

Abstract
Oxidative stress plays an important role in the pathogenesis of liver diseases. N-Acetyl-serotonin (NAS) has been reported to protect against oxidative damage, though the mechanisms by which NAS protects hepatocytes from oxidative stress remain unknown. To determine whether pretreatment with NAS could reduce hydrogen peroxide- (H2O2-) induced oxidative stress in HepG2 cells by inhibiting the mitochondrial apoptosis pathway, we investigated the H2O2-induced oxidative damage to HepG2 cells with or without NAS using MTT, Hoechst 33342, rhodamine 123, Terminal dUTP Nick End Labeling Assay (TUNEL), dihydrodichlorofluorescein (H2DCF), Annexin V and propidium iodide (PI) double staining, immunocytochemistry, and western blot. H2O2 produced dramatic injuries in HepG2 cells, represented by classical morphological changes of apoptosis, increased levels of malondialdehyde (MDA) and intracellular reactive oxygen species (ROS), decreased activity of superoxide dismutase (SOD), and increased activities of caspase-9 and caspase-3, release of cytochrome c (Cyt-C) and apoptosis-inducing factor (AIF) from mitochondria, and loss of membrane potential (ΔΨm). NAS significantly inhibited H2O2-induced changes, indicating that it protected against H2O2-induced oxidative damage by reducing MDA levels and increasing SOD activity and that it protected the HepG2 cells from apoptosis through regulating the mitochondrial apoptosis pathway, involving inhibition of mitochondrial hyperpolarization, release of mitochondrial apoptogenic factors, and caspase activity.
1. Introduction
It is now well established that oxidative stress, characterized by a cellular imbalance in the production and elimination of reactive oxygen species (ROS), plays an important role in the pathogenesis of various liver disorders, including liver inflammation [1–3], hepatic cirrhosis [4, 5], hepatoma [6], and hepatic ischemia-reperfusion (I/R) injury [7–9]. Hydrogen peroxide (H2O2) is an important cause of oxidative injury because its half-life is longer than that of other reactive oxygen species and it can easily transform into a hydroxyl radical, one of the most destructive free radicals [10]. Moreover, it is generated from nearly all sources of oxidative stress and can diffuse freely in and out of many kinds of cells and tissues. Furthermore, H2O2 has been used by other investigators to induce oxidative stress in myocytes [11], HT22 cells [12], retinal pigment epithelium cells [13], and BEL-7402 cells [14–16]. Therefore, we chose H2O2 to induce HepG2 cells death in our study because it has been reported to trigger apoptosis in hepatocytes, the main cause of hepatocyte death during hepatic I/R injury [17–22].
A large body of evidence indicates that though ROS are generated mainly as by-products of mitochondrial respiration, they are themselves extremely susceptible to oxidative damage. Thus, a feed-forward loop is set up whereby ROS-mediated oxidative damage to mitochondria favors more ROS generation, creating a vicious cycle [23]. The overproduction of ROS leads to lipid peroxidation, damage to the mitochondrial membrane, release of mitochondrial apoptogenic factors into the cytoplasm, followed by caspase activation and finally cellular apoptosis [24, 25]. It is becoming clear that mitochondria play a critical part in cellular bioenergetics through production of ROS and control of cell death. Therefore, modulation of mitochondrial function by antioxidants has become a novel therapeutic strategy to prevent oxidative damage, which is a key mechanism leading to liver inflammation, hepatic cirrhosis, hepatoma, and hepatic I/R injury.
Growing evidence supports the contention that H2O2-induced cell injury may be prevented by antioxidants such as N-acetyl-serotonin (NAS) and Vitamin E [26–28]. NAS, the immediate precursor of melatonin in the tryptophan metabolic pathway in the pineal gland, is a free radical scavenger. NAS has recently been shown to have various biologic activities, including memory-facilitating [29], hypothermic [30], analgesic [31], circadian rhythm adjustment [32], antidepressant [32], antiaging [33], lowering blood pressure [28], and antioxidative effects [28]. As described in our recent reports, NAS protects against acute hepatic I/R injury in mice by reversing the imbalance between oxidants and antioxidants and inhibiting hepatocyte apoptosis via the intrinsic apoptotic pathway [34] and offers neuroprotection through inhibiting mitochondrial death pathways in experimental models of ischemic stroke [35]. However, the mechanism by which NAS protects hepatocytes from oxidative stress remains unknown. Here we investigated the effects of NAS on oxidative HepG2 cells damage induced by H2O2 and whether mitochondria-dependent apoptosis was involved in the cytoprotective effect of NAS.
2. Materials and Methods
2.1. Chemicals
AIF (apoptosis-inducing factor) antibody, NAS, and 2,7-dichlorodihydrofluorescein diacetate were obtained from Sigma (St. Louis, MO). Cleaved caspase-3, cleaved caspase-9, Cyt-C, and β-actin antibodies were purchased from Cell Signaling Technology, Inc. (Beverly, MA). Cox-IV antibodies came from BD PharMingen (San Diego, CA). Horseradish-conjugated secondary anti-mouse antibody and anti-rabbit antibody were purchased from Amersham Pharmacia Biotech (Piscataway, NJ). FITC-conjugated secondary antibodies were purchased from Jackson Immunoresearch (West Grove, PA). Hoechst 33342, rhodamine 123, and tetramethylrhodamine methyl ester (TMRM) were purchased from Life Technologies Corporation (Grand Island, NY). The enhanced chemiluminescence (ECL) system was purchased from Amersham Pharmacia Biotech (Piscataway, NJ). MDA and SOD kits were obtained from the NanJing JianCheng Bioengineering Institute (Nanjing, China). The TUNEL apoptosis kits came from Boster Biotechnology (Wuhan, China). Annexin V-FITC/propidium iodide (PI) Apoptosis Detection Kit was purchased from Roche (Germany).
2.2. Cell Lines and Induction of Cell Death
The human hepatoma cell line HepG2 was kindly provided by Dr. Shujuan Liang. Originally derived from a hepatocellular carcinoma biopsy, this cell line synthesizes nearly all human plasma proteins. With high degree of differentiation, HepG2 cells retain many biological characteristics of hepatocytes and could synthesize nearly all human plasma proteins. It has been widely used as a cellular model to investigate the oxidative injury of hepatocytes elicited by H2O2 [36–40], arachidonic acid and iron [41], hepatocytes toxicity induced by alcohol [42] or CYP2E1 [43], and hepatocellular injury by Hepatitis C virus [44]. HepG2 cells were cultured in a humidified atmosphere of 95% air plus 5% CO2 in a 37°C incubator in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum, 100 μM streptomycin, and 100 U/mL penicillin. Cell death was induced by exposure to H2O2 as previously demonstrated by others [45, 46]. In brief, cells were challenged with H2O2 with or without NAS for 6 hours. The morphology of HepG2 cell nuclei was analyzed with Hoechst 33342 and Annexin V-FITC/PI double staining.
2.3. Determination of Cytotoxicity by MTT Assay
The MTT assay is based on the principle that viable cells convert MTT into an insoluble formazan salt. The absorbance at 490 nm was read in an ELISA plate reader. Briefly, HepG2 cells were cultured in 96-well microtiter plates in a final volume of 100 μL culture medium per well. After incubation for 24 hours at 37°C and 5% CO2, when a partial monolayer was formed, the supernatant was flicked off, washed once, 20 μL of MTT (5 mg/mL) in PBS solution was added to each well, and then the plate was further incubated for 4 h. Most of the medium was removed and 100 μL of DMSO was added into the wells to solubilize the crystals. Finally the optical density (OD) was measured by ELISA reader at wavelength of 490 nm. All cytotoxicity assays were performed in duplicate. The percentage growth inhibition was calculated using the following formula:
| (1) |
2.4. MDA and SOD Assays
MDA and SOD in HepG2 cells were measured with commercial kits according to the manufacturer's recommendations as previously described [23]. MDA levels and SOD activity were expressed as nmol/mg protein and U/mg protein, respectively.
2.5. Intracellular ROS Measurement
ROS was monitored using dihydrodichlorofluorescein (H2DCF) cell-permeant probe according to the manufacturer's recommendations. Briefly, the cells from different groups were collected and incubated for 20 min in the dark at 37°C in PBS containing 10 μM H2DCF. The levels of ROS in HepG2 cells were analyzed by measuring the mean fluorescence intensity (MFI) of DCF using a FACSCalibur flow cytometer (Becton-Dickinson, San Jose, CA, USA) with an excitation of 485 nm and emission of 530 nm. The results were expressed as the fold change of the percentage increase in DCF channel. The experiments were performed in triplicate and repeated three times.
2.6. Observation of Nuclear Damage by Hoechst 33342 Staining
HepG2 cells were exposed to 200 μM H2O2 with or without NAS for 6 hours. The cells were then incubated with Hoechst 33342 (1 : 10,000) for 5 minutes, and changes to cell nuclei were observed by fluorescence microscope (OLYMPUS, BX53F).
2.7. Terminal dUTP Nick End Labeling Assay (TUNEL)
HepG2 cells were fixed in 4% paraformaldehyde, permeabilized with 0.05% Triton X-100, and then subjected to a TUNEL assay with commercial kits according to the manufacturer's recommendations as previously described [23]. TUNEL-positive cells were colored using diaminobenzidine (DAB) and counterstained with hematoxylin.
2.8. Annexin V-FITC/Propidium Iodide Double Staining as a Measure of Cell Apoptosis
Briefly, cells were trypsinized following a wash in PBS and resuspended in 400 μL 1x binding buffer. Then 5 μL Annexin V-FITC and 5 μL PI (50 μM) working solution were added to every 200 μL of cell suspension. The cells were incubated at room temperature for 20 min in the dark and analyzed by flow cytometry. The Annexin V and PI emissions were detected in the FL1-H and FL2-H channels of a FACSVantage flow cytometer, using emission filters of 525 and 575 nm, respectively. The results were shown as quadrant dot plots with intact cells (Annextin V−/PI−), early apoptotic cells (Annextin V+/PI−), late apoptotic cells (Annextin V+/PI+), and necrotic cells (Annextin V−/PI+). The number of each kind of cells was expressed as percentages of the number of total stained cells. Data were acquired using Cellqest software and analyzed by ModFit software.
2.9. Isolation of Mitochondrial and Cytosolic Fractions and Western Blotting Analysis for Detection of Cyt-C and AIF
Cell cytosolic and mitochondrial fractionations were performed as described [47, 48]. Briefly, HepG2 cells (3 to 3.6 × 106) were homogenized using a Dounce homogenizer on ice in a homogenization buffer [10 mM HEPES, pH 7.4, 250 mM sucrose, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT plus protease inhibitor cocktail (Roche Molecular Biochemicals)], followed by 2000 ×g centrifugation for 5 minutes at 4°C; the supernatant was centrifuged at 15,000 ×g for 25 minutes at 4°C and used as the cytosolic component containing the released Cyt-C. The pellets were lysed with RIPA buffer for 10 minutes on ice, and supernatants were added to the sample buffer to obtain the mitochondria fraction. Protein concentration was determined by Lowry's method. Western blot was performed to identify the released Cyt-C and AIF. β-actin and COX-IV were used as cytosolic and mitochondrial component loading controls, respectively. Next, proteins (50 μg/sample) were run on 15% SDS-PAGE and transferred to PVDF membranes. The membranes were blocked with 5% nonfat milk in Tris-buffered saline and incubated with antibody to cleaved Cyt-C (1 : 500), AIF (1 : 500), β-actin (1 : 5000), and COX-IV (1 : 750), respectively. The reaction was followed by the addition of a horseradish-conjugated secondary anti-mouse antibody (1 : 2000) or anti-rabbit antibody (1 : 2000), and bands were visualized with an ECL system according to the manufacturer's instructions. Densitometric analyses of the western blot bands were performed using optical density scanning and ImageJ software. The ratio of target protein optical density values to those of the internal reference protein β-actin or COX-IV was determined for each group.
2.10. Western Blotting Analysis for Detection Activities of Caspase-9 and Caspase-3
HepG2 cells were exposed to 200 μM H2O2 with or without NAS. Cells were collected in lysis buffer [20 mM Tris, pH 8.0, 137 mM NaCl, 10% glycerol/1% Nonidet P-40, 2 mM EDTA with 5 mM Na2VO4, protease inhibitor mixture (Roche Molecular Biochemicals), 0.2 mM phenylmethylsulfonyl fluoride] on ice, centrifuged at 19,720 ×g for 10 minutes at 4°C, cytoplasmic proteins were extracted, and protein concentration was determined by Lowry's method. Samples of total protein lysates (50 μg/sample) were run on 10% SDS-PAGE gels and caspase-9 and caspase-3 were analyzed by western blot.
2.11. Immunocytochemistry
Cultured HepG2 cells were exposed to H2O2 with or without 10 μM NAS and then fixed in 4% paraformaldehyde for 15 minutes and permeabilized with 0.05% Triton X-100. After blocking with 5% BSA, cells were incubated with antibody to cleaved caspase-3 (1 : 200), with cleaved caspase-9 (1 : 200), and incubated with FITC-conjugated secondary antibodies (1 : 150), and these fluorescent slices were counterstained with Hoechst 33342 (1 : 10,000) for 5 minutes. These incubations were conducted in the dark so as not to bleach the fluorescent labels. As negative controls, we performed staining in the absence of each of the primary antibodies and added bovine serum albumin. Images were taken using a fluorescence microscope (OLYMPUS, BX53F).
2.12. Rhodamine 123 Staining
HepG2 cells were cultured in 6-well plates and pretreated with vehicle or 10 μM NAS for 2 hours and then treated with 200 μM H2O2 for 6 hours. Mitochondrial membrane potential was determined using rhodamine 123, a cationic fluorescent indicator that selectively accumulates within mitochondria in a membrane potential depending manner, as described previously [49, 50]. Briefly, the cultured cells were directly incubated with 2 μM rhodamine 123 for 25 minutes at room temperature in the dark, followed by rinsing with several changes of PBS (5 minutes per rinse). Images were taken using a fluorescence microscope (OLYMPUS, BX53F). A reduction in green rhodamine 123 fluorescence indicates dissipated ΔΨm.
Quantification of mitochondrial transmembrane potential (ΔΨm) was determined using tetramethyl rhodamine methyl ester (TMRM), a lipophilic cation that selectively accumulates within mitochondria in a membrane potential depending manner. Briefly, the cells from different groups were trypsinized following a wash in PBS. Then, add 15 μL of TMRM working solution to every 1000 μL of cell suspension (final concentration 150 nM of TMRM) for 25 minutes at 37°C, allowing mitochondria to load with TMRM fluorescent dye in proportion to mitochondrial membrane potential. Following washing 3 times with PBS gently, TMRM fluorescence was detected on a FACSCalibur flow cytometer.
2.13. Statistical Analysis
All data are expressed as mean ± standard deviation (SD) and were analyzed using SPSS 11.0 software (SPSS, Chicago, IL). P < 0.05 was considered statistically significant.
3. Results
3.1. NAS Inhibits H2O2-Induced Cell Death of HepG2 Cells
H2O2 has often been used in the oxidative stress injury model with hepatocytes as well as other cell types [17–22]. To determine the proper working concentrations of H2O2, we performed a series of dose-response assays. HepG2 cells were randomly assigned to nine groups: the untreated group and H2O2-treated groups with different concentrations (50, 100, 150, 200, 300, 500, 600, 800, or 1000 μM) for 6 hours. The viability of HepG2 cells in different groups was measured by MTT assay. We found that 200 μM H2O2 caused cell viability to decrease by about 50% (Figure 1(a)). Therefore we exposed HepG2 cells to concentration of 200 μM H2O2 for 6 hours to establish an oxidative stress injury model. To evaluate the most effective concentrations of NAS, H2O2-induced HepG2 cells were treated with different concentrations of NAS (from 1 to 100 μM). We found that NAS showed protective effects in a dose-dependent manner in the range 2.5–10 μM. There was no difference between 10 and 20 μM. Cell viability in the 30- and 100-μM groups was significantly lower than in the 20-μM group (Figure 1(b)). Therefore we used 10 μM in this study. As shown in Figure 1(c), the cell viability of HepG2 cells was significantly decreased after treatment with 200 μM H2O2 (n = 6, P < 0.01). However, NAS treatment effectively protected HepG2 cells from H2O2-induced cell death (n = 6, P < 0.01). The difference was statistically significant (P < 0.01). While NAS alone has no effect on the cell viability of HepG2 cells (Figure 1(c)), we further compared the effect of NAS with another well standardized antioxidant melatonin on cell viability of HepG2 cells and found that there was no difference between H2O2 + NAS group and H2O2 + melatonin group. The data indicate that NAS, similar with melatonin does, effectively protects HepG2 cells from H2O2-induced cell death.
Figure 1.
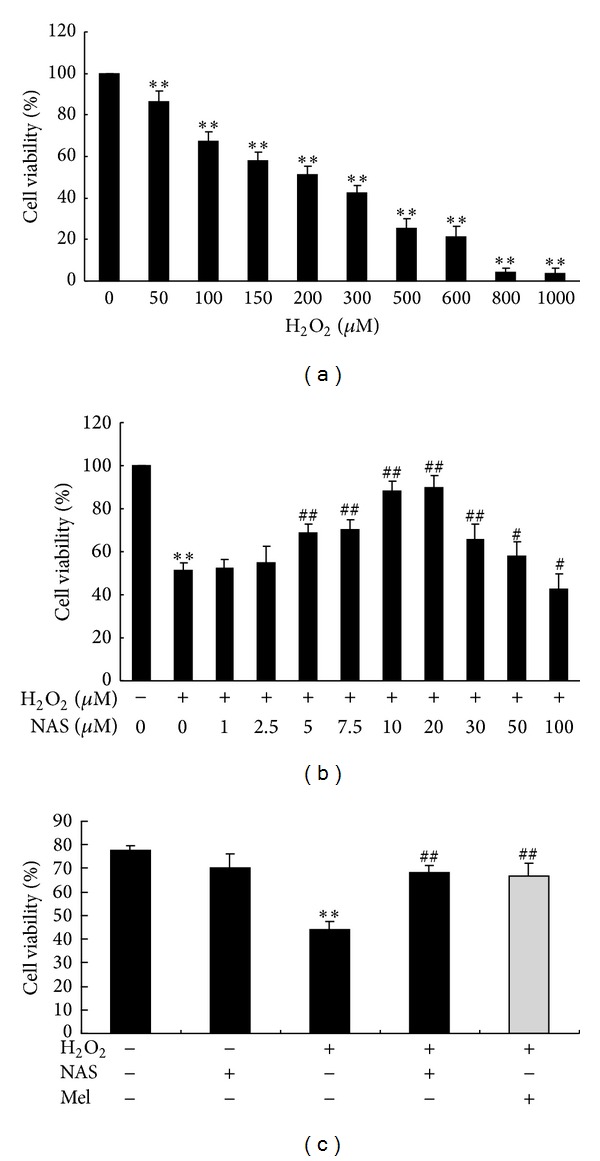
Cell viability of HepG2 cells assayed by MTT. The results are expressed as percentage of control, and each value represents the mean ± SD of six independent experiments. The annotation **indicates a P value < 0.01 versus untreated group. The annotation #indicates a P value < 0.05 versus H2O2 group. The annotation ##indicates a P value < 0.01 versus H2O2 group. (a) Cell viability of HepG2 cells following different concentrations of H2O2 exposure were measured by MTT assay. (b) Cytotoxicity of NAS to HepG2 cells was measured by MTT assay. HepG2 cells were treated by 200 μM H2O2 with or without a series of concentrations of NAS (1, 2.5, 5, 7.5, 10, 20, 30, 50, and 100 μM) for 6 hours. (c) The effect of NAS on HepG2 cell viability in response to H2O2 and comparing it with melatonin (grey bar), a well standardized antioxidant.
3.2. NAS Inhibits H2O2-Induced Reactive Oxygen Species Production in HepG2 Cells
To determine the effect of NAS on H2O2-induced intracellular ROS production, the intracellular ROS was visualized by detecting dichlorofluorescein (DCF) derived from the oxidation of H2DCF. The results show that untreated HepG2 cells had little basal intracellular ROS. However, after exposure to H2O2, cells had significantly increased intracellular ROS accumulation (P < 0.05). The intensity of the mean oxidized DCF in untreated and H2O2 group was (3.60 ± 1.02) and (34.85 ± 5.67), respectively. The intensity in NAS-treated group was (7.85 ± 2.93), which was significantly lower than in the H2O2 group. To determine whether the cytoprotective effect of NAS is a consequence of the breakdown of the endogenous antioxidant defense mechanisms, we investigated the levels of MDA, an end product of lipid peroxidation, and SOD, an oxygen radical scavenger [51]. As demonstrated in Figure 2, incubation of HepG2 cells with H2O2 caused a significant increase in MDA, intracellular ROS, and a marked decrease in SOD activity compared with untreated cells (P < 0.05). However, NAS significantly attenuated those changes of MDA, intracellular ROS, and SOD (P < 0.05). This observation suggests that H2O2 could induce ROS accumulation in HepG2 cells by breakdown of the balance of the endogenous antioxidant defense mechanisms and NAS effectively reduces H2O2-induced ROS production.
Figure 2.
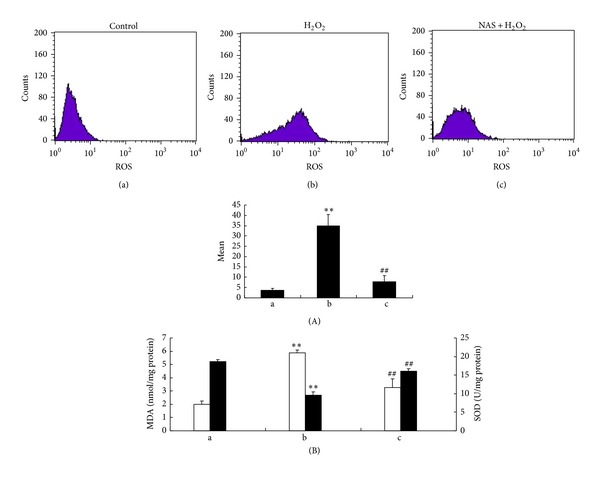
NAS reduces H2O2-induced reactive oxygen species production. (A) Intracellular ROSdetected by flow cytometry afterH2DCF staining. (B) MDA content (white bars) and SOD activity (black bars) were measured in HepG2 cell lysates. (a) Untreated group; (b) H2O2 group, HepG2 cells incubated with 200 μM H2O2 for 6 hours; (c) H2O2 + NAS group, 10 μM NAS was administered before subjecting to H2O2. The significant increase in MDA levels and decrease in SOD levels in the H2O2 group compared with untreated group are shown (P < 0.01). NAS attenuated the H2O2-induced changes in MDA and SOD (P < 0.01). Error bars represent SD (n = 3). The annotation **indicates a P value < 0.01 versus untreated group. The annotation ##indicates a P value < 0.01 versus H2O2 group. Experiments were performed at least three times.
3.3. NAS Inhibits H2O2-Induced HepG2 Cell Apoptosis
To evaluate the cytoprotective effect of NAS on H2O2-induced HepG2 cell apoptosis, three assays (Annexin V and PI double-staining, Hoechst 33342 staining and TUNEL staining) were conducted. The results of Annexin V and PI double staining demonstrated that an increase of apoptotic cells was observed in H2O2-treated group with a lower number of living cells. The apoptotic percentage was (3.07 ± 0.57)% in the untreated group, which was significantly lower than in the H2O2 group (7.61 ± 2.73)% (P < 0.01). Compared to H2O2 group, the apoptotic percentage in NAS-treated group (3.37 ± 0.56)% was decreased significantly (P < 0.01). The HepG2 cells in the untreated group showed normal shape with round intact nuclei (Figures 3(a) and 3(d)), whereas the H2O2-treated cells became more scarce and showed reduced nuclear size, extensive blebbing, strong fluorescent spot, and pyknotic nuclei (Figures 3(b) and 3(e)), indicating condensed chromatin and apoptotic bodies. In agreement with the results of Hoechst 33342 staining, there were occasional TUNEL-positive cells in the untreated group untreated group. Compared to the untreated group, the H2O2 group had significantly more TUNEL-positive cells. However, those changes in HepG2 cells were abrogated significantly by pretreatment with NAS (Figures 3(c) and 3(f)).
Figure 3.
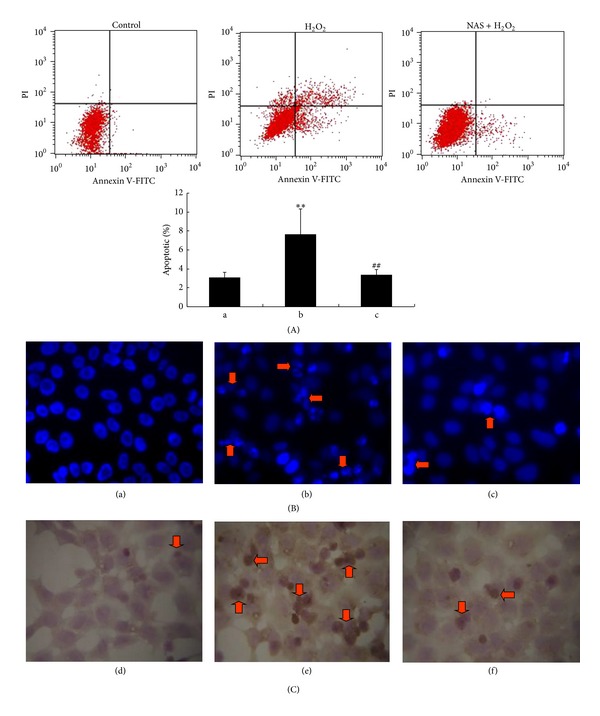
NAS inhibits H2O2-induced HepG2 cells apoptosis. (A) Apoptotic cells detected by flow cytometry after annexin V and PI double-staining. (B) (a, b, c) Morphologic changes in nuclei observed with Hoechst 33342 staining under fluorescence microscopy. (C) (d, e, f) Apoptotic cells detected by TUNEL staining. (a, d) Untreated group; (b, e) H2O2 group, HepG2 cells incubated with 200 μM H2O2 for 6 hours; (c, f) H2O2 + NAS group, 10 μM NAS was administered before subjecting to H2O2. Note that untreated group cells appeared to have a normal shape with a round intact nuclei; H2O2-treated cells showed reduced nuclear size, strong fluorescent spot, and pyknotic nuclei, indicating apoptotic nuclei; the strong fluorescent spots were almost completely abrogated by pretreatment with NAS. The arrows indicate apoptotic cells. Original magnification ×400.
3.4. NAS Inhibits H2O2-Induced Dissipation of ΔΨm of HepG2 Cells
Proper ΔΨm, the charge difference across a membrane that is permeable to ions, is critical for cellular bioenergetic homeostasis, and collapse of the ΔΨm is an important event associated with mitochondrial dysfunction, or even cell death [52]. Thus the ΔΨm assay was used as a more specific test for early mitochondrial injury. Both rhodamine 123 and TMRM are cationic fluorescent dye, which could enter the mitochondrial matrix and cause photoluminescent quenching dependent on ΔΨm. We therefore investigated possible loss of ΔΨm using rhodamine 123 and TMRM staining. The results showed that rhodamine 123 fluorescence in untreated group cells displayed a punctate staining pattern within the cell (Figures 4(a) and 4(d)). Following treatment with H2O2 for 6 hours, rhodamine 123 fluorescence became diffuse because of mitochondrial depolarization (Figures 4(b) and 4(e)), and NAS treatment preserved the punctate staining pattern of rhodamine 123 distribution in the presence of a cellular stress (Figures 4(c) and 4(f)). As shown in Figure 4(c), the fluorescence intensity of TMRM in H2O2 group decreased significantly compared to the untreated group. Cells of NAS group exhibited much more TMRM fluorescence intensity than H2O2 group. These data provide evidence that NAS-mediated protective effects not only involve rescue of nuclear morphology changes, but also prevent the dissipation of ΔΨm induced by H2O2.
Figure 4.
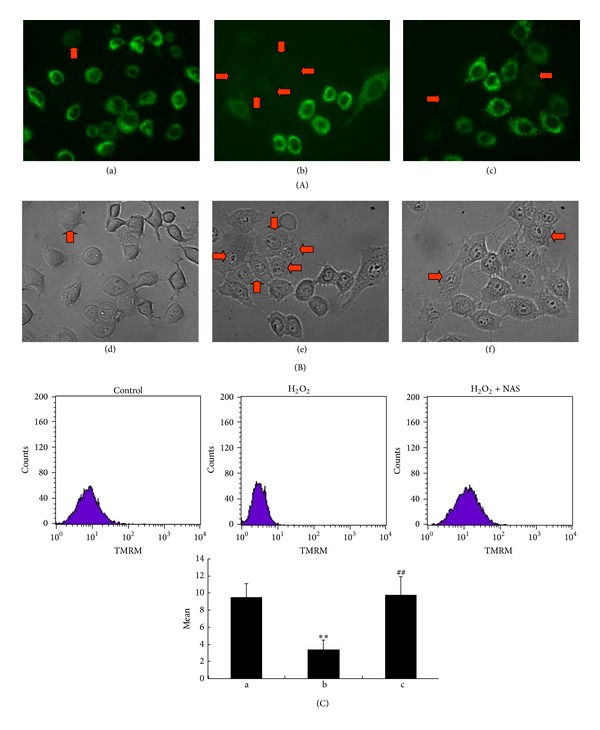
NAS inhibits the dissipation of ΔΨm of HepG2 cells induced by H2O2. (A) (a, b, c): ΔΨm observed with rhodamine 123 staining under fluorescence microscopy. (B) (d, e, f) Morphologic changes in nuclei observed under the inverted phase contrast microscope. (a, d) Untreated group; (b, e) H2O2 group, HepG2 cells incubated with 200 μM H2O2 for 6 hours; (c, f) H2O2 + NAS group, 10 μM NAS was administered before subjecting to H2O2. (C) ΔΨm detected by flow cytometry after TMRM staining. Note that untreated group cells display a granular pattern of TMRM fluorescence in cytoplasm; H2O2-treated cells showed diffuse pattern of TMRM fluorescence, indicating apoptotic nuclei; the dissipation of ΔΨm was almost completely abrogated by pretreatment with NAS. The arrows indicate the apoptotic cell. Original magnification ×400.
3.5. NAS Inhibits the Release of Mitochondrial Apoptogenic Factors from Mitochondria Induced by H2O2
Release of mitochondrial apoptogenic factors from the mitochondria into the cytoplasm is a common feature of cell death. We and other researchers previously reported that NAS effectively inhibited the release of Cyt-C and AIF from the mitochondria into the cytoplasm in apoptotic cells under oxidative stress injury induced by H2O2 and melatonin [35], while melatonin protected AIF-dependent cell death in a model of acute liver failure through its direct inhibition of hepatic RIP1 and subsequent JNK phosphorylation [53]. We therefore investigated the expression of Cyt-C and AIF in the cytosolic fractions and mitochondrial fractions of hepG2 cells by western blot analysis. As shown in Figures 5 and 6, H2O2 induced an increase of Cyt-C and AIF in the cytosolic fractions and a decrease in the mitochondrial fractions (P < 0.01). NAS effectively blocked leakage of AIF induced by H2O2 (P < 0.01).
Figure 5.
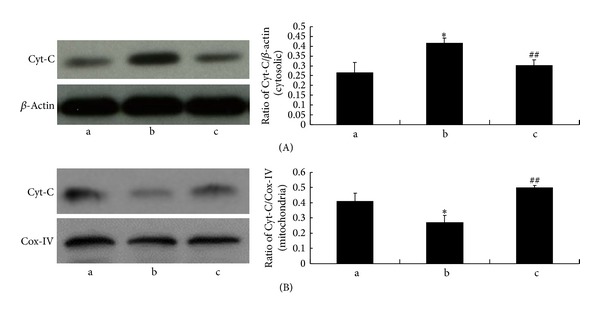
NAS reduces the release of Cyt-C from mitochondria in response to H2O2. The cells were treated with 200 μM H2O2 for 6 hours with or without NAS. Cytosolic and mitochondrial fractions were prepared, and the expression of Cyt-C was determined by western blot analysis. (A) Cytosolic fractions. (B) Mitochondrial fractions. (a) Untreated group; (b) H2O2 group, HepG2 cells incubated with 200 μM H2O2 for 6 hours; (c) H2O2 + NAS group, 10 μM NAS was administered before subjecting to H2O2. Note the significantly increased Cyt-C in cytosolic and decreased Cyt-C in mitochondria in H2O2 group compared with untreated group (P < 0.01). NAS attenuated the changes in Cyt-C. The results are expressed as ratio of Cyt-C optical density values to those of β-actin or COX-IV, and each value represents the mean ± SD of three independent experiments. The annotation *indicates a P value < 0.05 versus untreated group. The annotation ##indicates a P value < 0.01 versus H2O2 group.
Figure 6.
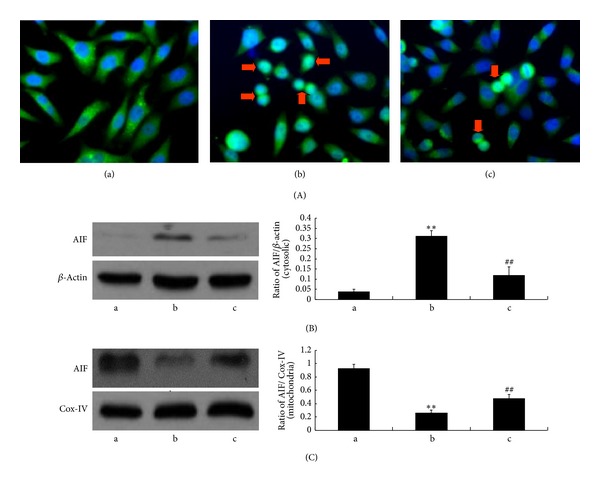
NAS inhibits the release of AIF from mitochondria in response to H2O2. (a) Untreated group; (b): H2O2 group, HepG2 cells incubated with 200 μM H2O2 for 6 hours; (c) H2O2 + NAS group, 10 μM NAS was administered before subjecting to H2O2. (A) The expression of AIF as assessed by immunocytochemistry ((a)–(c): 400x). (B, C): Cytosolic and mitochondrial fractions were prepared, and the expression of AIF was determined by western blot analysis. (B) Cytosolic fractions; (C) mitochondrial fractions. Note the AIF nuclear translocation and significantly increased AIF in cytosolic fractions and decreased AIF in mitochondrial fractions in the H2O2 group compared with untreated group (P < 0.01). NAS attenuated the changes in AIF. The results are expressed as ratio of AIF optical density values to those of β-actin or COX-IV, and each value represents the mean ± SD of three independent experiments. The annotation *indicates a P value < 0.05 versus untreated group. The annotation ##indicates a P value < 0.01 versus H2O2 group.
3.6. NAS Inhibits Activation of Caspases Induced by H2O2 in HepG2 Cells
Once Cyt-C and AIF trigger cytosolic reaction cascades encompassing caspases and regulatory factors, which eventually lead to caspase-dependent and caspase-independent cell death. As an important modulator of cell death, caspase activation was evaluated in our study. Because we were most interested in evaluating mitochondrial-dependent pathway events, we measured the activities of caspases-9 and caspase-3 by immunocytochemistry and western blot. As shown in Figures 7 and 8, caspase-9 and caspase-3 were activated by H2O2 insult, and NAS effectively inhibited their activation. These results, together with its ability to attenuate the release of Cyt-C and AIF from mitochondria, demonstrate that NAS prevents oxidant-induced apoptosis through inhibition of mitochondrial-dependent cell death pathways.
Figure 7.
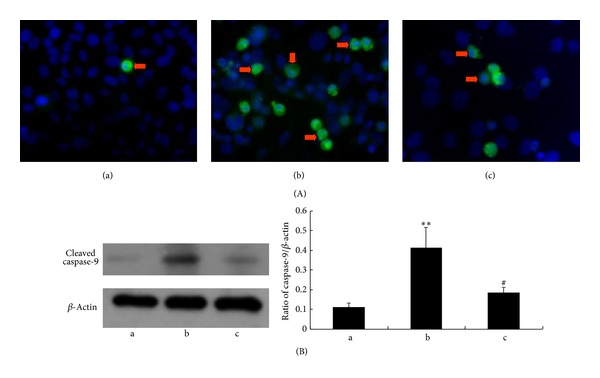
NAS prevents the activation of caspase-9. (A) Activities of caspase-9 as assessed by immunocytochemistry (400x). Cells were immunostained with active-caspase-9 antibody and stained with DAPI. (B) Activities of caspase-9 as assessed by western blot. (a) Untreated group; (b) H2O2 group, HepG2 cells incubated with 200 μM H2O2 for 6 hours; (c) H2O2 + NAS group, 10 μM NAS was administered before subjecting to H2O2. Note the increased activities of caspase-9 in the H2O2 group compared with the untreated group (P < 0.01) and the decreased activities in the H2O2 + NAS group compared with the H2O2 group (P < 0.01). The results are expressed as ratio of cleaved caspase-9 optical density values to those of β-actin, and each value represents the mean ± SD of three independent experiments. The annotation **indicates a P value <0.01 versus untreated group. The annotation #indicates a P value < 0.05 versus H2O2 group.
Figure 8.
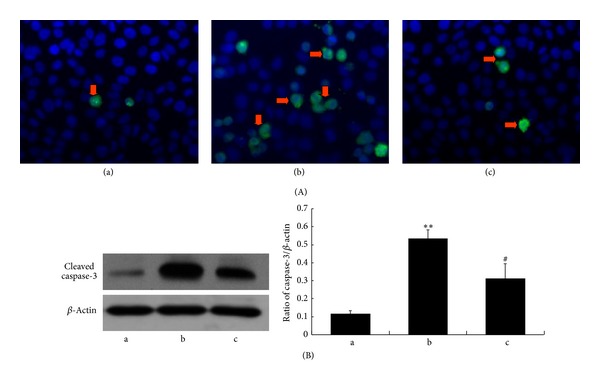
NAS prevents the activation of caspase-3. (A) Activities of caspase-3 as assessed by immunocytochemistry ((a)–(c): 400x). HepG2 cells were immunostained with active-caspase-3 antibody and FITC-conjugated secondary antibody and then stained with DAPI. (B) Activities of caspase-3 as assessed by western blot. (a) Untreated group; (b) H2O2 group, HepG2 cells incubated with 200 μM H2O2 for 6 hours; (c) H2O2 + NAS, 10 μM NAS was administered before exposure to H2O2. Note the activities of caspase-3 in the H2O2 group compared with the untreated group (P < 0.01) and the decreased activities in the H2O2 + NAS group compared with the H2O2 group (P < 0.01). The results are expressed as ratio of cleaved caspase-3 optical density values to those of β-actin, and each value represents the mean ± SD of three independent experiments. The annotation **indicates a P value < 0.01 versus untreated group. The annotation #indicates a P value < 0.05 versus H2O2 group.
4. Discussion
Hepatic I/R injury occurs in liver transplantation, hepatic resection, abdominal surgery with hepatic vascular occlusion, and coronary bypass surgery [54–59], which is a major cause of primary nonfunctioning graft. Oxidative stress associated with the formation of ROS plays an important role in the pathogenesis of hepatic I/R injury. As the primary cellular consumer of oxygen, together with multiple electron carriers and redox enzymes, mitochondria are considered a main source of ROS. However, mitochondria not only produce ROS, but are also targets of oxidative stress. In this way ROS-induced ROS release contributes to oxidative damage and mitochondrial dysfunction in a range of diseases.
Mitochondria not only play a crucial role in energy generation and intermediary metabolism, but are also known as gatekeepers in the intrinsic pathway or mitochondrial-mediated apoptosis [60]. Activation of the intrinsic pathway involves release of proapoptotic factors like Cyt-C, Smac/Diablo, Endonuclease G, and AIF from the mitochondrial intermembrane space into the cytosol. Among proapoptotic factors, Cyt-C has been the most intensively studied. Once released into the cytosol, Cyt-C triggers the formation of the apoptosome, an oligomeric complex of Cyt-C/Apaf-1/caspase-9, whose function is the recruitment and activation of caspase 9, which is the “initiator” caspase [61]. The cleaved caspase-9 targets and activates caspase-3, which is the “executioner” or “effector” caspases, catalyzing the final steps of the apoptotic signaling cascade [62]. Based on the important role of oxidative damage and mitochondrial dysfunction in a range of diseases, antioxidant and mitochondria-protective strategies will be of great importance in counteracting oxidative stress injury.
NAS is the immediate precursor of melatonin, the pineal gland indole. There are chemical and biological similarity between NAS and melatonin. Both of NAS and melatonin have the common indole ring, while hydroxy group of NAS is replaced by a methoxy at the same position of melatonin in the indole ring. Many of the biological effects of NAS including neuroprotection, antioxidant, circadian rhythm and sleep adjustment, antidepressant, antianxiety, antiaging, analgesic effects, and lowering blood pressure are similar to those produced by melatonin [28, 32, 35, 63, 64]. However, in-depth research eventually revealed that NAS has its own biological properties including its action as a potent TrkB receptor agonist and NAS is found in some areas of the brain where melatonin is absent [65]. In recent years, NAS has been reported to exert significant antioxidant properties both in vitro and in vivo. Using an erythrocyte oxidative damage model induced by cumene hydroperoxide and H2O2, Barsacchi found that 100–400 μM NAS, but not melatonin, has dose-dependent antioxidant effects [66]. Leaden and Catalá [67] and García et al. [68] found that NAS strengthened biological membranes against oxidative stress, which may be related to its ability to reduce lipid peroxidation. Additional studies have shown that NAS also exerts protective effects against peroxidative damage of neurons [69–71], lung epithelial cells [72], erythrocytes [73], testicular cells [74], retinal cells [75, 76], and lymphocytes [77]. The data imply that the antioxidant effect of NAS is independent of melatonin and stronger than that of melatonin [76, 78]. However, little is known about the effect of NAS on hepatic I/R injury.
In the present study, we show that H2O2 markedly decreases the viability of HepG2 cells, whereas pretreatment with NAS significantly inhibits cell injury, as demonstrated by MTT assay (Figure 1(c)). Our results indicate that while H2O2 can cause HepG2 cell death, NAS pretreatment effectively protects HepG2 cells from H2O2-induced damage.
Oxidative stress caused by ROS is responsible for a wide variety of cellular damage and is the most validated mechanism of secondary injury [79]. Following oxidative stress, the overproduction of ROS and subsequently the depletion of antioxidants resulted in the total breakdown of the endogenous antioxidant defense mechanisms, culminating in failure to protect cells from oxidative damage. Among biomarkers of oxidative stress, MDA and SOD are known as two sensitive indicators [48]. MDA is the end product of lipid peroxidation [80] and MDA levels reflect the extent of cell damage due to oxidative stress. SOD is an oxygen radical scavenger that scavenges superoxide radicals by converting them to hydrogen peroxide, which is then converted to water by catalase and GSH-Px [81]. In the present study, increased intracellular ROS and MDA levels and decreased SOD activity were noted in H2O2-induced HepG2 cells, suggesting that oxidative stress caused by ROS is involved in the pathogenesis of H2O2-induced HepG2 cellular injury. However, NAS reduced the decline in SOD activity and augmented MDA content (Figure 2). Our study demonstrates that NAS treatment significantly alleviates oxidative stress by reducing levels of ROS and MDA and increasing SOD activity in H2O2-induced HepG2 cells.
Through a variety of studies, we know that oxidants can not only stimulate inflammatory cytokine such as interleukin-8 [37, 82] in HepG2 cells and cause cellular senescence [83], but also induce apoptosis in HepG2 cells, [37]; certain apoptotic agents increase the production of ROS in mitochondria [84], and antioxidants such as N-acetylcysteine [85, 86], melatonin [87, 88], NAS [26], and Vitamin E [27] can prevent apoptosis. Such studies also confirmed the association between ROS-apoptosis. Several studies have shown that the level of apoptotic hepatocytes during hypoxia mirrored the level of intracellular ROS production. During the process of oxidative stress injury, mitochondria are both a significant source of ROS and also a target of damage, with a variety of consequences. Based on the prominent role of mitochondria in oxidative stress injury, many studies have clearly shown that ROS leads to cell apoptosis via the mitochondria-dependent apoptotic pathway, which involves damage to the mitochondrial membrane, release of Cyt-C into the cytoplasm followed by caspase-3 activation, and finally hepatocyte apoptosis [30]. The morphologic changes in nuclei, the dissipation of ΔΨm, the release of Cyt-C, and the expression of cleaved caspase-3 were employed as apoptotic biomarkers to elucidate the mechanisms underlying the antiapoptotic effects of NAS.
ΔΨm is a sensitive indicator of mitochondrial function. The decline of mitochondrial transmembrane potential leads to the release of caspase-activating proteins [89]. Increasing evidence suggests that the ΔΨm assay can be used as a more specific test for early mitochondrial injury. In this study, H2O2 group showed a significant decrease in mitochondrial membrane potential. Interestingly, NAS treatment preserved mitochondrial membrane potential, suggesting the inhibition of mitochondrial membrane permeability.
ROS can also induce the release of apoptogenic factors from mitochondria, which can trigger caspase cascade. Caspases are a family of cysteine proteases that cleave target proteins at specific residues. Among the more than ten members of the caspase family identified, the extensively studied caspase-3 (the “executor of apoptosis,”) plays a crucial role in cell death [90, 91]. In this study, upregulated cleaved caspase-9 and caspase-3, together with significantly decreased expression of Cyt-C and AIF in mitochondria and increased expression in the cytosol of H2O2-induced cells, indicate that the cells underwent apoptosis by a mitochondrial-dependent pathway; these parameters were lowered in the group pretreated with NAS. These findings imply that the ability of NAS to attenuate oxidative stress partly depends on inhibiting mitochondrial-related apoptosis.
In summary, the present study shows that H2O2 can damage HepG2 cells and cause cells apoptosis. NAS protects human HepG2 cells against H2O2-induced oxidative stress, as measured by cell viability, cell apoptosis, ROS activity, the dissipation of ΔΨm, release of mitochondrial apoptogenic factors, and activities of caspase-9 and caspase-3. NAS-mediated protection can be conferred by one or more of the following mechanisms: first, NAS could reduce the oxidative stress injury by restoration of endogenous antioxidation and the decrease of lipid peroxidation. Second, NAS could attenuate apoptosis through inhibiting the subsequent biochemical changes in the mitochondria-dependent apoptotic pathway, such as release of mitochondrial apoptogenic factors, activation of caspase family members, and alteration of mitochondrial membrane permeability. These data help explain the protective action of NAS against cell injuries involving the mitochondrial pathway.
Acknowledgments
The authors thank Dr. Shuanhu Zhou for technical assistance and Meina Xie for Flow Cytometric Analysis. The study was supported by the National Natural Science Foundation of China (30900775), the Natural Science Foundation of Shandong Province, China (ZR2010HM006 and ZR2011HM043), the Shandong Province Higher Educational Science and Technology Program (J11LF14), Science and Technology Program of Weifang Medical College (K1301001), the Bill & Melinda Gates Foundation, the Muscular Dystrophy Association (254530), the ALS Therapy Alliance, and the Shandong Province Taishan scholar project.
Conflict of Interests
The authors declare that they have no conflict of interests.
Authors' Contribution
Jiying Jiang and Shuna Yu wrote the paper. Jiying Jiang, Shuna Yu, Zhengchen Jiang, Cuihong Liang, Xin Wang, Wenbo Yu, Jin Li, Xiaodong Du, Hailiang Wang, and Xianghong Gao performed the studies. Jiying Jiang, Xin Wang, and Shuna Yu designed the study, reviewed the data, and revised the paper. Jiying Jiang and Shuna Yu contributed equally to this work.
References
- 1.Diesen DL, Kuo PC. Nitric oxide and redox regulation in the liver: part I. General considerations and redox biology in hepatitis. Journal of Surgical Research. 2010;162(1):95–109. doi: 10.1016/j.jss.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Das J, Ghosh J, Roy A, Sil PC. Mangiferin exerts hepatoprotective activity against D-galactosamine induced acute toxicity and oxidative/nitrosative stress via Nrf2-NFκB pathways. Toxicology and Applied Pharmacology. 2012;260(1):35–47. doi: 10.1016/j.taap.2012.01.015. [DOI] [PubMed] [Google Scholar]
- 3.Jaeschke H. Reactive oxygen and mechanisms of inflammatory liver injury: present concepts. Journal of Gastroenterology and Hepatology. 2011;26(supplement 1):173–179. doi: 10.1111/j.1440-1746.2010.06592.x. [DOI] [PubMed] [Google Scholar]
- 4.Sánchez-Valle V, Chávez-Tapia NC, Uribe M, Méndez-Sánchez N. Role of oxidative stress and molecular changes in liver fibrosis: a review. Current Medicinal Chemistry. 2012;19(28):4850–4860. doi: 10.2174/092986712803341520. [DOI] [PubMed] [Google Scholar]
- 5.Kim IH, Kim SW, Kim SH, et al. Parthenolide-induced apoptosis of hepatic stellate cells and anti-fibrotic effects in an in vivo rat model. Experimental and Molecular Medicine. 2012;44(7):448–456. doi: 10.3858/emm.2012.44.7.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farinati F, Piciocchi M, Lavezzo E, Bortolami M, Cardin R. Oxidative stress and inducible nitric oxide synthase induction in carcinogenesis. Digestive Diseases. 2010;28(4-5):579–584. doi: 10.1159/000320052. [DOI] [PubMed] [Google Scholar]
- 7.Sun K, Xie X, Liu Y, et al. Autophagy lessens ischemic liver injury by reducing oxidative damage. Cell and Bioscience. 2013;3(1, article 26) doi: 10.1186/2045-3701-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Golen RF, Reiniers MJ, Olthof PB, van Gulik TM, Heger M. Sterile inflammation in hepatic ischemia/reperfusion injury: present concepts and potential therapeutics. Journal of Gastroenterology and Hepatology. 2013;28(3):394–400. doi: 10.1111/jgh.12072. [DOI] [PubMed] [Google Scholar]
- 9.Boshra V, Moustafa AM. Effect of preischemic treatment with fenofibrate, a peroxisome proliferator-activated receptor-α ligand, on hepatic ischemia-reperfusion injury in rats. Journal of Molecular Histology. 2011;42(2):113–122. doi: 10.1007/s10735-011-9313-y. [DOI] [PubMed] [Google Scholar]
- 10.Clarkson PM, Thompson HS. Antioxidants: what role do they play in physical activity and health. The American Journal of Clinical Nutrition. 2000;72(2):637S–646S. doi: 10.1093/ajcn/72.2.637S. [DOI] [PubMed] [Google Scholar]
- 11.Liu Y, Zhang Y, Lin K, et al. Protective effect of piperine on electrophysiology abnormalities of left atrial myocytes induced by hydrogen peroxide in rabbits. Life sciences. 2014;94(2):99–105. doi: 10.1016/j.lfs.2013.10.024. [DOI] [PubMed] [Google Scholar]
- 12.Rao W, Zhang L, Su N, et al. Blockade of SOCE protects HT22 cells from hydrogen peroxide-induced apoptosis. Biochemical and Biophysical Research Communications. 2013;441(2):351–356. doi: 10.1016/j.bbrc.2013.10.054. [DOI] [PubMed] [Google Scholar]
- 13.Xu X, Hang L, Huang B, et al. Efficacy of ethanol extract of fructus iycii and its constituents lutein/zeaxanthin in protecting retinal pigment epithelium cells against oxidative stress: in vivo and in vitro models of age-related macular degeneration. Journal of Ophthalmology. 2013;2013:10 pages. doi: 10.1155/2013/862806.862806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu Y, Chen L, Yu H, Liu H, An W. Transfection of hepatic stimulator substance gene desensitizes hepatoma cells to H2O2-induced cell apoptosis via preservation of mitochondria. Archives of Biochemistry and Biophysics. 2007;464(1):48–56. doi: 10.1016/j.abb.2007.03.025. [DOI] [PubMed] [Google Scholar]
- 15.Li W, Zhang J, An W. The conserved CXXC motif of hepatic stimulator substance is essential for its role in mitochondrial protection in H2O2-induced cell apoptosis. FEBS Letters. 2010;584(18):3929–3935. doi: 10.1016/j.febslet.2010.08.043. [DOI] [PubMed] [Google Scholar]
- 16.Yang Z-C, Yang L, Zhang Y-X, Yu H-F, An W. Effect of heat and pH denaturation on the structure and conformation of recombinant human hepatic stimulator substance. Protein Journal. 2007;26(5):303–313. doi: 10.1007/s10930-007-9072-5. [DOI] [PubMed] [Google Scholar]
- 17.Chang G, Zhang D, Yu H, et al. Cardioprotective effects of exenatide against oxidative stress-induced injury. International Journal of Molecular Medicine. 2013;32(5):1011–1020. doi: 10.3892/ijmm.2013.1475. [DOI] [PubMed] [Google Scholar]
- 18.Small DM, Bennett NC, Roy S, Gabrielli BG, Johnson DW, Gobe GC. Oxidative stress and cell senescence combine to cause maximal renal tubular epithelial cell dysfunction and loss in an in vitro model of kidney disease. Nephron Experimental Nephrology. 2013;122(3-4):123–130. doi: 10.1159/000350726. [DOI] [PubMed] [Google Scholar]
- 19.Sun G-B, Qin M, Ye J-X, et al. Inhibitory effects of myricitrin on oxidative stress-induced endothelial damage and early atherosclerosis in ApoE-/- mice. Toxicology and Applied Pharmacology. 2013;271(1):114–126. doi: 10.1016/j.taap.2013.04.015. [DOI] [PubMed] [Google Scholar]
- 20.Tomassoni D, Amenta F, Amantini C, et al. Brain activity of Thioctic acid enantiomers: in vitro and in vivo studies in an animal model of cerebrovascular injury. International Journal of Molecular Sciences. 2013;14(3):4580–4595. doi: 10.3390/ijms14034580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu Y, Ruan S, Wu X, Chen H, Zheng K, Fu B. Autophagy and apoptosis in tubular cells following unilateral ureteral obstruction are associated with mitochondrial oxidative stress. International Journal of Molecular Medicine. 2013;31(3):628–636. doi: 10.3892/ijmm.2013.1232. [DOI] [PubMed] [Google Scholar]
- 22.Liu L, Duan JA, Tang YP, et al. The protective effects of the active fraction of Shaofu Zhuyu decoction on hydrogen peroxide-induced oxidative injury in vascular smooth muscle cells. Molecules. 2010;15(8):5066–5078. doi: 10.3390/molecules15085066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fiskum G, Rosenthal RE, Vereczki V, et al. Protection against ischemic brain injury by inhibition of mitochondrial oxidative stress. Journal of Bioenergetics and Biomembranes. 2004;36(4):347–352. doi: 10.1023/B:JOBB.0000041766.71376.81. [DOI] [PubMed] [Google Scholar]
- 24.Ghosh M, Manna P, Sil PC. Protective role of a coumarin-derived schiff base scaffold against tertiary butyl hydroperoxide (TBHP)-induced oxidative impairment and cell death via MAPKs, NF-κB and mitochondria-dependent pathways. Free Radical Research. 2011;45(5):620–637. doi: 10.3109/10715762.2011.564166. [DOI] [PubMed] [Google Scholar]
- 25.Dey S, Guha M, Alam A, et al. Malarial infection develops mitochondrial pathology and mitochondrial oxidative stress to promote hepatocyte apoptosis. Free Radical Biology and Medicine. 2009;46(2):271–281. doi: 10.1016/j.freeradbiomed.2008.10.032. [DOI] [PubMed] [Google Scholar]
- 26.Lezoualc’h F, Sparapani M, Behl C. N-acetyl-serotonin (normelatonin) and melatonin protect neurons against oxidative challenges and suppress the activity of the transcription factor NF-κB. Journal of Pineal Research. 1998;24(3):168–178. doi: 10.1111/j.1600-079x.1998.tb00530.x. [DOI] [PubMed] [Google Scholar]
- 27.Evans ZP, Mandavilli BS, Ellett JD, et al. Vitamin E succinate enhances steatotic liver energy status and prevents oxidative damage following ischemia/reperfusion. Transplantation Proceedings. 2009;41(10):4094–4098. doi: 10.1016/j.transproceed.2009.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oxenkrug G. Antioxidant effects of N-acetylserotonin: possible mechanisms and clinical implications. Annals of the New York Academy of Sciences. 2005;1053:334–347. doi: 10.1196/annals.1344.029. [DOI] [PubMed] [Google Scholar]
- 29.Satake N, Morton BE. Scotophobin A causes dark avoidance in goldfish by elevating pineal N-acetylserotonin. Pharmacology Biochemistry and Behavior. 1979;10(4):449–456. doi: 10.1016/0091-3057(79)90216-8. [DOI] [PubMed] [Google Scholar]
- 30.Morton DJ. Both hydroxy- and methoxyindoles modify basal temperature in the rat. Journal of Pineal Research. 1987;4(1):1–5. doi: 10.1111/j.1600-079x.1987.tb00835.x. [DOI] [PubMed] [Google Scholar]
- 31.Psarakis S, Brown GM, Grota LJ. Analgesia induced by N-acetylserotonin in the central nervous system. Life Sciences. 1988;42(10):1109–1116. doi: 10.1016/0024-3205(88)90567-x. [DOI] [PubMed] [Google Scholar]
- 32.Jang S-W, Liu X, Pradoldej S, et al. N-acetylserotonin activates TrkB receptor in a circadian rhythm. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(8):3876–3881. doi: 10.1073/pnas.0912531107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oxenkrug G, Requintina P, Bachurin S. Antioxidant and antiaging activity of N-acetylserotonin and melatonin in the in vivo models. Annals of the New York Academy of Sciences. 2001;939:190–199. doi: 10.1111/j.1749-6632.2001.tb03626.x. [DOI] [PubMed] [Google Scholar]
- 34.Yu S, Zheng J, Jiang Z, et al. Protective effect of N-acetylserotonin against acute hepatic ischemia-reperfusion injury in mice. International Journal of Molecular Sciences . 2013;14(9):17680–17693. doi: 10.3390/ijms140917680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou H, Wang J, Jiang J, et al. N-acetyl-serotonin offers neuroprotection through inhibiting mitochondrial death pathways and autophagic activation in experimental models of ischemic injury. The Journal of Neuroscience. 2014;34(8):2967–2978. doi: 10.1523/JNEUROSCI.1948-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dou X, Shen C, Wang Z, Li S, Zhang X, Song Z. Protection of nicotinic acid against oxidative stress-induced cell death in hepatocytes contributes to its beneficial effect on alcohol-induced liver injury in mice. Journal of Nutritional Biochemistry. 2013;24(8):1520–1528. doi: 10.1016/j.jnutbio.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deferme L, Briedé JJ, Claessen SMH, Jennen DGJ, Cavill R, Kleinjans JCS. Time series analysis of oxidative stress response patterns in HepG2: a toxicogenomics approach. Toxicology. 2013;306:24–34. doi: 10.1016/j.tox.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 38.Kondo Y, Ishitsuka Y, Kadowaki D, et al. Phosphoenolpyruvate, a glycolytic intermediate, as a cytoprotectant and antioxidant in ex-vivo cold-preserved mouse liver: a potential application for organ preservation. Journal of Pharmacy and Pharmacology. 2013;65(3):390–401. doi: 10.1111/j.2042-7158.2012.01602.x. [DOI] [PubMed] [Google Scholar]
- 39.Subramaniam SR, Ellis EM. Esculetin-induced protection of human hepatoma HepG2 cells against hydrogen peroxide is associated with the Nrf2-dependent induction of the NAD(P)H: quinone oxidoreductase 1 gene. Toxicology and Applied Pharmacology. 2011;250(2):130–136. doi: 10.1016/j.taap.2010.09.025. [DOI] [PubMed] [Google Scholar]
- 40.Cesaratto L, Vascotto C, D’Ambrosio C, et al. Overoxidation of peroxiredoxins as an immediate and sensitive marker of oxidative stress in HepG2 cells and its application to the redox effects induced by ischemia/reperfusion in human liver. Free Radical Research. 2005;39(3):255–268. doi: 10.1080/10715760400029603. [DOI] [PubMed] [Google Scholar]
- 41.Koo JH, Lee WH, Lee CG, Kim SG. Fyn inhibition by cycloalkane-fused 1,2-dithiole-3-thiones enhances antioxidant capacity and protects mitochondria from oxidative injury. Molecular Pharmacology. 2012;82(1):27–36. doi: 10.1124/mol.111.077149. [DOI] [PubMed] [Google Scholar]
- 42.Xu Y, Leo MA, Lieber CS. DLPC attenuates alcohol-induced cytotoxicity in HepG2 cells expressing CYP2E1. Alcohol and Alcoholism. 2005;40(3):172–175. doi: 10.1093/alcalc/agh142. [DOI] [PubMed] [Google Scholar]
- 43.Pérez MJ, Cederbaum AI. Antioxidant and pro-oxidant effects of a manganese porphyrin complex against CYP2E1-dependent toxicity. Free Radical Biology and Medicine. 2002;33(1):111–127. doi: 10.1016/s0891-5849(02)00865-1. [DOI] [PubMed] [Google Scholar]
- 44.Wen F, Brown KE, Britigan BE, Schmidt WN. Hepatitis C core protein inhibits induction of heme oxygenase-1 and sensitizes hepatocytes to cytotoxicity. Cell Biology and Toxicology. 2008;24(2):175–188. doi: 10.1007/s10565-007-9027-9. [DOI] [PubMed] [Google Scholar]
- 45.Miccadei S, Venere DD, Cardinali A, et al. Antioxidative and apoptotic properties of polyphenolic extracts from edible part of artichoke (Cynara scolymus L.) on cultured rat hepatocytes and on human hepatoma cells. Nutrition and Cancer. 2008;60(2):276–283. doi: 10.1080/01635580801891583. [DOI] [PubMed] [Google Scholar]
- 46.Rubin R, Farber JL. Mechanisms of the killing of cultured hepatocytes by hydrogen peroxide. Archives of Biochemistry and Biophysics. 1984;228(2):450–459. doi: 10.1016/0003-9861(84)90010-9. [DOI] [PubMed] [Google Scholar]
- 47.Wang X, Zhu S, Drozda M, et al. Minocycline inhibits caspase-independent and -dependent mitochondrial cell death pathways in models of Huntington’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(18):10483–10487. doi: 10.1073/pnas.1832501100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu S, Stavrovskaya IG, Drozda M, et al. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417(6884):74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]
- 49.Zhang W-H, Wang H, Wang X, et al. Nortriptyline protects mitochondria and reduces cerebral ischemia/hypoxia injury. Stroke. 2008;39(2):455–462. doi: 10.1161/STROKEAHA.107.496810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang X, Figueroa BE, Stavrovskaya IG, et al. Methazolamide and melatonin inhibit mitochondrial cytochrome C release and are neuroprotective in experimental models of ischemic injury. Stroke. 2009;40(5):1877–1885. doi: 10.1161/STROKEAHA.108.540765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tartibian B, Maleki BH. The effects of honey supplementation on seminal plasma cytokines, oxidative stress biomarkers, and antioxidants during 8 weeks of intensive cycling training. Journal of Andrology. 2012;33(3):449–461. doi: 10.2164/jandrol.110.012815. [DOI] [PubMed] [Google Scholar]
- 52.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407(6805):770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 53.Liang Y-L, Zhang Z-H, Liu X-J, et al. Melatonin protects against Apoptosis-Inducing Factor (AIF)-dependent cell death during acetaminophen-induced acute liver failure. PLoS ONE. 2012;7(12) doi: 10.1371/journal.pone.0051911.e51911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhai Y, Petrowsky H, Hong JC, Busuttil RW, Kupiec-Weglinski JW. Ischaemia-reperfusion injury in liver transplantation-from bench to bedside. Nature Reviews Gastroenterology and Hepatology. 2013;10(2):79–89. doi: 10.1038/nrgastro.2012.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zimmerman MA, Grenz A, Tak E, et al. Signaling through hepatocellular A2B adenosine receptors dampens ischemia and reperfusion injury of the liver. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(29):12012–12017. doi: 10.1073/pnas.1221733110. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 56.Elias-Miró M, Jiménez-Castro MB, Rodés J, Peralta C. Current knowledge on oxidative stress in hepatic ischemia/reperfusion. Free Radical Research. 2013;47(8):555–568. doi: 10.3109/10715762.2013.811721. [DOI] [PubMed] [Google Scholar]
- 57.Delgado C, Barturen F. Atrial tamponade causing acute ischemic hepatic injury after cardiac surgery. Clinical Cardiology. 1999;22(3):242–244. doi: 10.1002/clc.4960220319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Adluri RKP, Singh AV, Skoyles J, et al. The effect of fenoldopam and dopexamine on hepatic blood flow and hepatic function following coronary artery bypass grafting with hypothermic cardiopulmonary bypass. European Journal of Cardio-Thoracic Surgery. 2009;35(6):988–994. doi: 10.1016/j.ejcts.2009.01.024. [DOI] [PubMed] [Google Scholar]
- 59.Manfredi G, Xu Z. Mitochondrial dysfunction and its role in motor neuron degeneration in ALS. Mitochondrion. 2005;5(2):77–87. doi: 10.1016/j.mito.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 60.Hu Y, Ding L, Spencer DM, Núñez G. WD-40 repeat region regulates Apaf-1 self-association and procaspase-9 activation. Journal of Biological Chemistry. 1998;273(50):33489–33494. doi: 10.1074/jbc.273.50.33489. [DOI] [PubMed] [Google Scholar]
- 61.Henry-Mowatt J, Dive C, Martinou J-C, James D. Role of mitochondrial membrane permeabilization in apoptosis and cancer. Oncogene. 2004;23(16):2850–2860. doi: 10.1038/sj.onc.1207534. [DOI] [PubMed] [Google Scholar]
- 62.Simonneaux V, Ribelayga C. Generation of the melatonin endocrine message in mammals: a review of the complex regulation of melatonin synthesis by norepinephrine, peptides, and other pineal transmitters. Pharmacological Reviews. 2003;55(2):325–395. doi: 10.1124/pr.55.2.2. [DOI] [PubMed] [Google Scholar]
- 63.Oxenkrug GF, Bachurin SO, Prakhie IV, Zefirov NS. Quinone reductase 2 and antidepressant effect of melatonin derivatives. Annals of the New York Academy of Sciences. 2010;1199:121–124. doi: 10.1111/j.1749-6632.2009.05354.x. [DOI] [PubMed] [Google Scholar]
- 64.Wilhelmsen M, Amirian I, Reiter RJ, Rosenberg J, Gögenur I. Analgesic effects of melatonin: a review of current evidence from experimental and clinical studies. Journal of Pineal Research. 2011;51(3):270–277. doi: 10.1111/j.1600-079X.2011.00895.x. [DOI] [PubMed] [Google Scholar]
- 65.Raghavendra V, Kaur G, Kulkarni SK. Anti-depressant action of melatonin in chronic forced swimming-induced behavioral despair in mice, role of peripheral benzodiazepine receptor modulation. European Neuropsychopharmacology. 2000;10(6):473–481. doi: 10.1016/s0924-977x(00)00115-2. [DOI] [PubMed] [Google Scholar]
- 66.Barsacchi R, Kusmic C, Damiani E, Carloni P, Greci L, Donato L. Vitamin E consumption induced by oxidative stress in red blood cells is enhanced by melatonin and reduced by N-acetylserotonin. Free Radical Biology and Medicine. 1998;24(7-8):1187–1192. doi: 10.1016/s0891-5849(97)00431-0. [DOI] [PubMed] [Google Scholar]
- 67.Leaden PJ, Catalá A. Melatonin and N-acetyl serotonin inhibit selectively enzymatic and non-enzymatic lipid peroxidation of rat liver microsomes. Prostaglandins Leukotrienes and Essential Fatty Acids. 2007;77(1):29–35. doi: 10.1016/j.plefa.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 68.García JJ, Reiter RJ, Karbownik M, et al. N-acetylserotonin suppresses hepatic microsomal membrane rigidity associated with lipid peroxidation. European Journal of Pharmacology. 2001;428(2):169–175. doi: 10.1016/s0014-2999(01)01342-5. [DOI] [PubMed] [Google Scholar]
- 69.Tosini G, Ye K, Iuvone PM. N-acetylserotonin: neuroprotection, neurogenesis, and the sleepy brain. Neuroscientist. 2012;18(6):645–653. doi: 10.1177/1073858412446634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shen J, Ghai K, Sompol P, et al. N-acetyl serotonin derivatives as potent neuroprotectants for retinas. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(9):3540–3545. doi: 10.1073/pnas.1119201109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yoo DY, Nam SM, Kim W, et al. N-acetylserotonin increases cell proliferation and differentiating neuroblasts with tertiary dendrites through upregulation of brain-derived neurotrophic factor in the mouse dentate gyrus. Journal of Veterinary Medical Science. 2011;73(11):1411–1416. doi: 10.1292/jvms.11-0123. [DOI] [PubMed] [Google Scholar]
- 72.Yang S, Jan Y-H, Gray JP, et al. Sepiapterin reductase mediates chemical redox cycling in lung epithelial cells. Journal of Biological Chemistry. 2013;288(26):19221–19237. doi: 10.1074/jbc.M112.402164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sadowska-Woda I, Wójcik N, Karowicz-bilińska A, Bieszczad-bedrejczuk E. Effect of selected antioxidants in β-cyfluthrin-induced oxidative stress in human erythrocytes in vitro. Toxicology in Vitro. 2010;24(3):879–884. doi: 10.1016/j.tiv.2009.11.022. [DOI] [PubMed] [Google Scholar]
- 74.Gavazza MB, Català A. Protective effect of N-acetyl-serotonin on the nonenzymatic lipid peroxidation in rat testicular microsomes and mitochondria. Journal of Pineal Research. 2004;37(3):153–160. doi: 10.1111/j.1600-079X.2004.00150.x. [DOI] [PubMed] [Google Scholar]
- 75.Tang GY, Ip AK, Siu AW. Pinoline and N-acetylserotonin reduce glutamate-induced lipid peroxidation in retinal homogenates. Neuroscience Letters. 2007;412(3):191–194. doi: 10.1016/j.neulet.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 76.Guajardo MH, Terrasa AM, Catalá A. Protective effect of indoleamines on in vitro ascorbate-Fe 2+dependent lipid peroxidation of rod outer segment membranes of bovine retina. Journal of Pineal Research. 2003;35(4):276–282. doi: 10.1034/j.1600-079x.2003.00087.x. [DOI] [PubMed] [Google Scholar]
- 77.Fagali N, Catalá A. The antioxidant behaviour of melatonin and structural analogues during lipid peroxidation depends not only on their functional groups but also on the assay system. Biochemical and Biophysical Research Communications. 2012;423(4):873–877. doi: 10.1016/j.bbrc.2012.06.060. [DOI] [PubMed] [Google Scholar]
- 78.Wölfler A, Abuja PM, Schauenstein K, Liebmann PM. N-acetylserotonin is a better extra- and intracellular antioxidant than melatonin. FEBS Letters. 1999;449(2-3):206–210. doi: 10.1016/s0014-5793(99)00435-4. [DOI] [PubMed] [Google Scholar]
- 79.Wang C-C, Fang K-M, Yang C-S, Tzeng S-F. Reactive oxygen species-induced cell death of rat primary astrocytes through mitochondria-mediated mechanism. Journal of Cellular Biochemistry. 2009;107(5):933–943. doi: 10.1002/jcb.22196. [DOI] [PubMed] [Google Scholar]
- 80.Torun AN, Kulaksizoglu S, Kulaksizoglu M, Pamuk BO, Isbilen E, Tutuncu NB. Serum total antioxidant status and lipid peroxidation marker malondialdehyde levels in overt and subclinical hypothyroidism. Clinical Endocrinology. 2009;70(3):469–474. doi: 10.1111/j.1365-2265.2008.03348.x. [DOI] [PubMed] [Google Scholar]
- 81.Landis GN, Tower J. Superoxide dismutase evolution and life span regulation. Mechanisms of Ageing and Development. 2005;126(3):365–379. doi: 10.1016/j.mad.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 82.Dong W, Simeonova PP, Gallucci R, et al. Toxic metals stimulate inflammatory cytokines in hepatocytes through oxidative stress mechanisms. Toxicology and Applied Pharmacology. 1998;151(2):359–366. doi: 10.1006/taap.1998.8481. [DOI] [PubMed] [Google Scholar]
- 83.Chen J-H, Ozanne SE, Hales CN. Methods of cellular senescence induction using oxidative stress. Methods in Molecular Biology. 2007;371:179–189. doi: 10.1007/978-1-59745-361-5_14. [DOI] [PubMed] [Google Scholar]
- 84.Kroemer G, Reed JC. Mitochondrial control of cell death. Nature Medicine. 2000;6(5):513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- 85.Xue T, Luo P, Zhu H, et al. Oxidative stress is involved in Dasatinib-induced apoptosis in rat primary hepatocytes. Toxicology and Applied Pharmacology. 2012;261(3):280–291. doi: 10.1016/j.taap.2012.04.010. [DOI] [PubMed] [Google Scholar]
- 86.Chen C-H, Chen S-J, Su C-C, et al. Chloroacetic acid induced neuronal cells death through oxidative stress-mediated p38-MAPK activation pathway regulated mitochondria-dependent apoptotic signals. Toxicology. 2013;303:72–82. doi: 10.1016/j.tox.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 87.Mukherjee D, Ghosh AK, Bandyopadhyay A, et al. Melatonin protects against isoproterenol-induced alterations in cardiac mitochondrial energy-metabolizing enzymes, apoptotic proteins, and assists in complete recovery from myocardial injury in rats. Journal of Pineal Research. 2012;53(2):166–179. doi: 10.1111/j.1600-079x.2012.00984.x. [DOI] [PubMed] [Google Scholar]
- 88.Wang X. The antiapoptotic activity of melatonin in neurodegenerative diseases. CNS Neuroscience and Therapeutics. 2009;15(4):345–357. doi: 10.1111/j.1755-5949.2009.00105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang Y, Wang X, Baranov SV, et al. Dipyrone inhibits neuronal cell death and diminishes hypoxic/ischemic brain injury. Neurosurgery. 2011;69(4):942–956. doi: 10.1227/NEU.0b013e318222afb2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Reubold TF, Eschenburg S. A molecular view on signal transduction by the apoptosome. Cellular Signalling. 2012;24(7):1420–1425. doi: 10.1016/j.cellsig.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 91.Liu H, Xiao Y, Xiong C, Wei A, Ruan J. Apoptosis induced by a new flavonoid in human hepatoma HepG2 cells involves reactive oxygen species-mediated mitochondrial dysfunction and MAPK activation. European Journal of Pharmacology. 2011;654(3):209–216. doi: 10.1016/j.ejphar.2010.12.036. [DOI] [PubMed] [Google Scholar]