

Abstract
Background
The outcome of patients with metastatic colorectal carcinoma (mCRC) following first line therapy is poor, with median survival of less than one year. The purpose of this study was to identify candidate therapeutically targetable somatic events in mCRC patient samples by whole genome sequencing (WGS), so as to obtain targeted treatment strategies for individual patients.
Methods
Four patients were recruited, all of whom had received > 2 prior therapy regimens. Percutaneous needle biopsies of metastases were performed with whole blood collection for the extraction of constitutional DNA. One tumor was not included in this study as the quality of tumor tissue was not sufficient for further analysis. WGS was performed using Illumina paired end chemistry on HiSeq2000 sequencing systems, which yielded coverage of greater than 30X for all samples. NGS data were processed and analyzed to detect somatic genomic alterations including point mutations, indels, copy number alterations, translocations and rearrangements.
Results
All 3 tumor samples had KRAS mutations, while 2 tumors contained mutations in the APC gene and the PIK3CA gene. Although we did not identify a TCF7L2-VTI1A translocation, we did detect a TCF7L2 mutation in one tumor. Among the other interesting mutated genes was INPPL1, an important gene involved in PI3 kinase signaling. Functional studies demonstrated that inhibition of INPPL1 reduced growth of CRC cells, suggesting that INPPL1 may promote growth in CRC.
Conclusions
Our study further supports potential molecularly defined therapeutic contexts that might provide insights into treatment strategies for refractory mCRC. New insights into the role of INPPL1 in colon tumor cell growth have also been identified. Continued development of appropriate targeted agents towards specific events may be warranted to help improve outcomes in CRC.
Keywords: Metastatic colorectal cancer, Whole genome sequencing, KRAS mutations
Background
Colorectal cancer (CRC) is one of the most common cancers in the United States with an estimated 150,000 new cases and 50,000 deaths each year [1]. While early stage CRC (stage I and II) has a high cure rate after surgery, the recurrence rate is about 50% for stage III CRC after surgery alone and most patients with metastatic disease will ultimately succumb to their cancer [2]. Chemotherapy is the primary treatment for metastatic disease. Currently, there are roughly 5 classes of approved drugs for treating mCRC [3]. These agents include: (1) Fluoropyrimidines: 5-FU and capecitabine (2) Platinum derivative: oxaliplatin (3) Camptothecin derivative: irinotecan (4) EGFR inhibitors: cetuximab and panitumumab and (5) VEGF inhibitors: bevacizumab, afibercept and regorafenib. EGFR inhibitors represent “targeted agents” and their use is limited to about 60% of tumors, which have wild type KRAS genotype. These agents are given in combination, and ultimately patients with KRAS mutations run out of treatment options after 2–3 lines of therapy, with a commonly used sequence being a combination of 5-FU, oxaliplatin and bevacizumab (FOLFOX + bevacizumab) followed by 5-FU/irinotecan (FOLFIRI) with the addition of bevacizumab or afibercept, and the recently approved agent, regorafenib as a third line option in some patients [4].
A number of molecular targets and pathways have been described in CRC. Aberrations in chromosome instability and mismatch repair have been widely identified in a number of cases (85%) [5] with mutations in APC and MutL-homolog (MLH) genes. Mutations in TP53 have been found in about 50% of colorectal cancers globally [6] as p53 plays key roles in cell regulation, apoptosis, DNA repair and differentiation. KRAS mutations are also common in CRC, and occur at a frequency of ~ 40% [7]. Several other pathways which also trigger the malignant phenotype include the TGFβ signaling pathway mediated through downstream targets such as SMAD2 and SMAD4, and components of RAS/MAPK, JNK and PI3K/AKT pathways [8]. Studies of protein coding genomic sequences of colorectal cancers revealed that only a subset of these genes actually contribute to the process of carcinogenesis whereas a vast majority of them actually affect other cellular processes such as transcription, adhesion and invasion [9]. Sequencing studies of the mutational landscape of colorectal cancer revealed that the mutational spectrum is comprised of a limited number of frequently mutated genes and a large number of infrequently mutated genes [10]. Furthermore, a previous sequencing study of 9 colorectal cancers and matched normal tissues reported additional recurrent events, notably a VTI1A-TCF7L2 fusion gene present in ~ 3% of the patients [11].
Personalized CRC patient treatment based on characterizing the individual tumors by high throughput sequencing strategies has been attempted [12]. Most studies reporting sequencing data have been with panels of select genes, or with cell lines, patient derived xenografts, or primary tumors removed at surgery [13]. Our group has instituted a pilot program to sequence various solid tumors in patients with refractory solid tumors [14,15]. In obtaining clinically relevant information that can be of use by the treating physician, tumor biopsies are obtained in patients with advanced solid tumors refractory to approved therapies [16,17]. In our current study, we utilized next generation sequencing technologies (NGS) to identify potential biomarkers so as to identify treatment options for patients with mCRC.
Methods
Participants & samples
All patients signed an IRB approved consent form prior to participation at the Virginia G. Piper Cancer Center, Scottsdale Healthcare, Scottsdale, AZ. Fresh frozen tumor biopsy specimens were collected and quality assessed for tumor cellularity, necrosis, crush artifact, etc. A blood sample was also provided for the collection of constitutional genomic DNA. RNA and DNA were extracted from tumor biopsy specimens using the Qiagen All Prep kit (Qiagen, Germantown, MD). Germline DNA (Table 1) provides information regarding patients and samples.
Table 1.
Patient clinical information
| CLN2 | CLN3 | CLN4 | |
|---|---|---|---|
| Age at diagnosis |
73 |
45 |
50 |
| Gender |
male |
male |
male |
| Ethnicity |
Caucasian |
Caucasian |
Hispanic |
| Diagnosis |
Colon adenocarcinoma |
Rectal adenocarcinoma |
Colon adenocarcinoma |
| Tumor cellularity |
60% |
50% |
30% |
| Sequenced biopsy | Liver metastasis | Right gluteal mass | Right abdominal mass |
Genomic DNA isolation
Fresh frozen tissue
Tissue from the needle biopsy was disrupted and homogenized in Buffer RLT plus, Qiagen AllPrep DNA/RNA Mini Kit, using the Bullet Blender™, Next Advance. Specifically, tissue was transferred to a microcentrifuge tube containing 600 μl of Buffer RLT plus, and stainless steel beads. The tissue was homogenized in the Bullet Blender at room temperature. The sample was centrifuged at full speed and the lysate was transferred to the Qiagen AllPrep DNA spin column. Genomic DNA purification was conducted as directed by the AllPrep DNA/RNA Mini Handbook, Qiagen. DNA was quantified using the Nanodrop spectrophotometer and quality was accessed from 260/280 and 260/230 absorbance ratios.
Blood
The QIAamp DNA Blood Maxi Kit, Qiagen, was used to isolate DNA from 8–10 ml of whole blood. The protocol was conducted as written. Specifically, the buffy coat layer was isolated from whole blood by centrifugation. The volume of buffy coat was brought up to 5–10 ml with PBS and treated with Qiagen protease at 70°C. 100% ethanol was added and the sample was applied to a QIAamp Maxi column and centrifuged. Samples were then washed with buffers AW1 and AW2 and eluted in 1000 μl of Buffer AE. The Qubit 2.0 Fluorometer, Invitrogen, and the Nanodrop spectrophotometer, Thermo Scientific, were used to assess DNA quality and concentration.
Sequencing data analysis
Illumina whole genome sequencing
DNA libraries were prepared using the NEBNext DNA Sample Prep Master Mix Set (New England Biolabs, Ipswich, MA). For each sample library preparation, 1 μg of high molecular weight genomic DNA was fragmented using the Covaris S2 system. Fragmented samples were end repaired using T4 DNA polymerase, DNA polymerase I Klenow fragment, and T4 polynucleotide kinase. Samples were next adenylated using Klenow fragment 3′-5′ exo minus enzyme, ligated with Illumina adapters, size selected at 350-450 bp, and PCR amplified using Phusion High-Fidelity PCR Master Mix w/HF buffer (New England Biolabs). The DNA libraries were clustered onto flowcells using Illumina’s cBot and HiSeq Paired End Cluster Generation kits as per manufacturer protocol (Illumina, San Diego, CA). NGS of CLN2 and CLN3 samples were carried out using the Illumina HiSeq 2000 platform using the v1.5 chemistry reagents and flowcells. The CLN4 sample was sequenced on the Illumina HiSeq 2000 platform using the v3 chemistry. The total length of each paired end sequencing run was 200 cycles.
Single nucleotide variant detection
Somatic single nucleotide variant calling was performed using SolSNP [18] and Mutation Walker. SolSNP, an individual sample variant detector (classifier) implemented in Java does the variant calling based on a modified Kolmogorov-Smirnov like statistic, which incorporates base quality scores. The algorithm is non-parametric and makes no assumptions on the nature of the data. It compares the discrete sampled distribution, the pileup on each strand, to the expected distributions (according to ploidy). In case of diploid genome, both strands need to provide evidence for the variation. Zero quality bases are trimmed off the pileup before the comparisons. An important aspect of SolSNP that reduces overcalling inherent to the K-S statistic algorithm is that filters are included to reduce false positive rates, of which both strands must provide evidence for the variation.
While making somatic calls, SolSNP’s high quality genotype call is made for all callable loci of the normal sample. To reduce false negatives, variant loci in tumor samples are called with the Variant Consensus mode. Variant loci in tumor samples that exhibit a high quality homozygous reference genotype in the normal sample are considered as somatic. To call somatic variants, SolSNP is augmented by a Python script.
Mutation Walker, an in house tool developed in Java, utilizes the variant discovery tools from Genome Analysis Toolkit (GATK) [19] as a framework. SNPs that were called using both tools were compiled and further examined. Two sets of thresholds, strict and lenient, were enabled to reduce the false negative rate. Data from each of these two sets were visually examined for false positives to generate a final filtered list of true SNVs, which were annotated with GENCODE using an internal annotation script.
Copy number analysis
For copy number analysis, a custom tool was developed based on a sliding window comparison of coverage for tumor/normal. This method has been adopted by Liang et al. for their analysis [16]. Copy number gains and losses were calculated from log2 difference in normalized physical coverage between germline and tumor samples across a sliding window of 2 kb, where physical coverage was incremented for the length of the insert between the read pairs for insert sizes less than 3 standard deviations of the mean. High repeat regions such as centromeres were defined as regions where the log2 normalized coverage exceeded 3 in the germline sample and were thus excluded. Regions where the coverage was zero were replaced by 1 so that homozygous deletions avoid infinite values and are generally capped at approximately −3.
Indel (Insertion/deletion) detection
For detecting somatic indels we employed a two-step strategy. In the first step we removed from the tumor sample bam, reads whose insert size lay outside a 50 bp - 500 bp interval from the tumor bam files. GATK [19] is then used to generate a list of potential small indels from this bam. A customized perl script, which uses the Bio-SamTools library from BioPerl [20], takes these indel positions and for each of the indels looks at the region in the normal sample consisting of 5 bp upstream from the start and 5 bp downstream from the end of the indel. An indel is determined to be somatic only if there was no indel detected in the region under consideration in the normal DNA.
Translocation detection
A series of customized perl scripts are employed in the detection of translocation. These scripts use SAMtools [21] internally to access the bam files. The algorithm consists of two steps. The first is to detect potential translocation in both tumor and normal samples. The second is a comparison of potential translocations in tumor to those detected in the normal sample to weed out potential false positives. The detection of a potential translocation is an exercise in outlier detection. We take a sliding window of 2kbp and count the discordant reads, whose mates align on a different chromosome. We use 2kbp, as it is close to the mean of the estimated insert size distribution, and gives the best resolution for the detection of an interchromosomal translocation. For each window we choose the highest hit to be the chromosome to which mates of most of the discordant reads mapped. For purposes of brevity, we call the subset of discordant reads whose mate maps to the highest hit in the window as the hit discordant reads. We compare the ratios of the hit discordant reads, to the total aligned reads, across all the windows to detect potential outliers. Outlier detection is performed under the assumption that the distribution, of the proportion of hit discordant reads in a 2 kb window aggregates across the chromosome, and will follow a normal distribution. We then compute the mean of this distribution and choose a cutoff of 3 standard deviations. The window with a proportion of hit discordant reads, higher than this cutoff contains the region of potential translocation. The actual region of translocation is then determined by the span of the hit discordant reads in the window. For somatic translocations, the normal and the tumor sample are called separately and regions of overlap are eliminated. These regions were further inspected visually to reduce false positives to arrive at the most confident list.
SIFT analysis
Single nucleotide variations identified from paired tumor normal analysis were analyzed using the SIFT algorithm. Non-synonymous SNPs in the coding region were checked to see if an amino acid substitution in the protein could actually be damaging by altering the function of the protein [22].
Cell culture conditions and treatments
X-MAN HCT116 cell line was purchased from Horizon Discovery Ltd. (Cambridge, UK) and cultured in McCoy’s 5A modified media with 10% FBS (Life Technologies, Grand Island, NY). HEK293 cells were obtained from the American Type Culture Collection (ATCC) and passaged in RPMI with 10% FBS and 100 units insulin (Sigma-Aldrich, St. Louis, MO). Inhibition of INPPL1 expression was achieved through the use of siRNA sequences (Qiagen, Valencia, CA). siRNA transfections were performed with Lipofectamine 2000 (Life Technologies) and pooled INPPL1 (FlexiTube GeneSolution GS3636) siRNA sequences [Qiagen]. Non-silencing negative control and GFP siRNA sequences were utilized as negative controls (Qiagen). AllStars Hs Cell Death and UBBs1 sequences served as positive controls to assess transfection efficiency (Qiagen). Cells were seeded in 384-well pates containing siRNA sequences (i.e., reverse transfection) and proliferation was measured via CyQUANT Direct Cell Proliferation Assay (Life Technologies). Changes in proliferation were assessed based on comparison of non-silencing siRNA controls to target siRNA. Data shown for siRNA experiments was generated using Excel version 14.3.5 (Microsoft Corporation, Redmond, WA). For siRNA data, differences in proliferation were evaluated by T Test and p values ≤ 0.05 were considered statistically significant.
Cell lysates and immunoblotting
Untreated as well as transfected whole cell lysates were prepared using the cOmplete Lysis-M kit (Roche Applied Science, Indianapolis, IN). Protein samples were prepared by combining lysates with NuPAGE Sample Reducing Agent, NuPAGE LDS Sample Buffer, and nuclease-free water (Life Technologies) and then boiling for 5 minutes at 100°C. Samples were loaded onto NuPAGE 4-12% Bis Tris Gels (Life Technologies), separated by SDS-PAGE, and then transferred to PVDF membranes. Proteins were blocked in 5% non-fat milk and incubated with the appropriate antibody in 5% BSA (Sigma-Aldrich). The following antibodies were used for Western blot analyses: AKT (#9272), pAKT Thr308 (#4056), pAKT Ser473 (#4058), CASP7 (#9492), Fox01 (#2880), pFox01 Ser256 (#9461), GSK-3β (#9315), pGSK-3β Ser9 (#9336), INNPL1 (#2839), p70 S6 Kinase (#2708), PDK1 (#5662), pPDK1 Ser241 (#3438), PI3K p85 (#4292), and GAPDH (#2118) [Cell Signaling, Danvers MA]; and pGSK-3β Tyr216 (#75745) [Abcam Inc., Cambridge, MA]. Protein bands were visualized using Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific, Rockford, IL).
Results
Clinical history
Four patients were recruited for this study. In patient CLN1, the tumor sample was not considered adequate for NGS and results are not presented. Clinical information is provided in Table 1.
Patient CLN2 is a 73 year old Caucasian male first diagnosed with a T3N2 moderately differentiated adenocarcinoma of the ascending colon. He received adjuvant therapy with FOLFOX but had rapid recurrence of disease 5 months after initial surgery. The patient had subsequent therapy with irinotecan/bevacizumab and then gemcitabine, mitomycin and insulin potentiating therapy. At time of referral, there was metastatic disease in the pelvis and multiple omental lesions. A CT guided biopsy of a nodule in the right side of the abdomen was performed. The biopsy from this mass (60% tumor cellularity) was consistent with the colon primary.
Patient CLN3 is a 45 year old Caucasian male first diagnosed with a T2NO rectal cancer for which primary surgery was performed. The patient was found to have recurrent lung metastasis and a pelvic pre sacral mass 18 months after surgery. He also had a chronic draining fistula in the right buttock which developed soon after surgery. A hard mass developed in the right buttock and on biopsy was found to be another site of metastasis. Treatment for metastatic disease had included FOLFOX /bevacizumab, lung radiation and isolated pelvic chemotherapy perfusion. A biopsy of the right buttock mass was performed with pathology consistent with the rectal primary (50% tumor cellularity).
Patient CLN4 is a 50 year old Hispanic male diagnosed with hemicolectomy for a T3N1 right sided colon cancer. The patient was found to have synchronous liver metastasis at the same time. He had undergone prior therapy with FOLFOX, FOLFIRI/bevacizumab and yittum-90 therapy to the liver. The patient entered the study 3 years after first diagnosis of metastatic disease and had tumors in the liver and a soft tissue mass in the epigastric area of the abdomen. A biopsy from the abdominal mass was consistent with the colon primary (30% tumor cellularity).
Whole genome sequencing
For each patient tumor and germline DNA were sequenced to identify somatic alterations. WGS summary statistics are shown in Table 2. Aligned reads from both tumor and normal samples were evaluated for somatic events including non-synonymous single nucleotide variants (nsSNVs), indels, copy number variants (CNVs) and translocations. Circos plots in Additional files 1, 2 and 3 illustrate somatic events occurring in CLN2, CLN3 and CLN4 respectively [23].
Table 2.
Whole genome sequencing
| Participant code | Total reads sequenced | Total bases sequenced | Coverage | Number of germline variants | Percent dbSNP | Transition/transversion ratio |
|---|---|---|---|---|---|---|
| CLN2 Normal |
1.9 billion |
200 Gb |
50X |
2.3 million |
88% |
2.05 |
| CLN2 Tumor |
1.4 billion |
146 Gb |
42X |
— |
|
|
| CLN3 Normal |
2.0 billion |
210 Gb |
62X |
3.4 million |
88% |
2.06 |
| CLN3 Tumor |
1.96 billion |
204 Gb |
40X |
— |
|
|
| CLN4 Normal |
0.97 billion |
100 Gb |
28X |
3.3 million |
88% |
2.06 |
| CLN4 Tumor | 1.03 billion | 107 Gb | 31X | — |
The complete lists of somatic SNVs detected in the three evaluable specimens are provided in Additional file 4: SNVs and Indels. All three tumors contained KRAS mutations. Among the cancer genes mutated in patient CLN2 were APC, KRAS, PIK3CA, SMAD4, MYST4, HUNK, INPPL1, TGFβ3, and TCF7L2. Cancer genes mutated in CLN3 included KRAS, INPP4B, PTPRE, CARD16 and LRP2. Cancer gene mutations in CLN4 were detected in APC, KRAS, PIK3CA, KDR and AURKC (Table 3). Some of the genes which were known to have non-synonymous mutations in colon cancer genes in prior studies were also seen in these samples. They include APC, KCNQ5, KIAA1409, KRAS, LRP2, PIK3CA, SMAD4, TCF7L2 and UHRF2[9].
Table 3.
SNVs and indels in relevance to cancer
| Sample | Chr | Gene name | Position | Coding event | Sequence change | Substitution |
|---|---|---|---|---|---|---|
| CLN2 |
3 |
CPB1 |
150045158 |
SNV |
G/A |
R231Q |
| |
6 |
ESR1 |
152423875 |
SNV |
C/T |
T431I |
| |
10 |
TCF7L2 |
114907771 |
SNV |
G/A |
G424E |
| |
11 |
INPPL1 |
71621016 |
SNV |
A/G |
E567G |
| |
12 |
KRAS |
25289548 |
SNV |
C/T |
G13D |
| |
14 |
TGFβ3 |
75495381 |
SNV |
G/T |
Q381K |
| |
18 |
BCL2 |
59136317 |
SNV |
C/A |
W188L |
| |
18 |
PTPRM |
8384547 |
SNV |
G/A |
V1415M |
| |
10 |
MYST4 |
76460457 |
SNV |
C/T |
R1957W |
| |
18 |
SMAD4 |
46858795 |
SNV |
T/G |
L540R |
| |
19 |
SHANK1 |
55864301 |
SNV |
G/A |
R910C |
| |
21 |
HUNK |
32293208 |
SNV |
G/A |
R662Q |
| |
3 |
PIK3CA |
180399422 |
SNV |
G/A |
NA |
| |
1 |
PTPRC |
196985273 |
SNV |
G/A |
S852N |
| |
13 |
MLNR |
48693327 |
Indel |
cccgg−/−ccgcc |
Insertion |
| |
2 |
SLC4A10 |
162427730 |
Indel |
atcag−/−aaaa |
Insertion |
| |
6 |
UTRN |
145111149 |
Indel |
AAAT-g-GGAAA |
Frameshift |
| |
7 |
HNRNPA2B1 |
26202550 |
Indel |
CAGAT-cctcc-TCTAA |
Frameshift |
| CLN3 |
1 |
ARID1A |
26973896 |
SNV |
G/T |
E1531 |
| |
2 |
LRP2 |
169845248 |
SNV |
T/C |
N400S |
| |
3 |
MITF |
70011193 |
SNV |
C/G |
S92C |
| |
4 |
INPP4B |
143222686 |
SNV |
C/A |
E864 |
| |
5 |
GPR98 |
89974441 |
SNV |
G/T |
V787L |
| |
7 |
CYLN2 |
73409559 |
SNV |
C/A |
S344Y |
| |
10 |
PTPRE |
129758044 |
SNV |
G/A |
R369Q |
| |
12 |
KRAS |
25289551 |
SNV |
C/T |
G12D |
| |
4 |
MAML3 |
26202550 |
Indel |
AAAT-ctg-CTGCT |
AA_Deletion |
| |
6 |
PGC |
140871034 |
Indel |
CCTGC-aga-AGAGC |
AA_Deletion |
| CLN4 |
1 |
ARID4B |
233412380 |
SNV |
C/A |
R826M |
| |
3 |
PIK3CA |
180434779 |
SNV |
A/G |
H1048R |
| |
4 |
KDR |
55655861 |
SNV |
G/A |
R946C |
| |
5 |
APC |
112129944 |
SNV |
G/T |
G53V |
| |
5 |
APC |
112201150 |
SNV |
C/T |
Q654 |
| |
5 |
APC |
112203580 |
SNV |
G/T |
E1464 |
| |
5 |
APC |
112205360 |
SNV |
A/G |
K2057R |
| |
6 |
PTCRA |
42998820 |
SNV |
G/T |
V46F |
| |
12 |
KRAS |
25289552 |
SNV |
C/A |
G12C |
| 2 | HOXD9 | 176696536 | Indel | cagcc-/gcagc | Insertion |
Non-synonymous SNVs found in the coding regions of genes were analyzed using the SIFT (Sorting Tolerant From Intolerant) algorithm to determine if such mutations may affect protein function [22]. Genes identified to have damaging effects on the protein product are indicated in Additional file 5: SIFT Predictions for SNVs and Indels. Of the 115 coding variants in CLN2, 61 were predicted to be damaging (53%); CLN3 had 90 coding variants of which 38 were damaging (42%); and CLN4 had 44 coding variants of which 20 were damaging (47%).
Copy number analysis
Copy number analysis was performed using a sliding window comparison of coverage between tumor and normal using our in-house custom tool. Regions of gain or loss in the tumor samples are outputted as log2 ratios in Additional file 6: CNVs. CLN2 had whole chromosome copy number gain of chromosome 13, and chromosome 8q. Significant genes deleted in CLN2 include TP53, BCL2, PIK3CA, SMAD2, SMAD3, SMAD4, APC2, DCC, TGFβ1, TCF3, TCF4 and TCF12. CLN3 exhibited whole chromosome copy number gain of chromosomes 1 – 5. A significant amplification occurred at 1pter and encompassed NOCL2, PLEKHN1, SDF4, UBE2J2, CENTB5, CPSF3L, MXRA8, ATAD3B, ATAD3A, SSU7A, SLC35E2, NADK, GNB1, GABRD, and PRKCZ. A focal amplicon at 12p contains KRAS, which is also mutated in this patient’s tumor. An interstitial deletion of about 16 Mb was seen in chromosome 13, which encompasses the RB1 locus. Somatic copy number analysis in CLN4 was confounded by low tumor cellularity (30%); however we were able to detect several events including whole chromosome gain of chromosome 13.
Furthermore, several genes containing SNVs also mapped within regions of copy number change. A list of these genes has been included in Table 4. Notably in CLN2, SMAD4 was deleted and harbored an L540R somatic mutation. Additionally, PTPRM was deleted and contained a V1415M nsSNV.
Table 4.
Correlation of genes with SNVs and amplifications and deletions
| Sample | Chr | Gene name | SNV location | CNV variant type | Change associated with variant | Start | End | Correlation |
|---|---|---|---|---|---|---|---|---|
|
CLN2 |
8 |
ADAMDEC1 |
24312932 |
CNV-Loss |
-1.098323504 |
24298000 |
24318700 |
- |
| |
18 |
BCL2 |
59136317 |
CNV-Loss |
-1.142019005 |
58946900 |
59136800 |
- |
| |
17 |
C17orf39 |
17905985 |
CNV-Loss |
-1.086327397 |
17886900 |
17909200 |
- |
| |
13 |
DNAJC3 |
95127539 |
CNV-Gain |
1.034261883 |
95159600 |
95201700 |
+ |
| |
18 |
DOK6 |
65495961 |
CNV-Loss |
-1.080170349 |
65220300 |
65659400 |
- |
| |
19 |
EEF2 |
3929160 |
CNV-Loss |
-1.10433666 |
3927700 |
3935600 |
- |
| |
13 |
MED4 |
47567121 |
CNV-gain |
1.011225847 |
47549300 |
47562800 |
+ |
| |
15 |
MORF4L1 |
76965549 |
CNV-Loss |
-1.25642835 |
76953100 |
76976400 |
- |
| |
19 |
OR1M1 |
9065467 |
CNV-Loss |
-1.185833041 |
9065000 |
9065800 |
- |
| |
13 |
PCDH17 |
57105203 |
CNV-Gain |
1.158413612 |
57105000 |
57140500 |
+ |
| |
8 |
PI15 |
75900191 |
CNV-Gain |
1.077222314 |
75901700 |
75924000 |
+ |
| |
15 |
PML |
72124335 |
CNV-Loss |
-1.130706684 |
72074500 |
72122000 |
- |
| |
18 |
PTPRM |
8384547 |
CNV-Loss |
-1.028666588 |
7637600 |
8387100 |
- |
| |
19 |
PVRL2 |
50083243 |
CNV-Loss |
-1.070323897 |
50042300 |
50073700 |
- |
| |
19 |
SHANK1 |
55864301 |
CNV-Loss |
-1.135111902 |
55859100 |
55907600 |
- |
| |
18 |
SMAD4 |
46858795 |
CNV-Loss |
-1.077097357 |
46827500 |
46855600 |
- |
| |
15 |
THSD4 |
69844560 |
CNV-Loss |
-1.12539172 |
69808500 |
69856700 |
- |
| |
8 |
VPS13B |
100723477 |
CNV-Gain |
1.06690452 |
100098000 |
100957000 |
+ |
| |
19 |
XAB2 |
7594144 |
CNV-Loss |
-1.039001256 |
7596500 |
7599800 |
- |
| |
19 |
ZNF235 |
49483380 |
CNV-Loss |
-1.210566986 |
49486200 |
49499300 |
- |
| |
19 |
ZNF480 |
57510937 |
CNV-Loss |
-1.12807401 |
57496100 |
57517900 |
- |
| |
19 |
ZNF83 |
57808787 |
CNV-Loss |
-1.330073623 |
57808100 |
57809600 |
- |
|
CLN3 |
4 |
ANKRD17 |
74175875 |
CNV-Gain |
1.050578956 |
74176700 |
74257700 |
+ |
| |
2 |
APOB |
21086873 |
CNV-Gain |
1.381146277 |
21078200 |
21119700 |
+ |
| |
1 |
ARID1A |
26973896 |
CNV-Gain |
1.088551435 |
26707600 |
26977900 |
+ |
| |
3 |
ATP13A5 |
194544518 |
CNV-Gain |
1.074832063 |
194495900 |
194576400 |
+ |
| |
2 |
CLEC4F |
70897420 |
CNV-Gain |
1.474467603 |
70890000 |
70900800 |
+ |
| |
1 |
FNDC7 |
109063011 |
CNV-Gain |
1.060116535 |
109071800 |
109085900 |
+ |
| |
3 |
GADL1 |
30871204 |
CNV-Gain |
1.083837201 |
30750200 |
30822400 |
+ |
| |
4 |
GUCY1A3 |
156862650 |
CNV-Gain |
1.051035883 |
156845700 |
156866700 |
+ |
| |
4 |
INPP4B |
143222686 |
CNV-Gain |
1.071823665 |
143169400 |
143560600 |
+ |
| |
2 |
IWS1 |
127960697 |
CNV-Gain |
1.157172502 |
127955100 |
127999800 |
+ |
| |
3 |
KCNMB2 |
180028679 |
CNV-Gain |
1.036705549 |
180021600 |
180027700 |
+ |
| |
12 |
KRAS |
25289551 |
CNV-Gain |
1.035184393 |
25274800 |
25275900 |
+ |
| |
2 |
LRP2 |
169845248 |
CNV-Gain |
1.274350136 |
169693500 |
169926300 |
+ |
| |
3 |
LSG1 |
195872054 |
CNV-Gain |
1.046959963 |
195844100 |
195855800 |
+ |
| |
1 |
MACF1 |
39573148 |
CNV-Gain |
1.075279401 |
39322700 |
39722600 |
+ |
| |
3 |
MITF |
70011193 |
CNV-Gain |
1.055910933 |
70070200 |
70096500 |
+ |
| |
5 |
PAPD4 |
78951267 |
CNV-Gain |
1.023426699 |
78969500 |
78969700 |
+ |
| |
2 |
PCBP1 |
70168678 |
CNV-Gain |
1.147337373 |
70169200 |
70169400 |
+ |
| |
4 |
PDE5A |
120666238 |
CNV-Gain |
1.066043219 |
120655300 |
120742100 |
+ |
| |
1 |
PHTF1 |
114082912 |
CNV-Gain |
1.075512922 |
114060600 |
114098600 |
+ |
| |
3 |
RPSA |
39428561 |
CNV-Gain |
1.099952073 |
39424200 |
39428600 |
+ |
| |
4 |
SH3BP2 |
2805228 |
CNV-Gain |
1.208480128 |
2792500 |
2804000 |
+ |
| |
5 |
SLC6A19 |
1265596 |
CNV-Gain |
1.142273516 |
1257300 |
1274700 |
+ |
| |
3 |
SMC4 |
161633513 |
CNV-Gain |
1.02766824 |
161602500 |
161603100 |
+ |
| |
2 |
SMYD5 |
73305580 |
CNV-Gain |
1.59092836 |
73301300 |
73306500 |
+ |
| |
1 |
SPOCD1 |
32038250 |
CNV-Gain |
1.187067318 |
32029900 |
32040400 |
+ |
| |
1 |
ST6GALNAC3 |
76650550 |
CNV-Gain |
1.08116566 |
76355200 |
76864700 |
+ |
| |
1 |
TMEM39B |
32340671 |
CNV-Gain |
1.071050645 |
32318800 |
32339600 |
+ |
| |
2 |
VPS54 |
63993297 |
CNV-Gain |
1.158423232 |
63974100 |
64064600 |
+ |
| |
2 |
WDR35 |
20045755 |
CNV-Gain |
1.180181947 |
19976800 |
20052800 |
+ |
| |
5 |
WWC1 |
167824543 |
CNV-Gain |
1.083413977 |
167656200 |
167825900 |
+ |
| CLN4 |
16 |
ABCC12 |
46688204 |
CNV-Gain |
0.757287 |
46702800 |
46702800 |
+ |
| 13 | SPATA13 | 23761296 | CNV-Gain | 0.894588 | 23774800 | 23761296 | + |
Impact of INPPL1 silencing on cell proliferation in HCT116 cells
The PI3 kinase pathway has been previously associated with colorectal cancer, where PIK3CA mutations occur in approximately 15% of colorectal tumors [10,24]. Phosphatidyl-3,4,5-triphosphate (PI3,4,5P3), is a key phosphoinositide generated from PI3 kinase, which regulates PKB/AKT mediated cell survival and proliferation [25]. In our analysis, we identified a mutation in INPPL1 (inositol polyphosphate phosphatase-like 1), which encodes SHIP2, the phosphatase that plays an important role in the conversion of PI3,4,5P3 to PI3,4P2. The E567G mutation identified in the INPPL1gene was predicted to be damaging by SIFT, and validated by Sanger sequencing. Thus we investigated the impact of silencing INPPL1 by RNA inhibition on cell proliferation. For this study, we used small interfering RNA (siRNA). HCT116 cells were seeded into 384-well plates containing siRNA buffer (no transfection), control siRNA sequences, or INPPL1 siRNA sequences. At 72 hours post-transfection, cell proliferation was measured. Changes were measured based on differences between non-silencing control siRNA and INPPL1 siRNA sequences (Figure 1). At 72 hours post-transfection, we detected a 65% decrease in HCT116 cell proliferation, suggesting that INPPL1 may be required for CRC cell growth.
Figure 1.
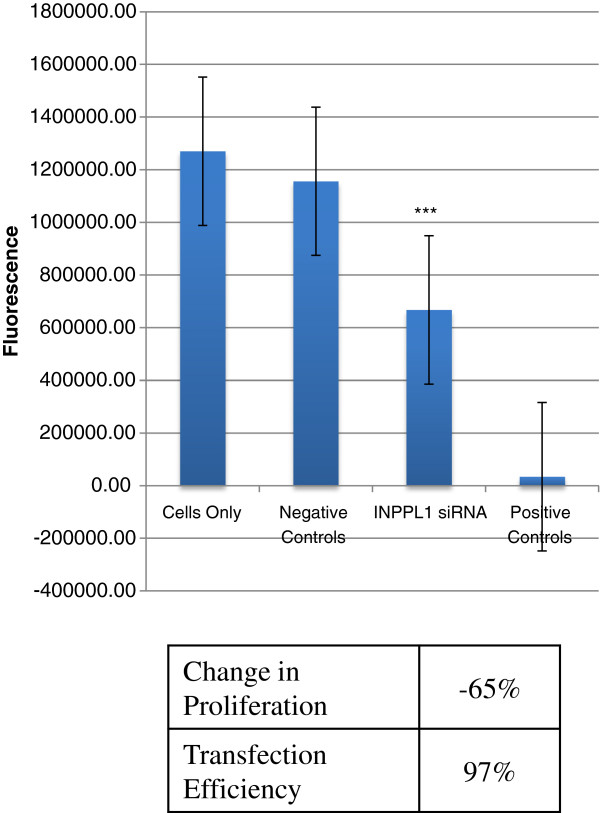
Effect of INPPL1 siRNA on cell proliferation in HCT116 cells. Changes in proliferation in HCT116 cells 72 hours post-transfection with pooled control or pooled INPPL1 sequences. Fluorescence data is shown for cells only (no siRNA sequences), negative controls (non-silencing siRNA sequences), INPPL1 siRNA, and positive controls (siRNA sequences targeting essential genes). The effects of INPPL1 siRNA are based on differences between average fluorescence signal generated by negative controls and INPPL1 siRNA sequences. Asterisks denotes statistical significance.
Impact of INPPL1 siRNA on protein expression of downstream signaling targets of PI3K -Western analysis
Since SHIP2 plays a role in PI3K/AKT signaling, INPPL1 siRNA transfected and untransfected HCT116 cell lysates were used for Western blots and probed with antibodies to several downstream PI3K/AKT pathway targets as illustrated in Figure 2. HEK293 cell lysates were used as a positive control in our western blots, due to the ease of maintenance, abundance of proteins, and known expression of multiple proteins. As expected, a reduction in INPPL1 protein levels was observed at 24, 48 and 72 hours post transfection. INPPL1 siRNA treated HCT116 cells showed no expression of PI3K as early as 24 hours post transfection. Hence we looked at the protein levels of a series of downstream signaling targets of PI3K. For PDK1, both pPDK1 (Ser 241) and PDK1 antibodies were used. In the untransfected cell lines, pPDK and PDK1 were expressed. Similar to PI3K, INPPL1 siRNA treated cells showed no expression of either the pPDK1 or PDK1 at 24 and 48 hours. AKT phosphorylation was detected at T308 and S473 in HEK293 cells, however, only phosphor-T308 was detected in HCT116 cells. Total AKT was detected at high levels in HCT116 cells. INPPL1 siRNA transfected cells showed a marked reduction in the phosphorylation of AKT as seen in 24 and 48 hours post transfection. Moreover, INPPL1 siRNA inhibited total AKT protein expression at 48 hours in HCT116 cells, with expression returning at 72 hours post transfection. INPPL1 siRNA transfection also led to reduced p70 S6 kinase in the HCT116 cells within 24 hours post transfection. Phosphorylated FOX01 (Ser256) was detected in untransfected cells, but was significantly diminished in HCT116 cells transfected with INPPL1 siRNA. Another downstream effector of AKT is GSK-3β, which upon phosphorylation by AKT at Ser9 becomes inactivated leading to increased cell cycle and β-catenin signaling. INPPL1 siRNA led to decreased total GSK-3β at 24 hours in HCT116 parental cell lines. Interestingly, INPPL1 knockdown in HCT116 cells led to increased phospho-(Ser9) GSK-3β in HCT116 cells at 48 hours. Finally, Caspase 7 activation, which is an indicator of apoptosis, also increased as indicated by the cleaved 35 kDa band in INPPL1 siRNA transfected HCT116 cells. These results support inactivation of the PI3K/AKT pathway upon INPPL1 knockdown, suggesting a growth promoting role for INPPL1 in colon cancer.
Figure 2.
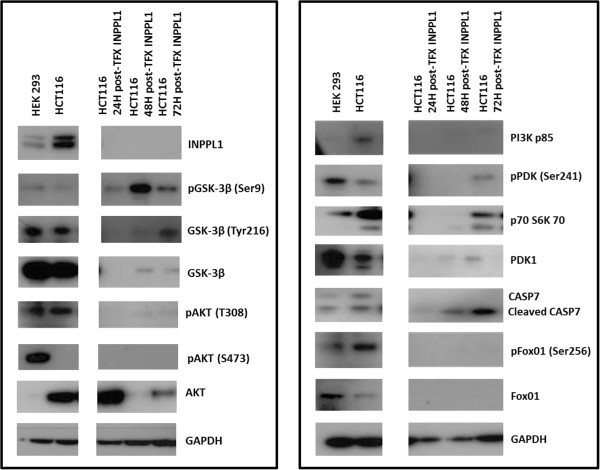
Western blots. Basal protein expression of HEK293 and HCT116 cells as well as changes in expression in HCT116 cells 24, 48, and 72 hours post-INPPL1 siRNA transfection. For post-transfection samples, cell lysates were treated with pooled INPPL1 siRNA sequences and harvested at each time point.
Discussion
It is now established that the key mediators of the cancer cell phenotype are single base mutations, copy number alterations, translocations/rearrangements and epigenetic modifications of genomic DNA. Only now with the advent of NGS and bioinformatics capabilities can the entire human genome be interrogated for these changes. Importantly, with the development of targeted therapies, somatic genome analysis of tumors can shed light on possible therapeutically relevant events that might help inform treatment recommendations for advanced cancers.
In our small sample size, all three tumors had mutations in KRAS. KRAS mutations have been observed in 33% of the CRCs and are crucial for the early progression of adenoma to carcinoma in these tumors. Activating KRAS mutations in CRC tumor samples are an early event in the progression of colon carcinoma and the only predictive molecular marker useful for treatment decision as EGFR directed therapy is ineffective in the context of concomitant KRAS mutation [26]. However, even in the presence of KRAS wild type, therapy with either cetuximab or panitumumab is only effective in about 30% of cases suggesting that there are other molecular determinants of resistance. Additional genes harboring somatic mutations that have been characterized as cancer genes in our study include, APC, TGFβ3, SMAD4, BCL2, INPPL1, INPP4B, PIK3CA, PTPRE and KDR. Mutations in the APC gene have been identified in sporadic cancers and play a role in the WNT/β-catenin signaling pathway. Loss of APC function leads to β-catenin accumulation and binding to TCF/LEF transcription factors thereby activating MYC and cyclin D1 leading to the transformation of the colon epithelium [27]. Thus APC mutations generally play a role in the initiation of colorectal cancers. APC also regulates cell proliferation of RAS induced ERK activation playing an important role in colorectal tumor suppression [28]. Colorectal tumors are known to have mismatch repair mutations that can increase the mutation rate in these tumors. However, one cannot also rule out the possibility that some of the mutations identified in these advanced cancers are not the result of previously administered chemo or radiation therapies.
Some notable CNVs were seen in these patients. CLN2 had amplification in the MYC oncogene. A number of known tumor suppressor genes were deleted in CLN2 such as TP53, SMAD2, SMAD3, SMAD4, BCL2 and TCF4. SMAD4 alterations occur in over 50% of CRC and are believed to occur later in the course of disease [29]. SMAD4 acts as a tumor suppressor to inhibit β-catenin [30] and targets the TGFβ signaling pathway to control epithelial cell growth [31]. Tumor CLN2 contained SMAD4 deletion and a concomitant SMAD4 mutation. Deletion of BCL2 leads to an increase in the relapse of stage II colon cancers and could be a likely biomarker for therapeutic decisions [32]. Importantly, CLN2 also contained a somatic mutation in the BCL2 gene. TCF4 has been found to be mutated in variety of tumor types such as renal cell carcinoma, gastric carcinoma and breast cancer [33,34]. TCF4 mutations have been reported in primary CRCs and its loss induces cell proliferation suggesting a possible role as a tumor suppressor [35-37]. CLN3 has a copy number gain encompassing the VEGFA locus, found in 3% of the cases and linked to higher tumor grade and vascular invasion and being an aggressive subgroup [38]. Copy number gain was also noted in KDR (VEGFR-2), a mediator of angiogenesis and its expression correlated with poor outcome in non-small cell lung carcinoma [39-42]. CLN4 had amplification of EGFR, CDK8, and MIRH1. EGFR mutations in the extracellular domain with gene amplifications are common in glioblastomas [43] and mutations in the tyrosine kinase domain with increased copy numbers are seen in lung carcinomas. However, amplification of EGFR seems to be an uncommon event in colorectal cancer [44,45]. CDK8, a cyclin dependent kinase amplified in CLN4, plays a major role in cell proliferation and PI3K inhibitors can be used in clinical trials for CDK8[46]. With regards to structural events in colon cancer, gene fusions of TCF7L2 with VTI1A have previously been observed in 3% of the colon cancers [11]. We did not detect this translocation event in any of our tumors due to our sample size, however we did detected a novel TCF7L2 (G424E) mutation in one of our tumors, suggesting that this gene can be perturbed by multiple mechanisms.
In our three CRC cases that underwent successful NGS, mutations were detected in genes and pathways that could possibly be therapeutically targetable. The PI3K pathway is recognized to be critical in cellular transformation, cell proliferation, adhesion, survival, and motility of cancer cells. In support of our observations, studies have shown that PIK3CA is mutated in up to 30% of CRC as well as other tumors such as breast, ovarian, and liver cancer [47] typically leading to activation of the PI3K/AKT/mTOR signaling pathway. Activated PI3K leads to an increase in the phosphoinositides PI3,4,5P3 and PI3,4P2 which in turn leads to phosphorylation of AKT. PI3,4,5P3 serves as a substrate for SHIP2 where it is converted to PI3,4P2. Both PI3,4,5P3 and PI3,4P2 have been shown to activate AKT phosphorylation [48]. However studies have shown that PI3,4P2 is more efficient than PI3,4,5P3 in binding to the PH domain of AKT, phosphorylating S473 and leading to membrane activation [25,49]. In vitro studies using phospholipid vesicles with PI3,4P2 alone were sufficient to activate AKT[50]. INPPL1, encodes SHIP2, the inositol phosphatase that converts PI3,4,5P3 to PI3,4P2. SHIP2 is also a negative regulator of insulin signaling, plays an important role in EGF receptor turnover, and also has been reported to negatively regulate the PI3K pathway [51]. INPPL1 has been shown to act as either a tumor suppressor or an oncogene in different tumor types. Furthermore, SHIP2 expression has been associated with metastasis in breast cancer [52]. INPPL1 mutations have been previously reported and occur in ~4% of colon tumors [53]. The INPPL1 E567G mutation discovered in tumor CLN2 resides within the catalytic inositol polyphosphate 5-phosphatase domain that is critical to inositol phosphatase activity, and is predicted to be damaging by SIFT. Thus we performed additional functional genomic and mechanistic studies using RNAi to better understand the role of INPPL1 in colon cancer cell growth and signaling. We used the HCT116 cell line model, which also harbors PIK3CA and KRAS mutations, similar to the patient containing the INPPL1 mutation. As knockdown of INPPL1 led to growth suppression, we hypothesize that this mutation may lead to a gain of function of INPPL1 thereby playing an oncogenic role in colon cancer as indicated by our in vitro studies. Upon INPPL1 knockdown, we observed significant negative changes in phopho-signaling of key effectors of the PI3K/AKT pathway that suggest INPPL1 might promote growth in colon cancer. This is an important insight as SHIP2 converts PI3,4,5P3 to PI3,4P2, which has been shown to directly activate AKT independent of PI3,4,5P3. Studies by Fuhler et al. also show that treatment of multiple myeloma cells with SHIP1/2 inhibitors causes cell arrest in the G2/M phase and induction of apoptosis via Caspase activation [54]. Therefore, additional studies of the role of SHIP2 in CRC are warranted, as this could provide alternative ways to approach inhibition of the PI3K/AKT axis as a means of treatment for a subset of colon cancer tumors.
Conclusion
This study provides insights into the mutational landscape of metastatic recurrent colorectal cancer. KRAS being the most frequently mutated in human cancers with ~30% in colorectal cancers are the hallmarks in all these tumor samples. Several inhibitors for the downstream signaling targets of RAS such as RAF and MEK have not been very successful. PI3K inhibitors have been used in phase II clinical trials but have also not been very promising. There remains an urgent need to develop KRAS inhibitors to enhance treatment options in mCRC patients with KRAS mutations. Using an in vitro model with a colon cancer cell line, we have identified that an effective way of activating AKT signaling could be through PI3,4P2 and INPPL1. The inhibition of INPPL1 may have a very significant role in cell proliferation and survival in colon cancer. And although specific recurrent mutations exist, our study further highlights therapeutically relevant contexts within metastatic colon tumors that might lead to new and improved ways to manage these difficult to treat tumors.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
VS Analysis and interpretation of data, and drafting and revising the content of the manuscript. RR Principal Investigator, acquisition of patient samples, designing treatment options based on the sequencing results, drafting the clinical content in the manuscript. NL Cell proliferation studies and Western analysis. SS Creating the pipeline for the sequencing analysis and running the initial analysis. MC Acquisition of patient’s sample and study design. WL Library preparation and Illumina sequencing of the tumor and normal samples, guidance and training of additional staff for library preparation and sequencing. AK, TI, AC Sequencing analysis pipeline support. HB, LP Library preparation and Illumina sequencing of the tumor and normal samples. AB DNA isolation from the patient blood samples and tumor specimens. CM Validation of sequencing data. GH Determined tumor cellularity in the tumor samples and chipped the tumor sample for DNA isolation. DVH, DC Contributed to intellectual content of the paper. JC Principal Investigator and contributing to the intellectual content of the paper. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Supplementary Material
CLN2 Circos Plot. Circos plot illustrating somatic events occurring in patient CLN2. Copy number changes are shown in the inner circle plot with red denoting copy number amplification and green denoting copy number deletion. Lines adjacent to gene names describe type of somatic event that a gene is involved in including somatic point mutation (blue), somatic indel (cyan).
{kind=link}
CLN3 Circos Plot. Circos plot illustrating somatic events occurring in patient CLN3. Copy number changes are shown in the inner circle plot with red denoting copy number amplification and green denoting copy number deletion. Lines adjacent to gene names describe type of somatic event that a gene is involved in including somatic point mutation (blue), somatic indel (cyan).
{kind=link}
CLN4 Circos Plot. Circos plot illustrating somatic events occurring in patient CLN4. Copy number changes are shown in the inner circle plot with red denoting copy number amplification and green denoting copy number deletion. Lines adjacent to gene names describe type of somatic event that a gene is involved in including somatic point mutation (blue), somatic indel (cyan).
{kind=link}
SNVs and Indels.
SIFT Predictions for SNVs and Indels.
CNVs.
Contributor Information
Vijayalakshmi Shanmugam, Email: viji@tgen.org.
Ramesh K Ramanathan, Email: rramanathan@tgen.org.
Nicole A Lavender, Email: nlavender@tgen.org.
Shripad Sinari, Email: shripad.sinari@gmail.com.
Manpreet Chadha, Email: mchadha@pvhomed.com.
Winnie S Liang, Email: wliang@tgen.org.
Ahmet Kurdoglu, Email: akurdoglu@tgen.org.
Tyler Izatt, Email: tizatt@tgen.org.
Alexis Christoforides, Email: achristoforides@tgen.org.
Hollie Benson, Email: hollieLbenson@gmail.com.
Lori Phillips, Email: lphillips@tgen.org.
Angela Baker, Email: abaker@tgen.org.
Christopher Murray, Email: cmurray@tgen.org.
Galen Hostetter, Email: ghostetter@tgen.org.
Daniel D Von Hoff, Email: dvh@tgen.org.
David W Craig, Email: dcraig@tgen.org.
John D Carpten, Email: jcarpten@tgen.org.
Acknowledgements
This work was funded by the Stardust Foundation (Scottsdale, AZ), and The Bernice E. Holland Foundation (Colorado Springs, CO).
References
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA: a cancer journal for clinicians. 2013;7:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- Cunningham D, Atkin W, Lenz HJ, Lynch HT, Minsky B, Nordlinger B, Starling N. Colorectal cancer. Lancet. 2010;7:1030–1047. doi: 10.1016/S0140-6736(10)60353-4. [DOI] [PubMed] [Google Scholar]
- Segal NH, Saltz LB. Evolving treatment of advanced colon cancer. Annu Rev Med. 2009;7:207–219. doi: 10.1146/annurev.med.60.041807.132435. [DOI] [PubMed] [Google Scholar]
- Grothey A, Van Cutsem E, Sobrero A, Siena S, Falcone A, Ychou M, Humblet Y, Bouche O, Mineur L, Barone C, Adenis A, Tabernero J, Yoshino T, Lenz HJ, Goldberg RM, Sargent DJ, Cihon F, Cupit L, Wagner A, Laurent D, Group CS. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;7:303–312. doi: 10.1016/S0140-6736(12)61900-X. [DOI] [PubMed] [Google Scholar]
- Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology. 2008;7:1079–1099. doi: 10.1053/j.gastro.2008.07.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern A, Taubert H, Scheele J, Rudroff C, Mothes H, Kappler M, Bartel F, Richter KK. Association of p53 mutations, microvessel density and neoangiogenesis in pairs of colorectal cancers and corresponding liver metastases. Int J Oncol. 2002;7:243–249. doi: 10.3892/ijo.21.2.243. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;7:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- Akhurst RJ. TGF beta signaling in health and disease. Nat Genet. 2004;7:790–792. doi: 10.1038/ng0804-790. [DOI] [PubMed] [Google Scholar]
- Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, Dawson D, Willson JK, Gazdar AF, Hartigan J, Wu L, Liu C, Parmigiani G, Park BH, Bachman KE, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;7:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, Kaminker JS, Zhang Z, Croshaw R, Willis J, Dawson D, Shipitsin M, Willson JK, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PV. et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;7:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- Bass AJ, Lawrence MS, Brace LE, Ramos AH, Drier Y, Cibulskis K, Sougnez C, Voet D, Saksena G, Sivachenko A, Jing R, Parkin M, Pugh T, Verhaak RG, Stransky N, Boutin AT, Barretina J, Solit DB, Vakiani E, Shao W, Mishina Y, Warmuth M, Jimenez J, Chiang DY, Signoretti S, Kaelin WG, Spardy N, Hahn WC, Hoshida Y, Ogino S. et al. Genomic sequencing of colorectal adenocarcinomas identifies a recurrent VTI1A-TCF7L2 fusion. Nat Genet. 2011;7:964–968. doi: 10.1038/ng.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roychowdhury S, Iyer MK, Robinson DR, Lonigro RJ, Wu YM, Cao X, Kalyana-Sundaram S, Sam L, Balbin OA, Quist MJ, Barrette T, Everett J, Siddiqui J, Kunju LP, Navone N, Araujo JC, Troncoso P, Logothetis CJ, Innis JW, Smith DC, Lao CD, Kim SY, Roberts JS, Gruber SB, Pienta KJ, Talpaz M, Chinnaiyan AM. Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci Transl Med. 2011;7:111ra121. doi: 10.1126/scitranslmed.3003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienstmann R, Serpico D, Rodon J, Saura C, Macarulla T, Elez E, Alsina M, Capdevila J, Perez-Garcia J, Sanchez-Olle G, Aura C, Prudkin L, Landolfi S, Hernandez-Losa J, Vivancos A, Tabernero J. Molecular profiling of patients with colorectal cancer and matched targeted therapy in phase I clinical trials. Mol Cancer Ther. 2012;7:2062–2071. doi: 10.1158/1535-7163.MCT-12-0290. [DOI] [PubMed] [Google Scholar]
- Craig DW, O’Shaughnessy JA, Kiefer JA, Aldrich J, Sinari S, Moses TM, Wong S, Dinh J, Christoforides A, Blum JL, Aitelli CL, Osborne CR, Izatt T, Kurdoglu A, Baker A, Koeman J, Barbacioru C, Sakarya O, De La Vega FM, Siddiqui A, Hoang L, Billings PR, Salhia B, Tolcher AW, Trent JM, Mousses S, Von Hoff D, Carpten JD. Genome and transcriptome sequencing in prospective metastatic triple-negative breast cancer uncovers therapeutic vulnerabilities. Mol Cancer Ther. 2013;7:104–116. doi: 10.1158/1535-7163.MCT-12-0781. [DOI] [PubMed] [Google Scholar]
- Weiss GJ, Liang WS, Demeure MJ, Kiefer JA, Hostetter G, Izatt T, Sinari S, Christoforides A, Aldrich J, Kurdoglu A, Phillips L, Benson H, Reiman R, Baker A, Marsh V, Von Hoff DD, Carpten JD, Craig DW. A pilot study using next-generation sequencing in advanced cancers: feasibility and challenges. PloS one. 2013;7:e76438. doi: 10.1371/journal.pone.0076438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang WS, Craig DW, Carpten J, Borad MJ, Demeure MJ, Weiss GJ, Izatt T, Sinari S, Christoforides A, Aldrich J, Kurdoglu A, Barrett M, Phillips L, Benson H, Tembe W, Braggio E, Kiefer JA, Legendre C, Posner R, Hostetter GH, Baker A, Egan JB, Han H, Lake D, Stites EC, Ramanathan RK, Fonseca R, Stewart AK, Von Hoff D. Genome-wide characterization of pancreatic adenocarcinoma patients using next generation sequencing. PloS one. 2012;7:e43192. doi: 10.1371/journal.pone.0043192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss GJ, Liang WS, Izatt T, Arora S, Cherni I, Raju RN, Hostetter G, Kurdoglu A, Christoforides A, Sinari S, Baker AS, Metpally R, Tembe WD, Phillips L, Von Hoff DD, Craig DW, Carpten JD. Paired tumor and normal whole genome sequencing of metastatic olfactory neuroblastoma. PloS one. 2012;7:e37029. doi: 10.1371/journal.pone.0037029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SolSNP-1.01. http://sourceforge.net/projects/solsnp/files/SolSNP-1.01/: SolSNP-1.01.
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;7:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stajich JE, Block D, Boulez K, Brenner SE, Chervitz SA, Dagdigian C, Fuellen G, Gilbert JG, Korf I, Lapp H, Lehvaslaiho H, Matsalla C, Mungall CJ, Osborne BI, Pocock MR, Schattner P, Senger M, Stein LD, Stupka E, Wilkinson MD, Birney E. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 2002;7:1611–1618. doi: 10.1101/gr.361602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. Genome Project Data Processing S. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;7:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;7:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;7:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;7:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- Ma K, Cheung SM, Marshall AJ, Duronio V. PI(3,4,5)P3 and PI(3,4)P2 levels correlate with PKB/akt phosphorylation at Thr308 and Ser473, respectively; PI(3,4)P2 levels determine PKB activity. Cell Signal. 2008;7:684–694. doi: 10.1016/j.cellsig.2007.12.004. [DOI] [PubMed] [Google Scholar]
- Normanno N, Tejpar S, Morgillo F, De Luca A, Van Cutsem E, Ciardiello F. Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat Rev Clin Oncol. 2009;7:519–527. doi: 10.1038/nrclinonc.2009.111. [DOI] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;7:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- Park KS, Jeon SH, Kim SE, Bahk YY, Holmen SL, Williams BO, Chung KC, Surh YJ, Choi KY. APC inhibits ERK pathway activation and cellular proliferation induced by RAS. J Cell Sci. 2006;7:819–827. doi: 10.1242/jcs.02779. [DOI] [PubMed] [Google Scholar]
- Maitra A, Molberg K, Albores-Saavedra J, Lindberg G. Loss of Dpc4 expression in colonic adenocarcinomas correlates with the presence of metastatic disease. The American journal of pathology. 2000;7:1105–1111. doi: 10.1016/S0002-9440(10)64625-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman TJ, Smith JJ, Chen X, Washington MK, Roland JT, Means AL, Eschrich SA, Yeatman TJ, Deane NG, Beauchamp RD. Smad4-mediated signaling inhibits intestinal neoplasia by inhibiting expression of beta-catenin. Gastroenterology. 2012;7:e562–571. doi: 10.1053/j.gastro.2011.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming NI, Jorissen RN, Mouradov D, Christie M, Sakthianandeswaren A, Palmieri M, Day F, Li S, Tsui C, Lipton L, Desai J, Jones IT, McLaughlin S, Ward RL, Hawkins NJ, Ruszkiewicz AR, Moore J, Zhu HJ, Mariadason JM, Burgess AW, Busam D, Zhao Q, Strausberg RL, Gibbs P, Sieber OM. SMAD2, SMAD3 and SMAD4 mutations in colorectal cancer. Cancer Res. 2013;7:725–735. doi: 10.1158/0008-5472.CAN-12-2706. [DOI] [PubMed] [Google Scholar]
- Poincloux L, Durando X, Seitz JF, Thivat E, Bardou VJ, Giovannini MH, Parriaux D, Barriere N, Giovannini M, Delpero JR, Monges G. Loss of Bcl-2 expression in colon cancer: a prognostic factor for recurrence in stage II colon cancer. Surg Oncol. 2009;7:357–365. doi: 10.1016/j.suronc.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Burwinkel B, Shanmugam KS, Hemminki K, Meindl A, Schmutzler RK, Sutter C, Wappenschmidt B, Kiechle M, Bartram CR, Frank B. Transcription factor 7-like 2 (TCF7L2) variant is associated with familial breast cancer risk: a case–control study. BMC Cancer. 2006;7:268. doi: 10.1186/1471-2407-6-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Kim SS, Ahn CH, Yoo NJ, Lee SH. Frameshift mutations of Wnt pathway genes AXIN2 and TCF7L2 in gastric carcinomas with high microsatellite instability. Hum Pathol. 2009;7:58–64. doi: 10.1016/j.humpath.2008.06.006. [DOI] [PubMed] [Google Scholar]
- Tang W, Dodge M, Gundapaneni D, Michnoff C, Roth M, Lum L. A genome-wide RNAi screen for Wnt/beta-catenin pathway components identifies unexpected roles for TCF transcription factors in cancer. Proc Natl Acad Sci U S A. 2008;7:9697–9702. doi: 10.1073/pnas.0804709105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;7:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angus-Hill ML, Elbert KM, Hidalgo J, Capecchi MR. T-cell factor 4 functions as a tumor suppressor whose disruption modulates colon cell proliferation and tumorigenesis. Proc Natl Acad Sci U S A. 2011;7:4914–4919. doi: 10.1073/pnas.1102300108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlajnic T, Andreozzi MC, Schneider S, Tornillo L, Karamitopoulou E, Lugli A, Ruiz C, Zlobec I, Terracciano L. VEGFA gene locus (6p12) amplification identifies a small but highly aggressive subgroup of colorectal cancer [corrected] patients. Mod Pathol. 2011;7:1404–1412. doi: 10.1038/modpathol.2011.96. [DOI] [PubMed] [Google Scholar]
- Ishii H, Yazawa T, Sato H, Suzuki T, Ikeda M, Hayashi Y, Takanashi Y, Kitamura H. Enhancement of pleural dissemination and lymph node metastasis of intrathoracic lung cancer cells by vascular endothelial growth factors (VEGFs) Lung Cancer. 2004;7:325–337. doi: 10.1016/j.lungcan.2004.02.021. [DOI] [PubMed] [Google Scholar]
- Ludovini V, Gregorc V, Pistola L, Mihaylova Z, Floriani I, Darwish S, Stracci F, Tofanetti FR, Ferraldeschi M, Di Carlo L, Ragusa M, Daddi G, Tonato M. Vascular endothelial growth factor, p53, Rb, Bcl-2 expression and response to chemotherapy in advanced non-small cell lung cancer. Lung Cancer. 2004;7:77–85. doi: 10.1016/j.lungcan.2004.03.018. [DOI] [PubMed] [Google Scholar]
- Seto T, Higashiyama M, Funai H, Imamura F, Uematsu K, Seki N, Eguchi K, Yamanaka T, Ichinose Y. Prognostic value of expression of vascular endothelial growth factor and its flt-1 and KDR receptors in stage I non-small-cell lung cancer. Lung Cancer. 2006;7:91–96. doi: 10.1016/j.lungcan.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Carrillo de Santa Pau E, Arias FC, Caso Pelaez E, Munoz Molina GM, Sanchez Hernandez I, Muguruza Trueba I, Moreno Balsalobre R, Sacristan Lopez S, Gomez Pinillos A, del Val Toledo Lobo M. Prognostic significance of the expression of vascular endothelial growth factors A, B, C, and D and their receptors R1, R2, and R3 in patients with nonsmall cell lung cancer. Cancer. 2009;7:1701–1712. doi: 10.1002/cncr.24193. [DOI] [PubMed] [Google Scholar]
- Frederick L, Wang XY, Eley G, James CD. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000;7:1383–1387. [PubMed] [Google Scholar]
- Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;7:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, Kris M, Varmus H. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;7:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markman B, Atzori F, Perez-Garcia J, Tabernero J, Baselga J. Status of PI3K inhibition and biomarker development in cancer therapeutics. Ann Oncol. 2010;7:683–691. doi: 10.1093/annonc/mdp347. [DOI] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;7:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Scheid MP, Huber M, Damen JE, Hughes M, Kang V, Neilsen P, Prestwich GD, Krystal G, Duronio V. Phosphatidylinositol (3,4,5)P3 is essential but not sufficient for protein kinase B (PKB) activation; phosphatidylinositol (3,4)P2 is required for PKB phosphorylation at Ser-473: studies using cells from SH2-containing inositol-5-phosphatase knockout mice. The Journal of biological chemistry. 2002;7:9027–9035. doi: 10.1074/jbc.M106755200. [DOI] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997;7:665–668. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- Klippel A, Kavanaugh WM, Pot D, Williams LT. A specific product of phosphatidylinositol 3-kinase directly activates the protein kinase Akt through its pleckstrin homology domain. Mol Cell Biol. 1997;7:338–344. doi: 10.1128/mcb.17.1.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagawa S, Soeda Y, Ishihara H, Oya T, Sasahara M, Yaguchi S, Oshita R, Wada T, Tsuneki H, Sasaoka T. Impact of transgenic overexpression of SH2-containing inositol 5′-phosphatase 2 on glucose metabolism and insulin signaling in mice. Endocrinology. 2008;7:642–650. doi: 10.1210/en.2007-0820. [DOI] [PubMed] [Google Scholar]
- Prasad NK, Tandon M, Badve S, Snyder PW, Nakshatri H. Phosphoinositol phosphatase SHIP2 promotes cancer development and metastasis coupled with alterations in EGF receptor turnover. Carcinogenesis. 2008;7:25–34. doi: 10.1093/carcin/bgm213. [DOI] [PubMed] [Google Scholar]
- Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, Teague JW, Campbell PJ, Stratton MR, Futreal PA. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;7:D945–950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhler GM, Brooks R, Toms B, Iyer S, Gengo EA, Park MY, Gumbleton M, Viernes DR, Chisholm JD, Kerr WG. Therapeutic potential of SH2 domain-containing inositol-5′-phosphatase 1 (SHIP1) and SHIP2 inhibition in cancer. Mol Med. 2012;7:65–75. doi: 10.2119/molmed.2011.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
CLN2 Circos Plot. Circos plot illustrating somatic events occurring in patient CLN2. Copy number changes are shown in the inner circle plot with red denoting copy number amplification and green denoting copy number deletion. Lines adjacent to gene names describe type of somatic event that a gene is involved in including somatic point mutation (blue), somatic indel (cyan).
CLN3 Circos Plot. Circos plot illustrating somatic events occurring in patient CLN3. Copy number changes are shown in the inner circle plot with red denoting copy number amplification and green denoting copy number deletion. Lines adjacent to gene names describe type of somatic event that a gene is involved in including somatic point mutation (blue), somatic indel (cyan).
CLN4 Circos Plot. Circos plot illustrating somatic events occurring in patient CLN4. Copy number changes are shown in the inner circle plot with red denoting copy number amplification and green denoting copy number deletion. Lines adjacent to gene names describe type of somatic event that a gene is involved in including somatic point mutation (blue), somatic indel (cyan).
SNVs and Indels.
SIFT Predictions for SNVs and Indels.
CNVs.