

Abstract
OBJECTIVE:
We tested the hypothesis that acquired small-fiber polyneuropathy (SFPN), previously uncharacterized in children, contributes to unexplained pediatric widespread pain syndromes.
METHODS:
Forty-one consecutive patients evaluated for unexplained widespread pain beginning before age 21 had medical records comprehensively analyzed regarding objective diagnostic testing for SFPN (neurodiagnostic skin biopsy, nerve biopsy, and autonomic function testing), plus histories, symptoms, signs, other tests, and treatments. Healthy, demographically matched volunteers provided normal controls for SFPN tests.
RESULTS:
Age at illness onset averaged 12.3 ± 5.7 years; 73% among this poly-ethnic sample were female (P = .001). Sixty-eight percent were chronically disabled, and 68% had hospitalizations. Objective testing diagnosed definite SFPN in 59%, probable SFPN in 17%, and possible SFPN in 22%. Only 1 of 41 had entirely normal SFPN test results. Ninety-eight percent of patients had other somatic complaints consistent with SFPN dysautonomia (90% cardiovascular, 82% gastrointestinal, and 34% urologic), 83% reported chronic fatigue, and 63% had chronic headache. Neurologic examinations identified reduced sensation in 68% and vasomotor abnormalities in 55%, including 23% with erythromelalgia. Exhaustive investigations for SFPN causality identified only history of autoimmune illnesses in 33% and serologic markers of disordered immunity in 89%. Treatment with corticosteroids and/or intravenous immune globulin objectively and subjectively benefited 80% of patients (12/15).
CONCLUSIONS:
More than half among a large series of patients with childhood-onset, unexplained chronic widespread pain met rigorous, multitest, diagnostic criteria for SFPN, which extends the age range of acquired SFPN into early childhood. Some cases appeared immune-mediated and improved with immunomodulatory therapies.
Keywords: peripheral nervous system disease, widespread chronic pain, dysautonomia
What’s Known on This Subject:
Acquired widespread pain syndromes of youth are prevalent, disabling, usually unexplained, and untreatable. Small-fiber polyneuropathy causes widespread pain and multisystem complaints in older adults. Some causes are treatable. Neurodiagnostic skin biopsy, autonomic function testing, and nerve biopsy permit objective diagnosis.
What This Study Adds:
It identifies definite (in 59%) and probable (in 17%) small-fiber polyneuropathy among 41 young patients with otherwise-unexplained, childhood-onset widespread pain. It characterizes this new disease’s clinical features, diagnostic, and treatment options. Some cases appeared immune mediated and responded to immunomodulatory therapies.
Syndromes involving unexplained chronic widespread pain (CWP) are prevalent and problematic in children and adults. The American College of Rheumatology defines CWP as pain affecting the axial, plus upper and lower body, plus the left- and right-side lasting ≥3 months.1 Uncertainty about the etiology and pathogenesis of CWP precludes effective treatment with disease-modifying therapies, and chronic administration of pain-palliating medications has medical and social adverse effects that are even less acceptable in children than in adults. These increasingly recognized juvenile CWP syndromes often disrupt entire families and interfere with children’s education and development.2–4 CWP syndromes often include other unexplained complaints including dizziness, fatigue, headache, and nausea. More than half of juvenile patients with dizziness attributed to postural orthostasis tachycardia syndrome (POTS) also had chronic headache, abdominal pain, and/or fatigue,5 suggesting a possible common etiology.
Small-fiber predominant polyneuropathy (SFPN) is a plausible biological substrate for CWP. SFPN refers to widespread damage predominantly affecting the small-diameter unmyelinated (C-fibers) or thinly myelinated (A-δ) peripheral axons that protect organisms by signaling pain upon harmful contact. SFPN usually begins in the longest axons, causing distal-onset or distal-predominant pain (in the feet). Occasional patients develop patchy or predominantly proximal symptoms attributed to proximal attack on the neuronal cell bodies (ganglionopathy/neuronopathy; Fig 2A).6 Because small fibers also densely innervate and regulate the tone of microvessels, abnormal appearance (eg, edema, color changes) is common in affected areas.7 Small-fibers were formerly dichotomized as somatic versus autonomic, a distinction blurred by recent discoveries that somatic/nociceptive axons have efferent and trophic effects formerly considered autonomic,8 and that axons innervating sweat glands and blood vessels express the TRPV1/capsaicin pain receptor.9 Diagnosing SFPN is difficult because familiar signs of large-fiber neuropathy are absent or minimal, and electrodiagnostic testing is insensitive. The tests recommended for objective SFPN diagnosis were applied here: specifically distal-leg skin biopsy immunolabeled to reveal nociceptive epidermal nerve-fibers (ENF)10 (level C recommendation by the American Academy of Neurology, level A recommendation by the European Federation of Neurologic Societies),11,12 and autonomic function testing (AFT). This consists of 4 validated tests of cardiovagal, adrenergic, and sudomotor small-fiber function (level B recommendation by the American Academy of Neurology).12,13
FIGURE 2.

A, Patient with delayed sweating on torso and arms during thermoregulatory sweat testing. This 20 year old had non–length-dependent erythromelalgia (pain worsened by heat, plus redness and swelling) that was worse in his cheeks and ears but also affected his hands and feet, labile heart rate and blood pressure, and diarrhea. His distal leg skin biopsy had minor abnormalities, and antinuclear antibodies were present at a titer of 1:80. Thermoregulatory sweat testing involves applying Alizarin red indicator to the skin before controlled heating; it is orange when dry and turns purple when wet. When performed at the Mayo Clinic this revealed delayed sweating on the torso and arms compared with the hands and thighs. This patient’s AFT results were normal at the Mayo Clinic but abnormal at Cleveland Clinic and at the National Institutes of Health, with reduced sweating at forearm and distal leg study sites. His patchy/proximal symptoms and test results were consistent with non–length-dependent neuronopathy/ganglionopathy. Image courtesy of Paola Sandroni, MD, PhD, Mayo Clinic. B, Gender- and site-specific comparison of acetylcholine-evoked sweat production in normal controls and patients. All values from patients (n = 33, 10 boys and 23 girls) were compared with all values from gender- and age range–matched controls (n = 38; 19 boys and 19 girls). Symbols depict mean sweat volumes at each study site ± SEM; lines connect same-site groups of controls and patients. Sweating was reduced in male patients versus male controls at the forearm (P = .0011) and proximal leg (P = .0017). Sweating was reduced in female patients versus female controls at the forearm (P = .024), proximal leg (P = .024), and foot (P = .009).
Polyneuropathy has been believed to be rare in children, consisting of occasional cases of acute Guillain-Barré syndrome (GBS) and chronic inflammatory demyelinating polyneuropathy (CIDP) caused by immune attack on large myelinated motor axons.14 SFPN, in which small-diameter C- and A-δ fibers are preferentially damaged, was little known in children except for vanishingly rare genetic cases.14,15 Isolated cases of erythromelalgia (aka erythermalgia), a historic phenotype comprising burning pain, redness, edema, and relief from cooling,16 have been described in children17 and some cases with prepubertal onset have been linked to sodium channel mutations.18,19 Skin biopsy was the key to diagnosing SFPN in a Japanese 12 year old with new widespread pain and gastroparesis.20 We and others have reported cases of teenagers with acute erythromelalgia and dysautonomia where skin biopsy confirmed severe SFPN, and corticosteroid treatment was curative.21,22 Similar steroid-responsive cases were then described in younger children.23,24 Children represent 4 of 21 cases of a recently characterized, acute small- plus large-fiber polyneuropathy,25 and recent studies link pediatric erythromelalgia in children to SFPN, just as in adults.26,27 Several new preliminary reports provide objective evidence that SFPN is prevalent in adult fibromyalgia, including our study identifying SFPN in 50% of fibromyalgia patients versus 0% of matched controls.28–31
Methods
Selection of Cases and Records
After Institutional Review Board approval, outpatient records were screened to select subjects. Inclusion criteria were medical care by author A.L.O. between April 2007 and April 2011 for widespread multifocal pain (present in >1 limb or body region) beginning before age 21. Exclusion criteria were an identified objective cause of the pain. We obtained and read all available records extracting all provider notes, laboratory, physiology, pathology, and radiology reports. All technically adequate test results from academic or commercial laboratories were included and interpreted as reported or by using age-normed standard reference ranges.32
Primary Outcome: Objective Diagnosis of SFPN
There are no consensus diagnostic criteria for SFPN in adults or children,33 so we integrated the results of all recommended objective diagnostic tests; PGP9.5-immunolabeled, distal-leg skin biopsy, AFT, and sensory nerve biopsy.11,12 Confirmed SFPN required ≥1 definite objective-test SFPN diagnosis. Probable SFPN required minor abnormalities on ≥2 different tests, and possible SFPN required ≥1 minor objective abnormality. Electrodiagnostic testing (electromyography and nerve conduction testing) was excluded because these tests do not capture small-fiber function, and quantitative sensory testing was excluded because it is a subjective test based on subject report.34 A definitively abnormal skin biopsy required diagnosis of SFPN in the report or meeting standard diagnostic criteria (density of epidermal nerve fibers [ENFs] ≤ fifth centile of laboratory norms). Minor skin biopsy abnormalities comprised borderline ENF densities (5.1–15th centile or report of excess axon swellings).35 A definitively abnormal AFT required SFPN diagnosis in the report or meeting standard diagnostic criteria (definite abnormalities in >1 domain). Minor AFT abnormalities required report of borderline, mild, minimal, or isolated abnormalities. A definitively abnormal nerve biopsy required SFPN diagnosis based on ultrastructural visualization of loss of unmyelinated axons.
Skin biopsy testing for SFPN has been standardized.12,36 One 2- to 3-mm-diameter skin biopsy punch is removed from an anesthetized site on the distal leg and then vertically sectioned and immunolabeled against PGP9.5, a pan-neuronal marker, to visualize ENF and permit measuring axon density (Fig 1).12,37 We report ENF densities per square millimeter of skin surface area to control for different laboratories’ varying skin-section thickness. Most skin biopsies were interpreted at the Massachusetts General Hospital, whose norms come from biopsying 240 screened normal volunteers 14 to 86 years old.38 Biopsies from patients <14 years old (n = 6) were normed to age 14 because of lack of norms for younger children. Performance of skin biopsies caused no adverse events.
FIGURE 1.

Loss of PGP 9.5 immunolabeled nerve fibers in vertical sections from distal leg skin biopsy.
A, Biopsy from a 19-year-old white male healthy control contains abundant innervation (675 ENF/mm2 skin surface area). B, Biopsy from a 19-year-old white male patient demonstrates reduced epidermal (155 ENF/mm2) and dermal nerve fibers. SFPN was confirmed by skin biopsy; ENF density = 1.3th percentile of laboratory norms. Bars represent 50 μm.
AFT, considered more sensitive than skin biopsy,39 has previously detected autonomic SFPN in half of adults with POTS.40 Because AFT norms come from adults,41 we obtained institutional review board permission to recruit age- and gender-matched normal individuals age ≥6 years to provide controls for pediatric study. Respondents with potentially neuropathic conditions were excluded, and those >18 years old underwent 2-hour oral glucose tolerance testing and were excluded for any abnormality. We used standard diagnostic methods, equipment (WR Medical Electronics, Stillwater, MN), and interpretations.12,41 We measured heart rate variability during deep breathing (6 breaths per minute while supine) and the Valsalva maneuver, hemodynamic responses during 80° head-up tilt, and acetylcholine-evoked sweating. Manufacturer-supplied reference ranges (RRs) defined normality of Valsalva responses. For heart rate variability, values <2.5th centile defined abnormal. For tilt testing, abnormal was a systolic blood pressure drop >20 mm Hg, diastolic blood pressure drop >10 mm Hg,42 and/or heart rate increase ≥40 (≤18 years) or ≥30 beats per minute (>18 years) within 3 minutes of tilt.43 For sweat testing, abnormality was defined as a value outside the 95% confidence interval of site-specific, gender- and age-adjusted norms.41 Performance of AFT caused no adverse events.
Statistical Analysis
Descriptive statistics, presented as means ± SDs, were used to compare attributes between groups. The χ2 statistic assessed between-group differences in proportions of abnormal results. Four-site sweat production was compared between patients and controls by using 2-tailed t tests with multiple-sample correction for type I error assuming α = 0.05 and r = 0.5; thus, P < .025 defined significance.
Results
Forty-one consecutive patients were eligible and were included. Mean age at symptom onset was 12.3 ± 5.7 years; age at presentation averaged 20.8 ± 9.1 years. Demographic characteristics were notable for female predominance (73%; P = .001) and ethnic and geographic diversity. Three children lived in Ecuador, Switzerland, and Trinidad, and one had developed symptoms before adoption from Estonia at age 6. Among American-born patients, 1 had 2 Korean parents, and 1 was half Lebanese and half white-American.
Outcome of Diagnostic Testing for SFPN
Overall, 59% (24/41) of patients met the criteria for definite SFPN, although none had undergone all of the 3 tests analyzed. Specifically, 30% (11/37) of skin biopsies, 100% (2/2) of nerve biopsies, and 53% (18/34) of AFT were definitively diagnostic for SFPN. Minor abnormalities reported in 81% of the remaining skin biopsies (22/26) and 93% of the remaining AFT (14/15) identified an additional 17% of patients with probable SFPN and 22% with possible SFPN. Only 1 of 41 patients had entirely normal results. Two of 2 nerve and muscle biopsies diagnosed SFPN based on Schwann cell stacks that were empty or contained isolated regenerating axons. The biopsied nerves lacked demyelination, loss of large fibers, inflammatory infiltrates, vasculitis, or amyloid. One muscle was normal, and the other had neuropathic and disuse changes.
Comparing AFT results between patients and normal controls revealed 27% of patients (vs 3% of controls; P < .001) with reduced heart rate variability during deep breathing, 42% of patients with abnormal cardiovascular responses to Valsalva (vs. 0% of controls; P < .001), and 75% with abnormal tilt table results (vs. 18% of controls; P < .001). Sweat production (Fig 2B), considered the most sensitive among the autonomic-function tests for diagnosing SFPN,44 was reduced at one or more among the 4 sites tested in 82% of patients (vs 34% of controls; P < .001).
Characterization of Medical Histories, Examinations, and Other Testing
Most patients were moderately or severely ill, explaining their extensive medical evaluations at leading academic medical centers. Sixty-eight percent had been hospitalized, and 68% had required leaves from school or work. No other pathogenic diagnoses explaining their widespread pain (eg, arthritis, myopathy) had been identified. The most common syndromic label at study entry was fibromyalgia; others included functional disorder, central sensitization, pain-amplification syndrome, chronic fatigue, myofascial pain syndrome, and seronegative chronic Lyme disease. Organ-specific syndromic labels included POTS, irritable bowel, functional dyspepsia, abdominal migraine, and chronic daily headache. Four patients also had polycystic ovarian syndrome, and 3 had Ehlers-Danlos syndrome. Psychiatric diagnoses (somatization, conversion) were often considered, although only 1 psychiatric illness had been documented, specifically major depression attributed to unremitting pain.
General examinations consistently identified only sinus tachycardia and abnormal blood pressures, usually orthostatic hypotension. Several patients documented episodic neurogenic blisters. These were often on the limbs, also intraorally and on the face. Occasional patients had sluggish pupillary reflexes.
Most tests other than for neuropathy were noncontributory. Cardiac monitoring revealed sinus tachycardia (5/5) with occasional intermittent bradycardia (consistent with neuropathic dysautonomia), and echocardiography was noncontributory. Electromyography (n = 20) was noncontributory, with only upper-limb denervation identified in 1 patient studied during brachial plexitis. Nerve conduction studies (n = 24) were noncontributory, with polyneuropathy identified in only 2 patients: one with long-standing type 1 diabetes and the other with renal failure from Goodpasture’s syndrome. Five patients had borderline or isolated sensory abnormalities that might represent subclinical large-fiber involvement. Gastrointestinal endoscopy was universally futile, but abdominal imaging and motility studies (ie, gastric emptying and Sitz marker studies) sometimes showed slowed motility consistent with neurogenic dysautonomia. Brain imaging by magnetic resonance (n = 23) and computerized axial tomography (n = 8) was nondiagnostic except in 1 patient with fatal hypertensive brain edema and hemorrhage associated with Goodpasture’s syndrome. Spinal imaging (n = 22) was noncontributory except for occasionally identifying obstipation. Sleep studies and electroencephalography were futile.
Evaluation for Causes of SFPN
Extensive evaluation for known causes of SFPN identified only 3 potentially causal or contributory systemic diagnoses: 2 patients had Sjögren’s spectrum disorders and 1 had type 1 diabetes.37 The diabetes was proven not to be the cause of this patient’s severe SFPN when she had immediate dramatic improvement after corticosteroid administration followed by sustained remission after intravenous immune globulin (IVIG) treatments. Patient histories were overall significant only for prevalence of autoimmune illnesses in 33%. These were usually organ specific and often autoantibody associated, including 6 patients with histories of autoimmune thyroiditis and 2 with Henoch-Schönlein purpura. One each had had episodes of brachial plexitis, type 1 diabetes, postviral arthritis, immune thrombocytopenic purpura, Crohn’s disease, autoimmune trochleitis, and Hashimoto’s encephalopathy. These, along with family histories of autoimmunity in 52%, suggested the possibility of dysimmune causes of juvenile-onset SFPN. There were no family histories of SFPN, although 1 patient had multiple family members labeled with fibromyalgia who subsequently received research diagnoses of SFPN from our laboratory, consistent with unrecognized familial SFPN.31
Sixty-one percent of patients/families attributed onset of their illness to preceding infections or injuries. Twenty-five percent had documented preceding infections including Mycoplasma pneumoniae, Bordatella pertussis, Mycobacterium tuberculosis, group A Streptococcus, mononucleosis, influenza, and parvovirus. Among the 11 documented injuries shortly preceding onset of widespread pain, 10 were limb injuries (eg, fractures, sprains, frostbite) featuring excess pain and swelling interpreted as complex regional pain syndrome. These had largely resolved and were distinct from the later widespread and multifocal pain illnesses studied here.
Among patients’ extensive body fluid testing (Tables 1 and 2), the only consistent abnormalities were immune related: elevated erythrocyte sedimentation rates (ESRs), antinuclear antibodies, and low levels of complement components C3 and C4. Overall, 89% of patients had ≥1 of these abnormalities. A 26% prevalence of high C-reactive protein was considered too nonspecific to analyze further. Type III cryoglobulinemia (IgG, IgM, and C3) without hepatitis was identified in 1 patient (5%) along with profound hypocomplementemia and high ESRs (Fig 3). These abnormalities resolved after treatment with prednisone. Scattered patients had been tested for autoantibodies associated with somatic/painful small-fiber polyneuropathies (peripherin, voltage-gated potassium channel complex, and N-type calcium channels)45–47: 1 of 15 tested had peripherin autoantibodies, 1 of 7 had voltage-gated potassium channel complex autoantibodies, and 1 of 11 had N-type calcium channel autoantibodies. Autoantibodies associated with large-fiber polyneuropathies and pure autonomic polyneuropathies (ie, ganglionic acetylcholine receptor autoantibody) were absent. Two patients’ skin biopsies underwent external dermatopathologic analysis including direct immunofluorescence (Fig 3). Both contained significant deposits of C4d, C5b-9, and IgM without inflammatory infiltrates; one also had IgA deposition.
TABLE 1.
Summary of Consistently Abnormal Medical Findings
| Percent (number) | |
|---|---|
| Most common symptoms | |
| Pain began or was predominant in legs | 76% (31/41) |
| Chronic disabling headaches | 63% (25/40) |
| Chronic disabling fatigue | 83% (24/29) |
| Any autonomic symptom | 98% (40/41) |
| Cardiovascular complaints | 90% (37/41) |
| Gastrointestinal complaints | 82% (32/39) |
| Sweating complaints | 63% (22/35) |
| Urological complaints | 34% (11/32) |
| All consistent abnormalities on neurologic examination | |
| Erythromelalgia (erythermalgia) | 23% (9/40) |
| Mild weakness of fingers or toes | 10% (4/41) |
| Dyscoordination of fingers or toes | 10% (4/41) |
| Sensory abnormalities | 68% (26/38) |
| Reduced distal cool sensation | 68% (25/37) |
| Reduced distal pinprick sensation | 61% (23/38) |
| Abnormal touch sensation (including allodynia) | 34% (14/41) |
| Reduced distal vibration sensation | 21% (8/38) |
| Reduced distal position sensation | 8% (3/38) |
| Reduced distal reflexes | 18% (7/40) |
| All consistently abnormal blood test results | |
| Elevated ESR (≥15 mm/h) | 37% (15/41) |
| Elevated antinuclear antibodies (≥1:80 dilution) | 45% (17/38) |
| Reduced C3 (<85 mg/dL) | 21% (6/29) |
| Reduced C4 (<20 mg/dL) | 46% (13/28) |
| Any 1 or more of the above blood test abnormalities | 89% (32/36) |
TABLE 2.
Summary of Consistently Normal/Negative/Noncontributory Test Results
| Cerebrospinal fluid tests: | All tests were normal in 11 patients |
| Blood tests: | Complete blood count, electrolytes including glucose, renal, liver, and thyroid function, hemoglobin A1c levels, lipids, vitamins, and immunoglobulins, serum protein immunofixation |
| Urine tests: | Heavy metals, protein immunofixation, porphyrins, amino and organic acids |
| Infectious tests: | Hepatitis C, syphilis, human immunodeficiency virus, deer-associated zoonotic infections including Lyme, babesiosis, and human monocytic ehrlichiosis |
| Immune tests: | Rheumatoid factor antibody, Sjögren’s autoantibodies (Ro/SS-A, La/SS-B), lupus autoantibodies (anti-dsDNA, Sm, RNP), antineutrophil cytoplasmic antibodies, total complement |
| Genetic tests: | Only occasionally performed: all tests for genetic neuropathy including Charcot-Marie-Tooth, Fabry, transthyretin, hereditary neuropathy with liability to pressure palsy, also familial hemiplegic migraine, cystic fibrosis |
FIGURE 3.
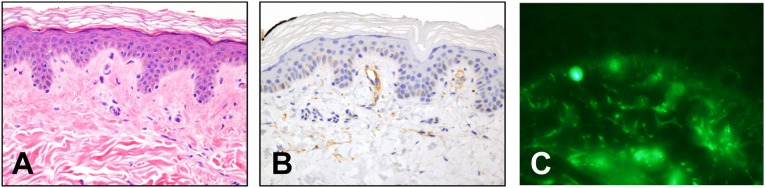
This 19 year old had widespread pain, tachycardia, orthostatic hypotension, headache, nausea, and obstipation since childhood. Blood tests identified type III cryoglobulinemia (IgG, IgM, C3), no hepatitis, low complement (C4 = 6, 7, 11; RR, 16–38 mg/dL; C3 = 35, 52; RR, 86–184 mg/dL; ESR = 20–26; RR, 0–17 mm/hour). SFPN was confirmed by skin biopsy; ENF density = 2.4th centile (not shown). Photomicrographs courtesy of Cynthia Magro, MD, Weill Cornell Medical College (vertical sections, 4 µm; 40× magnification). A, Hematoxylin and eosin staining depicting endothelial cell swelling and basement membrane thickening indicating chronic insidious microvasculopathy. B, Immunohistochemical reactivity of dermal microvessels for C4d, a stable component of classic complement activation and a likely correlate of her low serum C4. C, Direct immunofluorescent labeling of IgM deposits on dermal microvessels, a nonspecific marker of microvasculopathy consistent with deposition of the IgM immune complexes present in her blood.
Summary of Treatments for Symptoms of Neuropathic Dysautonomia and Pain
Diagnosing SFPN guided treatment of the patients’ symptoms. Hypotension was managed by advising more salt and fluid intake, avoiding rapid or prolonged standing, contracting leg muscles while standing, elevating the head of the bed, and avoiding potentiating medications. Refractory hypotension was treated with compressive stockings and abdominal binders, midodrine, or secondarily, fludrocortisone.48 Chronic tachycardia was generally well tolerated but if symptomatic was treated with calcium-channel blockers or β-blockers, taking care not to worsen hypotension. Gastrointestinal complaints were addressed with high-fiber diets, small meals, elevating the head of the bed, and avoiding postprandial recumbency. Constipation and nausea/vomiting usually required treatment with medications, and 3 patients required hospital admission for fecal disimpaction. For disabling CWP, gabapentin was most often prescribed, and secondary amine tricyclics (nortriptyline and desipramine) were considered after dysautonomia was excluded or controlled.49 Opioids were prescribed rarely for severe uncontrolled pain. The erythromelalgia phenotype prompted consideration of mexiletine.
Summary of Treatments With Immune-Modulating Therapies
Immunomodulation was considered for patients with disabling symptoms that were refractory to conservative management. Overall, 80% (12/15) among patients treated with corticosteroids and/or immune globulin improved. Lacking guidelines, doses were extrapolated from childhood GBS or CIDP and case reports of autoimmune SFPN.21,22,50,51 Short-term daily corticosteroids were considered first on the basis of low cost, wide availability, oral dosing, and low risk of several complications in young patients. Corticosteroids (effective for CIDP but not GBS52) were associated with documented sustained improvement in 67% (10/15) of patients and were associated with 1 major adverse event; the diabetic patient developed cataracts. Subsequent tapering of her prednisone caused a relapse of SFPN, so she was transitioned to IVIG with gradual sustained recovery as documented below. Among 5 severely ill hospitalized patients treated with intravenous methylprednisolone (1 g/day for 5 days) followed by a prednisone taper, the 2 acute cases (<3 months) including the index case22 had rapid, continuing, and objectively documented improvement (see below), whereas the 3 patients who had been ill for several years did not respond. Among clinic patients treated with oral prednisone (1 mg/kg/day for 4 weeks followed by rapid taper), 8 improved and 2 did not.
IVIG53 was tried in 11 patients who were unresponsive to corticosteroids or were steroid responsive but required long-duration treatment. Three had insufficient treatment for analysis (≤3 doses of 2 g/kg). Among the 8 treated with the standard regimen for autoimmune polyneuropathy (≥3 times with 2 g/kg/month), 3 (38%) did not respond and discontinued treatment, and 5 (62%) had documented significant improvement and continued treatment. Typical infusion-related symptoms responded to standard treatments. One significant adverse event (rash + deep vein thrombosis) resolved. One patient relapsed after IVIG taper and required a few additional treatments.
Objective testing was repeated to monitor treatment efficacy. Six of 6 repeat AFTs among patients treated with immunomodulatory therapies documented improvement, whereas 2 of 2 repeat AFTs in nonimmunomodulated patients did not improve. Two immunomodulated patients studied 3 times each had improved tilt table and sweating responses and heart rate variability during respiration (13.6 → 14.0 → 19.1 and 4.0 → 9.2 → 14.7 beats per minute, respectively). The 1 immunomodulated patient with a low Valsalva ratio at onset normalized (1.42 → 1.76 → 2.27). Four of 4 repeat skin biopsies among immunomodulated patients documented axonal regeneration, which lagged behind symptom improvement. The index patient with 0 ENF/mm2 skin surface area before corticosteroid treatment had 278 ENFs 4 years later.22 A 10 year old with 51 ENF/mm2 skin surface area before corticosteroid and then IVIG treatment had 226 ENFs 14 months later. The 19-year-old diabetic patient with 0 ENFs at baseline had 48 2 years later, after IVIG produced near-total resolution. In the fourth patient, 4 biopsies over 2.5 years corroborated corticosteroid inefficacy followed by IVIG efficacy.
Discussion
This case series provides new hypotheses about childhood-onset, unexplained, acquired CWP syndromes, implicating acquired SFPN, a biologically plausible diagnosis not previously recognized in children. The data suggest that SFPN can develop even in preschool-age children and that juvenile-onset SFPN can persist for decades into adulthood. The characteristic temporal (acute/fulminant versus chronic) and spatial (distal versus patchy/proximal) patterns characteristic of polyneuropathies were all identified.6 With ubiquitous dysautonomic symptoms, signs, and test abnormalities, juvenile SFPN integrates autonomic as well as neuropathic pain components. Unexpectedly, 63% of patients suffered chronic headaches. These could not be attributed to medication overuse, as few patients used any pain or headache medications. Chronic headache is not currently linked to hypotension, microvasculopathy, or polyneuropathy,54 but normalizing blood pressure sometimes resolved patients’ headaches, consistent with hemodynamic causality.55,56 The current study has the inherent limitation of case series from academic practices, namely referral bias, which likely elevated the prevalence of SFPN. This limitation should be addressed by prospective reevaluation of these retrospective findings in other settings.
These families' relentless testing for treatable causes of their children’s chronic pain generated the abundant data analyzed here. The current results may help to curtail unnecessary testing in other similar children. Imaging was futile, and body fluid testing excluded all common causes of adult-onset SFPN (Table 2).37,57,58 Laboratory testing implicated only specific serological markers associated with organ-specific dysimmunity, a known cause of large-fiber polyneuropathies. This study confirms and extends a 32-case series of pediatric erythromelalgia documenting similar results, namely SFPN in 59% of patients and antinuclear antibodies in 46%.27 That study did not measure complement and did not find high ESRs, conservatively defined as ≥30 mm/hour (Mark Davis, personal communication).27
In our study, the most informative serology was low C4 complement in 46% and low-normal levels in most other patients, consistent with innate and autoantibody-mediated immunity. This most likely involves the classic or lectin rather than the alternative complement pathway. Patients’ predominantly antibody-associated comorbid autoimmune illnesses and lack of cellular infiltrates within nerve biopsies also argued against T-cell involvement, although cellularity at onset cannot be excluded because nerve biopsies had been performed late in the disease course. Dermatopathological identification of complement (including C4) and immunoglobulin deposition without cellularity in 2 of 2 skin biopsies (Fig 3) provides additional evidence of a role for complement- and autoantibody-mediated dysimmunity in the pathogenesis of some cases of juvenile-onset SFPN.
Sixty-one percent of patients reported specific illness triggers including infections, which are well-known precipitants of organ-specific autoimmunity including neuropathy.14,59 Thirty-one percent reported a traumatic antecedent. Injury had only rarely been associated with autoimmune neuropathy60 until the recent seminal discovery of postsurgical autoimmune neuropathies.61 Most antecedent injuries among our patients initially caused focal pain syndromes labeled as complex regional pain syndrome, which has also been linked to small-fiber neuropathy and autoimmunity.62–64
Conclusions
This study analyzed the results of multiple objective tests to identify a potentially common cause for childhood-onset CWP syndromes: SFPN. It extends the range of SFPN into earliest childhood and characterizes the pediatric presentation. It offers preliminary evidence of disordered immunity in some patients, including hypocomplementemia among other serologic abnormalities, and responsiveness to corticosteroid and immunoglobulin therapy in some patients. Recognition of juvenile-onset SFPN should help patients, families, and clinicians by suggesting a rational pathway for diagnostic evaluation and treatment. Having a specific diagnosis to test for and treat when present may reduce ineffective, costly, and potentially harmful tests and treatments and permit objective testing and definitive treatment of some patients. Additionally, the results demonstrate the need for pediatric norms for tests of SFPN, and they provide new testable hypotheses for clinical and basic research study.
Acknowledgments
We thank the patients, families, and physicians for providing records; Heather Downs, BS, and Daniela Herzog, BA, for assistance with the normal subjects; Vanda Lennon, MD, PhD, for testing sera for autoantibodies; and Isabelle Rapin, MD, for helpful discussion. This work is dedicated to the memory of John W. Griffin, MD.
Glossary
- AFT
autonomic function testing
- CIDP
chronic inflammatory demyelinating polyneuropathy
- CWP
chronic widespread pain
- ENF
epidermal nerve fiber
- ESR
erythrocyte sedimentation rate
- GBS
Guillain-Barré syndrome, (acute inflammatory demyelinating polyneuropathy)
- IVIG
intravenous immune globulin
- POTS
postural orthostasis tachycardia syndrome
- RR
reference range (of normal values for laboratory tests)
- SFPN
small-fiber polyneuropathy
Footnotes
Dr Oaklander conceptualized and designed the study, obtained funding, extracted the data, participated in the data analysis, and drafted the initial manuscript and the rewrites. Dr Klein performed the autonomic function testing on the normal control subjects, participated in the data analysis, contributed to drafting and editing the figures, contributed to rewriting the manuscript, and approved the submitted and all revised versions of the manuscript.
This work was presented in abstract form to the American Neurologic Association (September 25–27, 2011; Manchester Grand Hyatt, San Diego, CA) and the Peripheral Nerve Society (June 25–29, 2011; Bolger Center, Potomac, MD).
FINANCIAL DISCLOSURE: The authors have indicated they have no financial relationships relevant to this article to disclose.
FUNDING: Supported in part by the Public Health Service (K24NS059892), the Department of Defense (GW093049), and the Bradley and Curvey Family Foundations.
References
- 1.Wolfe F, Smythe HA, Yunus MB, et al. Report of the Multicenter Criteria Committee . The American College of Rheumatology 1990 Criteria for the Classification of Fibromyalgia. Arthritis Rheum. 1990;33(2):160–172 [DOI] [PubMed] [Google Scholar]
- 2.Buskila D. Pediatric fibromyalgia. Rheum Dis Clin North Am. 2009;35(2):253–261 [DOI] [PubMed] [Google Scholar]
- 3.van Geelen SM, Bakker RJ, Kuis W, van de Putte EM. Adolescent chronic fatigue syndrome: a follow-up study. Arch Pediatr Adolesc Med. 2010;164(9):810–814 [DOI] [PubMed] [Google Scholar]
- 4.Mikkelsson M, El-Metwally A, Kautiainen H, Auvinen A, Macfarlane GJ, Salminen JJ. Onset, prognosis and risk factors for widespread pain in schoolchildren: a prospective 4-year follow-up study. Pain. 2008;138(3):681–687 [DOI] [PubMed] [Google Scholar]
- 5.Ojha A, Chelimsky TC, Chelimsky G. Comorbidities in pediatric patients with postural orthostatic tachycardia syndrome. J Pediatr. 2011;158(1):20–23 [DOI] [PubMed] [Google Scholar]
- 6.Gorson KC, Herrmann DN, Thiagarajan R, et al. Non-length dependent small fibre neuropathy/ganglionopathy. J Neurol Neurosurg Psychiatry. 2008;79(2):163–169 [DOI] [PubMed] [Google Scholar]
- 7.Novak V, Freimer ML, Kissel JT, et al. Autonomic impairment in painful neuropathy. Neurology. 2001;56(7):861–868 [DOI] [PubMed] [Google Scholar]
- 8.Holzer P. Efferent-like roles of afferent neurons in the gut: Blood flow regulation and tissue protection. Auton Neurosci. 2006;125(1-2):70–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gibbons CH, Wang N, Freeman R. Capsaicin induces degeneration of cutaneous autonomic nerve fibers. Ann Neurol. 2010;68(6):888–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCarthy BG, Hsieh ST, Stocks A, et al. Cutaneous innervation in sensory neuropathies: evaluation by skin biopsy. Neurology. 1995;45(10):1848–1855 [DOI] [PubMed] [Google Scholar]
- 11.Lauria G, Hsieh ST, Johansson O, et al. European Federation of Neurological Societies. Peripheral Nerve Society . European Federation of Neurological Societies/Peripheral Nerve Society Guideline on the use of skin biopsy in the diagnosis of small fiber neuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society. Eur J Neurol. 2010;17(7):903–912, e44–e49 [DOI] [PubMed] [Google Scholar]
- 12.England JD, Gronseth GS, Franklin G, et al. American Academy of Neurology . Practice Parameter: evaluation of distal symmetric polyneuropathy: role of autonomic testing, nerve biopsy, and skin biopsy (an evidence-based review). Report of the American Academy of Neurology, American Association of Neuromuscular and Electrodiagnostic Medicine, and American Academy of Physical Medicine and Rehabilitation. Neurology. 2009;72(2):177–184 [DOI] [PubMed] [Google Scholar]
- 13.Assessment: Clinical autonomic testing report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 1996;46(3):873–880 [PubMed] [Google Scholar]
- 14.McDonald CM. Peripheral neuropathies of childhood. Phys Med Rehabil Clin N Am. 2001;12(2):473–490 [PubMed] [Google Scholar]
- 15.Axelrod FB, Chelimsky GG, Weese-Mayer DE. Pediatric autonomic disorders. Pediatrics. 2006;118(1):309–321 [DOI] [PubMed] [Google Scholar]
- 16.Mitchell SW. On a rare vaso-motor neurosis of the extremities, and on the maladies with which it may be confounded. Am J Med Sci. 1878;151:17–36 [Google Scholar]
- 17.Rauck RL, Naveira F, Speight KL, Smith BP. Refractory idiopathic erythromelalgia. Anesth Analg. 1996;82(5):1097–1101 [DOI] [PubMed] [Google Scholar]
- 18.Drenth JP, Finley WH, Breedveld GJ, et al. The primary erythermalgia-susceptibility gene is located on chromosome 2q31-32. Am J Hum Genet. 2001;68(5):1277–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dib-Hajj SD, Rush AM, Cummins TR, et al. Gain-of-function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain. 2005;128(pt 8):1847–1854 [DOI] [PubMed] [Google Scholar]
- 20.Wakamoto H, Hirai A, Manabe K, Hayashi M. Idiopathic small-fiber sensory neuropathy in childhood: A diagnosis based on objective findings on punch skin biopsy specimens. J Pediatr. 1999;135(2 pt 1):257–260 [DOI] [PubMed] [Google Scholar]
- 21.Dabby R, Gilad R, Sadeh M, Lampl Y, Watemberg N. Acute steroid responsive small-fiber sensory neuropathy: a new entity? J Peripher Nerv Syst. 2006;11(1):47–52 [DOI] [PubMed] [Google Scholar]
- 22.Paticoff J, Valovska A, Nedeljkovic SS, Oaklander AL. Defining a treatable cause of erythromelalgia: acute adolescent autoimmune small-fiber axonopathy. Anesth Analg. 2007;104(2):438–441 [DOI] [PubMed] [Google Scholar]
- 23.Pfund Z, Stankovics J, Decsi T, Illes Z. Childhood steroid-responsive acute erythromelalgia with axonal neuropathy of large myelinated fibers: a dysimmune neuropathy? Neuromuscul Disord. 2009;19(1):49–52 [DOI] [PubMed] [Google Scholar]
- 24.Morales PS, Escobar RG, Lizama M, et al. Paediatric hypertension-associated erythromelalgia responds to corticosteroids and is not associated with SCN9A mutations. Rheumatology (Oxf). 2012;51(12):2295–2296 [DOI] [PubMed]
- 25.Koike H, Atsuta N, Adachi H, et al. Clinicopathological features of acute autonomic and sensory neuropathy. Brain. 2010;133(10):2881–2896 [DOI] [PubMed] [Google Scholar]
- 26.Davis MDP, Weenig RH, Genebriera J, Wendelschafer-Crabb G, Kennedy WR, Sandroni P. Histopathologic findings in primary erythromelalgia are nonspecific: special studies show a decrease in small nerve fiber density. J Am Acad Dermatol. 2006;55(3):519–522 [DOI] [PubMed] [Google Scholar]
- 27.Cook-Norris RH, Tollefson MM, Cruz-Inigo AE, Sandroni P, Davis MD, Davis DM. Pediatric erythromelalgia: A retrospective review of 32 cases evaluated at Mayo Clinic over a 37-year period. J Am Acad Dermatol. 2012;66(3):416–423 [DOI] [PubMed] [Google Scholar]
- 28.Üçeyler N, Kahn A-K, Zeller D, et al. Functional and morphological impairment of small nerve fibers in fibromyalgia syndrome. In: Proceedings from the 2012 Meeting of the International Association for the Study of Pain; August 27–31, 2012; Milan, Italy. Abstract PT102 [Google Scholar]
- 29.Solà R, Collado A, Antonelli F, Quiles C, Serra J. Is fibromyalgia a special type of small fiber neuropathy? A microneurography study. In: Proceedings from the 2012 Meeting of the International Association for the Study of Pain; August 27–31, 2012; Milan, Italy. Abstract PW100 [Google Scholar]
- 30.Shtein R, Hussain M, Hamid M, Raval N, Williams DA, Clauw DJ. In vivo corneal confocal microscopy and clinical correlations in fibromyalgia (FM). In: Proceedings from the 2012 Meeting of the International Association for the Study of Pain; August 27–31, 2012; Milan, Italy. Abstract PH149 [Google Scholar]
- 31.Oaklander AL, Herzog ZD, Downs HM, Napoleon S. Prevalence of small-fiber polyneuropathy in fibromyalgia. Ann Neurol. 2012 [Google Scholar]
- 32.Wallach J. Interpretation of Diagnostic Tests. 8th Ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2007 [Google Scholar]
- 33.Devigili G, Tugnoli V, Penza P, et al. The diagnostic criteria for small fibre neuropathy: from symptoms to neuropathology. Brain. 2008;131(pt 7):1912–1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Freeman R, Chase KP, Risk MR. Quantitative sensory testing cannot differentiate simulated sensory loss from sensory neuropathy. Neurology. 2003;60(3):465–470 [DOI] [PubMed] [Google Scholar]
- 35.Lauria G, Morbin M, Lombardi R, et al. Axonal swellings predict the degeneration of epidermal nerve fibers in painful neuropathies. Neurology. 2003;61(5):631–636 [DOI] [PubMed] [Google Scholar]
- 36.Lauria G, Cornblath DR, Johansson O, et al. European Federation of Neurological Societies . EFNS guidelines on the use of skin biopsy in the diagnosis of peripheral neuropathy. Eur J Neurol. 2005;12(10):747–758 [DOI] [PubMed] [Google Scholar]
- 37.Amato AA, Oaklander AL. Case records of the Massachusetts General Hospital. Case 16-2004. A 76-year-old woman with pain and numbness in the legs and feet. N Engl J Med. 2004;(350):2181–2189 [DOI] [PubMed] [Google Scholar]
- 38.Klein MM, Downs H, Oaklander AL. Normal innervation in distal-leg skin biopsies: evidence of superabundance in youth, subsequent axonal pruning, plus new diagnostic recommendations. Ann Neurol. 2010;68(Suppl S14):S68 [Google Scholar]
- 39.Stewart JD, Low PA, Fealey RD. Distal small fiber neuropathy: results of tests of sweating and autonomic cardiovascular reflexes. Muscle Nerve. 1992;15(6):661–665 [DOI] [PubMed] [Google Scholar]
- 40.Schondorf R, Low PA. Idiopathic postural orthostatic tachycardia syndrome: an attenuated form of acute pandysautonomia? Neurology. 1993;43(1):132–137 [DOI] [PubMed] [Google Scholar]
- 41.Low PA, Sletten DM. Laboratory evaluation of autonomic failure. In: Low PA, Benarroch EE, eds. Clinical Autonomic Disorders. 3rd ed. Baltimore, MD: Lippincott Williams & Wilkins; 2008:130–163 [Google Scholar]
- 42.Low PA. Prevalence of orthostatic hypotension. Clin Auton Res. 2008;18(suppl 1):8–13 [DOI] [PubMed] [Google Scholar]
- 43.Skinner JE, Driscoll SW, Porter CB, et al. Orthostatic heart rate and blood pressure in adolescents: reference ranges. J Child Neurol. 2010;25(10):1210–1215 [DOI] [PubMed] [Google Scholar]
- 44.Low VA, Sandroni P, Fealey RD, Low PA. Detection of small-fiber neuropathy by sudomotor testing. Muscle Nerve. 2006;34(1):57–61 [DOI] [PubMed] [Google Scholar]
- 45.Chamberlain JL, Pittock SJ, Oprescu AM, et al. Peripherin-IgG association with neurologic and endocrine autoimmunity. J Autoimmun. 2010;34(4):469–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dhamija R, Renaud DL, Pittock SJ, et al. Neuronal voltage-gated potassium channel complex autoimmunity in children. Pediatr Neurol. 2011;44(4):275–281 [DOI] [PubMed] [Google Scholar]
- 47.Kimpinski K, Iodice V, Vernino S, Sandroni P, Low PA. Association of N-type calcium channel autoimmunity in patients with autoimmune autonomic ganglionopathy. Auton Neurosci. 2009;150(1–2):136–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Low PA, Gilden JL, Freeman R, Sheng KN, McElligott MA, Midodrine Study Group . Efficacy of midodrine vs placebo in neurogenic orthostatic hypotension. A randomized, double-blind multicenter study. JAMA. 1997;277(13):1046–1051 [PubMed] [Google Scholar]
- 49.Dworkin RH, O’Connor AB, Backonja M, et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain. 2007;132(3):237–251 [DOI] [PubMed] [Google Scholar]
- 50.Teasley JE. Initial treatment of childhood chronic inflammatory demyelinating polyradiculoneuropathy. Muscle Nerve. 2008;38(6):1640–1643 [PubMed] [Google Scholar]
- 51.Sladky JT. What is the best initial treatment for childhood chronic inflammatory demyelinating polyneuropathy: corticosteroids or intravenous immunoglobulin? Muscle Nerve. 2008;38(6):1638–1643 [DOI] [PubMed] [Google Scholar]
- 52.Van den Bergh PY, Hadden RD, Bouche P, et al. European Federation of Neurological Societies. Peripheral Nerve Society . European Federation of Neurological Societies/Peripheral Nerve Society guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society - first revision. Eur J Neurol. 2010;17(3):356–363 [DOI] [PubMed] [Google Scholar]
- 53.Gelfand EW. Intravenous immune globulin in autoimmune and inflammatory diseases. N Engl J Med. 2012;367(21):2015–2025 [DOI] [PubMed] [Google Scholar]
- 54.Classification of the International Headache Society. The international classification of headache disorders, 2nd edition. Blackwell Publishing, Oxford, UK: Cephalalgia. 2004;24(suppl 1):1–160. [DOI] [PubMed] [Google Scholar]
- 55.Mokri B, Low PA. Orthostatic headaches without CSF leak in postural tachycardia syndrome. Neurology. 2003;61(7):980–982 [DOI] [PubMed] [Google Scholar]
- 56.Furlan JC. Headache attributed to autonomic dysreflexia: an underrecognized clinical entity. Neurology. 2011;77(8):792–798 [DOI] [PubMed] [Google Scholar]
- 57.England JD, Gronseth GS, Franklin G, et al. Practice parameter: evaluation of distal symmetric polyneuropathy: role of laboratory and genetic testing (an evidence-based review). Report of the AAN, AANEM, and AAPMR. Neurology. 2009;72(2):185–192 [DOI] [PubMed] [Google Scholar]
- 58.Cheng X, Dib-Hajj SD, Tyrrell L, Te Morsche RH, Drenth JP, Waxman SG. Deletion mutation of sodium channel Na(V)1.7 in inherited erythromelalgia: enhanced slow inactivation modulates dorsal root ganglion neuron hyperexcitability. Brain. 2011;134(Pt 7):1972–1986 [DOI] [PubMed] [Google Scholar]
- 59.McKhann GM, Cornblath DR, Griffin JW, et al. Acute motor axonal neuropathy: a frequent cause of acute flaccid paralysis in China. Ann Neurol. 1993;33(4):333–342 [DOI] [PubMed] [Google Scholar]
- 60.Beghi E, Kurland LT, Mulder DW, Nicolosi A. Brachial plexus neuropathy in the population of Rochester, Minnesota, 1970-1981. Ann Neurol. 1985;18(3):320–323 [DOI] [PubMed] [Google Scholar]
- 61.Staff NP, Engelstad J, Klein CJ, et al. Post-surgical inflammatory neuropathy. Brain. 2010;133(10):2866–2880 [DOI] [PubMed] [Google Scholar]
- 62.Oaklander AL, Fields HL. Is reflex sympathetic dystrophy/complex regional pain syndrome type I a small-fiber neuropathy? Ann Neurol. 2009;65(6):629–638 [DOI] [PubMed] [Google Scholar]
- 63.Kohr D, Tschernatsch M, Schmitz K, et al. Autoantibodies in complex regional pain syndrome bind to a differentiation-dependent neuronal surface autoantigen. Pain. 2009;143(3):246–251 [DOI] [PubMed] [Google Scholar]
- 64.Goebel A, Baranowski A, Maurer K, Ghiai A, McCabe C, Ambler G. Intravenous immunoglobulin treatment of the complex regional pain syndrome: a randomized trial. Ann Intern Med. 2010;152(3):152–158 [DOI] [PubMed] [Google Scholar]