

Abstract
Recent advances in three-dimensional (3D) tissue engineering have concomitantly generated a need for new methods to visualize and assess the tissue. In particular, methods for imaging intact volumes of whole tissue, rather than a single plane, are required. Herein, we describe the use of multiphoton microscopy, combined with optical clearing, to noninvasively probe decellularized lung extracellular matrix scaffolds and decellularized, tissue-engineered blood vessels. We also evaluate recellularized lung tissue scaffolds. In addition to nondestructive imaging of tissue volumes greater than 4 mm3, the lung tissue can be visualized using three distinct signals, combined or singly, that allow for simple separation of cells and different components of the extracellular matrix. Because the 3D volumes are not reconstructions, they do not require registration algorithms to generate digital volumes, and maintenance of isotropic resolution is not required when acquiring stacks of images. Once a virtual volume of tissue is generated, structures that have innate 3D features, such as the lumens of vessels and airways, are easily animated and explored in all dimensions. In blood vessels, individual collagen fibers can be visualized at the micron scale and their alignment assessed at various depths through the tissue, potentially providing some nondestructive measure of vessel integrity and mechanics. Finally, both the lungs and vessels assayed here were optically cleared, imaged, and visualized in a matter of hours, such that the added benefits of these techniques can be achieved with little more hassle or processing time than that associated with traditional histological methods.
Introduction
Cardiovascular system diseases account for nearly 1 million deaths per year in the United States, and aging demographics portend that this problem will only expand in the future. Likewise, lung disease is now the third largest killer of Americans.1 While lung transplant is the only curative solution for end-stage lung disease, it is severely hindered by lack of available tissue and poor outcomes 10 years postoperative.2 Available vascular interventions suffer from stenosis, occlusion, and infection.3 Tissue engineering/regenerative medicine approaches that can reconstruct functional healthy cardiovascular, pulmonary, or other tissues and organs would have a major impact on the health of the population. As regenerative medicine advances, tissue-engineered replacements and tissue-engineered constructs will continue to grow in structural complexity.3,4 As such, successful construction of such three-dimensional (3D) tissues and organs is only possible if the tissue engineer has a clear picture of all the individual components: cells, extracellular matrix, scaffold, and the biochemical and mechano-biological modulators of tissue growth that coordinate the structural and biological elements.
Currently, the standard method to identify structural cell–matrix relationships, cell–matrix integration, and tissue regeneration is via microtome sections (typically 5 μm thick) of a 3D engineered construct that may be many millimeters to several centimeters thick. Analyzing a handful of stained two-dimensional (2D) sections from a given sample may not provide sufficient information for evaluating 3D structures. For example, understanding changes in fibril orientation throughout an engineered vessel or observing the distribution of seeded cells throughout a 3D construct can be difficult and requires extrapolation. Visualizing entire constructs, as opposed to just a few sections, can also increase statistical significance of cell distribution measurements and reveal spatial inhomogeneity that might otherwise be missed. Although efforts have been made to reconstruct serial sections into 3D volumes, the process is computationally intensive, requires the laborious cutting, staining, and imaging of the entire sample, and is prone to suffer from cutting distortions and imperfect reconstruction.5 Furthermore, the entire sample must be processed to generate the 3D volume, leaving no remaining tissue for follow-up studies with specialized stains or immunohistochemistry. The field has not been able to image intact tissue-engineered constructs at depths of hundreds of microns beyond the surface of the tissue to provide an easily achieved global view of the tissue engineered organ while maintaining subcellular resolution.6–8
Laser scanning fluorescence microscopies, such as confocal or multiphoton microscopy (MPM), have the potential to circumvent these problems, providing high-resolution 3D image stacks with a large arsenal of fluorescent dyes available for staining. A significant advantage of MPM over standard wide-field and confocal microscopy is that it gives ready access to intrinsic signals, reducing the need for extrinsic dyes and in some cases obviating their use altogether. Intrinsic fluorescence, or autofluorescence, is usually considered background in conventional fluorescence microscopy. In the context of MPM, however, autofluorescence can, in fact, provide useful morphological detail. Sources of intrinsic fluorescence include NADH in cells and elastin and collagen in the extracellular matrix, as well as formalin-induced fluorescence. In addition, MPM can easily be extended to visualize second harmonic generation (SHG) from fibrillar collagen, which produces an exceptionally strong SHG signal. As a natural extension of MPM, SHG only requires appropriate filter selection and the use of transmitted light collection optics. However, even with these modalities, the use of fluorescence microscopy suffers from light scattering in most tissues that ultimately limits the imaging depth to less than ∼100 μm in fixed tissue.
Previous work in our laboratory9–11 used optical clearing, in which scattering of light in fixed tissue is drastically reduced by replacing water with a higher-refractive-index solvent, in combination with MPM to obtain large 3D image stacks from adult mouse organs with subcellular detail. A renewed interest in optical clearing has led to the discovery of new clearing chemicals and better compatibility of conventional protocols with exogenous fluorophores and additional imaging techniques.5,10–15 The use of optical clearing with a 1:2 ratio of benzyl alcohol to benzyl benzoate (BABB), for example, enables imaging to depths greater than 2 mm below the surface of tissue. Detailed tissue morphology was observed using only intrinsic fluorescence signals and SHG from fibrillar collagen. The use of MPM on cleared tissue is nondestructive, enabling the preservation of tissue postimaging for follow-up studies, including routine histology and immunohistochemistry.10,11,16
Herein, we demonstrate the use of optical clearing via BABB and the use of MPM to visualize both a whole decellularized and recellularized lung extracellular matrix scaffold and a decellularized tissue-engineered vascular conduit. We demonstrate imaging of the lung construct using intrinsically fluorescent collagen to visualize the intricate network of branching structures that make up the extracellular matrix scaffold. The use of fluorescently labeled cells also allows visualization of cells within the matrix. Three-dimensional imaging of the tissue-engineered vessel demonstrates the collagen alignment within bioreactor-grown vessels using SHG imaging on unstained tissue. In addition, we report an expedited 10-min BABB clearing protocol for decellularized tissue-engineered vessels and a total processing time for MPM imaging of only a few hours. Therefore, in a span of time comparable to the production of and analysis with frozen sections, a more complete spatial survey of the tissue can be achieved. Postimaging, both the lung and tissue-engineered vascular conduit were sectioned and examined via conventional hematoxylin and eosin (H&E) histology. Both histology and scanning electron microscopy images are shown along with cleared MPM images, for comparison.
Materials and Methods
Preparation of lung scaffolds
Lungs were decellularized as previously described.17 Briefly, the trachea, heart, and lungs were excised en bloc from adult (3–6 month old) rats and treated with decellularization solution containing 5 mM EDTA, 8 mM CHAPS, and 1 M NaCl in 1×PBS. After 2–3 h, lungs were treated with benzonase endonuclease, incubated for 1 h at 37°C, and rinsed extensively with PBS. Decellularized lungs were then rinsed with an antibiotic/antimycotic solution and stored at room temperature until seeding.
Seeding and culture of lung scaffolds
Two Tx150 flasks of confluent A549 cells, a human adenocarcinoma cell line, were labeled with diI membrane dye for 1 h at 37°C in media without serum. The 2.4 mg/mL stock solution of diI was prepared in DMSO and used at 1:1000 in the total labeling volume. Cells were rinsed, trypsinized, and pelleted in preparation for seeding. The pellet was resuspended in 3 mL of culture media (DMEM+10% FBS) and injected into the airway compartment of the lung via the trachea. The seeded lung scaffold was cultured for 2 days with pulsatile vascular perfusion and then fixed with 4% PFA for ∼12–16 h before dehydration and clearing.
Preparation of tissue-engineered vessels
Tissue-engineered vascular grafts (TEVGs) were grown as previously described.3 Briefly, SMCs were isolated from canine carotid arteries and were stored in liquid nitrogen vapor (−135°C) before use. Cells were seeded onto tubular PGA felt scaffolds (3-mm ID) and strained cyclically (2.5% at 2.75 Hz) in a bioreactor to produce grafts. The medium for the growth of TEVGs was low-glucose DMEM with 20% serum, bFGF (10 ng/mL), penicillin G (500 U/mL), copper sulfate (3 ng/mL), l-proline (50 ng/mL), l-alanine (20 ng/mL), and glycine (50 ng/mL) and was changed once per week. l-ascorbic acid was added thrice weekly. After 8 weeks of culture, TEVGs were decellularized as described previously,8 in PBS with 0.12 M sodium hydroxide, 1 M sodium chloride, and 25 mM EDTA, containing 8 mM CHAPS. TEVGs were exposed to detergent solution for 6 h at room temperature and were washed with PBS.
Preparation of tissues for imaging
Lungs
Fixed stained lung specimens were cleared by modifying a previously described method.10 At room temperature, whole lung specimens were rinsed twice with PBS and then dehydrated via a graded methanol assay (50%, 75%, 95%, 100%, 100%), with each step lasting 30 min. After each step, vials were drained of solution and refilled with the next solution in the series.
Dehydrated lungs were then cleared using a 1:2 BABB solution. After the second 100% methanol step, the organs were placed in a 1:1 solution by volume of methanol:BABB. After 30 min, the solution was drained and replaced with a 1:1 solution of methanol:BABB for an additional 30 min. The 1:1 solution was then replaced with 100% BABB solution. Lungs became noticeably clear after 10 min, and imaging was performed after 1 h in BABB. Total tissue processing time was 3.5 h.
Vessels
Vessels were cleared via an expedited 10-min protocol. At room temperature, intact vessels were rinsed twice with PBS. Next, vessels were cleared through a graded assay (50% methanol, 100% methanol, 1:1 methanol:BABB, 100% BABB, 100% BABB). After each step (2 min), vials were drained of solution and immediately refilled with the next solution in the assay. Native decellularized and engineered vessels appeared visibly clear after 1 min in BABB.
Imaging
Images were captured using a previously described custom-built multiphoton microscope.18 The excitation source was a 80 MHz pulsed Ti:Sapphire laser (Mai Tai; Spectra-Physics, Mountain View, CA), tunable between 710 and 990 nm. An excitation wavelength of 740 nm with a ∼100 fs pulse width was used. Based on the Olympus BX51 WI upright fluorescence microscope (Olympus America, Center Valley, PA), the multiphoton microscope was equipped with either a 5×Nikon objective lens with 0.5 numerical aperture (AZ Plan Fluor; Nikon Corp., Tokyo, Japan) or 4×Olympus with 0.28 numerical aperture. Samples were submerged in BABB and placed on a three-axis microscope stage (ASI Imaging, Eugene, OR).
Autofluorescence was filtered through a 460/80 bandpass filter, SHG was filtered through a 370/20 bandpass filter, and DiI was filtered through a 600/75 bandpass filter (Chroma Technologies, Rockingham, VT). One HC-125-02 and two H7422P-40 GaAsP photomultiplier tubes (Hamatsu, Bridgewater, NJ) were used for photon collection. Image z-stacks were collected and processed using SCANIMAGE software.19 Before capturing images, the stage was zeroed at the tissue's surface to quantify depth. Pseudocoloring and movies of rotating image stacks were created using ImageJ software.
Histology
Paraformaldehyde-fixed, BABB-cleared specimens were processed through conventional histology methods. After MPM/SHG imaging, specimens were placed in histology cassettes, embedded in paraffin wax using an unmodified automated progressive “dehydration” protocol (Shandom Excelsior Tissue Processor), cut into 5-μm-thick sections, and mounted on slides. H&E staining was performed using standard manual technique.
Results
Typical processing for biological tissue samples that are either harvested from experimental animals or are the product of tissue engineering efforts includes fixing a portion of the tissue and sending it to histology for processing and subsequent staining. Results from this approach are typified by the images in Figure 1A–C—thin sections that provide a snapshot of the tissue in one plane. When tissue samples are processed for clearing and optical sectioning, representative images at each focal plane bear a recognizable resemblance to traditional histological sections, with the added benefit that the assessment is nondestructive, and therefore, the investigator is able to query a significantly greater amount of tissue. In addition, distinct signals (that would otherwise both be encompassed by eosin staining) that denote the distinction between fibrillar collagens and basement membrane collagens (Fig. 1D–F) can be captured. The identification of these components is conventionally achieved by immunohistochemistry or immunofluorescence. This distinction is achieved by the simultaneous capture and combination of intrinsic fluorescence and SHG channels and visualized by the different colors along the branching airways and alveoli. When multiple 2D images are assembled into a composite image, nearly half a centimeter of tissue can be inspected at once (Fig. 2). With this broad perspective, patterns of cell seeding, in the case of lung, become more apparent in relation to the airways through which they were administered.
FIG. 1.

Traditional histological processing and hematoxylin and eosin staining (A–C) yields two-dimensional (2D) images of decellularized (B) and recellularized (C) lung tissue. Optically cleared tissue imaged with second harmonic generation and two-photon microscopy yields results similar to traditional histology (D–F), with the added differentiation between two different extracellular matrix components: those visualized by two-photon microscopy such as elastin and possibly collagen IV, and the fibrillar collagen signal captured by second harmonic generation (red, green, respectively, panels D–F). DAPI is used to identify the cells (blue, F). Scale bars=200 μm (A–C); 450 μm (D–F). Images (E, F) were taken at a depth of 650 μm below the surface of the tissue.
FIG. 2.
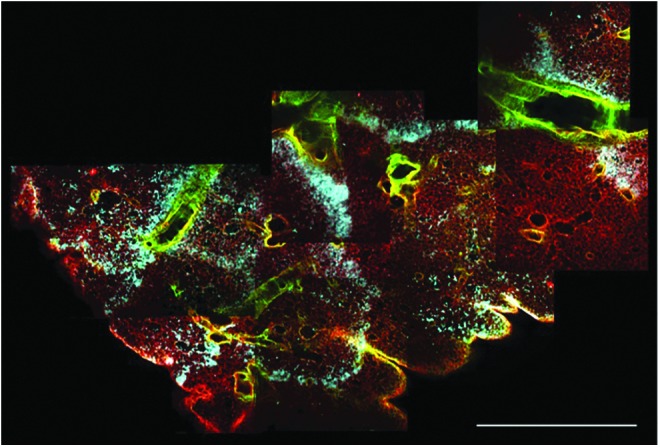
Multiple 2D multiphoton images can be compiled to obtain a high-resolution assessment of the entire width of the lobe of a rat lung. Separate channel colors for fibrillar collagens (green), cells (blue), and other extracellular matrix components (red) aide in visualizing seeding patterns and distribution of the cells within the extracellular matrix scaffold. Scale bar=1.2 mm.
A 3D volume of the BABB-cleared lung specimen provides an even more comprehensive view of the structural relationships between cell distribution (blue), airway branching (green), and alveoli (red) (Figure 3A–E). Not only are these different components characterized by distinct signals (that can then be pseudocolored), but the use of MPM with optically cleared tissue also improves the depth that is accessible for imaging 10-fold, from ∼100 μm (0.1 mm) to over 1 mm (Fig. 3A). Serial images of optically cleared, intact lung constructs are combined into multichannel 3D “optical biopsies” without the need for complex image registration algorithms (Fig. 3B and Supplementary Movie S1; Supplementary Data are available online at www.liebertpub.com/tec). Small branching airways traversing the XZ and YZ planes are readily identified even though isotropic resolution was not obtained (voxel size 0.9, 0.9, 10 μm; Supplementary Movie S2).
FIG. 3.

Different components of the tissue are characterized by distinct signals generated by multiphoton microscopy (MPM), second harmonic generation, and the use of a membrane incorporating dye (A, B). These signals can be pseudocolored and separated from one another in three dimensions to display only cells and fibrillar collagens (C), fibrillar collagens alone (D), or cells alone (E). Scale bars=450 μm (A, B); 250 μm (C–E).
Because of the relative density of alveoli relative to the larger branching structures, the optical volume of tissue is dominated by the alveoli (Fig. 3A, B). With traditional histology, alveoli and airways are visible concurrently (Fig. 1A–C), but the 3D nature of the tissue is lost. Because the MPM images are the combinations of images from multiple channels, however, it is possible to not only retain spatial information but also to separate the channels and select particular channels of interest for greater detail. Exclusion of the autofluorescence channel (red) shows cells organized in a distinctive billowing pattern, reminiscent of filled alveolar spaces, nested within the branched network of the conducting airways (Fig. 3C). Further image partitioning enables better visualization of airway branching and spatial distribution of the cells in three dimensions (Fig. 3D, E). Three-dimensional manipulation and animation allows the user to view the inner surfaces of an airway or medium to large vessel (Supplementary Movie S3).
The ability to manipulate the tissue in three dimensions allows efficient and comprehensive visualization of the various components of the decellularized tissue engineered vascular graft. Because the use of MPM with optically cleared tissue is not destructive to the tissue, multiple views of the tissue, from different directions, can even be obtained from the same tissue. Images of the whole tissue (Fig. 4A, B), sections through the entire vessel (Fig. 4C–E), or through a single wall of the vessel (Fig. 4F–H) can all provide unique information about the structure of the tissue. Furthermore, images can be obtained as longitudinal cross sections of the whole wall (Fig. 5B, C) or as parallel planes within the wall (Fig. 4F–H). Importantly, this dexterity does not compromise the resolution of the images. Indeed, this technique offers a unique means to closely examine the microstructure and organization of the collagen fibers at different depths within intact engineered vessels with a spatial resolution on the order of microns (Supplementary Movie S4). MPM shows that collagen fibers, which are actually quite challenging or impossible to identify in detail on traditional H&E sections (Fig. 5A), can be viewed in such fine detail as to illustrate that the fiber alignment changes with depth through the wall. In tissue-engineered vessels, for example, the fibers are aligned axially, helically, and helically circumferentially at the outer, middle, and inner layers, respectively (Fig. 4F–H). Individual collagen fibers can otherwise only be seen through the use of transmission or scanning electron microscopy (Fig. 5D, E). Previous work with MPM indicates that these three different collagen alignment families in engineered vessels are composed of≈33% axially, ≈37% circumferentially, and ≈30% helically oriented fibers.20 Interestingly, this change in collagen fiber organization is also found in native blood vessels where the collagen fibers are more longitudinally aligned in the adventitia layer and become circumferentially oriented at the media layer.21,22
FIG. 4.

Using MPM and optically cleared tissue, one can obtain a three-dimensional (3D) image of the vessel as a whole (A, B), as well as images of planes perpendicular to the radius; this technique allows effective imaging at depths great enough to traverse the vessel from one wall to the other, a distance of ∼2.5 mm (C–E). Planes within a single wall also reveal the changes in collagen fiber alignment that occur between the outer edge of the vessel and the luminal face, indicating that the ability to image large volumes of tissue does not sacrifice resolution to do so (F–H). Scale bars=1 mm (A–E); 100 μm (F–H).
FIG. 5.

MPM can be used to probe the organization of collagen fibers within the wall of the vessel. Although collagen fibers are difficult to identify in a longitudinal cross section of a tissue engineered vessel with traditional histology (A), MPM readily visualizes these fibers in optical sections (B, C). Although these images do not recapitulate all of the detail provided by traditional techniques such as scanning electron microscopy (D, E), the resulting images effectively convey important structural information related to fiber number and orientation, without the challenges of tissue processing. Scale bars=50 μm (A–C); 1 μm (D); 20 μm (E).
Discussion
Three-dimensional culture of cells and engineering of tissues is an increasingly important aspect of biomedical research. As tissue engineering in particular continues to progress and the tissues become more complex, additional tools will be required to characterize and explore the relationships that emerge between structural elements and function of the cells as well as the resulting tissue within the structure. Current radiological methods are typically not cost effective and lack the resolution for single cell visualization.
Histological processing, a mainstay of tissue study, provides thin 2D sectional analysis of 3D tissues. In addition, routine histology often provides information about only the first 50–100 μm of the tissue from the outer edge inward. Information about deeper portions of the tissue requires serial sectioning, possibly discarding intervening tissue or distorting structures. These additional steps not only increase the time required to obtain potentially vital information on tissue samples that may have already taken several months to generate (in the case of tissue engineered vessels in particular) but also increase the risk of losing or overlooking important information about local phenomena, especially in the case of engineered tissues and organs, which are often characterized by spatially heterogeneous cell seeding. By viewing the tissue in its entirety before sectioning, one could identify regions of interest based on cell density, organization, or structure and then concentrate additional studies on this population (Fig. 6). This would not only improve the efficiency of data collection but could also illuminate patterns associated with cell seeding or proliferation that might not be as prominent in two dimensions as in three. Initial cell engraftment, for example, might occur more readily or frequently in one region of the lung (for example, the center), whereas long-term survival during culture might favor peripheral locations. Additional characteristics of the scaffold, such as fluid flow through the vasculature might also be well defined by the methods described here.
FIG. 6.

Optical sections through a 3D volume (A, B) illustrate the ability to identify regions of interest for sectioning and further analysis noninvasively (C, D). Because the distribution of cells within the reseeded decellularized matrix scaffold is nonuniform, the ability to identify cutting depth a priori avoids loss of potentially important information. Depth into the tissue is 0, 160, and 760 μm, respectively (C–E). Scale bars=500 μm (A, B); 250 μm (C–E).
The collagen fiber structure in native blood vessels is also highly 3D, with fibers in native vessels characterized by circumferential, helical, and axial alignment. Visualization of this information, particularly with respect to depth, would not be possible without the use of optical clearing and is not even approximated by more rudimentary techniques. The combination of optically cleared engineered blood vessels and MPM, however, has demonstrated the ability to visualize collagen matrix alignment and orientation at the level of individual fibers, which range from 0.5 to 1.0 μm in diameter, with micron spatial resolution.23 This allows one to visualize the change in collagen fiber orientation and alignment across the entire vascular wall as well. Ultrastructure and organization of collagen fiber have a significant impact on the mechanical properties of both native and engineered vessels and can alter the ratio of forward- and backward-scattered SHG signal.20 Therefore, visualization of collagen orientation via SHG provides valuable information at the submicron level for understanding the vascular mechanics, which would have been difficult to achieve by other means.24
Importantly, the visualization of collagen fibers is independent of the detection of elastin. While collagen fibers produce an SHG signal, elastin emits a fluorescence signal under two-photon excitation. Because their signals inhabit different channels, collagen and elastin can easily be viewed together or separately. This distinction has been used successfully to image cross sections of elastic arterioles25 and could provide additional information about the functional capacity of lungs and vessels in the future; efforts to engineer these tissues thus far have had limited success in retaining or generating elastin.7,26,27
The ability to separate the elastin signal from the signal generated by nonfibrillar collagens, however, requires additional investigation. Imaging of the decellularized lung, for example, which exhibits loss of nearly 60% of the original elastin content,4 retains a fairly strong fluorescence signal. Coupled with the pattern of the signal (i.e., evenly distributed throughout the alveoli as a continuous boundary around the alveoli), this observation suggests that the two-photon fluorescence signal may in fact be a product of excitation of collagen IV, the predominant basement membrane protein in the alveoli.28
Overall, we have presented here a tool that overcomes many of the obstacles that are attendant to traditional histological examination of tissues, whether engineered or native. Optical clearing presents the opportunity to view a large amount of tissue rapidly, simultaneously, and in all three dimensions in a nondestructive way. As a result, this technique provides important structural information about the tissue overall and even about the fibers that make up these structures, easily surpassing the spatial resolution of either magnetic resonance imaging (MRI) or micro-computed tomography (CT), which fail to provide cell-level resolution.10 Finally, MPM and SHG can provide a wealth of information about the materials present in a construct since the signals of collagens and cells, for example, inhabit different channels, allowing for separation during analysis.
Although the application of this technique for the extraction of structural data still needs to be improved (airways and vessels in the lung, for example, are largely indistinguishable), we anticipate that improvements or modifications that allow greater focus on function will be even more valuable. Exploration of antibody staining, first demonstrated by Yokomizo in 2012,29 could not only provide a method to distinguish biochemically distinct but structurally similar structures such as vessels and airways, but would also open the door to exploring protein expression and cell function within the complicated 3D structures of whole organs. To avoid the added time and to circumvent diffusion limitations of staining a whole tissue with antibodies, the use of fluorescently labeled reporter lines could enhance the quantity and type of information that could be obtained. Cells that bear a constitutive or inducible marker of interest could be used to populate a decellularized matrix for example and track changes in phenotype in three dimensions. Alternatively, these cells could be used to probe the role of a particular cell type in a model of injury or repair in vivo. Administration of exogenous cells that either possess the reporter or are prelabeled with membrane-incorporating dyes, as described here (e.g., tagged neutrophils or progenitor cells), would allow these tagged cells to show up in the tissue that would then be harvested from the model animal assessed with these imaging techniques. Modalities not explored here that could also be useful in future tissue engineering applications include fluorescence lifetime imaging, which is a tool for identifying tissue components based on pH or oxygen saturation.10 Since these are critical factors that affect the growth and, in the case of stem cells, the differentiation of tissue, this may be a useful avenue to pursue.
Supplementary Material
Acknowledgments
Funding was provided by the National Science Foundation under grant NSF CAREER Award DBI-0953902 (Levene) and by the National Institutes of Health under grants R01 HL098220-01 (L.E.N.) and R01 HL083895-06A1 (L.E.N.).
Disclosure Statement
L.E.N. has a financial interest in Humacyte, Inc., a regenerative medicine company. Humacyte did not fund these studies, and Humacyte did not affect the design, interpretation, or reporting of any of the experiments herein.
References
- 1.Hoyert L.D., and Xu J.Deaths: preliminary data for 2011. Natl Vital Stat Rep 61,6, 2012 [PubMed] [Google Scholar]
- 2.Valapour M., et al. OPTN/SRTR 2011 annual data report: lung. Am J Transplant 13Suppl 1,149, 2013 [DOI] [PubMed] [Google Scholar]
- 3.Dahl S.L., et al. Readily available tissue-engineered vascular grafts. Sci Transl Med 3,68ra69, 2011 [DOI] [PubMed] [Google Scholar]
- 4.Petersen T.H., et al. Tissue-engineered lungs for in vivo implantation. Science 329,538, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hama H., et al. Scale: a chemical approach for fluorescence imaging and reconstruction of transparent mouse brain. Nat Neurosci 14,1481, 2011 [DOI] [PubMed] [Google Scholar]
- 6.Quint C., Arief M., Muto A., Dardik A, and Niklason L.E.Allogeneic human tissue-engineered blood vessel. J Vasc Surg 55,790, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petersen T.H., Calle E.A., Colehour M.B., and Niklason L.E.Matrix composition and mechanics of decellularized lung scaffolds. Cells Tissues Organs 195,222, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gong Z., and Niklason L.E.Use of human mesenchymal stem cells as alternative source of smooth muscle cells in vessel engineering. Methods Mol Biol 698,279, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parra S.G., Chia T.H., Zinter J.P., and Levene M.J.Multiphoton microscopy of cleared mouse organs. J Biomed Opt 15,036017, 2010 [DOI] [PubMed] [Google Scholar]
- 10.Vesuna S., Torres R., and Levene M.J.Multiphoton fluorescence, second harmonic generation, and fluorescence lifetime imaging of whole cleared mouse organs. J Biomed Opt 16,106009, 2011 [DOI] [PubMed] [Google Scholar]
- 11.Parra S.G., Vesuna S.S., Murray T.A., and Levene M.J.Multiphoton microscopy of cleared mouse brain expressing YFP. J Vis Exp e3848, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chung K., et al. Structural and molecular interrogation of intact biological systems. Nature 497,332, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ke M.T., Fujimoto S., and Imai T.SeeDB: a simple and morphology-preserving optical clearing agent for neuronal circuit reconstruction. Nat Neurosci 16,1154, 2013 [DOI] [PubMed] [Google Scholar]
- 14.Erturk A., et al. Three-dimensional imaging of solvent-cleared organs using 3DISCO. Nat Protoc 7,1983, 2012 [DOI] [PubMed] [Google Scholar]
- 15.Kuwajima T., et al. ClearT: a detergent- and solvent-free clearing method for neuronal and non-neuronal tissue. Development 140,1364, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Torres R., Vesuna S., and Levene M.J.High-resolution, 2- and 3-dimensional imaging of uncut, unembedded tissue biopsy samples. Arch Pathol Lab Med 2013[Epub ahead of print]; DOI: 10.5858/arpa.2013-0094-OA [DOI] [PubMed] [Google Scholar]
- 17.Calle E.A., Petersen T.H., and Niklason L.E.Procedure for lung engineering. J Vis Exp pii:, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zinter J.P., and Levene M.J.Maximizing fluorescence collection efficiency in multiphoton microscopy. Opt Express 19,15348, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pologruto T.A., Sabatini B.L., and Svoboda K.ScanImage: flexible software for operating laser scanning microscopes. Biomed Eng Online 2,13, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dahl S.L., Vaughn M.E., and Niklason L.E.An ultrastructural analysis of collagen in tissue engineered arteries. Ann Biomed Eng 35,1749, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bou-Gharios G., Ponticos M., Rajkumar V., and Abraham D.Extra-cellular matrix in vascular networks. Cell Prolif 37,207, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Timmins L.H., Wu Q., Yeh A.T., Moore J.E., Jr., and Greenwald S.E.Structural inhomogeneity and fiber orientation in the inner arterial media. Am J Physiol Heart Circ Physiol 298,19, 2010 [DOI] [PubMed] [Google Scholar]
- 23.Niklason L.E., et al. Enabling tools for engineering collagenous tissues integrating bioreactors, intravital imaging, and biomechanical modeling. Proc Natl Acad Sci U S A 107,3335, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams R.M., Zipfel W.R., and Webb W.W.Interpreting second-harmonic generation images of collagen I fibrils. Biophys J 88,1377, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zipfel W.R., et al. Live tissue intrinsic emission microscopy using multiphoton-excited native fluorescence and second harmonic generation. Proc Natl Acad Sci U S A 100,7075, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niklason L.E., et al. Morphologic and mechanical characteristics of engineered bovine arteries. J Vasc Surg 33,628, 2001 [DOI] [PubMed] [Google Scholar]
- 27.Niklason L.E., et al. Functional arteries grown in vitro. Science 284,489, 1999 [DOI] [PubMed] [Google Scholar]
- 28.Hinenoya N., et al. Type IV collagen alpha chains of the basement membrane in the rat bronchioalveolar transitional segment. Arch Histol Cytol 71,185, 2008 [DOI] [PubMed] [Google Scholar]
- 29.Yokomizo T., et al. Whole-mount three-dimensional imaging of internally localized immunostained cells within mouse embryos. Nat Protoc 7,421, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.