

Abstract
In plants, post-transcriptional gene silencing (PTGS) is mediated by DICER-LIKE1 (DCL1)-dependent miRNAs, that also trigger 21-nt secondary siRNA via RNA DEPENDENT RNA POLYMERASE6 (RDR6), DCL4, and ARGONAUTE1 (AGO1)1–3, while transcriptional gene silencing (TGS) of transposons is mediated by 24-nt heterochromatic (het)siRNA RDR2, DCL3 and AGO44. Transposons can also give rise to abundant 21-nt “epigenetically activated” small interfering RNAs (easiRNAs) in DECREASE IN DNA METHYLATION1 (ddm1) and DNA METHYLTRANSFERASE1 (met1) mutants, as well as in the vegetative nucleus of pollen grains5, and in dedifferentiated plant cell cultures6. Here we show that easiRNAs resemble secondary siRNAs, in that thousands of transposon transcripts are specifically targeted by more than fifty miRNAs for cleavage and processing by RDR6. Loss of RDR6, DCL4 or DCL1 in a ddm1 background results in loss of 21-nt easiRNA, and severe infertility, but 24-nt hetsiRNA are partially restored, supporting an antagonistic relationship between PTGS and TGS. Thus miRNA-directed easiRNA biogenesis is a latent mechanism that specifically targets transposon transcripts, but only when they are epigenetically reactivated during reprogramming of the germline. This ancient recognition mechanism may have been retained both by transposons to evade long-term heterochromatic silencing, and by their hosts for genome defence.
21-nt sRNAs, that we have termed easiRNAs, were previously found to accumulate from the 3′ UTR of ATHILA6 retrotransposons in wild type pollen and ddm1 inflorescence5, and to depend on RDR6, DCL4 and AGO17,8. To assess the origin of easiRNAs, we performed small RNA sequencing from inflorescence tissue, and we found that they accumulated in ddm1, but not in WT (Columbia-0) nor in ddm1 rdr6 double mutants7,8 (Fig. 1; Extended Data Fig. 1a). 21-nt easiRNAs in ddm1 mostly originated from ATGYPSY LTR retroelements (Supplementary Table 1) located in pericentromeric regions, especially the high copy but defective ATHILA retrotransposons, which are integrated into pericentromeric satellite repeats (Fig. 1). However, easiRNAs also arose from ATCOPIA families found in euchromatic regions, such as EVADE/ATCOPIA939 (Supplementary Table 1), and from specific VANDAL/MuDR, HAT and CACTA elements (such as AtEnSpm6), some of which are present in much lower copy number and known to transpose in ddm110. This raised the important question as to how specificity was conferred to these TEs such that they gave rise to easiRNA, when other transposons and genes did not. Genetically, easiRNA resemble 21-nt trans-acting (ta)siRNAs and other secondary siRNA, which are derived from non-coding RNA and mRNA respectively, and are triggered by miRNA bound by AGO711 and AGO12,3,12,13. miRNA were thus excellent candidates to confer transposon specificity.
Figure 1. miRNAs trigger RDR6-dependent 21-nt epigenetically activated (ea)siRNA biogenesis from reactivated transposons.

Whole-genome representation illustrating miRNAs triggering widespread easiRNA biogenesis at transposons in Arabidopsis. Outermost to innermost tracks depicting: miRNA abundance in ddm1-2 (Histogram); Arabidopsis Chromosome I – V, pericentromeric region (black) (Ideogram); Gene and transposon frequency, low density (red), high density (blue) (Heat-map); Retrotransposon derived 21-nt sRNAs (dark red = unique, light red = multiple mapping) and DNA transposon-derived 21-nt sRNAs (dark blue = unique, light blue = multiple mapping) 21-nt siRNAs in order of Col-0, ddm1-2, ddm1-2 rdr6-15, and ddm1-2 dcl1-11 (Histogram); miRNAs targeting transposons (Connectors); miR859a (Chr I, red), miR390a (Chr II, blue), miR172d (Chr III, red), eamiR2 ATHILAIV (Chr IV, grey) and miR172e (Chr V, blue). Transposons are post-transcriptionally targeted by 50 known miRNAs and newly discovered eamiRNAs (Table 1) giving rise to abundant RDR6-dependent 21-nt easiRNAs at transposons in Arabidopsis. (Refer to Extended Data Fig. 1; Supplementary Table 1; 3).
Extended Data Figure 1. 21-nt easiRNAs originate from transposons in ddm1 and are miRNA and RDR6-dependent.
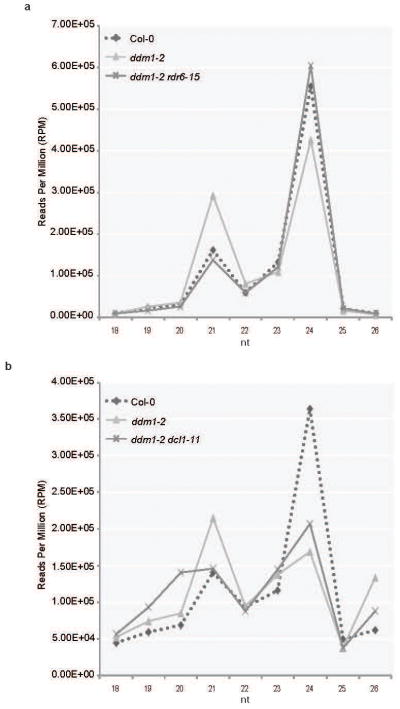
(a) 18-nt to 26-nt small RNA abundance in Col-0, ddm1-2, and ddm1-2 rdr6-15. Normalized reads per million (RPM). (b) 18-nt to 26-nt small RNA abundance in Col-0, ddm1-2, and ddm1-2 dcl1-11. Normalized reads per million (RPM).
miRNA biogenesis in Arabidopsis depends largely on DCL1, so that miRNA are greatly reduced in sterile dcl1-9, and partly reduced in fertile dcl1-1114. We were unable to recover ddm1 dcl1-9 double mutants, but easiRNA levels were reduced in ddm1 dcl1-11, consistent with a role for miRNA in targeting and easiRNA biogenesis (Fig. 1; Extended Data Fig. 1b). We identified miRNAs in our sRNA sequencing libraries from Col-0, ddm1 and ddm1 rdr6 inflorescence tissue. In addition to utilising a miRNA identification algorithm15, miRNAs were distinguishable from other 21-nt sRNAs that are RDR6-dependent, by comparing their abundance in ddm1 and ddm1 rdr6. We identified several known miRNAs, validating our algorithm (Extended Data Fig. 10; Supplementary Table 2) and genome-wide target prediction revealed that 3662 TEs are potentially targeted by these miRNAs (Supplementary Table 3). To determine whether miRNAs mediate targeting and cleavage of TE transcripts, we sequenced cleavage sites genome wide by Parallel Analysis of RNA Ends (PARE)11. In order to assess specificity, 5′ RNA ends from a 5-nt (s) window surrounding each predicted miRNA target site were compared to those from a larger 30-nt (l) window (Supplementary Table 3; for more specific 1-nt (ss) and 3-nt (sl) windows, refer to Supplementary Table 9). Significant enrichment of RNA ends at or near the target site was strong evidence for cleavage of the transposon transcripts guided by miRNA.
Extended Data Figure 10. Arabidopsis miRNAs target transposons.

Known Arabidopsis miRNAs, and novel epigenetically-activated (ea)miRNAs that arise in ddm1-2, are predicted to target transposon transcripts and confirmed to cleave transposon transcripts by PARE (Supplementary Table 3; Supplementary Methods). eamiRNAs, some known to be developmentally regulated, and TE-derived eamiRNAs that target specific transposon families. Transposons are identified by EVRY TE identifier (Supplementary Table 2, for further annotation refer to The Arabidopsis Information (TAIR10) annotation ORF ID). Transposon transcripts giving rise to 21-nt easiRNAs (bold); those that are targeted by multiple miRNA (*); and, those miRNAs that target multiple transposons of the same family (italics) are highlighted.
Approximately half of the 3662 predicted TE targets showed evidence of miRNA guided cleavage (Supplementary Table 3), and were targeted by more than fifty distinct miRNA (Extended Data Fig. 10), although some TEs had only one cleavage product in the target window (such as EVADE/ATCOPIA93, which is predicted to be cleaved by miR833). In some cases multiple miRNAs targeted single TEs (Extended Data Fig. 10), reminiscent of the “two-hit” model for tasiRNA biogenesis16. In other cases, TEs were targeted by longer forms of miRNA (22-nt), which are thought to promote secondary siRNA biogenesis3,17. Specifically, 1733 TE were predicted to be miRNA targets and generated easiRNAs (at least 10 reads). Of these, 1247 were detectably cleaved by PARE-seq. An additional 1929 TEs were predicted to be targeted by miRNAs but, did not generate easiRNAs, of these 442 were detectably cleaved by PARE-seq. Thus, more than half of the TEs targeted by miRNA also generated easiRNA in inflorescence tissue from ddm1. However, phasing of easiRNA, typical of other secondary siRNA, was not detected, likely due to the repetitive nature of the TE targets and their targeting by multiple miRNA (data not shown).
Interestingly, the miRNAs found to target TEs, were mostly known miRNAs (Extended Data Fig. 10), such as miR156, miR159, miR172 and miR859, which also generate secondary siRNAs from mRNA targets2,3. Many of these miRNAs were predicted to target ATHILA elements (Extended Data Fig. 1), generating abundant easiRNA corresponding to ATHILA ORF1, also known as TRANSCRIPTIONALLY SILENT INFORMATION (TSI) (Fig. 2a). ATHILA ORF1 contains a predicted target site for miR859, and PARE confirmed cleavage at this site (Fig. 2c, Supplementary Table 3). We further validated miR859-directed cleavage by modified 5′ RLM RACE PCR (Fig. 2e).
Figure 2. miRNA cleavage at ATHILA ORF1 and ATCOPIA43.

(a, b) 21-nt easiRNAs at ATHILA ORF1 (AT2G10280), in comparison to non-easiRNA generating ATCOPIA43 (AT1G36040) in track order Col-0 (i), ddm1-2 (ii), rdr6-15 (iii), ddm1-2 rdr6-15 (iv) and ddm1-2 dcl1-11 (v). (c, d) Parallel Analysis of RNA Ends (PARE) at ATHILA ORF1 and ATCOPIA43 in track order Col-0 (i), ddm1-2 (ii) and ddm1-2 rdr6-15 (iv). Individual reads, sense = red, antisense = blue. (e, f) 5′ RLM RACE-PCR products (arrows; black bars in a, b) from ATHILA ORF1 and ATCOPIA43 corresponding to cleaved mRNA fractionated by gel electrophoresis. Inset: miRNA alignment with 5′ RLM RACE-PCR cleavage sites, and frequencies of cleavage products indicated as fractions.
Despite cleavage of many TE families by multiple miRNAs (Extended Data Fig. 1), some did not generate easiRNAs. This was especially true of CACTA and ATCOPIA elements, for which most cleavage events were non-productive. For example, ATCOPIA43 elements were targeted by miR390, which targets non-coding RNA for tasiRNA production by the two-hit model16, but which did not generate easiRNAs in ddm1 (Fig. 2b). Instead, PARE detected uncapped degradation products from ATCOPIA43 indicating extensive secondary RNA decay (Fig. 2d), following miRNA cleavage (Fig. 2f), and similar mRNA decay patterns were found at many genes targeted by miRNA (Extended Data Fig. 2). In general, easiRNA-producing TEs were intact AtEnSpm2, AtEnSpm5, AtEnSpm6, ATCOPIA93 and ATCOPIA28 elements, while those that did not generate easiRNAs were non-autonomous elements (e.g. AtEnSpm1A), interrupted by insertion of other TEs, or otherwise truncated, and were subject to RNA decay. Occasionally, TEs were found to produce easiRNAs, and were predicted to be targeted by miRNAs, but cleavage could not be detected by PARE, which may be due to the underrepresentation of some miRNAs in inflorescence tissue (Supplementary Table 2). For example, EVADE/ATCOPIA93 produces abundant easiRNAs from the GAG gene, which is predicted to be targeted by several known miRNAs and eamiRNAs (Extended Data Fig. 3a), yet only miR833 shows evidence of cleavage by PARE, and this did not pass our cut-off for miRNA cleavage (Supplementary Table 3). EVADE/ATCOPIA93 is specifically expressed in only a subset of cells18.
Extended Data Figure 2. miRNA target genes and transposons that do not promote tasiRNA nor easiRNA, respectively, have degradation covering the entire region.

Read pattern distribution of 21-nt unique reads (represented as a histogram of read density (grey bars)) and PARE signatures at (a) TAS2 (AT2G39681), (b) RCC (AT3G02300), SEP2 (AT3G02310), (c) MEE58 (AT4G13940), and transposons (d) ATENSPM6 (AT2G06720), (e) ATLINE1_4 (AT2G15540), (f) ATCOPIA43 (AT3G0410), in track order Col-0, ddm1-2, rdr6-15, ddm1-2 rdr6-15 and ddm1-2 dcl1-11.
Extended Data Figure 3. Relationship between DNA methylation, easiRNA and hetsiRNA at transposons that miRNA are predicted to target.

DNA methylation, by CG (red), CHG (blue) and CHH+/− context (green) cytosine context (scale 1 = methylated cytosine, 0 = unmethylated cytosine) at (a) ATCOPIA93 (AT5G17125), (b) ATMU5 (AT4G08680) DNA transposon and the surrounding region, and (c) ATHILA6A (AT4TE15030) retrotransposon and the surrounding region, in Col-0, ddm1-2, rdr6-15 and ddm1-2 rdr6-15 (i). 21-nt and 24-nt siRNAs, represented as a histogram of read density (grey bars) in track order Col-0, ddm1-2, rdr6-15 and ddm1-2 rdr6-15 (ii). Key for sRNA reads, unique (U) mapping to one location of the genome, and multiple (M) mapping to more than one location of the genome. PARE read density in Col-0 and ddm1-2 (iii).
Two new classes of miRNAs were found in ddm1, namely those encoded by transposons, and those miRNA precursors silenced by DNA methylation. We have assigned these miRNAs as epigenetically-activated (ea)miRNAs, as alike easiRNAs, they are abundant in pollen and ddm1 (Extended Data Fig. 4; Extended Data Fig. 10; Supplementary Table 2). We also identified new miRNA isomers, from 21-nt to 22-nt and 24-nt sequence variants, originating from known miRNA precursors (Supplementary Table 2). 22-nt isoforms promote secondary siRNA biogenesis3,17 while 24-nt isoforms promote DNA methylation9,19. The newly identified eamiR2, originates from an immature precursor sequence within an ATHILA4 retroelement and is abundant in ddm1 only (Supplementary Table 2). PARE analysis of ddm1 confirmed release of this eamiRNA from its own ATHILA4 precursor (Supplementary Table 3), and cleavage of other ATHILA4 elements in trans (Fig. 1). However, this TE-derived eamiRNA does not appear to direct easiRNA biogenesis from its own precursor (Supplementary Table 1). Thus, the release of TE-producing eamiRNAs by DICER does not trigger easiRNA biogenesis per se, but only when the miRNA targets TE transcripts via AGO17.
Extended Data Figure 4. Epigenetically activated (ea)miRNA immature precursor sequence and predicted structure.

Epigenetically activated (ea)miRNAs immature precursor sequences have methylated cytosines (*) in Col-0, that are unmethylated in ddm1-2. Mature miRNA are underlined, and the putative stem-loop structures of the precursors are illustrated.
In order to test the requirement for miRNA in easiRNA biogenesis, we utilized miR845b, a 22-nt miRNA predicted to target several retroelements (Supplementary Table 3), but only in pollen where it is specifically expressed. We fused a GFP reporter gene to a ubiquitously expressed promoter, and to a 300 bp region of AtGP1 that included the predicted miR845b target site (Fig. 3a). Sequencing small RNA from pollen revealed novel 21-nt easiRNAs surrounding the miR845b target site (Fig. 3a) not found in constructs in which the target site was deleted (Fig. 3b). We confirmed by modified 5′ RNA Ligation-Mediated (RLM) RACE PCR that miR845b-directed cleavage products from GFP-AtGP1 transcripts accumulate exclusively in pollen (Fig. 3c). Thus, 21-nt easiRNA biogenesis at TEs depends on targeting by miRNA.
Figure 3. miR845b targets AtGP1 promoting easiRNA biogenesis.

(a) Transgenic line over-expressing AtGP1 containing the 20-nt miR845b target region (marked miR845) and novel overlapping 21-nt easiRNAs (star). Individual reads, sense = red, antisense = blue. (b) Transgenic line over-expressing AtGP1 that did not contain the 20-nt miR845b predicted target region, with no 21-nt easiRNAs overlapping this site. (c) Frequencies of 5′ RLM RACE-PCR miR845b AtGP1 cleavage products indicated as fractions.
24-nt hetsiRNAs guide asymmetric CHH methylation at TEs and are RDR2-dependent4. We profiled 24-nt hetsiRNAs in Col-0, ddm1, rdr6, ddm1 rdr6, and ddm1 dcl1-11 (Supplementary Table 1; Fig. 4). In Col-0, 24-nt hetsiRNAs target LTR retrotransposons, and most DNA transposons (Extended Data Fig. 5). In ddm1, we found a slight increase of multiple mapping 24-nt hetsiRNAs from ATGYPSY retrotransposons that were saturated by easiRNA biogenesis (Supplementary Table 1), but an overall general loss from DNA transposons and other individual retrotransposons, so that less than half of these retained 24-nt siRNA in ddm1 (Extended Data Fig. 5). Many of these TEs gained 24-nt hetsiRNAs in ddm1 rdr6 and ddm1 dcl1 (Supplementary Table 1; Extended Data Fig. 5) and, furthermore, hundreds of TEs that did not have detectable hetsiRNAs in Col-0 or ddm1, gained them in ddm1 rdr6 (Fig. 4a; Extended Data Fig. 6). We hypothesized that miRNA targeting and easiRNA biogenesis might inhibit 24-nt hetsiRNA production in ddm1, and we found that almost all TEs cleaved by miRNA either gained 21-nt easiRNAs in ddm1, lost 24-nt hetsiRNA in ddm1, or gained 24-nt hetsiRNA in ddm1 rdr6 (Fig. 4b, c; Extended Data Fig. 6). This enrichment was even greater for those TEs targeted by more than one miRNA (Extended Data Fig. 6c).
Figure 4. Transposons targeted by miRNA, gain hetsiRNA when the easiRNA pathway is lost.

(a) Overlap of transposons targeted by hetsiRNAs in Col-0, ddm1-2 and ddm1-2 rdr6-15. (b) Overlap of transposons targeted by miRNAs, that have lost hetsiRNAs in ddm1-2, gained hetsiRNAs in ddm1-2 rdr6-15, and those whose transcripts undergo productive miRNA cleavage resulting in easiRNA biogenesis in ddm1-2. 21-nt easiRNAs, 24-nt hetsiRNAs and PARE degradome at easiRNA-generating (c) ATHILA (AT3G32118) in comparison to non-easiRNA generating (d) ATENSPM1 (AT4G02314). Individual reads, sense = red, antisense = blue in track order Col-0 (i), ddm1-2 (ii) rdr6-15 (iii), and ddm1-2 rdr6-15 (iv).
Extended Data Figure 5. 24-nt hetsiRNAs at transposons in Col-0, are lost in ddm1, and gained in ddm1 rdr6 and ddm1 dcl1.
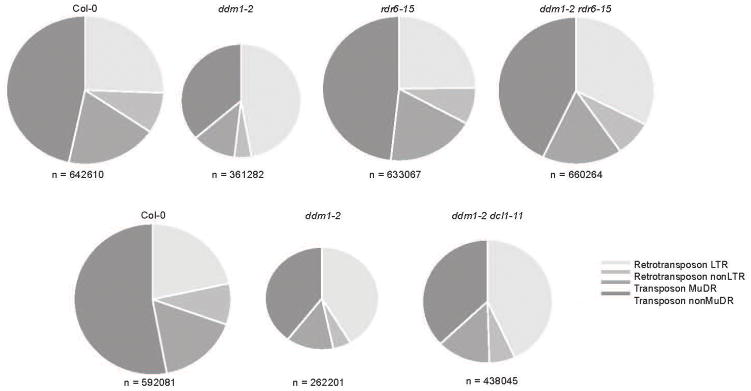
24-nt hetsiRNAs by transposon class in Col-0, ddm1-2, rdr6-15, ddm1-2 rdr6-15, and ddm1-2 dcl1-11. Normalized reads per million (RPM).
Extended Data Figure 6. Overlap of TEs that undergo easiRNA biogenesis, hetsiRNA loss and miRNAs targeting.

Individual transposons were grouped depending on small RNA abundance in each genotype. (a) TEs that lose 24-nt hetsiRNAs in ddm1-2, gain 21-nt easiRNAs in ddm1-2 overlap with those that gain 24-nt hetsiRNAs in ddm1-2 rdr6-15. (b) TEs that are targeted and cleaved by miRNAs overlap with those that gain 21-nt easiRNAs in ddm1-2 and 24-nt hetsiRNAs in ddm1-2 rdr6-15. (c) TEs that are targeted and cleaved by two or more miRNAs overlap with those that gain 21-nt easiRNAs in ddm1-2, and those that gain 24-nt hetsiRNAs in ddm1-5 rdr6-15. (d) TEs that are predicted to be targeted by miRNAs, but without supporting PARE cleavage data, also overlap with those that gain 21-nt easiRNAs in ddm1-2 and 24-nt hetsiRNAs in ddm1-2 rdr6-15.
Thus miRNA cleavage appears to inhibit 24-nt siRNA biogenesis in a manner that depends on RDR6, whether or not easiRNA accumulate (Fig. 4b; Supplementary Table 3). This response was observed when miRNA cleavage sites were on the antisense or occasionally on the sense (mRNA) strand (Supplementary Table 3). It is possible, therefore, that miRNA-directed cleavage of antisense Pol IV/Pol V transcripts, and processing by RDR6 rather than RDR2, might prevent the formation of 24-nt siRNAs, without necessarily generating 21-nt easiRNAs (that are Pol II dependent8). Thus, RDR6 partially antagonizes RDR2 activity, inhibiting 24-nt hetsiRNA biogenesis at TEs. Likewise, RDR2 partially antagonizes RDR6 activity at transgenes subject to PTGS20.
Symmetric CG and CHG methylation contexts are maintained by DNA methyltransferases and histone modifications, while CHH methylation is associated with 24-nt hetsiRNA guided RdDM4. Methylation mediates transcriptional silencing of TE promoters21, found near the TIR of most DNA transposons and in the LTR of retrotransposons, as well as internally in the case of the ATHILA ORF1 and some DNA transposons. As expected, whole-genome sequencing of bisulphite-treated DNA from inflorescence tissue showed global loss of DNA methylation from all classes of transposons in ddm1 when compared to wildtype, Col-0 (Extended Data Fig. 7a, b). As previously reported, most of this loss occurred in the symmetric CG and CHG contexts, rather than the asymmetric CHH context, consistent with retention of 24-nt hetsiRNA (Supplementary Table 4)22. By whole genome bisulphite sequencing we measured levels of DNA methylation in ddm1 rdr6 F3 progeny from two independent double mutants, relative to ddm1 (Extended Data Fig. 7a). A modest difference in DNA methylation is expected due to inbreeding of the ddm1 parents23, and was observed in one ddm1 rdr6 replicate, however, methylation at TEs substantially increased in the other replicate (Extended Data Fig. 7e, f; Supplementary Table 4). These results suggest that elevated levels of 24-nt siRNA could only guide remethylation of TEs stochastically in ddm1 rdr6 (Extended Data Fig. 7g, h), likely reflecting the epigenetic inheritance of unmethylated and methylated TEs from the ddm1 and rdr6 parents, respectively, as previously observed24.
Extended Data Figure 7. Loss of methylation at transposons in ddm1 is partially restored in ddm1 rdr6.

(a) Transposon methylation in Col-0, ddm1-2, rdr6-15 and ddm1-2 rdr6-15 replicates. Scale 1 = methylated cytosine, 0 = unmethylated cytosine. Total DNA methylation at transposons, grouped by superfamily in (b) Col-0, (c) ddm1-2, (d) rdr6-15 and (e, f) ddm1-2 rdr6-15 replicates. Key (1) LTR Retrotransposons ATGYPSY, (2) LTR Retrotransposons ATCOPIA, (3) NonLTR Retrotransposons ATLINE, (4) nonLTR Retrotransposons TSCL, (5) TIR DNA Transposons MuDR, (6) non-TIR DNA Transposons MuDR, (7) DNA Transposons EnSpM, (8) DNA Transposons Helitron and (9) Other Repeats. Total methylation by total converted cytosine to thymine and non-converted cytosine counts (at least 10 reads per cytosine). Scale 0 = unmethylated, 1 = methylated. Boxplots indicate median, range and standard deviations (box). DNA methylation, by CG (red), CHG (blue) and CHH+/− context (Green) at (g) ATHILA ORF1 (AT2G10280) and (h) ATCOPIA43 (AT1G36040). Track order Col-0 (i), ddm1-2 (ii), rdr6-15 (iii) and ddm1-2 rdr6-15 (iv).
In order to assess whether the loss of the easiRNA pathway leads to a further outburst of transposon reactivation in ddm1 rdr6, we utilized Affymetrix ATH1 microarrays to analyse the transcriptome. A few hundred representative TEs are included on Affymetrix ATH1 microarrays, and can be used to assess TE reactivation5. We found that most of these TE transcripts were abundantly and similarly expressed in both ddm1 and ddm1 rdr6 (Supplementary Table 5; Extended Data Fig. 8), indicating that easiRNA biogenesis does not reduce TE transcript levels in ddm1. In fact, consistent with our findings of sporadic TE methylation and gain of hetsiRNAs in ddm1 rdr6, several TEs had reduced expression in ddm1 rdr6, including ATHILA and ATCOPIA elements, as well as TEs of the MuDR Superfamily (Supplementary Table 5). Therefore, the loss of both DDM1 and RDR6 does not enhance transcriptional reactivation of TEs, but instead, some TEs become transcriptionally repressed in ddm1 rdr6 (Extended Data Fig. 8; Supplementary Table 5), and elevation in methylation level is observed at least for some of these TEs, for example, ATCOPIA22 and ARNOLD3, in ddm1 rdr6 (Supplementary Table 4).
Extended Data Figure 8. Transposon transcript abundance in ddm1-2 and ddm1 rdr6.

(a, b, c) Ath1 Affymetrix microarray expression (log2 signal intensity) in Col-0 in comparison to ddm1-2, rdr6-15 and ddm1-2 rdr6-15. (d) TEs upregulated in ddm1-2 were not further upregulated in ddm1-2 rdr6-15. Key: red = transposons, black = genes.
We have found that loss of TE methylation results in epigenetic activation and consequent transcription, whereby transposon mRNA become preferentially targeted by more than 50 miRNAs bound to AGO1. Productive cleavage of transposon transcripts usually engages RDR6, allowing DCL4 to generate 21-nt epigenetically-activated (ea)siRNAs from transposon open reading frames, in a PTGS mechanism (Extended Data Fig. 9). About half of these transposons belong to the LTR/ATHILA element family (Extended Data Fig. 10), which occur in high copy number at pericentromeric repeats, and are targeted by multiple miRNAs (Extended Data Fig. 3c; Fig. 2). Several known MIRNA genes, for example miR843, are methylated, and only target transposon transcripts when unmethylated and expressed: in pollen, and in ddm1 mutants (Supplementary Table 2; Extended Data Fig. 4). Other miRNAs are developmentally regulated, for example miR156, which is required to maintain the juvenile phase of plant development, and miR172, which is required for the transition to the adult phase25, accounting perhaps for tissue-specific silencing of TEs during different phases of plant development26–28.
Extended Data Figure 9. miRNA-directed easiRNA biogenesis from activated transposons.
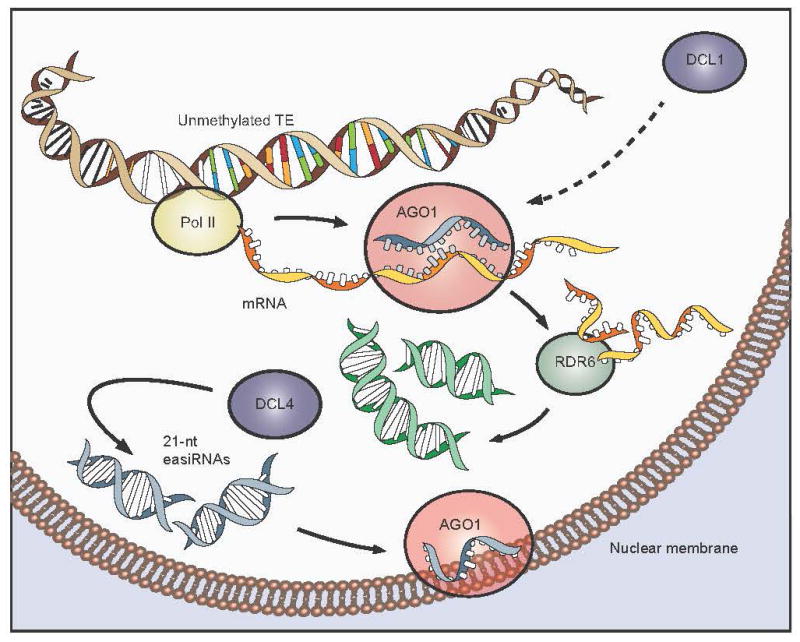
When TEs are epigenetically activated, through the loss of DNA methylation and/or heterochromatin, transposon mRNA transcripts become preferentially targeted by miRNAs (DCL1-dependent) bound by AGO1. Productive cleavage of transposon transcripts engages RDR6 and DCL4, which generate 21-nt epigenetically-activated (ea)siRNAs from transposon open reading frames, in a post-transcriptional gene silencing (PTGS) mechanism, that are then loaded into AGO1, and thus, prevents engagement of RDR2 and RdDM. This antagonism accounts for the retention of miRNA binding sites by transposons, to evade long-term heritable silencing, elicited by DNA methylation via RDR2. This model also accounts for the retention of the miRNA-directed mechanism by the host organism, in order to generate easiRNAs to silence TEs when they are epigenetically reprogrammed in the germline.
TEs are targeted by the same conserved miRNA (miR845) in rice and Arabidopsis leading to the idea that they may have retained miRNA-binding sites despite selection against them. Furthermore, some of the newly identified miRNAs are themselves encoded by TEs (Extended Data Fig. 10). Why would transposons retain miRNA sites that could potentially silence them post-transcriptionally? One explanation lies in the preferential processing of targeted TE transcripts via RDR6, that appears to act antagonistically to RDR2, preventing 24-nt hetsiRNA biogenesis (Extended Data Fig. 9)20. We found a striking overlap between TEs that produced easiRNAs in ddm1, with those that gain 24-nt hetsiRNAs in ddm1 rdr6 (Fig. 4b, Extended Data Fig. 6). 24-nt hetsiRNA promote RNA dependent DNA methylation (RdDM), and it is likely, therefore, that TEs tolerate developmentally regulated PTGS by miRNA and easiRNAs to avoid long-term TGS by DNA methylation. Given the likely evolution of miRNA precursors from TEs and other inverted repeats21, miRNAs may have arisen in ancient eukaryotes to target retrotransposons and other TEs rather than genes.
ddm1 mutants are remarkably normal, despite the heritable loss of heterochromatin. In contrast, ddm1 rdr6, ddm1 dcl4 and ddm1 dcl1 have a wide range of developmental phenotypes and heritable epigenetic defects, including widespread infertility in subsequent generations (K.M. Creasey and R.A. Martienssen, unpublished data). We, therefore, postulate that the function of easiRNAs for the host is to protect the genome from TE-mediated epigenomic instability via PTGS. Consistent with this idea, easiRNAs specifically target the ATHILA ORF1 gene29, thought to be involved in retrovirus-like particle formation, as well as the ATCOPIA integrase gene and the CACTA transposase gene, required for transposition (Extended Data Fig. 3b; Fig. 4c). RDR6-directed PTGS could be dangerous to the host by presenting an opportunity to the TE for evasion and transposition, while RDR2-directed TGS is a safer silencing strategy not requiring transcription. An important developmental stage during which PTGS is deployed is in the pollen grain, when pericentromeric retrotransposons, and some DNA transposons, lose CHH methylation and RdDM in the male germline30. Many of these same TEs accumulate easiRNAs in sperm, which are generated by TE activation in the companion vegetative nucleus, accompanied by the loss of DDM15. Transient loss of RdDM in sperm is restored after fertilization by maternal 24-nt hetsiRNAs30,31 allowing an opportunity to distinguish “self” from “non-self” pollen upon fertilization, according to whether the transposon load is sufficiently foreign not to be recognized by maternal small RNAs. PTGS, mediated by easiRNAs, would provide a backup silencing mechanism during this critical but potentially dangerous window. A similar secondary siRNA transposon control mechanism, triggered by piRNA rather than miRNA, has recently been described in C. elegans and may have similar roles32,33.
Supplementary Methods
Biological Plant Materials, DNA and RNA isolation
Genomic DNA and Total RNA were isolated from inflorescence tissue collected from Columbia-0 wildtype (WT) and loss of function lines in ddm1-2, rdr6-15, ddm1-2 rdr6-15, and ddm1-2 dcl1-11 in a Columbia-0 background, grown under standard long-day growth conditions. The ddm1-2 line used in this study was in the fifth generation of inbreeding and used for the genetic cross rdr6-15. Double heterozygous lines were selected, selfed and second generation ddm1-2 rdr6-15 were isolated for sequencing in this study. DNA was isolated from pooled inflorescence tissue (n = 3) collected phenol/chloroform extraction. Total RNA was isolated from pooled inflorescence tissue (n = 3) and TRIzol (Invitrogen) extraction following manufacturers instructions.
Small RNA Sequencing Library Construction and Analysis
Pooled inflorescence tissue between stage 9 and 11 (unopened/just opened flowers) (n = 3). Col-0, ddm1-2, rdr6-15 and ddm1-2 rdr6-15 TruSeq small RNA sequencing libraries, sequenced on Illumina HiSeq. For the Col-0, ddm1-2 replicates, and ddm1-2 dcl1-11 small RNA sequencing libraries were prepared by NEBNext multiplex small RNA libraries, sequenced on Illumina MiSeq. Mapping sRNA-Seq was performed using Bowtie from the open-source Tuxedo suite, allowing for both unique (U) and multiple (M) mapping reads, normalized by reads per million (RPM). Library read count given in Supplementary Table 1.
Whole-Genome Bisulphite Sequencing Library Construction and Analysis
Library construction and bisulphite conversion were carried out, essentially, as described in34. For each library, 1–5 μg of genomic DNA was sheared using a Covaris S220 Adaptive Focused Acoustics ultra sonicator. Libraries were constructed following standard protocol using the NEB Next DNA Sample Prep Master Mix Set 1 (NEB E6040) and Illumina-compatible paired-end adaptors, which had all cytosines, methylated. 50 ng of each library was treated with sodium bisulphite using EZ DNA Methylation-Gold™ Kit (Zymo Research D5005) according to the protocol provided by the manufacturer. For each purified library 5–10 ng of purified library was amplified using Expand High Fidelity PLUS PCR system (Roche 03300242001), which is capable of efficiently amplifying uracil-containing templates. 50 μl reactions contained 200 μM each dNTP, 1 μM primer, 2.5 mM MgCl2 and 2.5 U Expand HiFi enzyme, and were performed according to the manufacturer’s instructions for 18 cycles. Amplified libraries were ran on 2% MetaPhor® agarose (Lonza 50108) gel. Fragments of 220–350 bp were excised from gel and purified using the QIAquick PCR Purification kit (Qiagen 28104). DNA concentrations were quantified on Bioanalyzer (Agilent), diluted to 10 nM and loaded on flow cells to generate clusters. Libraries were sequenced on Illumina GAII or HiSeq2000 machines using the paired-end 50 cycles protocol. Mapping of BIS-seq libraries performed utilizing Bismark35. Library read count given in Supplementary Table 8. For statistical analysis of TE methylation in ddm1 rdr6 replicates compared to ddm1, we performed Binomial Exact Test, alternative = two-sided, taking the total methylation per transposon superfamily and asking the number of TEs that are more methylated in ddm1 rdr6 compared to ddm1, and the number of TEs less methylated, p-value < 2.2e-16 alternative hypothesis: true probability of success is not equal to 0.5, 95 % confidence interval, sample estimates: probability of success given (Supplementary Table 10).
miRNA Identification
Our miRNA prediction algorithm36 was utilized to identify miRNAs. In total, 128 precursors were identified, representing 96 unique miRNA candidates. Among these 96 sequences, 50 were annotated as Ath miRNA in miRBase v17. Abundances of these sequences are shown Supplementary Table 2. t/r/sn/snoRNA related reads were filtered and the miRNA abundance to be at least 10 (normalized abundance) in one or more libraries and with genome hits no more than 20. The sRNAs passed this filter were mapped to the genome and precursor structure was checked by miREAP, developed by BGI, http://sourceforge.net/projects/mireap/). Each precursor was defined by extending from the abundant tag (as potential miRNA) on either side, up to 200-nt to the potential miRNA-star site. After this, two additional filters (strand bias and top1 + top2 ratio) were applied to distinguish miRNA from siRNA loci. Strand bias was calculated by the sum of sRNA abundance on sense strand divided by the total abundance on both strands. Top1 + top2 is the proportion of the abundance of top2 abundant tags, also referred as distribution filter. The cut-off applied was based on known miRNAs from Ath and Osa. The number of known Ath-miRNAs remaining served as an indicator of the efficiency of the filtering. CentroidFold was used with default settings to visualize the overall miRNA precursor structure for manual evaluation. miRNA target prediction was performed using the Noble sRNA targeting resource http://plantgrn.noble.org/psRNATarget/.
Whole Genome PARE-Sequencing Library Construction and Analysis
mRNA purification using Invitrogen Dynabeads mRNA purification kit. 5′ Adaptor ligation. Reverses Transcription for PARE following first strand cDNA synthesis, (short PCR) 7 cycles, (long PCR) 35 cycles. cDNA purification by AMPure XP. Ligation of 3′ double-stranded DNA adaptor overnight. PCR amplification of PARE library (long PCR) 15 cycles followed by PAGE purification. 5′-PARE RNA adaptor 5′-GUUCAGAGUUCUACAGUCCGAC-3′. Target RT-primer: 5′ CGA GCA CAG AAT TAA TAC GAC TTT TTT TTT TTT TTT TTT. Short PCR primer: 5′-adapter primer 5′-GTTCAGAGTTCTACAGTCCGAC-3′ 3′-adapter primer 5′-CGAGCACAGAATTAATACGACT-3′ dsDNA adapter (PARE TruSeq Duplex) (PAGE purified): dsDNA_2_Top: 5′-TGG AAT TCT CGG GTG CCA AGG dsDNA_2_Bottom: (5′Phos) 5′-CCT TGG CAC CCG AGA ATT CCA NN Final PCR primer (P2 sRNA long primer) 5′: AATGATACGGCGACCACCGACAGGTTCAGAGTTCTACAGTCCGA. Indexed Truseq 3′sRNA primer, Index 1~24.
PARE-Sequencing analysis performed by defining two flanking windows at each predicted target site. The sum of abundance of PARE tags matching to (1) a small window (WS) of 5-nt (cleavage site ± 2-nt), and (2) a long window (WL) of 31-nt (cleavage site ±15 nt) was calculated. Cleavage sites were filtered to retain those for which WS/WL ≥ 0.5 in the PARE-seq libraries; sites failing this criteria were considered background mRNA cleavage levels. Some chloroplast contamination can account for the distribution of raw read count abundances in our libraries. However, when comparing to the normalization basis, and the mapping of PARE-tags to multiple loci, these reads appear to be of chloroplast mRNA in origin.
The miRNA target lists were filtered by combining both miRNA target penalty score (≤ 7) with PARE-seq data filters of WS/WL ≥ 0.3 and WS ≥ 3 (strict), or relaxed; miRNA target penalty score (≤ 7) with PARE-seq data filters of WS/W ≥ 0.3 and WS ≥ 2. Library information given in Supplementary Table 8. For statistical significance we performed the Chi-square Goodness of Fit Test, calculating the p-value for observed value given the expected value. If we assume PARE tags fall randomly on all the positions in the long window (31-nt), the expected frequency for PARE reads fall into the small window (5-nt) would be 5/31, therefore, the expected frequency would be would 26/31 for other regions. AtGRF8, target of miR396, in Col-0, we observe 603 in the small window, and, 614 in the long window (614 − 603 = 11 in other region), the Chi-squared test for given probabilities c(603, 11) X-squared = 3057.857, df = 1, p-value < 2.2e-16 (Supplementary Table 3). For specific miRNA targeting, the sum of abundance of PARE tags matching to (1) a shorter small window (WSs) of 1-nt (cleavage site ± 0-nt), and (2) a shorter long window (WsL) of 3-nt (cleavage site ±1nt) was calculated (Supplementary Table 9).
5′ RLM RACE-PCR
As per instructions 5′ RLM-RACE mapping the cleavage of miRNA FirstChoice (Ambion). 5′ RLM-RACE Adapter ligation followed by RT-PCR, followed by two PCRs (outer and inner), cloning and sequencing. 5′ RLM-RACE adapter: 5′-GCUGAUGGCGAUGAAUGAACACUGCGUUUGCUGGCUUUGAUGAAA-3′. 5′ RLM-RACE outer primer: 5′-GCTGATGGCGATGAATGAACACTG-3′. 5′ RLM- RACE inner primer: 5′-CGCGGATCCGAACACTGCGTTTGCTGGCTTTGATG-3′.
Supplementary Material
sRNA-sequencing for 21-nt and 24-nt sRNAs at transposons in Col-0, ddm1-2, rdr6-15, ddm1-2 rdr6-15 (HiSeq) and Col-0, ddm1-2 and ddm1-2 dcl1-11 (MiSeq). Normalized reads per million (RPM) for sRNA reads, unique (U) mapping to one location of the genome, and multiple (M) mapping to more than one location of the genome.
Exact binomial two-sided test given for transposon methylation in ddm1-2 rdr6-15 replicates as compared to ddm1-2. p-value < 2.2e-16 alternative hypothesis: true probability of success is not equal to 0.5, 95 % confidence interval, sample estimates: probability of success given.
Known and predicted miRNA abundance, sequence, length (nt) and RNA type indicating if new miRNA length at known immature miRNA precursor for Col-0, ddm1-2, rdr6-15 and ddm1-2 rdr6-15 sRNA-seq libraries.
Analysis of PARE libraries by utilizing the predicted miRNA binding site within each transposon-coded open-reading frame (ORF). miRNA sequence and length are shown for each target ORF (ATX) along with the co-ordinate and genomic DNA strand (+/−) of the predicted cleavage site. PARE normalized read abundance ending within small 5-nt (s) and long 31-nt (l) windows at the miRNA binding site are shown in Col-0, ddm1-2, rdr6-15 and ddm1-2 rdr6-15 (Refer to Supplementary Methods for details), followed by the genomic co-ordinates and strand (+/−) of the target ORF. Cleavage products were detected from the same strand or different strand as the target ORF, depending whether cleavage was of the sense or antisense transcript, respectively. For statistical significance of the PARE reads at the miRNA cleavage site, we performed the Chi-square Goodness of Fit Test, calculating the p-value for observed value given the expected value. If we assume PARE tags fall randomly on all the positions in the long window (31-nt), the expected frequency for PARE reads fall into the small window (5-nt) would be 5/31, therefore, the expected frequency would be would 26/31 for other regions. (For example of the statistical test, refer to Supplementary Methods).
DNA methylation by cytosine context (CG, CHG and CHH) calculated per transposon by overall count of cytosine to thymine at each cytosine position (1 = methylated cytosine, 0 = unmethylated cytosine) for each bisulphite sequencing library: Col-0, ddm1-2, rdr6-15 and two replicates of ddm1-2 rdr6-15.
Expression of TEs by fold change normalized to Col-0 WT, in ddm1-2, rdr6-15 and ddm1-2 rdr6-15 by log2 Affymetrix microarray signal intensity.
For known miRNAs targeting TEs passing stringent criteria based on PARE, confirming targeting, both abundance and strand orientation is given at transposons. (Refer to Methods Summary, to accompany Table 1, adapted from Supplementary Table 3). 86/92 TEs with no uniquely mapping easiRNA are cleaved on the antisense strand.
Epigenetically-activated miRNAs with relaxed criteria for PARE cleavage data, confirming targeting, with both abundance and strand orientation given at TEs. (Refer to Methods Summary, to accompany Table 1, adapted from Supplementary Table 3).
Read count for sRNA-seq, BIS-seq and PARE libraries used in this study, mapped to the Col-0 reference genome; TAIR10 (Refer to Methods Summary for computational details).
Analysis of PARE libraries by utilizing the predicted miRNA binding site within each transposon-coded open-reading frame (ORF). miRNA sequence and length are shown for each target ORF along with the co-ordinate and genomic DNA strand (+/−) of the predicted cleavage site. PARE normalized read abundance ending within the shorter small 1-nt (ss) and shorter long 3-nt (sl) windows at the miRNA binding site are shown in Col-0, ddm1-2, rdr6-15 and ddm1-2 rdr6-15 (Refer to Supplementary Methods for details), followed by the genomic coordinates and strand (+/−) of the target ORF. Cleavage products were detected from the same strand or different strand as the target ORF, depending whether cleavage was of the sense or antisense transcript, respectively.
Acknowledgments
We thank Vincent Colot, Oiliver Voinnet and Alexis Sarazin for sharing unpublished data and for discussions, Joseph Simorowski for plant genetics, and Jude Kendall for computational advice. We thank Keith Slotkin for sharing data before publication. K.C. and M. R. were supported, in part, by a research collaboration with DuPont Pioneer. F.V. was supported by a fellowship from the Belgian American Educational Foundation (B.A.E.F). J.Z. was supported by a University of Delaware Graduate fellowship. Research in the Martienssen Laboratory is supported by a grant from NIH (RO1GM067014 to R.M.) and by the Howard Hughes Medical Institute and Gordon and Betty Moore Foundation. The authors acknowledge this work was performed with assistance from the Cold Spring Harbor Laboratory Shared Resources, which are funded, in part, by the Cancer Center Support Grant (5PP30CA045508) and declare no competing interests.
Footnotes
References
- 1.Allen E, Xie Z, Gustafson AM, Carrington JC. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell. 2005;121:207–221. doi: 10.1016/j.cell.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 2.Ronemus M, Vaughn MW, Martienssen RA. MicroRNA-targeted and small interfering RNA-mediated mRNA degradation is regulated by argonaute, dicer, and RNA-dependent RNA polymerase in Arabidopsis. Plant Cell. 2006;18:1559–1574. doi: 10.1105/tpc.106.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cuperus JT, et al. Unique functionality of 22-nt miRNAs in triggering RDR6-dependent siRNA biogenesis from target transcripts in Arabidopsis. Nat Struct Mol Biol. 2010;17:997–1003. doi: 10.1038/nsmb.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11:204–220. doi: 10.1038/nrg2719. rg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Slotkin RK, et al. Epigenetic Reprogramming and Small RNA Silencing of Transposable Elements in Pollen. Cell. 2009;136:461–472. doi: 10.1016/j.cell.2008.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanurdzic M, et al. Epigenomic Consequences of Immortalized Plant Cell Suspension Culture. PLoS Biology. 2008;6:e302. doi: 10.1371/journal.pbio.0060302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCue AD, Nuthikattu S, Reeder SH, Slotkin RK. Gene Expression and Stress Response Mediated by the Epigenetic Regulation of a Transposable Element Small RNA. PLoS Genetics. 2012;8:e1002474. doi: 10.1371/journal.pgen.1002474.t001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nuthikattu S, et al. The Initiation of Epigenetic Silencing of Active Transposable Elements Is Triggered by RDR6 and 21–22 Nucleotide Small Interfering RNAs. PLANT PHYSIOLOGY. 2013;162:116–131. doi: 10.1104/pp.113.216481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mirouze M, et al. Selective epigenetic control of retrotransposition in Arabidopsis. Nature. 2009;461:427–U130. doi: 10.1038/nature08328. [DOI] [PubMed] [Google Scholar]
- 10.Miura A, et al. Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature. 2001;411:212–214. doi: 10.1038/35075612. [DOI] [PubMed] [Google Scholar]
- 11.German MA, Luo S, Schroth G, Meyers BC, Green PJ. Construction of Parallel Analysis of RNA Ends (PARE) libraries for the study of cleaved miRNA targets and the RNA degradome. Nat Protoc. 2009;4:356–362. doi: 10.1038/nprot.2009.8. papers2://publication/doi/ [DOI] [PubMed] [Google Scholar]
- 12.Hsieh TF, et al. Genome-wide demethylation of Arabidopsis endosperm. Science. 2009;324:1451–1454. doi: 10.1126/science.1172417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gehring M, et al. DEMETER DNA glycosylase establishes MEDEA polycomb gene self-imprinting by allele-specific demethylation. Cell. 2006;124:495–506. doi: 10.1016/j.cell.2005.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwab R, Speth C, Laubinger S, Voinnet O. Enhanced microRNA accumulation through stemloop-adjacent introns. EMBO reports. 2013;14:615–621. doi: 10.1038/embor.2013.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jeong DH, et al. Massive Analysis of Rice Small RNAs: Mechanistic Implications of Regulated MicroRNAs and Variants for Differential Target RNA Cleavage. THE PLANT CELL ONLINE. 2012;23:4185–4207. doi: 10.1105/tpc.111.089045. papers2://publication/doi/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Axtell MJ, Bartel DP. Antiquity of microRNAs and their targets in land plants. The Plant cell. 2005;17:1658–1673. doi: 10.1105/tpc.105.032185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen HM, et al. 22-Nucleotide RNAs trigger secondary siRNA biogenesis in plants. Proc Natl Acad Sci U S A. 2010;107:15269–15274. doi: 10.1073/pnas.1001738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mari-Ordonez A, et al. Reconstructing de novo silencing of an active plant retrotransposon. Nat Genet. 2013;45:1029–1039. doi: 10.1038/ng.2703. [DOI] [PubMed] [Google Scholar]
- 19.Wu L, et al. DNA Methylation Mediated by a MicroRNA Pathway. Molecular cell. 2010;38:465–475. doi: 10.1016/j.molcel.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 20.Jauvion V, Rivard M, Bouteiller N, Elmayan T, Vaucheret H. RDR2 partially antagonizes the production of RDR6-dependent siRNA in sense transgene-mediated PTGS. PLoS ONE. 2012;7:e29785. doi: 10.1371/journal.pone.0029785. papers2://publication/uuid/722C12F4-2345-4441-A712-569DBA2D0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Slotkin RK, Freeling M, Lisch D. Heritable transposon silencing initiated by a naturally occurring transposon inverted duplication. Nature Genetics. 2005;37:641–644. doi: 10.1038/ng1576. papers2://publication/doi. [DOI] [PubMed] [Google Scholar]
- 22.Stroud H, Greenberg MVC, Feng S, Bernatavichute YV, Jacobsen SE. Comprehensive Analysis of Silencing Mutants Reveals Complex Regulation of the Arabidopsis Methylome. Cell. 2013;152:352–364. doi: 10.1016/j.cell.2012.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeddeloh JA, Stokes TL, Richards EJ. Maintenance of genomic methylation requires a SWI2/SNF2-like protein. Nature Genetics. 1999;22:94–97. doi: 10.1038/8803. [DOI] [PubMed] [Google Scholar]
- 24.Teixeira FK, et al. A role for RNAi in the selective correction of DNA methylation defects. Science. 2009;323:1600–1604. doi: 10.1126/science.1165313. [DOI] [PubMed] [Google Scholar]
- 25.Poethig RS. Small RNAs and developmental timing in plants. Current opinion in genetics & development. 2009;19:374–378. doi: 10.1016/j.gde.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martienssen R, Barkan A, Taylor WC, Freeling M. Somatically heritable switches in the DNA modification of Mu transposable elements monitored with a suppressible mutant in maize. Genes Dev. 1990;4:331–343. doi: 10.1101/gad.4.3.331. [DOI] [PubMed] [Google Scholar]
- 27.Martienssen R, Baron A. Coordinate suppression of mutations caused by Robertson’s mutator transposons in maize. Genetics. 1994;136:1157–1170. doi: 10.1093/genetics/136.3.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poethig RS. Phase change and the regulation of shoot morphogenesis in plants. Science. 1990;250:923–930. doi: 10.1126/science.250.4983.923. [DOI] [PubMed] [Google Scholar]
- 29.Wright DA, Voytas DF. Athila4 of Arabidopsis and Calypso of soybean define a lineage of endogenous plant retroviruses. Genome research. 2002;12:122–131. doi: 10.1101/gr.196001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Calarco JP, et al. Reprogramming of DNA methylation in pollen guides epigenetic inheritance via small RNA. Cell. 2012;151:194–205. doi: 10.1016/j.cell.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mosher RA, et al. Uniparental expression of PolIV-dependent siRNAs in developing endosperm of Arabidopsis. Nature. 2009;460:283–286. doi: 10.1038/nature08084. [DOI] [PubMed] [Google Scholar]
- 32.Bagijn MP, et al. Function, targets, and evolution of Caenorhabditis elegans piRNAs. Science (New York, NY) 2012;337:574–578. doi: 10.1126/science.1220952. papers2://publication/doi/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shirayama M, et al. piRNAs initiate an epigenetic memory of nonself RNA in the C. elegans germline. Cell. 2012;150:65–77. doi: 10.1016/j.cell.2012.06.015. papers2://publication/doi/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Regulski M, et al. The maize methylome influences mRNA splice sites and reveals widespread paramutation-like switches guided by small RNA. Genome Res. 2013;23:1651–1662. doi: 10.1101/gr.153510.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhai J, et al. MicroRNAs as master regulators of the plant NB-LRR defense gene family via the production of phased, trans-acting siRNAs. Genes & Development. 2011;25:2540–2553. doi: 10.1101/gad.177527.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
sRNA-sequencing for 21-nt and 24-nt sRNAs at transposons in Col-0, ddm1-2, rdr6-15, ddm1-2 rdr6-15 (HiSeq) and Col-0, ddm1-2 and ddm1-2 dcl1-11 (MiSeq). Normalized reads per million (RPM) for sRNA reads, unique (U) mapping to one location of the genome, and multiple (M) mapping to more than one location of the genome.
Exact binomial two-sided test given for transposon methylation in ddm1-2 rdr6-15 replicates as compared to ddm1-2. p-value < 2.2e-16 alternative hypothesis: true probability of success is not equal to 0.5, 95 % confidence interval, sample estimates: probability of success given.
Known and predicted miRNA abundance, sequence, length (nt) and RNA type indicating if new miRNA length at known immature miRNA precursor for Col-0, ddm1-2, rdr6-15 and ddm1-2 rdr6-15 sRNA-seq libraries.
Analysis of PARE libraries by utilizing the predicted miRNA binding site within each transposon-coded open-reading frame (ORF). miRNA sequence and length are shown for each target ORF (ATX) along with the co-ordinate and genomic DNA strand (+/−) of the predicted cleavage site. PARE normalized read abundance ending within small 5-nt (s) and long 31-nt (l) windows at the miRNA binding site are shown in Col-0, ddm1-2, rdr6-15 and ddm1-2 rdr6-15 (Refer to Supplementary Methods for details), followed by the genomic co-ordinates and strand (+/−) of the target ORF. Cleavage products were detected from the same strand or different strand as the target ORF, depending whether cleavage was of the sense or antisense transcript, respectively. For statistical significance of the PARE reads at the miRNA cleavage site, we performed the Chi-square Goodness of Fit Test, calculating the p-value for observed value given the expected value. If we assume PARE tags fall randomly on all the positions in the long window (31-nt), the expected frequency for PARE reads fall into the small window (5-nt) would be 5/31, therefore, the expected frequency would be would 26/31 for other regions. (For example of the statistical test, refer to Supplementary Methods).
DNA methylation by cytosine context (CG, CHG and CHH) calculated per transposon by overall count of cytosine to thymine at each cytosine position (1 = methylated cytosine, 0 = unmethylated cytosine) for each bisulphite sequencing library: Col-0, ddm1-2, rdr6-15 and two replicates of ddm1-2 rdr6-15.
Expression of TEs by fold change normalized to Col-0 WT, in ddm1-2, rdr6-15 and ddm1-2 rdr6-15 by log2 Affymetrix microarray signal intensity.
For known miRNAs targeting TEs passing stringent criteria based on PARE, confirming targeting, both abundance and strand orientation is given at transposons. (Refer to Methods Summary, to accompany Table 1, adapted from Supplementary Table 3). 86/92 TEs with no uniquely mapping easiRNA are cleaved on the antisense strand.
Epigenetically-activated miRNAs with relaxed criteria for PARE cleavage data, confirming targeting, with both abundance and strand orientation given at TEs. (Refer to Methods Summary, to accompany Table 1, adapted from Supplementary Table 3).
Read count for sRNA-seq, BIS-seq and PARE libraries used in this study, mapped to the Col-0 reference genome; TAIR10 (Refer to Methods Summary for computational details).
Analysis of PARE libraries by utilizing the predicted miRNA binding site within each transposon-coded open-reading frame (ORF). miRNA sequence and length are shown for each target ORF along with the co-ordinate and genomic DNA strand (+/−) of the predicted cleavage site. PARE normalized read abundance ending within the shorter small 1-nt (ss) and shorter long 3-nt (sl) windows at the miRNA binding site are shown in Col-0, ddm1-2, rdr6-15 and ddm1-2 rdr6-15 (Refer to Supplementary Methods for details), followed by the genomic coordinates and strand (+/−) of the target ORF. Cleavage products were detected from the same strand or different strand as the target ORF, depending whether cleavage was of the sense or antisense transcript, respectively.