement with several other similar studies done in transgenic AD models and further supports the hypothesis of HPA axis dysfunction in AD (Pedersen et al., 1999; Touma et al., 2004; Green et al., 2006; Pedersen et al., 2006; Dong et al., 2008). AD pathology in the hippocampus can cause hyperactivation of the HPA axis due to disinhibition leading to an increase in circulating glucocorticoids; however, glucocorticoids can also exacerbate AD pathology in the hippocampus, and the time course of these effects is not yet clear. In at least one study, accumulations of Aβ in the hippocampus preceded elevations in basal levels of plasma corticosterone in 3xTgAD mice, implying that the observed alteration in the HPA axis in AD may be due to hippocampal damage from AD neuropathology (Green et al., 2006). Our results, however, are not in agreement with the latter sequence of events. Data presented here show little Aβ accumulation in 3xTgAD mice that did not undergo stress, despite elevations in basal corticosterone levels. This implies that the dysfunction of the HPA axis may actually precede the formation of extracellular Aβ plaques and, potentially, the onset of cognitive symptoms. In fact, our results show an increase in levels of Aβ oligomers in AD during 6 weeks of chronic stress, an effect that could potentially be due to the observed increase in circulating glucocorticoids from chronic stress. Further, even in the absence of stress, these 3xTgAD mice will develop Aβ plaques (Oddo et al., 2003), a process that could potentially be promoted elevated circulating glucocorticoids, even in naïve un-stressed AD mice. While it is not clear what is causing hyperactivation of the HPA axis and the associated increase in basal corticosterone, literature indicates a role for glucocorticoids in modifying the production and metabolism of Aβ in the brain. The glucorticoid response element is present in the APP gene and at least one study demonstrated an increase in APP mRNA after dexamethasone treatment, further lending support to the hypothesis that dysregulation of the HPA axis precedes AD pathology (Lahiri 2004; Green et al., 2006). Increases in glucocorticoid levels seem to promote accelerated Aβ production and a decrease in Aβ degradation in AD (Green et al., 2006; Sayer et al., 2008) which further supports the hypothesis that the HPA axis has an important role in the pathogenesis of AD. Because Aβ oligomers (an aggregating form of Aβ that generates reactive oxygen species; Mattson, 2011) can impair synaptic plasticity (Wang et al., 2002) and inhibit BDNF expression (Tong et al., 2001 ), there is likely a role for increased levels of Aβ oligomers in the effects of chronic social stress on BDNF levels and behavior in the 3xTgAD mice documented in the present study.
We found that social stress had no significant effect on the level of ptau in hippocampal neurons of 3xTgAD mice. Previous studies have found that more severe chronic stressors and administration of high levels of glucocorticoids to rodents or cultured neurons can elicit antigenic changes in tau similar to those seen in neurofibrillary tangles. For example, exposure of rats to a battery of stressors, continuously for 3 days, enhanced AD-like tau pathology in hippocampal pyramidal neurons induced by the excitotoxin kainic acid (Stein-Beherens et al., 1994); administration of corticosterone to rats had a similar effect on tau immunoreactivity (Elliott et al., 1993). Glucocorticoids increase tau phosphorylation in PC12 cells that express human tau, demonstrating a direct effect of glucocorticoids on neurons (Sotiropoulos et al., 2008). Results presented here indicate little change in ptau or total tau between stressed and unstressed subjects, for both strain of mice tested (Fig 3), implying that the social stress used, and the observed rise in corticosterone, were not sufficient to induce changes in tau expression or processing. It is possible that the threshold for the induction of changes in Aβ processing is much lower than that for changes in tau processing, and further, that changes in tau may not be required for the high anxiety phenotype of 3xTgAD mice.
We observed changes in Aβ levels the CA1 region of the hippocampus, whereas the CA3 region exerts inhibitory control over the HPA axis and is the site of dendritic remodeling following stress (Watanabe et al., 1992; Rothman and Mattson 2010). While the effects of stress on the hippocampus are more widely studied in the CA3 region, recent work showed that the developing CA1 region undergoes changes in dendritic plasticity following corticosterone administration (Alfarez, et al., 2009). Further, the CA1 region is also the site of expression of the glucocorticoid receptor (Nishi et al., 2007). But perhaps most interestingly, glucocorticoids have a role in neuronal cell death in the CA1 region following ischemia. Administration of glucocorticoids after transient ischemia exacerbates neuronal loss in the CA1 region and dosage of either metyrapone (the glucocorticoid synthesis inhibitor) or RU38486 (a glucocorticoid receptor antagonist) increased neuronal survival in CA1 (Smith-Swintosky et al., 1996; Antonawich et al., 1999). These effects are likely a direct result of the high expression of GR in CA1 and support the hypothesis that glucocorticoids could mediate cellular responses in CA1 in AD.
It is possible that the greater susceptibility of 3xTgAD mice to social stress, with regards to the anxiety phenotype, is related to the drop in hippocampal BDNF levels in 3xTgAD mice. BDNF is a neurotrophin that is beneficial for cognition and is abundant in the hippocampus, and its levels are modulated by exposure to stress or application of glucocorticoids. Some mild stressors, such as exercise and caloric restriction, have beneficial effects on cognition and cause an increase in BDNF levels (Khabour 2010; Stranahan 2009). Adverse stressors, such as restraint stress, or the application of corticosterone can cause a decrease in hippocampal BDNF mRNA in vivo (Smith and Cizza, 1996; Schaaf et al., 2000). BDNF protein levels are also affected by glucocorticoids; just one injection of 1 mg/kg of corticosterone can cause a decrease in BDNF protein at just 4 hours (Schaaf et al., 1998). We found that 6 weeks of mild social stress, a protocol that resulted in periodic moderate increases and decreases in circulating corticosterone levels, caused a decrease in hippocampal BDNF protein levels in 3xTgAD mice, but not in control mice. The increased sensitivity of BDNF expression to stress in 3xTgAD mice may be due to the stress-induced increase in the accumulation of Aβ in the hippocampus. Indeed, previous studies have shown that exposure of cultured neurons to subtoxic concentrations of Aβ1-42 results in a reduction in BDNF expression (Tong et al., 2001).
Interestingly, we found that overnight fasting causes a dramatic and significant rise in corticosterone levels in non-stressed 3xTgAD and WT mice (Fig 1A), but not for socially-stressed mice. Overnight fasting is commonly used in many protocols animal model and human research, particularly when investigating metabolic outcomes. Two major points are raised by our data. First, it appears that overnight fasting is highly stressful for mice and may be confounding results obtained in some studies. This is of particular importance because elevated glucocorticoids may cause elevated blood glucose levels due to increases in insulin resistance and gluconeogenesis. Second, mice that underwent social stress did not display rises in circulating corticosterone after an overnight fast, implying that this type of social stress has a hormetic effect (Mattson, 2008); mice were able to withstand a second stressor (fasting) as the result of an adaptive response to the first stressor (social stress). Future work will be required to determine the cellular pathways involved is such an adaptive stress response, but it is noteworthy that this response was noted for both strains of mice studied. Finally, as shown in Figure 1, there is no difference in plasma corticosterone between AD and wild-type controls at the morning non-fasting time point, however at the 2 hour time point AD mice display a rise in corticosterone despite being unstressed, whereas wild-type mice do not experience this increase. Plasma corticosterone levels vary diurnally, with levels normally reaching their peak in the afternoon, which could explain the rise in corticosterone in the control AD mice between the morning time point and 2 hours after the beginning of the experimental period. Although it appears that the corresponding wild-type mice do not display this rise, it is possible that the AD mice reach their peak in corticosterone levels at a different time of day due to alterations in circadian rhythmicity in this model of AD (Sterniczuk et al., 2010).
Our findings advance an understanding of how alterations in the HPA axis contribute to a distinct stress response in AD. Indeed, an age-related alteration in the HPA axis is noted experimentally (Dalm et al., 2005) suggesting that the stress response plays an important role in aging and age-related disease processes. The social stress used in the current study was very mild, only 2–3 sessions of stress per week and each consisting of only 6 hours with new cage mates followed by a return to their home cage. It is unsurprising, therefore, that wild type mice did not display major changes in behavior or brain biochemistry for the outcomes probed. Yet, the 3xTgAD mice did experience significant alterations in behavioral and biochemical outcomes, indicating a high sensitivity to stress for these mice and suggesting that AD patients and individuals at risk for AD may also be susceptible to even mild stressors. We further hypothesize that chronic stress may worsen the progression of cognitive deficits and even accelerate their onset in AD.
Acknowledgments
The authors thank Dr. Henriette van Praag for the use of imaging equipment and Catherine Crews for assistance with maintenance for the in vivo portion of the study. This research was supported by the Intramural Research Program of the National Institute on Aging.
Footnotes
Disclosure Statement
The authors report no conflicts of interest.
References
- Akram A, Christoffel D, Rocher AB, Bouras C, Kövari E, Perl DP, Morrison JH, Herrmann FR, Haroutunian V, Giannakopoulos P, Hof PR. Stereologic estimates of total spinophilin-immunoreactive spine number in area 9 and the CA1 field: relationship with the progression of Alzheimer’s disease. Neurobiol Aging. 2008;29:1296–1307. doi: 10.1016/j.neurobiolaging.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfarez DN, De Simoni A, Velzing EH, Bracey E, Joëls M, Edwards FA, Krugers HJ. Corticosterone reduces dendritic complexity in developing hippocampal CA1 neurons. Hippocampus. 2009;19:828–836. doi: 10.1002/hipo.20566. [DOI] [PubMed] [Google Scholar]
- Alzoubi KH, Abdul-Razzak KK, Khabour OF, Al-Tuweiq GM, Alzubi MA, Alkadhi KA. Adverse effect of combination of chronic psychosocial stress and high fat diet on hippocampus-dependent memory in rats. Behav Brain Res. 2009;204:117–123. doi: 10.1016/j.bbr.2009.05.025. [DOI] [PubMed] [Google Scholar]
- Antonawich FJ, Miller G, Rigsby DC, Davis JN. Regulation of ischemic cell death by glucocorticoids and adrenocorticotropic hormone. Neuroscience. 1999;88:319–325. doi: 10.1016/s0306-4522(98)00213-9. [DOI] [PubMed] [Google Scholar]
- Arabadzisz D, Diaz-Heijtz R, Knuesel I, Weber E, Pilloud S, Dettling AC, Feldon J, Law AJ, Harrison PJ, Pryce CR. Primate early life stress leads to long-term mild hippocampal decreases in corticosteroid receptor expression. Biol Psychiat. 2010;67:1106–1109. doi: 10.1016/j.biopsych.2009.12.016. [DOI] [PubMed] [Google Scholar]
- Baran SE, Campbell AM, Kleen JK, Foltz CH, Wright RL, Diamond DM, Conrad CD. Combination of high fat diet and chronic stress retracts hippocampal dendrites. Neuroreport. 2005;16:39–43. doi: 10.1097/00001756-200501190-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brett LP, Chong GS, Coyle S, Levine S. The pituitary–adrenal response to novel stimulation and ether stress in young adult and aged rats. Neurobiol Aging. 1983;4:133–138. doi: 10.1016/0197-4580(83)90037-4. [DOI] [PubMed] [Google Scholar]
- Breyhan H, Wirths O, Duan K, Marcello A, Rettig J, Bayer TA. APP/PS1KI bigenic mice develop early synaptic deficits and hippocampus atrophy. Acta Neuropathol. 2009;117:677–685. doi: 10.1007/s00401-009-0539-7. [DOI] [PubMed] [Google Scholar]
- Catania C, Sotiropoulos I, Silva R, Onofri C, Breen KC, Sousa N, Almeida OF. The amyloidogenic potential and behavioral correlates of stress. Mol Psychiat. 2009;14:95–105. doi: 10.1038/sj.mp.4002101. [DOI] [PubMed] [Google Scholar]
- Cerqueira JJ, Mailliet F, Almeida OF, Jay TM, Sousa N. The prefrontal cortex as a key target of the maladaptive response to stress. J Neurosci. 2007;27:2781–2787. doi: 10.1523/JNEUROSCI.4372-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JX, Tang YT, Yang JX. Changes of glucocorticoid receptor and levels of CRF mRNA, POMC mRNA in brain of chronic immobilization stress rats. Cell Mol Neurobiol. 2008;28:237–244. doi: 10.1007/s10571-007-9170-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csernansky JG, Dong H, Fagan AM, Wang L, Xiong C, Holtzman DM, Morris JC. Plasma cortisol and progression of dementia in subjects with Alzheimer-type dementia. Am J Psychiat. 2006;163:2164–2169. doi: 10.1176/appi.ajp.163.12.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalm S, Enthoven L, Meijer OC, van der Mark MH, Karssen AM, de Kloet ER, Oitzl MS. Age-related changes in hypothalamic-pituitary-adrenal axis activity of male C57BL/6J mice. Neuroendocrinol. 2005;81:372–380. doi: 10.1159/000089555. [DOI] [PubMed] [Google Scholar]
- Davis KL, Davis BM, Greenwald BS, Mohs RC, Mathe AA, Johns CA, Horvath TB. Cortisol and Alzheimer’s disease, I: Basal studies. Am J Psychiat. 1986;143:300–305. doi: 10.1176/ajp.143.3.300. [DOI] [PubMed] [Google Scholar]
- de Kloet ER, Oitzl MS, Joëls M. Stress and cognition: are corticosteroids good or bad guys? Trends Neurosci. 1999;22:422–426. doi: 10.1016/s0166-2236(99)01438-1. [DOI] [PubMed] [Google Scholar]
- Deltheil T, Guiard BP, Cerdan J, David DJ, Tanaka KF, Repérant C, Guilloux JP, Coudoré F, Hen R, Gardier AM. Behavioral and serotonergic consequences of decreasing or increasing hippocampus brain-derived neurotrophic factor protein levels in mice. Neuropharmacol. 2008;55:1006–1014. doi: 10.1016/j.neuropharm.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Dong H, Goico B, Martin M, Csernansky CA, Bertchume A, Csernansky JG. Modulation of hippocampal cell proliferation, memory, and amyloid plaque deposition in APPsw (Tg2576) mutant mice by isolation stress. Neuroscience. 2004;127:601–609. doi: 10.1016/j.neuroscience.2004.05.040. [DOI] [PubMed] [Google Scholar]
- Dong H, Yuede CM, Yoo HS, Martin MV, Deal C, Mace AG, Csernansky JG. Corticosterone and related receptor expression are associated with increased β-amyloid plaques in isolated Tg2576 mice. Neuroscience. 2008;155:154–163. doi: 10.1016/j.neuroscience.2008.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott EM, Mattson MP, Vanderklish P, Lynch G, Chang I, Sapolsky RM. Corticosterone exacerbates kainate-induced alterations in hippocampal tau immunoreactivity and spectrin proteolysis in vivo. J Neurochem. 1993;61:57–67. doi: 10.1111/j.1471-4159.1993.tb03537.x. [DOI] [PubMed] [Google Scholar]
- España J, Giménez-Llort L, Valero J, Miñano A, Rábano A, Rodriguez-Alvarez J, LaFerla FM, Saura CA. Intraneuronal beta-amyloid accumulation in the amygdala enhances fear and anxiety in Alzheimer’s disease transgenic mice. Biol Psychiat. 2010;67:513–521. doi: 10.1016/j.biopsych.2009.06.015. [DOI] [PubMed] [Google Scholar]
- Gerges NZ, Stringer JL, Alkadhi KA. Combination of hypothyroidism and stress abolishes early LTP in the CA1 but not dentate gyrus of hippocampus of adult rats. Brain Res. 2001;922:250–260. doi: 10.1016/s0006-8993(01)03181-x. [DOI] [PubMed] [Google Scholar]
- Green KN, Billings LM, Roozendaal B, McGaugh JL, LaFerla FM. Glucocorticoids increase amyloid-β and tau pathology in a mouse model of Alzheimer’s disease. J Neurosci. 2006;26:9047–9056. doi: 10.1523/JNEUROSCI.2797-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RB, Zhou J, Youngblood BD, Rybkin II, Smagin GN, Ryan DH. Effect of repeated stress on body weight and body composition of rats fed low- and high-fat diets. Am J Physiol. 1998;275:R1928–R1938. doi: 10.1152/ajpregu.1998.275.6.R1928. [DOI] [PubMed] [Google Scholar]
- Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer Disease in the US Population: Prevalence Estimates Using the 2000 Census. Arch Neurol. 2003;60:1119–1122. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- Herman JP, Schäfer MK, Young EA, Thompson R, Douglass J, Akil H, Watson SJ. Evidence for hippocampal regulation of neuroendocrine neurons of the hypothalamo-pituitary-adrenocortical axis. J Neurosci. 1989;9:3072–3082. doi: 10.1523/JNEUROSCI.09-09-03072.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson L, Sapolsky R. The role of the hippocampus in feedback regulation of the hypothalamic-pituitary-adrenocortical axis. Endocr Rev. 1991;12:118–134. doi: 10.1210/edrv-12-2-118. [DOI] [PubMed] [Google Scholar]
- Jeong YH, Park CH, Yoo J, Shin KJ, Ahn SM, Kim HS, Lee SH, Emson PC, Suh YH. Chronic stress accelerates learning and memory impairments and increases amyloid deposition in APPV7171-CT100 transgenic mice, an Alzheimer’s disease model. FASEB. 2006;20:729–731. doi: 10.1096/fj.05-4265fje. [DOI] [PubMed] [Google Scholar]
- Kang JE, Cirrito JR, Dong H, Csernansky JG, Holtzman DM. Acute stress increases interstitial fluid amyloid-beta via corticotropin-releasing factor and neuronal activity. PNAS. 2007;104:10673–10678. doi: 10.1073/pnas.0700148104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapogiannis D, Mattson MP. Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer’s disease. Lancet Neurol. 2011;10:187–198. doi: 10.1016/S1474-4422(10)70277-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khabour OF, Alzoubi KH, Alomari MA, Alzubi MA. Changes in spatial memory and BDNF expression to concurrent dietary restriction and voluntary exercise. Hippocamp. 2010;20:637–645. doi: 10.1002/hipo.20657. [DOI] [PubMed] [Google Scholar]
- Krugers HJ, Douma BR, Andringa G, Bohus B, Korf J, Luiten PG. Exposure to chronic psychosocial stress and corticosterone in the rat: effects on spatial discrimination learning and hippocampal protein kinase Cgamma immunoreactivity. Hippocampus. 1997;7:427–436. doi: 10.1002/(SICI)1098-1063(1997)7:4<427::AID-HIPO8>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Kudielka BM, Buske-Kirschbaum A, Hellhammer DH, Kirschbaum C. HPA axis responses to laboratory psychosocial stress in healthy elderly adults, younger adults, and children: impact of age and gender. Psychoneuroendocrinol. 2004;29:83–98. doi: 10.1016/s0306-4530(02)00146-4. [DOI] [PubMed] [Google Scholar]
- Kulstad JJ, McMillan PJ, Leverenz JB, Cook DG, Green PS, Peskind ER, Wilkinson CW, Farris W, Mehta PD, Craft S. Effects of chronic glucocorticoid administration on insulin-degrading enzyme and amyloid-beta peptide in the aged macaque. J Neuropathol Exp Neurol. 2005;64:139–146. doi: 10.1093/jnen/64.2.139. [DOI] [PubMed] [Google Scholar]
- Lahiri DK. Functional characterization of amyloid beta precursor protein regulatory elements: rationale for the identification of genetic polymorphism. Ann N Y Acad Sci. 2004;1030:282–288. doi: 10.1196/annals.1329.035. [DOI] [PubMed] [Google Scholar]
- Landfield PW, Waymire JC, Lynch G. Hippocampal aging and adrenocorticoids: quantitative correlations. Science. 1978;202:1098–1102. doi: 10.1126/science.715460. [DOI] [PubMed] [Google Scholar]
- Lee KW, Kim JB, Seo JS, Kim TK, Im JY, Baek IS, Kim SH, Lee JK, Han PL. Behavioral stress accelerates plaque pathogenesis in the brain of Tg2576 mice via generation of metabolic oxidative stress. J Neurochem. 2009;108:165–175. doi: 10.1111/j.1471-4159.2008.05769.x. [DOI] [PubMed] [Google Scholar]
- Li S, Wang C, Wang W, Dong H, Hou P, Tang Y. Chronic mild stress impairs cognition in mice: from brain homeostasis to behavior. Life Sci. 2008;82:934–942. doi: 10.1016/j.lfs.2008.02.010. [DOI] [PubMed] [Google Scholar]
- Liang KY, Mintun MA, Fagan AM, Goate AM, Bugg JM, Holtzman DM, Morris JC, Head D. Exercise and Alzheimer’s disease biomarkers in cognitively normal older adults. Ann Neurol. 2010;68:311–318. doi: 10.1002/ana.22096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löckenhoff CE, Terracciano A, Patriciu NS, Eaton WW, Costa PT., Jr Self-reported extremely adverse life events and longitudinal changes in five-factor model personality traits in an urban sample. J Trauma Stress. 2009;22:53–59. doi: 10.1002/jts.20385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Hormesis defined. Ageing Res Rev. 2008;7:1–7. doi: 10.1016/j.arr.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Commentary: proteooxidotoxic process of aggregation. Neuromolecular Med. 2011;13:91–92. doi: 10.1007/s12017-011-8146-x. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Effects of adverse experiences for brain structure and function. Biol Psychiat. 2000;48:721–731. doi: 10.1016/s0006-3223(00)00964-1. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Plasticity of the hippocampus: adaptation to chronic stress and allostatic load. Ann NY Acad Sci. 2001;933:265–277. doi: 10.1111/j.1749-6632.2001.tb05830.x. [DOI] [PubMed] [Google Scholar]
- Mejía S, Giraldo M, Pineda D, Ardila A, Lopera F. Nongenetic factors as modifiers of the age of onset of familial Alzheimer’s disease. Int Psychogeriatr. 2003;15:337–349. doi: 10.1017/s1041610203009591. [DOI] [PubMed] [Google Scholar]
- Mitra R, Sapolsky RM. Acute corticosterone treatment is sufficient to induce anxiety and amygdaloid dendritic hypertrophy. PNAS. 2008;105:5573–5578. doi: 10.1073/pnas.0705615105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moles A, Bartolomucci A, Garbugino L, Conti R, Caprioli A, Coccurello R, Rizzi R, Ciani B, D’Amato FR. Psychosocial stress affects energy balance in mice: modulation by social status. Psychoneuroendocrinology. 2006;31:623–633. doi: 10.1016/j.psyneuen.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Mooring MS, Patton ML, Lance VA, Hall BM, Schaad EW, Fetter GA, Fortin SS, McPeak KM. Glucocorticoids of bison bulls in relation to social status. Horm Behav. 2006;49:369–375. doi: 10.1016/j.yhbeh.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Mueller SG, Schuff N, Yaffe K, Madison C, Miller B, Weiner MW. Hippocampal atrophy patterns in mild cognitive impairment and Alzheimer’s disease. Hum Brain Mapp. 2010;31:1339–1347. doi: 10.1002/hbm.20934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munck A, Guyre PM. Glucocorticoid physiology, pharmacology and stress. Adv Exp Med Biol. 1986;196:81–96. doi: 10.1007/978-1-4684-5101-6_6. [DOI] [PubMed] [Google Scholar]
- Myers B, Greenwood-Van Meerveld B. Corticosteroid receptor-mediated mechanisms in the amygdala regulate anxiety and colonic sensitivity. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1622–G1629. doi: 10.1152/ajpgi.00080.2007. [DOI] [PubMed] [Google Scholar]
- Nelson DH. Regulation of glucocorticoid release. Am J Med. 1972;53:590–594. doi: 10.1016/0002-9343(72)90155-6. [DOI] [PubMed] [Google Scholar]
- Nishi M, Usuku T, Itose M, Fujikawa K, Hosokawa K, Matsuda KI, Kawata M. Direct visualization of glucocorticoid receptor positive cells in the hippocampal regions using green fluorescent protein transgenic mice. Neurosci. 2007;146:1555–1560. doi: 10.1016/j.neuroscience.2007.03.020. [DOI] [PubMed] [Google Scholar]
- Nuntagij P, Oddo S, LaFerla FM, Kotchabhakdi N, Ottersen OP, Torp R. Amyloid deposits show complexity and intimate spatial relationship with dendrosomatic plasma membranes: an electron microscopic 3D reconstruction analysis in 3xTg-AD mice and aged canines. J Alzheimers Dis. 2009;16:315–323. doi: 10.3233/JAD-2009-0962. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Patel PD, Katz M, Karssen AM, Lyons DM. Stress-induced changes in corticosteroid receptor expression in primate hippocampus and prefrontal cortex. Psychoneuroendocrinology. 2008;33:360–367. doi: 10.1016/j.psyneuen.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen WA, Culmsee C, Ziegler D, Herman JP, Mattson MP. Aberrant stress response associated with severe hypoglycemia in a transgenic mouse model of Alzheimer’s disease. J Mol Neurosci. 1999;13:159–165. doi: 10.1385/JMN:13:1-2:159. [DOI] [PubMed] [Google Scholar]
- Pedersen WA, McMillan PJ, Kulstad JJ, Leverenz JB, Craft S, Haynatzki GR. Rosiglitazone attenuates learning and memory deficits in Tg2576 Alzheimer mice. Exp Neurol. 2006;199:265–273. doi: 10.1016/j.expneurol.2006.01.018. [DOI] [PubMed] [Google Scholar]
- Pereira AM, Tiemensma J, Romijn JA. Neuropsychiatric disorders in Cushing’s syndrome. Neuroendocrinology. 2010;92:65–70. doi: 10.1159/000314317. [DOI] [PubMed] [Google Scholar]
- Rasmuson S, Andrew R, Näsman B, Seckl JR, Walker BR, Olsson T. Increased glucocorticoid production and altered cortisol metabolism in women with mild to moderate Alzheimer’s disease. Biol Psychiat. 2001;49:547–552. doi: 10.1016/s0006-3223(00)01015-5. [DOI] [PubMed] [Google Scholar]
- Ray B, Gaskins DL, Sajdyk TJ, Spence JP, Fitz SD, Shekhar A, Lahiri DK. Restraint stress and repeated corticotrophin-releasing factor receptor activation in the amygdala both increase amyloid-β precursor protein and amyloid-β peptide but have divergent effects on brain-derived neurotrophic factor and pre-synaptic proteins in the prefrontal cortex of rats. Neuroscience. 2011;184:139–150. doi: 10.1016/j.neuroscience.2011.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reul JM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology. 1985;117:2505–2511. doi: 10.1210/endo-117-6-2505. [DOI] [PubMed] [Google Scholar]
- Romeo RD, Ali FS, Karatsoreos IN, Bellani R, Chhua N, Vernov M, McEwen BS. Glucocorticoid receptor mRNA expression in the hippocampal formation of male rats before and after pubertal development in response to acute or repeated stress. Neuroendocrinology. 2008;87:160–167. doi: 10.1159/000109710. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Phillips RG, Power AE, Brooke SM, Sapolsky RM, McGaugh JL. Memory retrival impairment induced by hippocampal CA3 lesions is blocked by adrenocortical suppression. Nat Neurosci. 2001;4:1169–1171. doi: 10.1038/nn766. [DOI] [PubMed] [Google Scholar]
- Rothman SM, Mattson MP. Adverse stress, hippocampal networks, and Alzheimer’s disease. Neuromol Med. 2010;12:56–70. doi: 10.1007/s12017-009-8107-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapolsky RM. The influence of social hierarchy on primate health. Science. 2005;308:648–652. doi: 10.1126/science.1106477. [DOI] [PubMed] [Google Scholar]
- Sayer R, Robertson D, Balfour DJ, Breen KC, Stewart CA. The effect of stress on the expression of the amyloid precursor protein in rat brain. Neurosci Lett. 2008;431:197–200. doi: 10.1016/j.neulet.2007.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf MJ, de Jong J, de Kloet ER, Vreugdenhil E. Downregulation of BDNF mRNA and protein in the rat hippocampus by corticosterone. Brain Res. 1998;813:112–120. doi: 10.1016/s0006-8993(98)01010-5. [DOI] [PubMed] [Google Scholar]
- Schaaf MJ, de Kloet ER, Vreugdenhil E. Corticosterone effects on BDNF expression in the hippocampus. Implications for memory formation. Stress. 2000;3:201–208. doi: 10.3109/10253890009001124. [DOI] [PubMed] [Google Scholar]
- Shoji H, Mizoguchi K. Acute and repeated stress differentially regulates behavioral, endocrine, neural parameters relevant to emotional and stress response in young and aged rats. Behav Brain Res. 2010;211:169–177. doi: 10.1016/j.bbr.2010.03.025. [DOI] [PubMed] [Google Scholar]
- Simard M, Hudon C, van Reekum R. Psychological distress and risk for dementia. Curr Psychiatry Rep. 2009;11:41–47. doi: 10.1007/s11920-009-0007-z. [DOI] [PubMed] [Google Scholar]
- Smith MA, Cizza G. Stress-induced changes in brain-dervied neurotrophic factor expression are attenuated in aged Fischer 344/N rats. Neurobiol Aging. 1996;17:859–864. doi: 10.1016/s0197-4580(96)00066-8. [DOI] [PubMed] [Google Scholar]
- Smith-Swintosky VL, Pettigrew LC, Sapolsky RM, Phares C, Craddock SD, Brooke SM, Mattson MP. Metyrapone, an inhibitor of glucocorticoid production, reduces brain injury induced by focal and global ischemia and seizures. J Cereb Blood Flow Metab. 1996;16:585–598. doi: 10.1097/00004647-199607000-00008. [DOI] [PubMed] [Google Scholar]
- Sotiropoulos I, Catania C, Riedemann T, Fry JP, Breen KC, Michaelidis TM, Almeida OF. Glucocorticoids trigger Alzheimer disease-like pathobiochemistry in rat neuronal cells expressing human tau. J Neurochem. 2008;107:385–397. doi: 10.1111/j.1471-4159.2008.05613.x. [DOI] [PubMed] [Google Scholar]
- Srivareerat M, Tran TT, Alzoubi KH, Alkadhi KA. Chronic psychosocial stress exacerbates impairment of cognition and long-term potentiation in b-amyloid rat model of Alzheimer’s disease. Biol Psychiat. 2009;65:918–926. doi: 10.1016/j.biopsych.2008.08.021. [DOI] [PubMed] [Google Scholar]
- Stein-Behrens B, Mattson MP, Chang I, Yeh M, Sapolsky R. Stress exacerbates neuron loss and cytoskeletal pathology in the hippocampus. J Neurosci. 1994;14:5373–5380. doi: 10.1523/JNEUROSCI.14-09-05373.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterniczuk R, Antle MC, Laferla FM, Dyck RH. Characterization of the 3xTg-AD mouse model of Alzheimer’s disease: part 2. Behavioral and cognitive changes. Brain Res. 2010;1348:149–155. doi: 10.1016/j.brainres.2010.06.011. [DOI] [PubMed] [Google Scholar]
- Stranahan AM, Arumugam TV, Cutler RG, Lee K, Egan JM, Mattson MP. Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat Neurosci. 2008;11:309–317. doi: 10.1038/nn2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranahan AM, Lee K, Martin B, Maudsley S, Golden E, Cutler RG, Mattson MP. Voluntary exercise and caloric restriction enhance hippocampal dendritic spine density and BDNF levels in diabetic mice. Hippocampus. 2009;19:951–961. doi: 10.1002/hipo.20577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong L, Thornton PL, Balazs R, Cotman CW. Beta-amyloid-(1-42) impairs activity-dependent cAMP-response element-binding protein signaling in neurons at concentrations in which cell survival is not compromised. J Biol Chem. 2001;276:17301–17306. doi: 10.1074/jbc.M010450200. [DOI] [PubMed] [Google Scholar]
- Touma C, Ambrée O, Görtz N, Keyvani K, Lewejohann L, Palme R, Paulus W, Schwarze-Eicker K, Sachser N. Age- and sex-dependent development of adrenocortical hyperactivity in a transgenic mouse model of Alzheimer’s disease. Neurobiol Aging. 2004;25:893–904. doi: 10.1016/j.neurobiolaging.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Umegaki H, Ikari H, Nakahata H, Endo H, Suzuki Y, Ogawa O, Nakamura A, Yamamoto T, Iguchi A. Plasma cortisol levels in elderly female subjects with Alzheimer’s disease: a cross-sectional and longitudinal study. Brain Res. 2000;881:241–243. doi: 10.1016/s0006-8993(00)02847-x. [DOI] [PubMed] [Google Scholar]
- Van Cauter E, Leproult R, Kupfer DJ. Effects of gender and age on the levels and circadian rhythmicity of plasma cortisol. J Clin Endocrinol Metab. 1996;81:2468–2473. doi: 10.1210/jcem.81.7.8675562. [DOI] [PubMed] [Google Scholar]
- Van Den Heuvel C, Thornton E, Vink R. Traumatic brain injury and Alzheimer’s disease: a review. Prog Brain Res. 2007;161:303–316. doi: 10.1016/S0079-6123(06)61021-2. [DOI] [PubMed] [Google Scholar]
- van Leeuwen SD, Bonne OB, Avraham Y, Berry EM. Separation as a new animal model for self-induced weight loss. Physiol Behav. 1997;62:77–81. doi: 10.1016/s0031-9384(97)00144-3. [DOI] [PubMed] [Google Scholar]
- Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, Viola KL, Klein WL, Stine WB, Krafft GA, Trommer BL. Soluble oligomers of beta amyloid (1-42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002;924:133–140. doi: 10.1016/s0006-8993(01)03058-x. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Gould E, McEwen BS. Stress induces atrophy of apical dendrites of hippocampal CA3 pyramidal neurons. Brain Res. 1992;588:341–345. doi: 10.1016/0006-8993(92)91597-8. [DOI] [PubMed] [Google Scholar]
- Watson D, Clark LA. Negative affectivity: the disposition to experience aversive emotional states. Psychol Bull. 1984;96:465–490. [PubMed] [Google Scholar]
- Weiss A, Sutin AR, Duberstein PR, Friedman B, Bagby RM, Costa PT., Jr The personality domains and styles of the five-factor model are related to incident depression in Medicare recipients aged 65 to 100. Am J Geriatr Psychiat. 2009;17:591–601. doi: 10.1097/jgp.0b013e31819d859d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RS, Arnold SE, Schneider JA, Kelly JF, Tang Y, Bennett DA. Chronic psychological distress and risk of Alzheimer’s disease in old age. Neuroepidemiology. 2006;27:143–153. doi: 10.1159/000095761. [DOI] [PubMed] [Google Scholar]
- Wilson RS, Arnold SE, Schneider JA, Li Y, Bennett DA. Chronic distress, age-related neuropathology, and late-life dementia. Psychosom Med. 2007;69:47–53. doi: 10.1097/01.psy.0000250264.25017.21. [DOI] [PubMed] [Google Scholar]
- Wilson RS, Begeny CT, Boyle PA, Schneider JA, Bennett DA. Vulnerability to stress, anxiety, and development of dementia in old age. Am J Geriatr Psychiatry. 2011;19:327–34. doi: 10.1097/JGP.0b013e31820119da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf OT, Minnebusch D, Daum I. Stress impairs acquisition of delay eyeblink conditioning in men and women. Neurobiol Learn Mem. 2009;91:431–436. doi: 10.1016/j.nlm.2008.11.002. [DOI] [PubMed] [Google Scholar]