

Abstract
Posterior polymorphous corneal dystrophy (PPCD) is a dominantly inherited disorder of the corneal endothelium that has been associated with mutations in the zinc-finger E-box binding homeobox 1 gene (ZEB1) gene in approximately one-third of affected families. While the corneal dystrophies have traditionally been considered isolated disorders of the corneal endothelium, we have recently identified two cases of maldevelopment of the corpus callosum in unrelated individuals with PPCD. The proband of the first family was diagnosed shortly after birth with agenesis of the corpus callosum and several other developmental abnormalities. Karyotype, FISH and whole genome copy number variant analyses were normal. She was subsequently diagnosed with PPCD, prompting screening of the ZEB1 gene, which identified a novel deletion (c.449delG; p.(Gly150Alafs*36)) present in the heterozygous state that was not identified in either unaffected parent. The proband of the second family was diagnosed several months after birth with thinning of the corpus callosum and PPCD. Whole genome copy number variant analysis revealed a 1.79 Mb duplication of 17q12 in the proband and her father and brother, neither of whom had PPCD. ZEB1 sequencing identified a novel deletion (c.1913-1914delCA; p.(Ser638Cysfs*5)) present in the heterozygous state, which was also identified in the proband’s affected mother. Thus, we report the first two cases of the association of PPCD with a developmental abnormality of the brain, in this case maldevelopment of the corpus callosum. Keywords: agenesis, corpus callosum, PPCD3, ZEB1, ZEB2, corneal dystrophy
1. INTRODUCTION
Agenesis of the corpus callosum (ACC), describes the complete or partial absence of the sheet of nerve fiber bundles derived from neural crest tissue that connect the right and left hemispheres of the brain, and is estimated to occur in approximately 1 in 4000–5000 individuals in the general population (Paul et al., 2007). Abnormal development of the corpus callosum (CC) is accompanied by variable degrees of cognitive dysfunction, such as problem solving skills, that parallel the severity of aberrant corpus callosum development (Paul et al., 2007). Investigations into the genetic cause of ACC have demonstrated evidence of linkage to 12 genomic loci (O’Driscoll et al., 2010). ACC typically is associated with an autosomal recessive or an X-linked recessive inheritance, although autosomal dominant inheritance has been reported (Inbar et al., 1997). While a variety of ocular abnormalities have been previously associated with various syndromes that arise from an abnormally developed CC, such as Aicardi Syndrome, Septo-optic Dysplasia, Mowat-Wilson Syndrome, and Menkes Syndrome, none have previously included an inherited disorder of the cornea (Ariss et al., 2012; Ferreira et al., 1998; Fruhman et al., 2012; Goyal, Watts, & Hourihan, 2010; Masri et al., 2011).
Posterior polymorphous corneal dystrophy (PPCD) is a dominantly inherited disorder of the neural crest-derived corneal endothelium that is associated with characteristic corneal endothelial morphologic abnormalities (Shimizu et al., 2004). While corneal dystrophies are typically considered isolated disorders of the cornea without associated ocular or extraocular manifestations, PPCD has been associated with a variety of other ocular and nonocular disorders, including keratoconus, non-keratometric corneal steepening, Alport syndrome, inguinal hernia and hydrocele formation (Aldave et al., 2007; Cremona et al., 2009; Krafchak et al., 2005; Raber et al., 2011; Teekhasaenee et al., 1991). In approximately 1/3 of affected probands screened to date, mutations have been identified in the zinc-finger E-box binding homeobox 1 gene (ZEB1) gene, which is the genetic feature that defines PPCD3 (Aldave et al., 2007; Bakhtiari et al., 2013; Krafchak et al., 2005; Lechner et al., 2013; Liskova et al., 2013; Liskova et al., 2007; Nguyen et al., 2010; Vincent et al., 2009). ZEB1 is a transcription factor that has also been implicated in the pathogenesis of Fuchs endothelial corneal dystrophy (Mehta et al., 2008; Riazuddin et al., 2010). ZEB1 also plays a significant role in the development of the neural tube and its derivatives, as an absence of ZEB1 gene expression is associated with severe defects in neurodevelopment, such as exencephaly and a failure of neural tube closure in a mouse model (Liu et al., 2008). ZEB1 is also expressed in cerebral progenitor cells, regions of the lateral ventricles and other areas of the brain (Liu et al., 2008), indicating that ZEB1 may play a role in cell differentiation and regional brain development. We report one such developmental anomaly, agenesis and hypoplasia of the corpus callosum, in two individuals with PPCD3.
2. MATERIALS AND METHODS
Study approval was obtained from the Institutional Review Board at the University of California, Los Angeles (UCLA IRB # 94-07-243-(33A and 34)).
2.1 Patient identification/DNA collection and preparation
A slit lamp examination was performed of the proband of each family and available family members to determine their affected status. The diagnosis of PPCD was based upon the presence of characteristic clinical features in each cornea (Aldave et al., 2007). After obtaining informed consent from the parents of each proband and assent from one of the probands (Case 1), blood was obtained from each individual and DNA extraction was performed. For each enrolled individual, genomic DNA was prepared from the peripheral blood leukocytes and buccal epithelial cells using the FlexiGene DNA and QIAamp DNA Blood Mini Kits (Qiagen, Valencia, CA), respectively.
2.2 PCR Amplification
Each of the nine coding exons of ZEB1 was amplified using the previously described primers and conditions, (Aldave et al., 2007; Krafchak et al., 2005) with the exception of exon 1, which was amplified using custom-designed oligonucleotide primers (Yellore et al., 2012). Thermal cycling was performed in an iCycler Thermal Cycler (Bio-Rad, Hercules, CA).
2.3 DNA Sequencing
Purification of the PCR products and sequencing reactions were performed using previously described reagents and conditions (Aldave et al., 2007). Nucleotide sequences, read manually and with Mutation Surveyor v2.2 (Softgenetics, State College, PA), were compared to the published ZEB1 RefSeqGene sequence (GenBank accession number NG_017048.1).
3. RESULTS
3.1 Case 1
A 14 year old girl of mixed European descent, with Italian, Swedish, Irish, and Dutch ancestry, was diagnosed with congenital anomalies affecting a number of organ systems, including: HLA-B27 related spondyloarthropathy and hypermotile joints; a bicornuate uterus; and agenesis of the corpus callosum and colpocephaly, which were diagnosed on a postnatal CT scan (Figure 1). The girl’s parents denied a family history of any corneal disorders or the other conditions that their daughter demonstrated. An ocular examination performed at age 15 revealed visual acuities of 20/20 OD and 20/40 OS. Slit lamp examination revealed bilateral scattered, small annular and discoid gray opacities in the corneal endothelium, with smaller “vesicles” noted within the discoid opacities (Figure 2). No edema was noted in the corneal epithelium or stroma, and the remainder of the ocular examination was unremarkable. Central corneal pachymetry measured 624 um in the right eye and 626 um in the left eye. Specular microscopic imaging of the central corneal endothelium revealed estimated corneal endothelial cell densities of 2392 cells/mm2 OD and 2288 cells/mm2 OS. The girl’s mother and father had normal slit lamp examinations.
Figure 1.

MRI images of individuals with PPCD3 and unaffected control individual. Case 1. Axial (top left) and sagittal (bottom left) T1-weighted MRI images performed for proband 1 at 10 years of age demonstrate agenesis of the corpus callosum. Case 2. Axial (top middle) and sagittal (bottom middle) T1-weighted MRI images performed for proband 5 months of age demonstrate thinning of the corpus callosum compared to the control images (top and bottom right). Arrows superimposed on the control images designate the location of the corpus callosum. Case 1 images courtesy of Dr. Elliot Sherr. Control images obtained from the McConnell Brain Imaging Center of the Montreal Neurological Institute (Colin 27 Average Brain, Stereotaxic Registration Model; Copyright (C) 1993–2009 Louis Collins, McConnell Brain Imaging Centre, Montreal Neurological Institute, McGill University).
Figure 2.
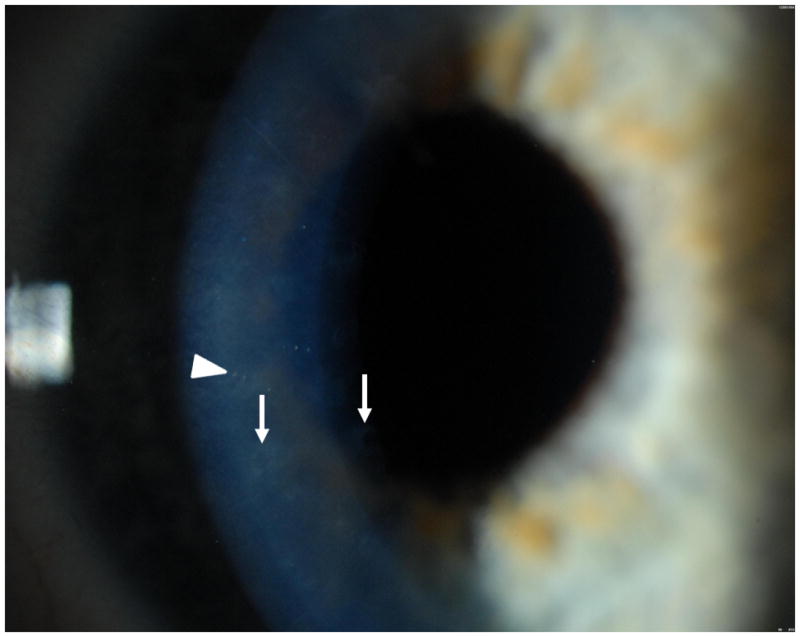
Case 1. Slit lamp photograph of 14 year old girl demonstrates scattered, small annular (arrows) and discoid (arrowhead) gray opacities in the corneal endothelium, with smaller “vesicles” noted within the discoid opacities. Image courtesy of Dr. Christopher Rapuano.
Karyotype, subtelomere FISH and whole genome copy number variant analyses performed to identify an explanation for the constellation of disorders identified in the proband were unremarkable. Screening of the ZEB1 gene in the proband demonstrated a frameshift mutation in exon 4 (c.449delG; p.(Gly150Alafs*36)), which was not identified in either of the proband’s parents (Figure 3A). Paternity testing confirmed that proband’s father was indeed the biologic father, indicating that the identified mutation was most likely spontaneous.
Figure 3.

Pedigrees and results of ZEB1 sequencing in families with a proband affected by PPCD and agenesis of the corpus callosum (A - Case 1; B – Case 2). Filled symbols represent affected individuals and open symbols represent unaffected individuals. The proband is indicated by a black arrowhead. Below the symbol of each individual in whom ZEB1 sequencing was performed, the wild type allele (designated by a + symbol) and/or the mutant allele is shown. Chromatograms beneath each pedigree depict the wild-type (WT) and mutant (MU) DNA sequences.
3.2 Case 2
A 4 year old Caucasian girl with a history of developmental delay and possible seizures during the first few months of life underwent MRI imaging of the brain at 5 months of life. Thinning of the corpus callosum was noted, was also seen on subsequent MRI examinations performed at 23 and 44 months of life (Figure 1). The proband’s father also underwent MRI imaging of the brain, which was unremarkable. Whole genome copy number variant analysis performed in the proband, her parents and siblings revealed a 1.79 Mb duplication of 17q12 in the proband (Figure 3B, II-3), her father (I-1) and one sibling (II-1), who also had been diagnosed with autism. The proband’s mother (I-2) and twin sister (II-2) were found to have a 1.5 Mb deletion of 1q21.1. The patient was referred for a complete ocular examination, which revealed corrected distance visual acuity (CDVA) of 20/30 OD and 20/25 OS. Slit lamp examination demonstrated bilateral, diffusely scattered endothelial vesicles, without evidence of epithelial or stromal edema. The remainder of the ocular examination was unremarkable. However, the central corneal thickness was increased in each eye, measuring 671 μm in the right eye and 676 μm in the left eye. Slit lamp examination of the proband’s mother revealed corneal endothelial changes characteristic of PPCD, which were not identified in either of the proband’s siblings.
After informed consent was obtained from the proband’s mother, a saliva sample was obtained from her and each of her three children as a source of genomic DNA. Screening of the ZEB1 gene in the proband demonstrated a single nucleotide deletion (c.1913-1914delCA; p.(Ser638Cysfs*5)) in exon 7 that was also present in the proband’s affected mother but absent in her father and siblings (Figure 3B).
4. DISCUSSION
Although this represents the initial report of the association of maldevelopment of the corpus callosum with a corneal dystrophy, ACC has been reported previously in association with a variety of inherited retinal disorders. A well-recognized syndrome involving ACC and ocular maldevelopment is Aicardi’s syndrome, a dominantly-inherited X-linked disorder characterized by partial or complete ACC (Fruhman et al., 2012). The pathognomonic ocular abnormality associated with Aicardi syndrome is chorioretinal lucinae, which is a condition affecting the retina and is characterized by clusters of punched out lesions around the optic disk in which areas of the choroidal and retinal pigment epithelium have atrophied (Fruhman et al., 2012; Paul et al., 2007).
The ZEB family of transcription factors is comprised of two members, ZEB1 and the zinc finger E-box binding homeobox 2 gene (ZEB2), which regulate various cell functions involved in development and disease (Postigo et al., 2003). The role of ZEB proteins in the development of the vertebrate CNS is evident from the fact that murine ZEB2 protein is present at the neural crest, while ZEB1 protein is primarily in neural tissues derived from the neural crest (Takagi et al., 1998). In addition, gene expression arrays show that ZEB1 and ZEB2 are expressed in several vertebrate tissues including the brain (Su et al., 2004; Wu et al., 2009). However, while ZEB1 protein has been demonstrated in human corneal endothelium, unpublished RNA microarray analysis results obtained from 11 corneal endothelium samples (donors between 4 and 70 years old) consistently demonstrated the absence of ZEB2 expression (Yellore et al., 2012). Although ZEB2 is unlikely to play a role in the post-natal endothelium, ZEB2 expression has been distinctly (compared to adult endothelium) observed in a layer of cuboidal epithelial cells that line the posterior surface of the early human fetal cornea (Chen et al., 2013). These early cells are anatomically consistent with, but morphologically and functionally distinct from, adult corneal endothelial cells.
Mowat-Wilson Syndrome, which is characterized by presence of ACC and in at least one case, bilateral microphthalmia, cataract, and retinal aplasia, is associated with over 100 unique mutations in ZEB2 (Saunders, Zhao, & Ardinger, 2009). Here, we report two frameshift mutations in ZEB1 in individuals that have been diagnosed with hypoplasia and agenesis of the corpus callosum. Taken together, the similar functional properties and expression patterns for ZEB1 and ZEB2 and their apparent association with ACC and ocular abnormalities suggest that the ZEB family of transcription factors may play a role in neural and ocular development. However, we have identified mutations in ZEB1 in 12 other PPCD3 probands without developmental delay and other associated clinical features that characterize ACC. Since many genes and certain environmental factors influence the development of the corpus callosum, the presence of a compensatory mechanism that limits the deleterious effects of the majority of ZEB1 mutations on neural tissue development may be at play (Paul et al., 2007).
4.1 Conclusion
PPCD3 has previously been described in association with a number of other ocular and extraocular abnormalities. We report the first association of PPCD3 with a developmental abnormality of the brain, agenesis of the corpus callosum. Screening of the ZEB1 gene in two affected individuals demonstrated frameshift mutations, predicted to result in protein truncation, confirming the diagnosis of PPCD3. Due to the similar functional properties and expression patterns for ZEB1 and ZEB 2, and the previous association of mutations within the ZEB2 gene and ACC, we suggest that the ZEB family of transcription factors may play a role in both neural and ocular development.
Highlights.
Posterior polymorphous corneal dystrophy is linked to agenesis of the corpus callosum
ZEB1 frameshift mutations are present in individuals with PPCD and ACC
ZEB1 and ZEB2, which has been linked ACC, have similar functional properties
The ZEB family of transcription factors may influence neural and ocular development
Acknowledgments
Support provided by National Eye Institute grants 1R01 EY022082 (A.J.A.), P30 EY000331 (core grant), an unrestricted grant from Research to Prevent Blindness and a grant from the Gerald Oppenheimer Family Foundation Center for the Prevention of Eye Disease (A.J.A.).
We would like to thank Drs. Christopher Rapuano, Elliot Sherr and John Sutphin for providing patient information and clinical images.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aldave AJ, Yellore VS, Yu F, Bourla N, Sonmez B, Salem AK, Rayner SA, Sampat KM, Krafchak CM, Richards JE. Posterior polymorphous corneal dystrophy is associated with TCF8 gene mutations and abdominal hernia. Am J Med Genet A. 2007;143A(21):2549–2556. doi: 10.1002/ajmg.a.31978. [DOI] [PubMed] [Google Scholar]
- Ariss M, Natan K, Friedman N, Traboulsi EI. Ophthalmologic abnormalities in Mowat-Wilson syndrome and a mutation in ZEB2. Ophthalmic Genet. 2012;33(3):159–160. doi: 10.3109/13816810.2011.610860. [DOI] [PubMed] [Google Scholar]
- Bakhtiari P, Frausto RF, Roldan AN, Wang C, Yu F, Aldave AJ. Exclusion of pathogenic promoter region variants and identification of novel nonsense mutations in the zinc finger E-box binding homeobox 1 gene in posterior polymorphous corneal dystrophy. Mol Vis. 2013;19:575–580. [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Huang K, Nakatsu MN, Xue Z, Deng SX, Fan G. Identification of novel molecular markers through transcriptomic analysis in human fetal and adult corneal endothelial cells. Hum Mol Genet. 2013;22(7):1271–1279. doi: 10.1093/hmg/dds527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremona FA, Ghosheh FR, Rapuano CJ, Eagle RC, Jr, Hammersmith KM, Laibson PR, Ayres BD, Cohen EJ. Keratoconus associated with other corneal dystrophies. Cornea. 2009;28(2):127–135. doi: 10.1097/ICO.0b013e3181859935. [DOI] [PubMed] [Google Scholar]
- Ferreira RC, Heckenlively JR, Menkes JH, Bateman JB. Menkes disease. New ocular and electroretinographic findings. Ophthalmology. 1998;105(6):1076–1078. doi: 10.1016/S0161-6420(98)96010-9. [DOI] [PubMed] [Google Scholar]
- Fruhman G, Eble TN, Gambhir N, Sutton VR, Van den Veyver IB, Lewis RA. Ophthalmologic findings in Aicardi syndrome. J AAPOS. 2012;16(3):238–241. doi: 10.1016/j.jaapos.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal R, Watts P, Hourihan M. Ocular findings in pediatric patients with partial agenesis of corpus callosum. J Pediatr Ophthalmol Strabismus. 2010;47(4):236–241. doi: 10.3928/01913913-20090918-06. [DOI] [PubMed] [Google Scholar]
- Inbar D, Halpern GJ, Weitz R, Sadeh M, Shohat M. Agenesis of the corpus callosum in a mother and son. Am J Med Genet. 1997;69(2):152–154. [PubMed] [Google Scholar]
- Krafchak CM, Pawar H, Moroi SE, Sugar A, Lichter PR, Mackey DA, Mian S, Nairus T, Elner V, Schteingart MT, Downs CA, Kijek TG, Johnson JM, Trager EH, Rozsa FW, Mandal MN, Epstein MP, Vollrath D, Ayyagari R, Boehnke M, Richards JE. Mutations in TCF8 cause posterior polymorphous corneal dystrophy and ectopic expression of COL4A3 by corneal endothelial cells. Am J Hum Genet. 2005;77(5):694–708. doi: 10.1086/497348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner J, Dash DP, Muszynska D, Hosseini M, Segev F, George S, Frazer DG, Moore JE, Kaye SB, Young T, Simpson DA, Churchill AJ, Heon E, Willoughby CE. Mutational spectrum of the ZEB1 gene in corneal dystrophies supports a genotype-phenotype correlation. Invest Ophthalmol Vis Sci. 2013;54(5):3215–3223. doi: 10.1167/iovs.13-11781. [DOI] [PubMed] [Google Scholar]
- Liskova P, Palos M, Hardcastle AJ, Vincent AL. Further genetic and clinical insights of posterior polymorphous corneal dystrophy 3. JAMA Ophthalmol. 2013;131(10):1296–1303. doi: 10.1001/jamaophthalmol.2013.405. [DOI] [PubMed] [Google Scholar]
- Liskova P, Tuft SJ, Gwilliam R, Ebenezer ND, Jirsova K, Prescott Q, Martincova R, Pretorius M, Sinclair N, Boase DL, Jeffrey MJ, Deloukas P, Hardcastle AJ, Filipec M, Bhattacharya SS. Novel mutations in the ZEB1 gene identified in Czech and British patients with posterior polymorphous corneal dystrophy. Hum Mutat. 2007;28(6):638. doi: 10.1002/humu.9495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, El-Naggar S, Darling DS, Higashi Y, Dean DC. Zeb1 links epithelial-mesenchymal transition and cellular senescence. Development. 2008;135(3):579–588. doi: 10.1242/dev.007047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masri AT, Abu-Libdeh AM, Ababneh OH, Al-Hadidy AM. Septo-optic dysplasia syndrome with schizencephaly and sudden visual loss. A new observation. Neurosciences (Riyadh) 2011;16(3):281–282. [PubMed] [Google Scholar]
- Mehta JS, Vithana EN, Tan DT, Yong VH, Yam GH, Law RW, Chong WG, Pang CP, Aung T. Analysis of the posterior polymorphous corneal dystrophy 3 gene, TCF8, in late-onset Fuchs endothelial corneal dystrophy. Invest Ophthalmol Vis Sci. 2008;49(1):184–188. doi: 10.1167/iovs.07-0847. [DOI] [PubMed] [Google Scholar]
- Nguyen DQ, Hosseini M, Billingsley G, Heon E, Churchill AJ. Clinical phenotype of posterior polymorphous corneal dystrophy in a family with a novel ZEB1 mutation. Acta Ophthalmol. 2010;88(6):695–699. doi: 10.1111/j.1755-3768.2009.01511.x. [DOI] [PubMed] [Google Scholar]
- O’Driscoll MC, Black GC, Clayton-Smith J, Sherr EH, Dobyns WB. Identification of genomic loci contributing to agenesis of the corpus callosum. Am J Med Genet A. 2010;152A(9):2145–2159. doi: 10.1002/ajmg.a.33558. [DOI] [PubMed] [Google Scholar]
- Paul LK, Brown WS, Adolphs R, Tyszka JM, Richards LJ, Mukherjee P, Sherr EH. Agenesis of the corpus callosum: genetic, developmental and functional aspects of connectivity. Nat Rev Neurosci. 2007;8(4):287–299. doi: 10.1038/nrn2107. [DOI] [PubMed] [Google Scholar]
- Postigo AA, Depp JL, Taylor JJ, Kroll KL. Regulation of Smad signaling through a differential recruitment of coactivators and corepressors by ZEB proteins. EMBO J. 2003;22(10):2453–2462. doi: 10.1093/emboj/cdg226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raber IM, Fintelmann R, Chhabra S, Ribeiro MP, Eagle RC, Jr, Orlin SE. Posterior polymorphous dystrophy associated with nonkeratoconic steep corneal curvatures. Cornea. 2011;30(10):1120–1124. doi: 10.1097/ICO.0b013e3182114452. [DOI] [PubMed] [Google Scholar]
- Riazuddin SA, Zaghloul NA, Al-Saif A, Davey L, Diplas BH, Meadows DN, Eghrari AO, Minear MA, Li YJ, Klintworth GK, Afshari N, Gregory SG, Gottsch JD, Katsanis N. Missense mutations in TCF8 cause late-onset Fuchs corneal dystrophy and interact with FCD4 on chromosome 9p. Am J Hum Genet. 2010;86(1):45–53. doi: 10.1016/j.ajhg.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders CJ, Zhao W, Ardinger HH. Comprehensive ZEB2 gene analysis for Mowat-Wilson syndrome in a North American cohort: a suggested approach to molecular diagnostics. Am J Med Genet A. 2009;149A(11):2527–2531. doi: 10.1002/ajmg.a.33067. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Krafchak C, Fuse N, Epstein MP, Schteingart MT, Sugar A, Eibschitz-Tsimhoni M, Downs CA, Rozsa F, Trager EH, Reed DM, Boehnke M, Moroi SE, Richards JE. A locus for posterior polymorphous corneal dystrophy (PPCD3) maps to chromosome 10. Am J Med Genet A. 2004;130A(4):372–377. doi: 10.1002/ajmg.a.30267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, Cooke MP, Walker JR, Hogenesch JB. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A. 2004;101(16):6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi T, Moribe H, Kondoh H, Higashi Y. DeltaEF1, a zinc finger and homeodomain transcription factor, is required for skeleton patterning in multiple lineages. Development. 1998;125(1):21–31. doi: 10.1242/dev.125.1.21. [DOI] [PubMed] [Google Scholar]
- Teekhasaenee C, Nimmanit S, Wutthiphan S, Vareesangthip K, Laohapand T, Malasitr P, Ritch R. Posterior polymorphous dystrophy and Alport syndrome. Ophthalmology. 1991;98(8):1207–1215. doi: 10.1016/s0161-6420(91)32152-3. [DOI] [PubMed] [Google Scholar]
- Vincent AL, Niederer RL, Richards A, Karolyi B, Patel DV, McGhee CN. Phenotypic characterisation and ZEB1 mutational analysis in posterior polymorphous corneal dystrophy in a New Zealand population. Mol Vis. 2009;15:2544–2553. [PMC free article] [PubMed] [Google Scholar]
- Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S, Hodge CL, Haase J, Janes J, Huss JW, 3rd, Su AI. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10(11):R130. doi: 10.1186/gb-2009-10-11-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellore VS, Rayner SA, Nguyen CK, Gangalum RK, Jing Z, Bhat SP, Aldave AJ. Analysis of the role of ZEB1 in the pathogenesis of posterior polymorphous corneal dystrophy. Invest Ophthalmol Vis Sci. 2012;53(1):273–278. doi: 10.1167/iovs.11-8038. [DOI] [PMC free article] [PubMed] [Google Scholar]