

Abstract
Although roles of the metabolic stress in organ ischemia reperfusion injury (IRI) have been well recognized, the question of whether and how these stress responses regulate innate immune activation against IR remains unclear. In a murine liver partial warm ischemia mode, we showed that prolonged ischemia triggered endoplasmic reticulum (ER) stress response, particularly, the ATF6 branch, in liver Kupffer cells and altered their responsiveness against TLR stimulation. Ischemia-primed cells increased pro-, but decreased anti-, inflammatory cytokine productions. Alleviation of ER stress in vivo by small chemical chaperon 4-phenylbutyrate or ATF6 siRNA diminished the pro-inflammatory priming effect of ischemia in KCs, leading to the inhibition of liver immune response against IR and protection of livers from IRI. In vitro, ATF6 siRNA abrogated the ER stress-mediated pro-inflammatory enhancement of macrophage TLR4 response, by restricting NF-kB and restoring Akt activations. Thus, ischemia primes liver innate immune cells by ATF6-mediated ER stress response. The IR-induced metabolic stress and TLR activation function in synergy to activate tissue inflammatory immune response.
Keywords: ER stress, TLR activation, ischemia reperfusion injury, Kupffer cells, ATF6
Introduction
Liver ischemia-reperfusion injury (IRI) remains the major cause of liver dysfunction and failure after hepatic trauma, resection and transplantation (1, 2). Tissue inflammatory immune response triggered by innate immune receptor activation, such as TLR4, plays a key role in the pathogenesis of liver IRI (3, 4). Initial tissue damage and stress prompt the release of danger-associated molecular pattern (DAMP), such as HMGB1, which activate pattern recognition receptors, such as TLR4, to trigger production of cytokines and chemokines (5). During the reperfusion stage, inflammatory cytokines drive further hepatocellular damages (6). Thus, inhibition of immune activation is the most effective approaches to alleviate experimental organ IRI. However, TLR activation by itself is not sufficient to facilitate IRI, as a low dose endotoxin, which triggers a strong inflammatory immune response in livers, causes minimal hepatocellular injuries. One potential explanation is that TLR activation induces both pro- and anti-inflammatory gene programs, which is self-limiting and minimal pathogenicity. Thus, it is important to identify additional pro-inflammatory mechanisms that may function in synergy with TLR activation, leading to the development of IRI.
Metabolic disturbance due to hypoxia and nutrient deficiency is the primary consequence of the ischemia process. Redox alteration and ATP deficiency cause dysfunction of key intracellular organelles, such as mitochondria and endoplasmic reticulum (ER), and trigger cellular stress responses. Although their roles in direct hepatocellular damage is well established (7), the potential interplay between stress responses and innate immune activations has only recently been revealed. Increasing evidence from chronic vascular disease and diabetes models indicates that metabolic stress may constitute a direct activating mechanism of tissue inflammation (8). ER stress due to the accumulation of unfolded proteins in ER lumen represents one of cellular stress responses against IR. Cells develop an evolutionarily conserved adaptive response called Unfolded Protein Response (UPR) to resolve ER stress (9). Recent studies have documented that liver IR does indeed trigger UPR (10-12)whereas small molecule chaperons, such as tauroursodeoxycholic acid (TUDCA) or 4-phenyl butyric acid (PBA)that reduce ER stress, do protect livers from IR-damage. Although IR-induced parenchymal cell necrosis/apoptosis was proposed as the primary targets of these chaperon in vivo, liver inflammatory immune activation was also affected by the ER stress inhibition (13, 14). The question is whether it regulates immune responses directly or indirectly via parenchymal cell death, which releases DAMP to trigger sterile inflammation.
Direct regulation of innate immune activation by ER-stress induced UPR signaling molecules has been demonstrated in vitro (15-17). To determine whether similar mechanism operates in liver innate immune responses at the organ level would require cell-type specific experiments. Here, we isolated KCs from normal vs. ischemic livers and compared their immune functions in vitro. The relevance of ER stress in ischemia regulation of liver immune response was determined by in vivo targeting of ER stress with PBA, and more specifically with ATF6 siRNA. In parallel, immune regulatory functions of ER stress and ATF6 were studied in vitro in macrophage cultures. Our results provide direct evidence that ischemia-induced ER stress: 1/ constitutes a pro-inflammatory activation mechanism in innate immune cells in synergy with TLR activation, and 2/ is essential in liver inflammatory immune response against IR, at least in part via ATF6 activation.
Materials and Methods
Animals
Male wide-type (WT) C57BL/6 mice (8-12 weeks old) were purchased from the Jackson Laboratory (Bar Harbor, ME). Animals were housed in the UCLA animal facility under specific pathogen-free conditions, and received human care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the National Institute of Health.
Mouse liver IRI model
As described (18), an atraumatic clip was used to interrupt the arterial and portal venous blood supply to the cephalad liver lobes for 90min. Sham controls underwent the same procedure, but without vascular occlusion. PBA was administered 1h prior to the ischemia at 100mg/kg, i.p.(Sigma, St. Luis, MO). Mice were sacrificed after 0, 1 or 6 h and liver and serum samples were collected. Serum alanine aminotransferase (sALT) levels were measured with an auto analyzer by ANTECH Diagnostics (Los Angeles, CA). Part of liver specimens were fixed in 10% buffered formalin and embedded in paraffin. Liver sections were stained with hematoxylin and eosin (HE). The severity of liver IRI was graded blindly using Suzuki's criteria on a scale from 0 to 4 (19).
Cell cultures
KCs were isolated as follows: livers were perfused in situ via the portal vein with calcium- and magnesium-free HBSS supplemented with 2% heat-inactivated FBS, followed by 0.27% collagenase IV (Sigma, St Louis, MO). Perfused livers were dissected, and teased through 70μm nylon mesh cell strainers (BD Biosciences, San Diego, CA). Non-parenchymal cells (NPCs) were separated from hepatocytes by centrifuging at 50g x2min for 3 times. NPCs were then suspended in HBSS and layered onto a 50%/25% two-step percoll gradient (Sigma, St Louis, MO) in a 50-ml conical centrifuge tube and centrifuged at 1400g, 4°C for 20 minutes. KCs in the middle layer were collected and allowed to attach onto cell culture plates in DMED with 10% FBS for 15 minutes at 37°C. Nonadherent cells were removed by replacing the culture medium. The purity of KCs (.80%) was assessed via staining with immunofluorescence-labeled anti-F4/80 Ab.
Bone marrow-derived macrophages (BMMs), obtained from femoral bones of 6-10-week old C57B/6 mice, were cultured in DMEM w/ 10% FBS and 20% L929 conditioned medium for 6 days. The cell purity was assayed to be 94-99% CD11b+.
Cells were stimulated with LPS (1μg/ml, Invivogen, San Diego, CA). ER-stressed macrophages were prepared by pre-incubating cells with Tunicamycin (Tm; 1μg/ml) for 6h, or Thapsigargin (Tg; 1μM/ml) for 1h (all from Sigma, St. Luis, MO), and washed with fresh warm media prior to LPS stimulation. No significant cell death is detected in cell cultures (>90% viable).
ATF6 knockdown
The siRNA sequences of mouse ATF6 are as follows: 5′-AAGGATCATCAGCGGAACCAA-3′; nonspecific (NS): 5′-CGAATCCACAAAGCGCGCTT-3′. In vitro, BMMs were transiently transfected with siRNA using Genmute™ Reagent (SignaGen Laboratories, MD, USA) according to the manufacturer's protocol. In vivo, siRNA were mixed with mannose-conjugated polymers (Polyplus transfection™, France) at a ratio specified by the manufacturer, and administered by tail vein injection (siRNA 2mg/kg) at 4h prior to the onset of liver ischemia.
Quantitative RT-PCR
Total RNA (2.5 μg) was reverse-transcribed into cDNA using SuperScriptTM III System (Invitrogen, Carlsbad, CA). Quantitative-PCR was performed using SuperMix (Platinum SYBR Green qPCR Kit, Invitrogen, Carlsbad, CA) in the DNA Engine with Chromo 4 Detector (MJ Research, Waltham, MA) as described previously (20). We used the comparative CT method (21) to quantitate gene expression levels, and target gene expressions were normalized using the HPRT gene expression level of the same samples.
Western blots
Tissue or cellular proteins were extracted with ice cold lysis buffer (1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 10% glycerol, 137mM sodium chloride, 20mM Tris, pH 7.4). Proteins (20 μg) were separated by 12% SDS-PAGE electrophoresis and transferred to PVDF nitrocellulose membrane. Western blot antibodies include: ATF4, pAkt, IkB, pNF-kBp65, pErk and β-actin (Cell Signaling Technology, San Diego, CA), XBP1 and ATF6 (Abcam, Cambrige, MA) and HRP-conjugated secondary antibody (Cell Signaling Technology, San Diego, CA). SuperSignal® West Pico Chemiluminescent Substrates (Thermo Fisher Scientific, Rockford, IL) were used for chemo-luminescence development.
The quantitation of Western blots was performed by using ImageJ software. Quantities of target proteins were normalized to that of β-actin of the same sample.
ELISA
Cytokine (TNF-α, L-6 and IL-10) levels in cell culture supernatants or serum was measured according to the manufacturer's protocols (eBioscience, San Diego, CA), Results were obtained using a Multiscan FC plate reader with SkanIt software (Thermo Scientific).
Statistical analysis
Results are shown as mean±SD. One-way ANOVA test was used in comparisons of three groups (Fig.1 and 2c, 0m, 30m and 90m ischemia at 0h post-reperfusion). Unpaired t-test was used for comparison of two groups. A two-tailed P value less than 0.05 was considered to be statistically significant.
Figure 1.

Prolonged liver ischemia primes KCs. KCs were isolated from livers after 0, or 30m or 90m ischemia at 0h post-reperfusion, and cultured overnight ±LPS stimulation. TNF-α, IL-6 and IL-10 levels in culture supernatants were measured by ELISA. Representative experiment of 2; n=3 mice/group/expt., ANOVA (—) or t-test (
 ): *p<0.05 **p<0.01.
): *p<0.05 **p<0.01.
Figure 2.
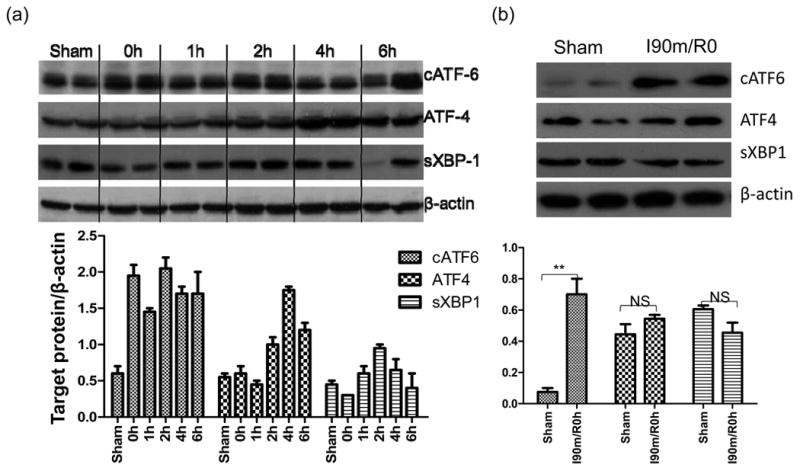

ER stress mediates the pro-inflammatory priming of ischemia in liver innate immune cells. (a) Western blots of ATF4, spliced XBP1, cleaved ATF6, and β actin in sham or ischemic livers, harvested at 0, 1, 4, 6h post-reperfusion after 90min ischemia. Quantitation of target protein vs. b-actin was plotted. (b) Western blot and quantitative plot of cleaved ATF6, ATF4, and spliced XBP1 in liver NPCs isolated from sham-operated or 90m-ischemic livers at 0h post-reperfusion. (c) Expressions of ER stress-induced genes in liver NPCs. Cells were isolated from livers after 0, 30m or 90m (with or without PBA treatment) of ischemia. Gene expressions were measured by qRT-PCR and ratios of target gene/HPRT were shown. (d) Effects of in vivo PBA treatment on ischemia-primed KCs. KCs isolated from groups of sham-operated, ischemic, or PBA-treated ischemic livers, as described in Material and Methods, were cultured in vitro ±LPS for 24h. Cytokine levels in culture supernatants, measured by ELISA, were shown. Note: LPS stimulated TNF- α and IL-6 levels were plotted on the right Y axis. Representative of 2 separate experiments; n=2-3 mice/group/expt., ANOVA (—) or t-test (
): *p<0.05 **p<0.01.
Results
Prolonged ischemia primed KCs to enhance the pro-inflammatory nature of their TLR response
To determine the immune regulatory effects of ischemia, we cultured KCs isolated from either sham-operated livers or those subjected to 30 or 90 min. of warm ischemia (harvested at 0h of reperfusion). Their productions of inflammatory cytokines, constitutive or after stimulation with LPS, were measured by ELISA. Clearly, prolonged ischemia activated KCs, as evidenced by significantly increased TNF-α, IL-6 and IL-10 secretion by cells from livers after 90m, but not 30m, of ischemia in vivo (Fig.1a), as compared with sham controls. More significantly, these cells became more responsive to TLR stimulation that they produced much higher levels of TNF-α and IL-6 than those from sham or short ischemic livers. Interestingly, IL-10 production was selectively downregulated upon in vitro stimulation in KCs by ischemia. Thus, prolonged ischemia not only activates KCs, but also enhances the pro-inflammatory property of their TLR response.
ER stress response mediated the pro-inflammatory priming of ischemia in liver innate immune cells
As ER stress represents a key cellular stress response against IR, we determined the activation of UPR signaling pathways in ischemic livers after various reperfusion time. As shown by Western blot results, all three UPR signature signaling molecules: ATF4, spliced (s) XBP1, and cleaved (c) ATF6 were upregulated by IR with distinctive kinetics (Fig.2a). In particular, cATF6 level was increased at 0h post reperfusion and sustained throughout the reperfusion period, while ATF4 and sXBP1 levels were increased transiently between 2-4h post reperfusion and returned to baseline at 6h. The induction of cATF6, but not ATF4 and sXBP1, was also detected specifically in liver NPCs isolated from 90m ischemic livers (at 0h post reperfusion), as compared with those from sham (Fig.2b). Simultaneously, the expression of ER stress associated genes, represented by CHOP, Grp78 and Xbp1, were also increased in these NPCs by ischemia and their levels were correlated with liver ischemia times in vivo (Fig.2c).
To test functional roles of ER stress in ischemia priming of liver NPCs, we administrated small chemical chaperon 4-PBA in vivo prior to liver ischemia, which has been shown to alleviated ER stress in vivo and protected livers from IRI in our previous study (13). Indeed, 4-PBA treatment prevented the induction of ER stress genes in liver NPCs (Fig.2c). More importantly, KCs isolated from PBA-treated ischemic livers produced significantly less TNF-α and IL-6, but more IL-10, both constitutively and after stimulation with LPS in vitro (Fig.2d). PBA treatment by itself did not directly suppress macrophage TLR activation in vitro and in vivo (data not shown). Thus, ER stress is critically involved in the priming of liver NPCs by ischemia to facilitate their pro-inflammatory immune activation.
ATF6 played a key role in ischemia priming of liver innate immune cells and the development of IRI
To study the mechanism of ER stress regulation of liver immune activation, we tested the functional role of ATF6 in KCs using its specific siRNA. The knock-down (KD) efficacy of ATF6 expressions by our siRNA construct was validated in vitro in macrophage cultures at both protein and mRNA levels. The Tm-induced ATF6 upregulation, both of its cleaved protein measured by Western blot (Fig.4b) and gene expression measured by qRT-PCR (Supplement 1a), were significantly inhibited in the ATF6-, but not non-specific (NS), siRNA transfected cells. Tm-induced expressions of other ER stress-associated genes, including XBP1, CHOP and Grp78, were not much different between these transfected cells. To target KCs in livers, we utilized mannose-conjugated polymers to deliver siRNA in vivo. The bio-distribution pattern of fluorescence in spleen indicated that phagocytes (expressing mannose receptors) were transfected by the fluorescence-labeled siRNA/polymers (Supplement 2, upper panel). In livers, fluorescent cells merged with CD68 positive cells, whereas hepatocytes were mostly negative for any fluorescence signals (Supplement 2, lower panel). Four experimental groups, including sham and IR with either NS or ATF6 siRNA, were set up to test ATF6 functions in the ischemia priming of liver KCs (0h), as well as the activation of liver immune response and the development of IRI (6h). Although ATF6KD did not affect constitutive cytokine levels in KCs, it abrogated the pro-inflammatory priming effect of ischemia: cells from ATF6 siRNA-treated ischemic livers produced significantly less TNF-a and IL-6, but more IL-10, than those from NS siRNA treated ischemic livers upon in vitro LPS stimulation (Fig.3a). At the organ level, liver TNF- α and IL-6 gene expressions were reduced, but IL-10 was selectively increased in ATF6 siRNA-treated mice 6h post reperfusion, as compared with those in NS siRNA-treated ones (Fig.3b). This translated into liver protection from IRI that serum ALT levels were significantly lower by ATF6 KD, with better preserved liver tissue architectures and lower Suzuki scores than those in controls (Fig.3c). Thus, ATF6 played a key role in liver inflammatory immune response against IR by mediating a pro-inflammatory priming effect of ischemia in liver innate immune cells.
Figure 4.

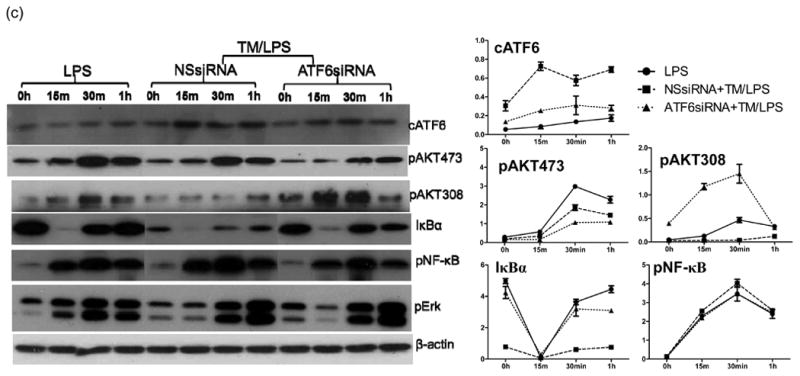
ATF6 regulates TLR response in ER stressed macrophages in vitro. (a) Chemical-induced ER stress primes macrophages. BMMs were pre-treated with Tm or Tg, followed by LPS stimulations for 24h, as described in Material and Methods. Cytokine levels in culture supernatants were measured by ELISA. (b) ATF6 regulates cytokine productions in ER-stressed macrophages. The siRNA transfected BMMs were pre-treated with Tm, followed by stimulations with LPS, as described in Material and Methods. Cells were harvested at 4 to measure gene expressions by qRT-PCR or culture supernatants at or 24h to measure cytokines by ELISA. (c) ER stress regulates TLR4 intracellular signaling pathways via ATF6. BMMs were transfected with NS or ATF6 siRNA, followed by sequential treatment of Tm and LPS and harvested at different time points, as described in Material and Methods. The expression of ATF6 and activation of intracellular signaling pathways were analyzed by Western blots. Quantitation of target protein vs. β-actin at different time points was plotted. Representative of at least 2 separate experiments, n=3 replicates/group/expt, t-test, *p<0.05 **p<0.01.
Figure 3.
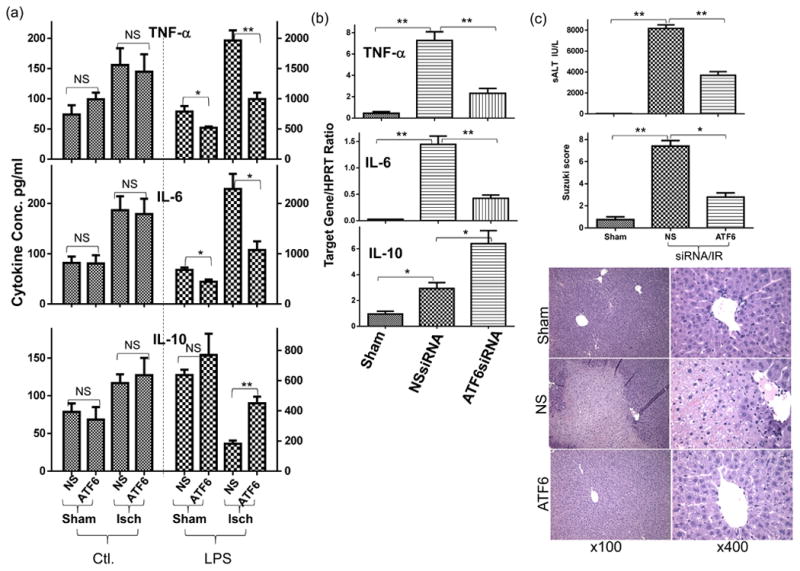
ATF6 regulates KC activation and liver IRI. (a) ATF6 KD diminishes ischemia-induced pro-inflammatory priming of liver innate immune cells. KCs, isolated from groups of sham-operated or ischemic livers treated with NS or ATF6 siRNA, as described in Material and Methods, were cultured in vitro ±LPS stimulation for 24h. Cytokine levels in culture supernatants, measured by ELISA, were shown. (b) ATF6 KD inhibits liver inflammatory immune activation by IR. Groups of mice were treated with NS or ATF6 siRNA, followed by liver IR, as described in Material and Methods. Liver inflammatory gene levels were measured by qRT-PCR at 6h post reperfusion. (c) ATF6 KD alleviates liver IRI. Serum ALT levels and liver histological analysis (H/E staining scored by the Suzuki standard) from the same groups of mice as in (b) at 6h post reperfusion were shown. Representative at least two independent experiments. n=3-5 mice/group/expt., t-test, *p<0.05 **p<0.01.
ER stress-induced ATF6 inhibited AKT-Gsk3b but enhanced NF-kB signaling
To dissect molecular mechanisms of ATF6 regulation of innate immune response, we studied TLR responses in vitro in ER stressed macrophages induced by chemicals. Bone-marrow-derived macrophages were pre-treated with either Tm or Tg, followed by TLR stimulation (Tm/Tg was removed prior to the stimulation). ER-stressed macrophages did not produce cytokines spontaneously. However, they became more responsive to LPS stimulation, by producing significantly higher levels of TNF-α and IL-6, but much lower level of IL-10, as measured by ELISA (Fig.4a). Interestingly, the IP-10 gene induction was not altered significantly by ER stress (Supplement 1b). Similar pro-inflammatory effects were also observed in macrophage responses against other TLR ligands (data not shown). Thus, similar to ischemia, chemical-induced ER stress had a pro-inflammatory priming effect in macrophages in vitro.
To determine the role of ATF6 in ER stress priming of macrophages, siRNA-mediated gene KD was performed prior to the induction of ER stress in macrophages. ATF6KD resulted in significant less productions of TNF-α and IL-6, but more IL-10 in ER stressed macrophages in response to TLR4 activation, at both transcriptional and translational levels (Fig.4b), as compared with the NS siRNA transfection. ATF6 KD had a similar, but less prominent anti-inflammatory effect, in normal macrophages as well, indicating a role of endogenous ATF6 in regulating TLR response. ATF6 or NS siRNA did not alter macrophage IP-10 response regardless of ER stress status (Supplement 1c).
Activations of intracellular signaling pathways by LPS were compared between normal vs. ER-stressed macrophages. Western blot results (Fig.4c) showed that AKT phosphorylation at Ser308 was decreased most significantly in ER-stressed cells. ATF6 KD restored/enhanced this phosphorylation. Meanwhile, although Thr473 phosphorylation of AKT was also slight inhibited in ER stressed cells, ATF6 KO further decreased the phosphorylation at this site. Although NF-kB phosphorylation was not affected by ER stress, IkB recovery from the TLR activation-induced degradation was inhibited in ER stressed cells (Fig.4c). ATF6 KD facilitated the IkB recovery. Meanwhile, extracellular signal-regulated kinase-1 (ERK1) activations were similar between normal and ER stressed cells, and ATF6 KD had no impact on this kinase activation. Thus, ER stress-induced ATF6 activation enhances the pro-inflammatory nature of TLR4 response by both inhibiting Akt and enhancing NF-kB signaling.
Discussion
Our current data documents that prolonged ischemia not only activated liver innate immune cells but also altered their responsiveness against subsequent TLR stimulation by increasing its pro-inflammatory property. Importantly, we found that ER stress and its specific signaling pathway mediated by ATF6 constituted a key intracellular mechanism of ischemia in regulating innate immune cells, which inhibited the anti-inflammatory Akt, but enhanced the NF-kB, activations. While the latter has a broad effect to promote inflammatory gene expression, the Akt inhibition would limit the induction of IL-10. These results provide us definitive evidence in a clinically relevant disease model that ER stress directly regulates innate immunity in vivo.
The involvement of ER stress in IRI has been documented in various animal models, including liver, kidney, heart and brain (22-28). The mechanism, however, was explored mostly in cell death pathways with contradictory results. As ER stress-induced UPR is an adaptive response, inhibition of UPR often decreases cell survival against mild or transient ER stress, as demonstrated in series of studies in cardiac myocyte ischemic cultures and heart ischemia models (23, 29-31). Overexpression of ATF6 in myocardium increased Grp78 induction against ex vivo IR and reduced cell death, while ATF6 KD did the opposite. Similar cytoprotective effects were observed with ER stress-induced Xbp1 in vitro (32-34). Despite its adaptive nature, prolonged UPR resulted from unresolved/lethal ER stress could lead to cell death. C/EBP homologous protein (CHOP) plays a key role in ER stress-induced cellular damage and its genetic deletion attenuated myocardial and brain IRI in vivo (26, 35), as well as hepatocyte death in vitro (36). Alleviation of ER stress by chemical chaperons, therefore, effectively protected organs from IRI (14, 25, 27, 28). Although several studies, including our own, have demonstrated a potential role of ER stress in regulating inflammatory immune response against IR (13, 28, 35), the underlying mechanism was unclear. By isolating liver innate immune cells from ischemic livers, we not only confirmed the induction of UPR in these particular cells, but also revealed its functional significance in regulating immune responses of these cells. Our finding that ischemia-induced ER stress differentially regulates TLR-triggered pro- and anti-inflammatory gene production programs is of high significance for us to further understand the mechanism of liver inflammatory injuries.
The ER stress-induced UPR, in fact, shares multiple signaling components with innate immune responses, leading to the activation of MAP kinases, NF-kB and JNK (17, 37). Thus, structurally, synergistic interactions between ER stress and innate immune activation are conceivable. It has been postulated that the transient global inhibition of protein translation mediated by the PERK pathway of UPR could activate NF-kB. Indeed, we have observed a significant delay of IkB recovery from degradation in ER-stressed macrophages, which would leads to prolonged NF-kB activation. The specific UPR signaling pathway XBP1 has been demonstrated closely involved in innate immune activation. Its deletion in macrophages reduces IL-6, TNF-α and IL-1β gene induction by LPS (15). It has been shown that XBP1 binds to a subset of inflammatory gene promoters. Relevant to our experimental setting, XBP1 activation by chemical ER stress inducers enhances TLR activation, which is abrogated by IRE1 α KD (15). Our own preliminary data using Xbp1siRNA also indicted its role in ER stress pro-inflammatory priming of macrophages (data not shown). Furthermore, ER stress-induced XBP1 activation was shown to play a key role in the synergy of LPS and ER stress in macrophage IFN-β induction. Thus, its relevance in IR-induced inflammatory immune response is conceivable. Systemic biology studies of ER stressed cells have resulted in the identification of a novel NLRP3 inflammasome activation pathway mediated by IRE1 α (upstream of XBP1), which promoted β cell death in diabetes (38, 39). Although the direct connection of ATF4 to inflammatory innate immune activation has yet to be established, a recent study revealed its involvement in saturated fatty acid induced IL-6 production in macrophages (40). Although CHOP downstream of ATF4 has been firmly linked to ER stress-induced cell death, it has been shown to regulate IL-23 expression in ER stressed dendritic cells (41).
ATF6 has been shown to become readily activated by ischemia in cardiac myocytes in vitro and in heart ischemia model in vivo (23, 30). Its functional roles, however, were limited to protect cells from death. Additionally, ATF6 was found to be involved in ER stress-induced liver steatosis (42, 43). Our study is the first to document the role of ATF6 in acute liver IRI and its direct involvement in innate immune regulation, both in vivo and in vitro. ATF6 KO significantly reduced TNF-a/IL-6 and enhanced IL-10 production by ER-stressed macrophages in vitro and in ischemic livers in vivo. Mechanistically, we have observed that the anti-inflammatory signaling pathway AKT was inhibited significantly in ER-stressed cells and ATF6 KD could restored its activation by LPS. Akt inhibition or genetic deletion has been shown to enhance the pro-inflammatory property of TLR response via either Gsk3b or microRNAs (44, 45). Thus, the ER stress-induced ATF6 signaling prohibits Akt activation upon TLR stimulation, resulting in a more pro-inflammatory response. Consistent with our in vitro finding, we have observed that prolonged ischemia inactivated liver Akt (dephosphorylation) (data not shown). Interestingly, it has been reported that the activation of ATF6 in macrophages by bacterial toxin subtilase triggered a transient AKT phosphorylation leading to NF-kB activation (46). This indicates us a possibility that Akt might become refractory to subsequent TLR stimulation after initial ER stress induced activation in ER-stressed macrophages.
In summary, this study provides us a novel mechanistic insight of liver inflammatory immune activation in the course of IR, and documents a direct immune regulatory role of ischemia via ER stress induced ATF6 activation. The functional synergy between IR-induced metabolic stress and TLR activation is critical for tissue inflammatory immune activation.
Supplementary Material
Figure S1. Effects of ATF6 siRNA.(A) ATF6 siRNA selectively inhibited the ATF6 expression in ER-stressed macrophages. The ATF6- or NS-siRNA transfected BMMs were pretreated with Tm, followed by stimulations with LPS, as described in Material and Methods. Cells were harvested at 4 to measure gene expressions of ATF6, XBP1, CHOP and Grp78 by qRT-PCR. (B) ER stress did not alter IP-10 expressions. BMMs were pretreated with Tm or Tg, followed by LPS stimulations for 4h, as described in Material and Methods. IP-10 gene expressions were measured by qRT-PCR. (C) ATF6 siRNA did not regulate macrophage IP-10 gene expression. As described in (A), IP-10 expression in siRNA transfected macrophages under ER stress was measured by qRT-PCR. Results are representative of at least 2 separate experiments, n=3 replicates/group/expt, t-test, *p<0.05.
Figure S2. Targeted delivery of siRNA in vivo by mannose-conjugated polymers. AlexaFluor488-labeled siRNA (green) were mixed with mannose-conjugated polymers and injected via tail vein, as described in Material and Methods. Spleen and livers were collected at 4 hours post injection. Tissue specimens were fixed with 10% neutral formaldehyde, embedded and frozen in OTC. Liver sections (4μm) were stained with anti-CD68 antibody (Santa Cruz Biotechnology, Inc., San Diego, CA) and Cy3-conjugated AffiniPure goat anti-mouse IgG antibody (red, Jackson Immunoresearch, West Grove, PA). DAPI (blue) was used for nuclear counterstaining. The biodistribution of green fluoresence positive cells in spleen were indicative of phagocytes (expressing mannose receptors) being transfected. In livers, these positive cells merged with red immunofluorescence-stained CD68 positive cells.
Acknowledgments
The study is supported by NIH Grants RO1 DK083408 (YZ), The Dumont Research Foundations; and grants from National Nature Science Foundation of China 81100270, 1310108001, 81210108017 (LL)
Abbreviations
- PBA
4-Phenylbutyric acid
- ATF4
Activating transcription factor 4
- ATF6
Activating transcription factor 6
- BMMs
Bone-marrow-derived macrophages
- ER
Endoplasmic reticulum
- IRI
Ischemia/Reperfusion Injury
- LPS
Lipopolysaccharide
- NPCs
Non-parenchymal cells
- sALT
Serum alanine aminotransferase
- siRNA
Small interfering RNA
- Tg
Thapsigargin
- Tm
Tunicamycin
- TLR
Toll-Like-Receptor
- UPR
Unfolded Protein Response
- XBP1
X-box binding protein 1
Footnotes
Disclosures: The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
Supporting Information: Additional Supporting Information is available in the online version of this article.
References
- 1.Henderson JM. Liver transplantation and rejection: an overview. Hepato-Gastroenterology. 1999;46(Suppl 2):1482–1484. [PubMed] [Google Scholar]
- 2.Howard TK, Klintmalm GB, Cofer JB, Husberg BS, Goldstein RM, Gonwa TA. The influence of preservation injury on rejection in the hepatic transplant recipient. Transplantation. 1990;49(1):103–107. doi: 10.1097/00007890-199001000-00023. [DOI] [PubMed] [Google Scholar]
- 3.Zhai Y, Busuttil RW, Kupiec-Weglinski JW. Liver ischemia and reperfusion injury: new insights into mechanisms of innate-adaptive immune-mediated tissue inflammation. Am J Transplant. 2011;11(8):1563–1569. doi: 10.1111/j.1600-6143.2011.03579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.King TD, Song L, Jope RS. AMP-activated protein kinase (AMPK) activating agents cause dephosphorylation of Akt and glycogen synthase kinase-3. Biochem Pharmacol. 2006;71(11):1637–1647. doi: 10.1016/j.bcp.2006.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gill R, Tsung A, Billiar T. Linking oxidative stress to inflammation: Toll-like receptors. Free Radic Biol Med. 2010;48(9):1121–1132. doi: 10.1016/j.freeradbiomed.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwabe RF, Brenner DA. Mechanisms of Liver Injury. I. TNF-alpha-induced liver injury: role of IKK, JNK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol. 2006;290(4):G583–589. doi: 10.1152/ajpgi.00422.2005. [DOI] [PubMed] [Google Scholar]
- 7.Gu T, Zhang Z, Wang J, Guo J, Shen WH, Yin Y. CREB is a novel nuclear target of PTEN phosphatase. Cancer Res. 2011;71(8):2821–2825. doi: 10.1158/0008-5472.CAN-10-3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hasnain SZ, Tauro S, Das I, Tong H, Chen AC, Jeffery PL, et al. IL-10 promotes production of intestinal mucus by suppressing protein misfolding and endoplasmic reticulum stress in goblet cells. Gastroenterology. 2013;144(2):357–368. doi: 10.1053/j.gastro.2012.10.043. e359. [DOI] [PubMed] [Google Scholar]
- 9.Todd DJ, Lee AH, Glimcher LH. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat Rev Immunol. 2008;8(9):663–674. doi: 10.1038/nri2359. [DOI] [PubMed] [Google Scholar]
- 10.Tsaytler P, Bertolotti A. Exploiting the selectivity of protein phosphatase 1 for pharmacological intervention. FEBS J. 2013;280(2):766–770. doi: 10.1111/j.1742-4658.2012.08535.x. [DOI] [PubMed] [Google Scholar]
- 11.Liu TF, Brown CM, El Gazzar M, McPhail L, Millet P, Rao A, et al. Fueling the flame: bioenergy couples metabolism and inflammation. J Leukoc Biol. 2012;92(3):499–507. doi: 10.1189/jlb.0212078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clavarino G, Claudio N, Couderc T, Dalet A, Judith D, Camosseto V, et al. Induction of GADD34 is necessary for dsRNA-dependent interferon-beta production and participates in the control of Chikungunya virus infection. PLoS Pathog. 2012;8(5):e1002708. doi: 10.1371/journal.ppat.1002708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu J, Ren F, Cheng Q, Bai L, Shen X, Gao F, et al. Endoplasmic reticulum stress modulates liver inflammatory immune response in the pathogenesis of liver ischemia and reperfusion injury. Transplantation. 2012;94(3):211–217. doi: 10.1097/TP.0b013e318259d38e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson CD, Upadhya G, Conzen KD, Jia J, Brunt EM, Tiriveedhi V, et al. Endoplasmic reticulum stress is a mediator of posttransplant injury in severely steatotic liver allografts. Liver Transpl. 2011;17(2):189–200. doi: 10.1002/lt.22220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. 2010;11(5):411–418. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith JA, Turner MJ, DeLay ML, Klenk EI, Sowders DP, Colbert RA. Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic IFN-beta induction via X-box binding protein 1. Eur J Immunol. 2008;38(5):1194–1203. doi: 10.1002/eji.200737882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martinon F, Glimcher LH. Regulation of innate immunity by signaling pathways emerging from the endoplasmic reticulum. Curr Opin Immunol. 2010 doi: 10.1016/j.coi.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen XD, Ke B, Zhai Y, Amersi F, Gao F, Anselmo DM, et al. CD154-CD40 T-cell costimulation pathway is required in the mechanism of hepatic ischemia/reperfusion injury, and its blockade facilitates and depends on heme oxygenase-1 mediated cytoprotection. Transplantation. 2002;74(3):315–319. doi: 10.1097/00007890-200208150-00005. [DOI] [PubMed] [Google Scholar]
- 19.Suzuki S, Toledo-Pereyra LH, Rodriguez FJ, Cejalvo D. Neutrophil infiltration as an important factor in liver ischemia and reperfusion injury. Modulating effects of FK506 and cyclosporine. Transplantation. 1993;55(6):1265–1272. doi: 10.1097/00007890-199306000-00011. [DOI] [PubMed] [Google Scholar]
- 20.Zhai Y, Shen XD, Gao F, Zhao A, Freitas MC, Lassman C, et al. CXCL10 regulates liver innate immune response against ischemia and reperfusion injury. Hepatology. 2008;47(1):207–214. doi: 10.1002/hep.21986. [DOI] [PubMed] [Google Scholar]
- 21.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 22.Bailly-Maitre B, Fondevila C, Kaldas F, Droin N, Luciano F, Ricci JE, et al. Cytoprotective gene bi-1 is required for intrinsic protection from endoplasmic reticulum stress and ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2006;103(8):2809–2814. doi: 10.1073/pnas.0506854103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martindale JJ, Fernandez R, Thuerauf D, Whittaker R, Gude N, Sussman MA, et al. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ Res. 2006;98(9):1186–1193. doi: 10.1161/01.RES.0000220643.65941.8d. [DOI] [PubMed] [Google Scholar]
- 24.Mizukami T, Orihashi K, Herlambang B, Takahashi S, Hamaishi M, Okada K, et al. Sodium 4-phenylbutyrate protects against spinal cord ischemia by inhibition of endoplasmic reticulum stress. Journal of vascular surgery : official publication, the Society for Vascular Surgery [and] International Society for Cardiovascular Surgery, North American Chapter. 2010;52(6):1580–1586. doi: 10.1016/j.jvs.2010.06.172. [DOI] [PubMed] [Google Scholar]
- 25.Qi X, Hosoi T, Okuma Y, Kaneko M, Nomura Y. Sodium 4-phenylbutyrate protects against cerebral ischemic injury. Molecular Pharmacology. 2004;66(4):899–908. doi: 10.1124/mol.104.001339. [DOI] [PubMed] [Google Scholar]
- 26.Tajiri S, Oyadomari S, Yano S, Morioka M, Gotoh T, Hamada JI, et al. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 2004;11(4):403–415. doi: 10.1038/sj.cdd.4401365. [DOI] [PubMed] [Google Scholar]
- 27.Vilatoba M, Eckstein C, Bilbao G, Smyth CA, Jenkins S, Thompson JA, et al. Sodium 4-phenylbutyrate protects against liver ischemia reperfusion injury by inhibition of endoplasmic reticulum-stress mediated apoptosis. Surgery. 2005;138(2):342–351. doi: 10.1016/j.surg.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 28.Ben Mosbah I, Alfany-Fernandez I, Martel C, Zaouali MA, Bintanel-Morcillo M, Rimola A, et al. Endoplasmic reticulum stress inhibition protects steatotic and non-steatotic livers in partial hepatectomy under ischemia-reperfusion. Cell Death Dis. 2010;1:e52. doi: 10.1038/cddis.2010.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Belaidi E, Decorps J, Augeul L, Durand A, Ovize M. Endoplasmic reticulum stress contributes to heart protection induced by cyclophilin D inhibition. Basic Res Cardiol. 2013;108(4):363. doi: 10.1007/s00395-013-0363-z. [DOI] [PubMed] [Google Scholar]
- 30.Doroudgar S, Thuerauf DJ, Marcinko MC, Belmont PJ, Glembotski CC. Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response. J Biol Chem. 2009;284(43):29735–29745. doi: 10.1074/jbc.M109.018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vekich JA, Belmont PJ, Thuerauf DJ, Glembotski CC. Protein disulfide isomerase-associated 6 is an ATF6-inducible ER stress response protein that protects cardiac myocytes from ischemia/reperfusion-mediated cell death. J Mol Cell Cardiol. 2012;53(2):259–267. doi: 10.1016/j.yjmcc.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupta S, Deepti A, Deegan S, Lisbona F, Hetz C, Samali A. HSP72 protects cells from ER stress-induced apoptosis via enhancement of IRE1alpha-XBP1 signaling through a physical interaction. PLoS Biol. 2010;8(7):e1000410. doi: 10.1371/journal.pbio.1000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ibuki T, Yamasaki Y, Mizuguchi H, Sokabe M. Protective effects of XBP1 against oxygen and glucose deprivation/reoxygenation injury in rat primary hippocampal neurons. Neurosci Lett. 2012;518(1):45–48. doi: 10.1016/j.neulet.2012.04.053. [DOI] [PubMed] [Google Scholar]
- 34.Thuerauf DJ, Marcinko M, Gude N, Rubio M, Sussman MA, Glembotski CC. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ Res. 2006;99(3):275–282. doi: 10.1161/01.RES.0000233317.70421.03. [DOI] [PubMed] [Google Scholar]
- 35.Miyazaki Y, Kaikita K, Endo M, Horio E, Miura M, Tsujita K, et al. C/EBP homologous protein deficiency attenuates myocardial reperfusion injury by inhibiting myocardial apoptosis and inflammation. Arterioscler Thromb Vasc Biol. 2011;31(5):1124–1132. doi: 10.1161/ATVBAHA.111.224519. [DOI] [PubMed] [Google Scholar]
- 36.Pfaffenbach KT, Gentile CL, Nivala AM, Wang D, Wei Y, Pagliassotti MJ. Linking endoplasmic reticulum stress to cell death in hepatocytes: roles of C/EBP homologous protein and chemical chaperones in palmitate-mediated cell death. Am J Physiol Endocrinol Metab. 2010;298(5):E1027–1035. doi: 10.1152/ajpendo.00642.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hasnain SZ, Lourie R, Das I, Chen AC, McGuckin MA. The interplay between endoplasmic reticulum stress and inflammation. Immunol Cell Biol. 2012;90(3):260–270. doi: 10.1038/icb.2011.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A, et al. IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012;16(2):250–264. doi: 10.1016/j.cmet.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oslowski CM, Hara T, O'Sullivan-Murphy B, Kanekura K, Lu S, Hara M, et al. Thioredoxin-interacting protein mediates ER stress-induced β cell death through initiation of the inflammasome. Cell Metab. 2012;16(2):265–273. doi: 10.1016/j.cmet.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iwasaki Y, Suganami T, Hachiya R, Shirakawa I, Kim-Saijo M, Tanaka M, et al. Activating transcription factor 4 links metabolic stress to interleukin-6 expression in macrophages. Diabetes. 2014;63(1):152–161. doi: 10.2337/db13-0757. [DOI] [PubMed] [Google Scholar]
- 41.Goodall JC, Wu C, Zhang Y, McNeill L, Ellis L, Saudek V, et al. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc Natl Acad Sci U S A. 2010;107(41):17698–17703. doi: 10.1073/pnas.1011736107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cinaroglu A, Gao C, Imrie D, Sadler KC. Activating transcription factor 6 plays protective and pathological roles in steatosis due to endoplasmic reticulum stress in zebrafish. Hepatology. 2011;54(2):495–508. doi: 10.1002/hep.24396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamamoto K, Takahara K, Oyadomari S, Okada T, Sato T, Harada A, et al. Induction of liver steatosis and lipid droplet formation in ATF6alpha-knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol Biol Cell. 2010;21(17):2975–2986. doi: 10.1091/mbc.E09-02-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Androulidaki A, Iliopoulos D, Arranz A, Doxaki C, Schworer S, Zacharioudaki V, et al. The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity. 2009;31(2):220–231. doi: 10.1016/j.immuni.2009.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005;6(8):777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harama D, Koyama K, Mukai M, Shimokawa N, Miyata M, Nakamura Y, et al. A subcytotoxic dose of subtilase cytotoxin prevents lipopolysaccharide-induced inflammatory responses, depending on its capacity to induce the unfolded protein response. J Immunol. 2009;183(2):1368–1374. doi: 10.4049/jimmunol.0804066. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Effects of ATF6 siRNA.(A) ATF6 siRNA selectively inhibited the ATF6 expression in ER-stressed macrophages. The ATF6- or NS-siRNA transfected BMMs were pretreated with Tm, followed by stimulations with LPS, as described in Material and Methods. Cells were harvested at 4 to measure gene expressions of ATF6, XBP1, CHOP and Grp78 by qRT-PCR. (B) ER stress did not alter IP-10 expressions. BMMs were pretreated with Tm or Tg, followed by LPS stimulations for 4h, as described in Material and Methods. IP-10 gene expressions were measured by qRT-PCR. (C) ATF6 siRNA did not regulate macrophage IP-10 gene expression. As described in (A), IP-10 expression in siRNA transfected macrophages under ER stress was measured by qRT-PCR. Results are representative of at least 2 separate experiments, n=3 replicates/group/expt, t-test, *p<0.05.
Figure S2. Targeted delivery of siRNA in vivo by mannose-conjugated polymers. AlexaFluor488-labeled siRNA (green) were mixed with mannose-conjugated polymers and injected via tail vein, as described in Material and Methods. Spleen and livers were collected at 4 hours post injection. Tissue specimens were fixed with 10% neutral formaldehyde, embedded and frozen in OTC. Liver sections (4μm) were stained with anti-CD68 antibody (Santa Cruz Biotechnology, Inc., San Diego, CA) and Cy3-conjugated AffiniPure goat anti-mouse IgG antibody (red, Jackson Immunoresearch, West Grove, PA). DAPI (blue) was used for nuclear counterstaining. The biodistribution of green fluoresence positive cells in spleen were indicative of phagocytes (expressing mannose receptors) being transfected. In livers, these positive cells merged with red immunofluorescence-stained CD68 positive cells.