

Abstract
Abstract
Background
Immunohistochemical staining for mismatch repair proteins is efficient and widely used to identify mismatch repair defective tumors. The tumors typically show uniform and widespread loss of MMR protein staining. We identified and characterized colorectal cancers with alternative, heterogenous mismatch repair protein staining in order to delineate expression patterns and underlying mechanisms.
Methods
Heterogenous staining patterns that affected at least one of the mismatch repair proteins MLH1, PMS2, MSH2 and MSH6 were identified in 14 colorectal cancers. Based on alternative expression patterns macro-dissected and micro-dissected tumor areas were separately analyzed for microsatellite instability and MLH1 promoter methylation.
Results
Heterogenous retained/lost mismatch repair protein expression could be classified as intraglandular (within or in-between glandular formations), clonal (in whole glands or groups of glands) and compartmental (in larger tumor areas/compartments or in between different tumor blocks). These patterns coexisted in 9/14 tumors and in the majority of the tumors correlated with differences in microsatellite instability/MLH1 methylation status.
Conclusions
Heterogenous mismatch repair status can be demonstrated in colorectal cancer. Though rare, attention to this phenomenon is recommended since it corresponds to differences in mismatch repair status that are relevant for correct classification.
Virtual Slides
The virtual slide(s) for this article can be found here: http://www.diagnosticpathology.diagnomx.eu/vs/1771940323126788
Keywords: Mismatch repair, immunohistochemistry, heterogeneity, MLH1, MSH2, MSH6, PMS2
Background
Mismatch repair (MMR) defects characterize 2-4% of colorectal cancers linked to Lynch syndrome and 15% of sporadic colorectal cancers caused by epigenetic MLH1 promoter methylation. Various strategies can be used to preselect colorectal cancers for MMR protein testing, e.g. clinical guidelines for hereditary cancer, MMR prediction models that combine clinical and pathological information and potentially novel biomarker-based strategies [1-5].
Universal assessment of immunohistochemical MMR staining is increasingly applied in colorectal cancer diagnostics in order to identify cases suspected of Lynch syndrome for further molecular diagnostics and to obtain treatment-predictive information linked to somatic methylation of MLH1[6].
The monoclonal antibodies used for immunohistochemical MMR protein staining generally result in stable and consistent staining patterns with retained staining or loss of staining. Functional interaction between the MLH1/PMS2 and the MSH2/MSH6 proteins implies that the expression pattern of the heterodimerizing protein partner may be used to direct mutation analysis. Aberrant MMR function typically leads to complete loss of nuclear staining in the tumor cells, particularly when linked to MLH1 promoter hypermethylation that leads to complete gene silencing [7]. In Lynch syndrome, the multitude of disease-predisposing mutations may have variable effects on epitope expression, from complete loss to weak or retained expression for one or both heterodimerizing proteins [8,9]. Variable epitope expression may also result in alternative expression patterns, e.g. cytoplasmic staining and perinuclear staining, which are typically present throughout the tumor [10]. MMR protein immunostainings are generally stable and relatively easy to interpret; though challenges and pitfalls have been reported with false positive as well as false negative interpretations [10-12]. Most commonly, these observations relate to technical artifacts caused by suboptimal fixation or paraffin-embedding, necrotic areas, sample storage, antibody specificity, clone selection or staining conditions [13,14]. Also, neoadjuvant chemotherapy and radiotherapy may influence the results with a particular effect on MSH2/MSH6 staining [15,16]. Heterogenous expression patterns with retained staining in the adenomatous part and loss of staining in a smaller, invasive part of the tumor have been reported but their relevance is uncertain [17]. We systematically collected colorectal cancers with heterogenous MMR protein staining patterns for detailed analysis with correlations e.g. to MSI status and MLH1 promoter methylation.
Methods
Materials
Colorectal cancers with heterogenous MMR protein expression were identified during evaluations at the Departments of Pathology, Helsingborg Hospital, Sweden and Hvidore Hospital, Denmark. Following the first observation of heterogenous MMR protein staining in 2007, two gastrointestinal pathologists (PJ and SH) collected all such cases identified at these two institutions during 5 years. In total, 14 colorectal cancers with heterogenous MMR protein expression were identified for in-depth analysis (Table 1). The materials consisted of resection specimens from 12 colon cancers and 2 rectal cancers. None of the patients had received neoadjuvant radiotherapy or chemotherapy. All cases were histologically re-evaluated by one pathologist (P.J.). Tumor stage was determined according to the American Joint Cancer Committee/Union Internationale Contre le Cancer (AJCC/UICC) staging system and the grade according to the WHO system. Mucinous cancers were considered poorly differentiated. A tumor was classified as mucinous cancer if more than 50% of the tumor area showed such differentiation [18]. Tumors with mucinous components that encompassed &50% of the area were classified as having a mucinous component, though not fulfilling the criteria for mucinous tumors [19]. Mucinous cancer was observed in 6 cases. MMR gene mutation testing had been performed in 8 cases, 4 of which carried disease-predisposing mutations. Ethical approval for the study was granted from the ethical committees at Lund University, Sweden and at the Capital Region, Copenhagen.
Table 1.
Summary of clinical and pathological data
| Case | Sex/Age | Tumor location | Histologic type | Differentiation | pTNM | Stage | MMR gene mutation |
|---|---|---|---|---|---|---|---|
| 1 |
F/57 |
Cecum |
Adca |
Moderate |
T2N0MX |
I |
Not tested |
| 2 |
F/33 |
Cecum |
Mucinous adca |
Poor |
T2N0MX |
I |
MSH2 |
| 4 |
M/41 |
Rectum |
Adca |
Moderate |
T3N0MX |
II |
MSH6 |
| 5 |
F/63 |
Transverse colon |
Mucinous adca |
Moderate |
T4N1MX |
III |
Not tested |
| 6 |
F/82 |
Rectum |
Mucinous adca |
Moderate |
T2N0MX |
I |
Not identified |
| 8 |
M/85 |
Cecum |
Adca |
Poor |
T4N0MX |
II |
Not identified |
| 9 |
M/71 |
Ascending colon |
Mucinous adca |
Poor |
T3N0MX |
II |
Not tested |
| 10 |
F/66 |
Ascending colon |
Mucinous adca |
Poor |
T3N0MX |
II |
Not identified |
| 11 |
F/57 |
Descending colon |
Adca |
Moderate |
T3N0MX |
II |
MSH6 |
| 12 |
F/55 |
Ascending colon |
Adca |
Poor |
T3N0MX |
II |
MLH1 |
| 14 |
M/82 |
Ascending colon |
Adca |
Moderate |
T3N0MX |
II |
Not tested |
| 15 |
M/83 |
Ascending colon |
Adca |
Moderate |
T3N0MX |
II |
Not tested |
| 16 |
M/74 |
Transverse colon |
Mucinous adca |
Poor |
T3N1MX |
III |
Not tested |
| 17 | F/74 | Cecum | Adca | Poor | T3N1MX | III | Not identified |
F: female; M: Male; Adca: adenocarcinoma; MMR: mismatch repair.
MMR protein immunostaining
Sections from all tumor blocks (n = 4-11) from the 14 cases were subjected to independent MMR protein staining using alternative MMR protein antibodies from other manufacturers (Table 2). Fresh 4-μm sections from formalin-fixed, paraffin-embedded tumors were mounted on Dako REAL™ Capillary gap microscope slides (Dako, Glostrup, Denmark). The slides were dried overnight at room temperature and thereafter at 60°C for 1–2 hours. The tissue was deparaffinized in xylene for two times 5 min, followed by 5 min each in 99.5% and 95% ethanol and 5 min in distilled water. Heat-induced epitope retrieval was achieved by pressure boiler-treatment in ethylene diamine tetraacetic acid (EDTA)-Tris buffer (1:10 mM, pH 9.0) for 20 min. Hereafter, the slides were cooled for 20 min and rinsed in distilled water. Immunostaining was performed using the Dako Autostainer and the EnVision™ visualization method (Dako, Glostrup, Denmark). Endogenous peroxidase activity was blocked for 5 min and primary mouse monoclonal IgG antibodies were used (Table 2). Following primary antibody incubation, the slides were incubated with EnVision™/horseradish peroxidase (HRP) rabbit/mouse (Dako) and stained using the EnVision™ Detection System peroxidase/DAB rabbit/mouse (Dako). The immunohistochemical stainings were classified as retained, lost or reduced, i.e. a weaker than expected staining in the tumor cells compared to the stromal cells. The areas of the respective expression patterns were estimated in each block end expressed in percentage.
Table 2.
Information on the MMR protein antibodies used
| MMR protein | Lab | Supplier | Clone | Dilution | Immunogen/epitope |
|---|---|---|---|---|---|
| MLH1 |
L |
BD Pharmingen |
G168-15 |
1:100 |
Full-length |
| |
C |
DAKO |
ES05 |
RTU |
Recombinant protein, 210 aa |
| |
L |
DAKO |
ES05 |
1:100 |
Recombinant protein, 210 aa |
| PMS2 |
L |
BD Pharmingen |
A16-4 |
1:300 |
aa 431–862, C-terminal |
| |
H |
Ventana (Cell Marque) |
EPR3947 |
RTU |
100 aa, C-terminal |
| |
C |
Epitomics |
EPR3947 |
1:50 |
100 aa, C-terminal |
| MSH2 |
L |
Calbiochem |
FE11 |
1:100 |
C-terminal |
| |
H |
Ventana (Cell Marque) |
G219-1129 |
RTU |
Full-length |
| |
C |
Novocastra |
25D12 |
1:50 |
Full-length |
| MSH6 |
C |
Epitomics |
EP49 |
1:100 |
Synthetic peptide, N-terminal |
| |
L |
Epitomics |
EP3945 |
1:100 |
Synthetic peptide, N-terminal |
| L | BD Transduction Lab. | 44 | 1:500 | Synthectic peptide aa 225-333 |
MMR: mismatch repair; L: Lund; H Helsingborg; C: Copenhagen; RTU: ready to use.
Microsatellite instability analysis
To assess the impact from heterogenous MMR protein stainings on MMR protein function, the tumors were subjected analysis of microsatellite instability (MSI). Depending on the extent of the area involved, microdissection or macrodissction was used to obtain material from areas with the respective expression patterns (Table 3). Laser capture micro-dissection was performed using Polyethylene Teraphthalate (PET)-membrane FrameSlides (Carl Zeiss MicroImaging, Germany). In tumors where larger areas showed variable expression patterns, macro-dissection was performed. Tissue from 2–6 tumor areas with the respective MMR protein stained patterns were collected from 10-μm tissue sections. DNA extraction was performed using QIAamp® DNA micro kit (Qiagen) for laser micro-dissected tissues and using the QIAcube machine (Qiagen) or the QIAamp® DNA FFPE tissue kit (Qiagen) for macro-dissected tissues or whole tumor sections. MSI analyses were performed using the MSI Analysis System, Version 1.2 (Promega, Madison, WI), PCR products were size separated on a 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA). The results were evaluated using the GeneMapper® software Version 4.0 (Applied Biosystems, Foster City, CA). The analysis included the 5 mononucleotide markers BAT-25, BAT-26, NR-21, NR-24, and MONO-27 (Promega, MSI Analysis System, Version 1.2, Madison, WI). Tumors with instability for 1 marker were classified as MSI low, tumors with instability for ≥2 markers were classified as MSI-high (MSI-H), and tumors with stability for all markers were classified as microsatellite stable (MSS).
Table 3.
Summary of MMR heterogeneity
| Case | Block no. | Hetero-geneity | Pattern | Involved area (%) |
MMR protein immunostaining |
Fraction of MSI in markers | MLH1 promoter methylation | |||
|---|---|---|---|---|---|---|---|---|---|---|
| MLH1 | PMS2 | MSH2 | MSH6 | |||||||
| 1 |
1A¤/* |
+ |
CL |
15 |
+/− |
+/− |
+ |
+ |
0/5 and 4/5 |
+/− |
| |
1B |
+ |
CL |
5 |
- |
- |
+/R |
+/− |
|
|
| |
1C |
- |
H |
0 |
- |
- |
+ |
+ |
|
|
| |
1D |
- |
H |
0 |
- |
- |
+ |
+ |
|
|
| |
1E¤ |
+ |
CL, IG |
70 / 10 |
+/− |
+/− |
+/R |
+/− |
0/3 vs 4/5 |
|
| 2 |
24 |
- |
H |
0 |
+ |
+ |
+ |
+ |
|
- |
| |
25* |
- |
H |
0 |
+ |
+ |
+ |
+ |
0/5 |
|
| |
26 |
- |
H |
0 |
+ |
+ |
+ |
+ |
|
|
| |
27* |
- |
H |
0 |
+ |
+ |
+ |
+ |
0/5 |
|
| |
28 |
+ |
CL, IG |
15 |
+/− |
+/− |
+ |
+ |
|
|
| |
29* |
+ |
CL, IG |
40 |
+/− |
+/− |
+ |
+ |
5/5 |
|
| |
30 |
+ |
IG |
10 |
+/− |
+/− |
+ |
+ |
|
|
| |
31 |
+ |
IG |
5 |
+/− |
+/− |
+ |
+ |
|
|
| |
32 |
+ |
IG |
5 |
+/− |
+/− |
+ |
+ |
|
|
| 4 |
2 |
+ |
IG |
97 |
+ |
+ |
+/− |
R/- |
|
NE |
| |
3¤/* |
+ |
CL, IG |
95 |
+ |
+ |
+/− |
R/- |
3/3 vs 3/5 |
|
| |
4 |
+ |
CL, IG |
80 |
+ |
+ |
+/− |
R/- |
|
|
| |
5* |
+ |
IG |
95 |
+ |
+ |
+/− |
R/- |
5/5 |
|
| 5 |
20* |
+ |
CL, IG |
50 / 100 |
+ |
+/− |
- |
R/- |
5/5 |
- |
| |
21 |
+ |
IG |
100 |
+ |
+ |
- |
R/- |
|
|
| |
23 |
+ |
IG |
100 |
+ |
+ |
- |
R/- |
|
|
| |
24 |
+ |
IG |
100 |
+ |
+ |
- |
R/- |
|
|
| |
25 |
+ |
COM, IG |
100 |
+ |
- |
- |
R/- |
|
|
| |
26 |
+ |
COM, IG |
100 |
+ |
- |
- |
R/- |
|
|
| |
27 |
+ |
COM IG |
100 |
+ |
- |
- |
R/- |
|
|
| |
28 |
+ |
COM, IG |
100 |
+ |
- |
- |
R/- |
|
|
| |
29 |
+ |
COM, IG |
100 |
+ |
- |
- |
R/- |
|
|
| |
30 |
+ |
COM, IG |
100 |
+ |
- |
- |
R/- |
|
|
| |
31 |
+ |
COM, IG |
100 |
+ |
- |
- |
R/- |
|
|
| 6 |
5¤ |
+ |
IG |
100 |
+ |
R/- |
+ |
+ |
0/5 vs 0/4 |
NE |
| |
6* |
+ |
IG |
100 |
+ |
R/- |
+ |
+ |
0/5 |
|
| |
7* |
- |
H |
0 |
+ |
+ |
+ |
+ |
0/5 |
|
| |
8* |
+ |
IG |
100 |
+ |
R/- |
+ |
+ |
0/5 |
|
| |
9* |
+ |
IG |
95 |
+ |
R/- |
+ |
+ |
0/5 |
|
| |
10 |
+ |
IG |
95 |
+ |
R/- |
+ |
+ |
|
|
| |
11* |
+ |
IG |
100 |
+ |
R/- |
+ |
+ |
0/5 |
|
| 8 |
5* |
- |
H |
0 |
- |
- |
+ |
+ |
5/5 |
+ |
| |
6 |
+ |
CL, IG |
60 |
- |
- |
+ |
R/- |
|
|
| |
7 |
+ |
CL |
20 |
- |
- |
+ |
+/− |
|
|
| |
8 |
- |
H |
0 |
- |
- |
+ |
+ |
|
|
| |
9 |
- |
H |
0 |
- |
- |
+ |
+ |
|
|
| |
10 |
+ |
CL, IG |
20 |
- |
- |
+/R |
+/− |
|
|
| |
11 |
- |
H |
0 |
- |
- |
+ |
+ |
|
|
| |
12 |
+ |
CL |
20 |
- |
- |
+/− |
+/− |
|
|
| 9 |
16 |
- |
H |
0 |
+ |
+ |
+ |
+ |
|
+ |
| |
19 |
- |
H |
0 |
+ |
+ |
+ |
+ |
|
|
| |
20* |
- |
H |
0 |
+ |
+ |
+ |
+ |
0/5 |
|
| |
21* |
- |
COM |
100 |
- |
- |
+ |
+ |
5/5 |
|
| |
22 |
- |
H |
0 |
+ |
+ |
+ |
+ |
|
|
| |
40 |
- |
H |
0 |
+ |
+ |
+ |
+ |
|
|
| |
44 |
- |
H |
0 |
+ |
+ |
+ |
+ |
|
|
| 10 |
6 |
+ |
CL |
50 |
- |
- |
+/− |
+/− |
|
+ |
| |
7 |
- |
H |
0 |
- |
- |
+ |
+ |
|
|
| |
8 |
- |
H |
0 |
- |
- |
+ |
+ |
|
|
| |
9* |
- |
H |
0 |
- |
- |
+ |
+ |
5/5 |
|
| |
10 |
- |
H |
0 |
- |
- |
+ |
+ |
|
|
| |
11 |
- |
H |
0 |
- |
- |
+ |
+ |
|
|
| 11 |
3 |
- |
H |
0 |
- |
- |
+ |
+ |
|
+ |
| |
4 |
+ |
CL, IG |
5 |
- |
- |
+/− |
+/− |
|
|
| |
5 |
- |
H |
0 |
- |
- |
+ |
+ |
|
|
| |
6* |
- |
H |
0 |
- |
- |
+ |
+ |
5/5 |
|
| |
7 |
- |
H |
0 |
- |
- |
+ |
+ |
|
|
| |
8* |
+ |
CL, IG |
30 |
- |
- |
+/R |
+/− |
5/5 |
|
| 12 |
3* |
+ |
CL |
5 |
- |
- |
+/− |
+/− |
5/5 |
+ |
| |
4 |
+ |
CL, IG |
90 |
- |
- |
+/− |
+/− |
|
|
| |
5 |
+ |
CL, IG |
80 |
- |
- |
+/− |
+/− |
|
|
| |
6 |
- |
H |
100 |
- |
- |
- |
- |
|
|
| |
7 |
+ |
CL |
60 |
- |
- |
+/− |
+/− |
|
|
| |
8 |
+ |
CL, IG |
90 |
- |
- |
+/− |
+/− |
|
|
| |
9* |
+ |
CL, IG |
60 |
- |
- |
+/− |
+/− |
5/5 |
|
| 14 |
1A* |
- |
H |
0 |
- |
- |
+ |
+ |
5/5 |
+ |
| |
1B* |
+ |
CL |
5 |
- |
- |
+/R |
+/− |
5/5 |
|
| |
1C* |
+ |
CL, IG |
30 |
- |
- |
R/- |
+/− |
5/5 |
|
| |
1D* |
+ |
CL |
40 |
- |
- |
+/R |
+/− |
5/5 |
|
| |
1E* |
+ |
CL, IG |
40 |
- |
- |
R/- |
+/− |
5/5 |
|
| 15 |
4A* |
+ |
IG |
100 |
+ |
+ |
+/R |
R/- |
5/5 |
NE |
| |
4B* |
+ |
IG |
100 |
+ |
+ |
+ |
R/- |
5/5 |
|
| |
4C |
+ |
IG |
100 |
+ |
+ |
+/R |
R/- |
|
|
| |
4D |
+ |
IG |
100 |
+ |
+ |
+ |
R/- |
|
|
| |
4E |
+ |
IG |
100 |
+ |
+ |
+/R |
R/- |
|
|
| 16 |
6A |
+ |
IG |
15 |
- |
- |
R |
+/− |
|
+ |
| |
6B* |
+ |
CL |
3 |
- |
- |
+/R |
+/− |
5/5 |
|
| |
6C |
+ |
CL |
10 |
- |
- |
+/R |
+/− |
|
|
| |
6D |
+ |
IG |
5 |
- |
- |
R |
+/− |
|
|
| |
6E |
+ |
IG |
5 |
- |
- |
+/R |
+/− |
|
|
| 17 |
18* |
+ |
CL |
55 |
- |
- |
+/− |
+/− |
5/5 |
+ |
| |
19 |
- |
H |
0 |
- |
- |
+ |
+ |
|
|
| |
20 |
+ |
CL |
8 |
- |
- |
+/− |
+/− |
|
|
| |
21 |
- |
H |
0 |
- |
- |
+ |
+ |
|
|
| 22 | - | H | 0 | - | - | + | + | |||
microdissection; *: macrodissection; CL: clonal, IG: intraglandular; COM: compartmental; H: homogenous; R: reduced staining; +/-: heterogenous staining; MMR: mismatch repair; MSI: microsatellite instability; NE: not evaluated.
Methylation-specific PCR analysis
Extracted DNA was treated with bisulfite using the EZ DNA Methylation-Lightning™ Kit (Zymo Research, CA, USA) according to the manufacturer’s instructions. MLH1 promoter methylation status was analyzed by means of a fluorescence-based, real-time methylation-specific PCR assay, as described previously [20]. Two sets of primers and probes, designed specifically for bisulfite-converted DNA, were used: MLH1-M2B for the methylation-specific reaction [21] and ALU-C4 for the methylation independent control reaction used to measure the amount of bisulfite-converted input DNA [22]. Amplification was performed on QuantStudio™ 12 K Flex Real-Time PCR System (Life Technologies). Samples were run in duplicate, including positive and negative controls.
Flow cytometry
Flow cytometric DNA analysis was performed as previously described [23,24]. The separated cells were then treated with ribonuclease (Sigma-Aldrich, Stockholm, Sweden), incubated with trypsin for 48 h (Merck, Darmstadt, Germany), and stained with propidium iodide (Sigma-Aldrich, Stockholm). Flow cytometric DNA analysis was performed in a FACS Caliber (Becton, Dickinson, BD Biosciences, USA). Up to 20,000 nuclei were analysed from each sample. The DNA histograms obtained were automatically processed using Modfit LT 3.3™ software. The DNA index (DI) was calculated as the ratio of the respective modal channel values of the non-diploid and the diploid G0/G1 peaks. The S-phase fraction (Spf) was estimated assuming that the S-phase compartment constituted a rectangular distribution between the modal values of the G0/G1 and G2 peaks.
Results
Immunohistochemical staining using alternative MMR protein antibodies confirmed heterogenous MMR protein expression in all 14 tumors. Heterogenous expression affected MLH1/PMS2 in 3 tumors, PMS2 in 2 tumors, MSH2/MSH6 in 10 tumors (of which two also expressed heterogeneity for MLH1/PMS2) and MSH6 only in 1 tumor (in which one block also expressed heterogeneity for PMS2). Areas with alternative expression patterns were well demarcated and appeared in three distinct patterns: “intraglandular” (retained/lost staining within or in between glandular formations), “clonal” (retained/lost staining in whole glands or groups of glands) and “compartmental” (retained/lost staining in larger tumor areas/compartments leading to retained/lost staining in between different tumor blocks) (Figure 1, Table 3). Various heterogenous expression patterns co-existed in 9/14 tumors, most commonly as intraglandular and clonal heterogeneity (figure 1c). The heterogenous staining patterns were present in 3-100% of the examined tumor area. In 4/14 cases, all tumor blocks showed heterogeneity, whereas the remaining tumors showed heterogeneity in a variable fraction of the tumor blocks (Table 3).
Figure 1.
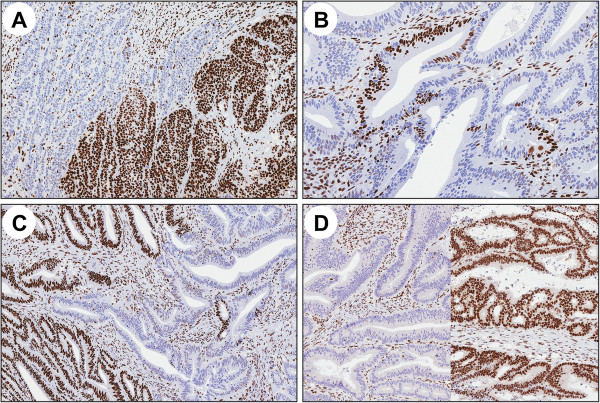
Examples of the different MMR protein staining patterns. A) clonal loss, B) intraglandular loss, C) co-existence of clonal and intraglandular loss and D) compartmental loss with different patterns in two separate tumor blocks.
MSI was demonstrated in 13/14 tumors. Intra-tumor differences in MMR status, i.e.MSI versus MSS, in line with MMR protein staining expression was observed in 3 tumors (Table 3; case 1, 2 and 9). Non-consistent, homogenous MSI status in tumors with heterogenous MMR protein expression was observed in 2 cases (Table 3; cases 4 and 6). MLH1 promoter methylation was demonstrated in all 7 cases with complete (non-heterogenic) loss of MLH1/PMS2. In 2 cases (Table 3; cases 1 and 9) heterogenous MMR protein staining for MLH1/PMS2 correlated with heterogenous MLH1 promoter methylation, i.e. tumor areas with retained MLH1 expression did not show MLH1 methylation, whereas areas with loss of MLH1 expression showed MLH1 methylation. Concordant immunostaining and methylation status suggest functional intratumoral heterogeneity (Figure 2). DNA flow cytometric analysis was performed in one tumor (case 1) and demonstrated differences in DNA content within the heterogenous areas, which had DNA indices of 1.13 and 1.57, respectively (Figure 2).
Figure 2.

An adenocarcinoma (case 1) with 4 different expression patterns and various combinations of heterogeneity, loss of MLH1/PMS2 and heterogeneity/retained expression for MSH2/MSH6. A) clonal loss of MLH1 staining. B) MSI corresponding to loss of staining, C) MSS corresponding to retained MMR protein staining. Methylation analysis revealed D) presence and E) absence, respectively, of MLH1 promoter methylation, which verifies clonal MLH1 methylation status. Flow cytometric analysis showing different DNA indices, i.e. F) 1.13 in the MSI area and G) 1.57 in the MSS area.
Discussion
Heterogeneous MMR protein expression is a rare phenomenon, but corresponds to differences in MMR status within the tumor and is therefore important to recognize to prevent false-positive or false-negative evaluations. We identified three distinct patterns of heterogeneity, i.e. intraglandular, clonal and compartmental heterogenous MMR protein expression. The different patterns co-existed within the same tumor and the extent of the tumor involved varied. In-depth analysis suggests that multiple causes may apply, e.g. variable epitope expression, expression related to variable differentiation, second hit mutations or methylation in selected tumor clones and possibly influence from factors linked to the tumor microenvironment such as hypoxia and oxidative stress [25].
Intraglandular and/or clonal heterogeneity throughout the tumor, which may be caused by variable epitope expression, was identified in 4 tumors (cases 4, 5, 15 and 16, Table 3). Homogenous loss of MLH1/PMS2 and heterogenous expression of MSH2/MSH6 was identified in 7 tumors that were consistently MSI and showed MLH1 promoter methylation (Table 3). This expression pattern has previously been observed and may relate either to a germline MSH2/MSH6 mutation that allows for partial epitope binding in the presence of somatic MLH1 methylation or to secondary MSH2/MSH6 inactivation [25-27]. Heterogenous MLH1 and/or PMS2 expression, suggestive of variable MLH1 methylation/second hit mutations was observed in 2 tumors (case 2 and 6, Table 3). Case 1 showed a more complex pattern of MMR protein expression and intra-tumor differences in MSI, MLH1 promoter methylation and DNA content, suggestive of a tumor composed of two distinct clones (case 1, Figure 2).
Variable MMR status did in some tumors correspond to variable differentiation, e.g. mucinous areas (cases 5 and 15), poor differentiation (cases 16 and 17) or adenomatous components (case 2, Figure 3). Correlation between MSI status and expression also of other molecular markers has been described [28,29]. Different, through homogenous, MMR protein expression patterns in distinct tumor compartments were observed in a mucinous adenocarcinoma (case 9) with loss of MLH1/PMS2 expression, MSI and MLH1 methylation in 1/7 tumor blocks that corresponded to an adenomatous tumor component (Figure 1d). Sample mix was excluded through histologic review and penta-D marker fragment analysis (data not shown). Homogenous loss/reduced staining of MSH2/MSH6 throughout the tumor and additional loss of PMS2 in 7/10 tumor blocks was observed in a mucinous adenocarcinoma (case 5). This case most likely reflects how the mucinous tumor component progressed in another line than the non-mucinous tumor component. Though compartmental loss of MMR protein expression is rare, this observation motivates thorough evaluation of different tumor compartments, particularly when areas with variable expression are identified.
Figure 3.

Variable MMR protein expression in relation to tumor differentiation. A) case 5 with retained expression for PMS2 in a mucinous tumor component and loss of PMS2 expression in a non-mucinous component. B) case 2 with clonal and intraglandular heterogeneity for MLH1 in the adenomatous component of the tumor, whereas the remaining tumors showed retained expression for MLH1. C-D) case 17 with clonal heterogeneity for MLH6 in a poorly differentiated tumor component and homogenous expression in a well-differentiated tumor component.
Limitations to our study include analysis based on surgical specimens though biopsy material may produce stainings of better technical quality [30-33]. At the same time use of biopsy material implies analysis of a restricted tumor area that may not capture areas with alternative expression. Also, information on MMR gene mutation status was not available in all cases. The 4 tumors from Lynch syndrome mutation carriers, however, expressed heterogeneity in between different tumor blocks, which showed homogenous as well as heterogenous loss in clonal and intraglandular patterns. Recognition of so-called “patchy” MMR protein staining has been reported and were also considered herein (Table 3). This phenomenon differs from the heterogenous staining patterns described herein in that it primarily relates to MSH6 stainings, neoadjuvant treatment [15,16] or represents a weak or cytoplasmic staining rather than the distinct and well-demarcated areas of retained staining and loss of staining described herein.
Conclusions
Our study verifies heterogenous MMR status in a subset of colorectal cancer. Heterogenous MMR protein expression appears in three major forms, which frequently co-exist and correlate to differences in MMR status. We suggest that variable MMR protein staining patterns should be considered and when observed linked to extended analysis in order to ensure correct classification of MMR status.
Abbreviations
MMR: Mismatch repair; MSI: Microsatellite instability; MSI-H: MSI-high; MSS: Microsatellite stable.
Competing interests
The author’s declare that they have no competing interests.
Authors' contributions
PJ: corresponding author, study design, review of pathology samples, data analysis and manuscript writing, NV: data collection, macrodissection, MSI analysis, SH: identification of relevant cases, histopathologic review, LK: identification of relevant cases, histopathologic review, AB/MH: MLH1 methylation analysis, BB: DNA ploidy analysis, ER: sample collection, MMR protein immunostaining, MN: study design, data analysis, MMR evaluation, manuscript review. All authors read and approved the final manuscript.
Contributor Information
Patrick Joost, Email: patrick.joost@med.lu.se.
Nynke Veurink, Email: nveurink@hotmail.com.
Susanne Holck, Email: susanne.holck@regionh.dk.
Louise Klarskov, Email: louise.klarskov@dadlnet.dk.
Anders Bojesen, Email: anders.bojesen@rsyd.dk.
Maria Harbo, Email: maria.harbo@rsyd.dk.
Bo Baldetorp, Email: bo.baldetorp@med.lu.se.
Eva Rambech, Email: eva.rambech@med.lu.se.
Mef Nilbert, Email: mef.nilbert@med.lu.se.
Acknowledgments
Financial support was granted from the Swedish Cancer Society, the Nilsson Cancer Foundation, the Kamprad Cancer Foundation and through an ALF grant from the Lund University Medical Faculty, Sweden.
References
- Kastrinos F, Balmana J, Syngal S. Prediction models in Lynch syndrome. Fam Cancer. 2013;12:217–228. doi: 10.1007/s10689-013-9632-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinke V, Holzapfel S, Loeffler M, Holinski-Feder E, Morak M, Schackert HK, Gorgens H, Pox C, Royer-Pokora B, von Knebel-Doeberitz M, Buttner R, Propping P, Engel C. Evaluating the performance of clinical criteria for predicting mismatch repair gene mutations in Lynch syndrome: a comprehensive analysis of 3,671 families. Int J Cancer. 2014;135:69–77. doi: 10.1002/ijc.28650. [DOI] [PubMed] [Google Scholar]
- Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, Hamilton SR, Hiatt RA, Jass J, Lindblom A, Lynch HT, Peltomaki P, Ramsey SD, Rodriguez-Bigas MA, Vasen HF, Hawk ET, Barrett JC, Freedman AN, Srivastava S. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96:261–268. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116:1453–1456. doi: 10.1016/s0016-5085(99)70510-x. [DOI] [PubMed] [Google Scholar]
- Yatabe J, Yatabe MS, Ishibashi K, Nozawa Y, Sanada H. Early detection of colon cancer by amino acid profiling using AminoIndex Technology: a case report. Diagn Pathol. 2013;8:203. doi: 10.1186/1746-1596-8-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira L, Balaguer F, Lindor N, de la Chapelle A, Hampel H, Aaltonen LA, Hopper JL, Le Marchand L, Gallinger S, Newcomb PA, Haile R, Thibodeau SN, Gunawardena S, Jenkins MA, Buchanan DD, Potter JD, Baron JA, Ahnen DJ, Andreu M, Ponz de Leon M, Rustgi AK, Castells A. Identification of Lynch syndrome among patients with colorectal cancer. JAMA. 2012;308:1555–1565. doi: 10.1001/jama.2012.13088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Yao X, Wang Y, Hu F, Wang F, Jiang L, Liu Y, Wang D, Sun G, Zhao Y. MLH1 promoter methylation frequency in colorectal cancer patients and related clinicopathological and molecular features. PLoS One. 2013;8:e59064. doi: 10.1371/journal.pone.0059064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangold E, Pagenstecher C, Friedl W, Fischer HP, Merkelbach-Bruse S, Ohlendorf M, Friedrichs N, Aretz S, Buettner R, Propping P, Mathiak M. Tumours from MSH2 mutation carriers show loss of MSH2 expression but many tumours from MLH1 mutation carriers exhibit weak positive MLH1 staining. J Pathol. 2005;207:385–395. doi: 10.1002/path.1858. [DOI] [PubMed] [Google Scholar]
- Shia J, Klimstra DS, Nafa K, Offit K, Guillem JG, Markowitz AJ, Gerald WL, Ellis NA. Value of immunohistochemical detection of DNA mismatch repair proteins in predicting germline mutation in hereditary colorectal neoplasms. Am J Surg Pathol. 2005;29:96–104. doi: 10.1097/01.pas.0000146009.85309.3b. [DOI] [PubMed] [Google Scholar]
- Muller A, Giuffre G, Edmonston TB, Mathiak M, Roggendorf B, Heinmoller E, Brodegger T, Tuccari G, Mangold E, Buettner R, Ruschoff J. Challenges and pitfalls in HNPCC screening by microsatellite analysis and immunohistochemistry. J Mol Diagn. 2004;6:308–315. doi: 10.1016/S1525-1578(10)60526-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klarskov L, Ladelund S, Holck S, Roenlund K, Lindebjerg J, Elebro J, Halvarsson B, von Salome J, Bernstein I, Nilbert M. Interobserver variability in the evaluation of mismatch repair protein immunostaining. Hum Pathol. 2010;41:1387–1396. doi: 10.1016/j.humpath.2010.03.003. [DOI] [PubMed] [Google Scholar]
- Overbeek LI, Ligtenberg MJ, Willems RW, Hermens RP, Blokx WA, Dubois SV, van der Linden H, Meijer JW, Mlynek-Kersjes ML, Hoogerbrugge N, Hebeda KM, van Krieken JH. Interpretation of immunohistochemistry for mismatch repair proteins is only reliable in a specialized setting. Am J Surg Pathol. 2008;32:1246–1251. doi: 10.1097/pas.0b013e31816401bb. [DOI] [PubMed] [Google Scholar]
- Engel KB, Moore HM. Effects of preanalytical variables on the detection of proteins by immunohistochemistry in formalin-fixed, paraffin-embedded tissue. Arch Pathol Lab Med. 2011;135:537–543. doi: 10.5858/2010-0702-RAIR.1. [DOI] [PubMed] [Google Scholar]
- Shia J, Ellis NA, Klimstra DS. The utility of immunohistochemical detection of DNA mismatch repair gene proteins. Virchows Arch. 2004;445:431–441. doi: 10.1007/s00428-004-1090-5. [DOI] [PubMed] [Google Scholar]
- Bao F, Panarelli NC, Rennert H, Sherr DL, Yantiss RK. Neoadjuvant therapy induces loss of MSH6 expression in colorectal carcinoma. Am J Surg Pathol. 2010;34:1798–1804. doi: 10.1097/PAS.0b013e3181f906cc. [DOI] [PubMed] [Google Scholar]
- Radu OM, Nikiforova MN, Farkas LM, Krasinskas AM. Challenging cases encountered in colorectal cancer screening for Lynch syndrome reveal novel findings: nucleolar MSH6 staining and impact of prior chemoradiation therapy. Hum Pathol. 2011;42:1247–1258. doi: 10.1016/j.humpath.2010.11.016. [DOI] [PubMed] [Google Scholar]
- Giuffre G, Muller A, Brodegger T, Bocker-Edmonston T, Gebert J, Kloor M, Dietmaier W, Kullmann F, Buttner R, Tuccari G, Ruschoff J. Microsatellite analysis of hereditary nonpolyposis colorectal cancer-associated colorectal adenomas by laser-assisted microdissection: correlation with mismatch repair protein expression provides new insights in early steps of tumorigenesis. J Mol Diagn. 2005;7:160–170. doi: 10.1016/S1525-1578(10)60542-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton SR, Bosman FT, Boffetta P, Ilyas M, Morreau H, Nakamura SI, Quirke P, Riboli E, Sobin LH. In: WHO Classification of Tumours of the Digestive System. Volume 3. 4. Bosman FT, Carneiro F, Hruban RH, Theise ND, editor. Lyon, France: IARC Press; 2010. Carcinoma of the colon and rectum; pp. 134–146. [Google Scholar]
- Halvarsson B, Anderson H, Domanska K, Lindmark G, Nilbert M. Clinicopathologic factors identify sporadic mismatch repair-defective colon cancers. Am J Clin Pathol. 2008;129:238–244. doi: 10.1309/0PP5GDRTXUDVKAWJ. [DOI] [PubMed] [Google Scholar]
- Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, Danenberg PV, Laird PW. MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000;28:E32. doi: 10.1093/nar/28.8.e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiegl H, Gattringer C, Widschwendter A, Schneitter A, Ramoni A, Sarlay D, Gaugg I, Goebel G, Muller HM, Mueller-Holzner E, Marth C, Widschwendter M. Methylated DNA collected by tampons–a new tool to detect endometrial cancer. Cancer Epidemiol Biomarkers Prev. 2004;13:882–888. [PubMed] [Google Scholar]
- Weisenberger DJ, Campan M, Long TI, Kim M, Woods C, Fiala E, Ehrlich M, Laird PW. Analysis of repetitive element DNA methylation by MethyLight. Nucleic Acids Res. 2005;33:6823–6836. doi: 10.1093/nar/gki987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldetorp B, Dalberg M, Holst U, Lindgren G. Statistical evaluation of cell kinetic data from DNA flow cytometry (FCM) by the EM algorithm. Cytometry. 1989;10:695–705. doi: 10.1002/cyto.990100605. [DOI] [PubMed] [Google Scholar]
- Schutte B, Reynders MM, Bosman FT, Blijham GH. Flow cytometric determination of DNA ploidy level in nuclei isolated from paraffin-embedded tissue. Cytometry. 1985;6:26–30. doi: 10.1002/cyto.990060106. [DOI] [PubMed] [Google Scholar]
- Shia J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I. The utility of immunohistochemistry. J Mol Diagn. 2008;10:293–300. doi: 10.2353/jmoldx.2008.080031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapusot C, Martin L, Bouvier AM, Bonithon-Kopp C, Ecarnot-Laubriet A, Rageot D, Ponnelle T, Laurent Puig P, Faivre J, Piard F. Microsatellite instability and intratumoural heterogeneity in 100 right-sided sporadic colon carcinomas. Br J Cancer. 2002;87:400–404. doi: 10.1038/sj.bjc.6600474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson N, Grieu F, Morris M, Harvey J, Stewart C, Schofield L, Goldblatt J, Iacopetta B. Heterogeneous staining for mismatch repair proteins during population-based prescreening for hereditary nonpolyposis colorectal cancer. J Mol Diagn. 2007;9:472–478. doi: 10.2353/jmoldx.2007.060162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nodin B, Johannesson H, Wangefjord S, O’Connor DP, Lindquist KE, Uhlen M, Jirstrom K, Eberhard J. Molecular correlates and prognostic significance of SATB1 expression in colorectal cancer. Diagn Pathol. 2012;7:115. doi: 10.1186/1746-1596-7-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangefjord S, Brandstedt J, Lindquist KE, Nodin B, Jirstrom K, Eberhard J. Associations of beta-catenin alterations and MSI screening status with expression of key cell cycle regulating proteins and survival from colorectal cancer. Diagn Pathol. 2013;8:10. doi: 10.1186/1746-1596-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadhil W, Ilyas M. Immunostaining for mismatch repair (MMR) protein expression in colorectal cancer is better and easier to interpret when performed on diagnostic biopsies. Histopathology. 2012;60:653–655. doi: 10.1111/j.1365-2559.2011.04021.x. [DOI] [PubMed] [Google Scholar]
- Kumarasinghe AP, de Boer B, Bateman AC, Kumarasinghe MP. DNA mismatch repair enzyme immunohistochemistry in colorectal cancer: a comparison of biopsy and resection material. Pathology. 2010;42:414–420. doi: 10.3109/00313025.2010.493862. [DOI] [PubMed] [Google Scholar]
- Shia J, Stadler Z, Weiser MR, Rentz M, Gonen M, Tang LH, Vakiani E, Katabi N, Xiong X, Markowitz AJ, Shike M, Guillem J, Klimstra DS. Immunohistochemical staining for DNA mismatch repair proteins in intestinal tract carcinoma: how reliable are biopsy samples? Am J Surg Pathol. 2011;35:447–454. doi: 10.1097/PAS.0b013e31820a091d. [DOI] [PubMed] [Google Scholar]
- Warrier SK, Trainer AH, Lynch AC, Mitchell C, Hiscock R, Sawyer S, Boussioutas A, Heriot AG. Preoperative diagnosis of Lynch syndrome with DNA mismatch repair immunohistochemistry on a diagnostic biopsy. Dis Colon Rectum. 2011;54:1480–1487. doi: 10.1097/DCR.0b013e318231db1f. [DOI] [PubMed] [Google Scholar]