

Summary
Malaria is a major cause of morbidity worldwide with reports of over 200–500 million infected individuals and nearly 1 million deaths each year. Antibodies have been shown to play a critical role in controlling the blood stage of this disease; however, in malaria-endemic areas antibody immunity is slow to develop despite years of exposure to Plasmodium spp. the causative parasite. Using rodent P. yoelii YM, we provide evidence that malarial infections result in a decrease in the proportion of dendritic cells (DCs) that express the B-cell survival factor, BAFF, resulting in a decreased ability of these DCs to support memory B cell (MBC) differentiation into antibody secreting cells (ASCs) and/or the survival of ACSs. Further, compared with infected WT mice, ASC numbers were significantly increased in malaria-infected transgenic mice that either over-expressed BAFF or mice with BAFF-independent B-cell survival (B cell-restricted TRAF3 deletion). Remarkably, BAFF- over-expressing mice were protected from lethal malaria infections, indicating the significance of the role BAFF plays in determining the outcome of malaria infections. These findings describe a previously unappreciated mechanism by which Plasmodium spp. can depress the generation of protective antibody responses.
Keywords: Memory B cells, Malaria, BAFF, Dendritic cells
Introduction
Adults in malaria-endemic areas will at best only develop non-sterile immunity, which protects against clinical symptoms but does not efficiently clear the parasite. Antibodies protect against malaria [1] but antibody responses in malaria-endemic areas are short-lived [2]. A longitudinal study undertaken in malaria-endemic Mali, with intense seasonal transmission of this disease, found that P. falciparum-specific memory B cells (MBCs) were generated after acute malaria episodes during the wet season [3]. However, similar to short-lived antibody levels [2], the levels of MBCs diminished considerably within 6 months to slightly higher than pre-infection levels despite ongoing malaria episodes [3]. Similarly, a Phase 1 clinical trial of the apical membrane antigen-1 (AMA-1) malaria vaccine, also undertaken in Mali, found that following the second vaccination antibody levels decreased to baseline levels within one year, at the time of the third vaccination [4]. The third dose induced little or no increase in antibody levels. This suggests that MBC responses were lost after the second vaccination and thus the third dose was unable to boost antibody levels. In contrast, studies from Madagascar [5] and Thailand [6] with only sporadic malaria transmission found that Plasmodium-specific MBCs survived for several years in individuals who had not been exposed to malaria in the interim. Thus, on-going malarial infections may cause loss of MBC responses not seen with sporadic transmission of malaria.
MSP119, a leading malaria vaccine candidate was tested in clinical trials in Kenya with holoendemic transmission of P. falciparum, and was found to generate very high titers of antibodies but did not protect against infection [7]. An evaluation of this vaccine using an experimental mouse model found that vaccination with MSP119 generated MSP119-specific MBCs which when transferred to naïve mice secreted antibody in response to MSP119, but not infection [8]. It was determined that infectious challenge activated MSP119-specific MBCs, which then underwent proliferation followed by apoptosis, thereby resulting in reduced numbers of MSP119-specific antibody-secreting cells (ASCs). Furthermore, this study also showed that the vaccine generated long-lived plasma cells (LLPCs), which secrete high levels of antibody that protect mice against a lethal infection. However, the LLPCs also underwent apoptosis within a few days of infection in this model. Therefore, considering these observations in an experimental model, we propose that with continual exposure to malaria parasites, Plasmodium-specific MBCs may also undergo deletion in malaria-endemic areas. Apoptosis of MBC may explain why antibody responses against the AMA-1 vaccine were not boosted in Mali with ongoing exposure to malaria [4]. The loss of Plasmodium-specific MBCs following malaria infection would be a potential problem for all antibody-based malarial vaccines. For rational design of a malaria vaccine we need a better understanding of the fate of MBCs in individuals exposed to malaria.
There is growing evidence that DCs play a substantial role in mediating B-cell immunity. DCs can activate naïve B cells and MBCs by presentation of unprocessed antigen directly to B cells [9–11], provide proliferation, isotype switch and survival signals [12–15] and promote survival in the bone marrow [16]. Furthermore, conventional DCs (cDC) were shown to mediate survival of plasmablasts in spleen and lymph nodes [17]. Most significantly, DCs express BAFF which up-regulates anti- apoptotic genes in B cells [18] for the survival of ASCs that have differentiated from antigen- activated MBCs [12, 19, 20]. We thus questioned whether altered DC function during malarial infection may contribute to poor survival of vaccine-specific MBCs given that in previous studies, we observed that DC taken from mice infected with lethal P. yoelii YM malaria were unable to secrete IL-12 and prime T cells [21]. This was also shown in other studies using different rodent parasite species and strains [21–25] and with human DCs [26].
In this current study, we used mouse models to measure the contribution of DCs and BAFF to loss of MBC responses against a malaria vaccine (MSP119) during malaria. The study design was based on the principles that mouse memory B and T cells survive >10 weeks after generation, while primary immune responses have subside by this time [27]. Additionally, MBC function is characterized by the production of high titers of IgG antibody within 4–5 days of exposure to antigen, whereas primary B-cell responses require 8–14 days for IgG production. The assays were designed so that similar T-cell help was available to test and control groups, and only differences in MBC responses were measured. Finally, antigen-pulsed DCs were transferred into vaccinated mice to activate MBC responses in vivo as demonstrated for naïve B cells [11]. These approaches were utilized to demonstrate that low BAFF expression on DCs limited MBC responses following malarial infections.
Results
DCs from malaria-infected mice are inefficient at supporting survival of MSP119-specific ASC
We previously showed that MSP119-specific MBCs were activated by experimental malaria challenge but following activation, the resultant ASC underwent apoptosis within 4 days [8]. To determine if a defect in DCs prevented survival of these ASC, we isolated DCs from naïve, P. yoelii YM (day 7) or P. yoelii 17XNL (day 10) infected mice, pulsed them in vitro with MSP119, and transferred them into MSP119-vaccinated (12–17 weeks after immunization) or naive mice (Figure 1A). After 5 days, we enumerated ASCs in the spleen of recipient mice (Figure 1B). Previous studies have established that antigen-pulsed DCs take at least 10 days to generate IgG secreting ASC in naive mice, and that within the 5 day window of the assay used here, only MBC could generate MSP119-specific IgG ASC [11]. The various combinations tested are labeled a–e in Figure 1B.
Figure 1. DCs from P. yoelii-infected mice are inefficient at supporting survival of MSP119- specific MBC responses.

(A) A representation of the protocol used to show that DCs from infected mice are inefficient at supporting malaria vaccine (MSP119)-specific MBC responses. (B) Numbers of IgG MSP119-specific ASCs/spleen in MSP119-vaccinated mice or naïve mice following transfer of MSP119-pulsed DCs from naïve or infected mice, measured by ELISPOT. The un-pulsed DCs from naïve mice show the background levels of ASCs remaining from vaccination. Data are shown as mean ± SEM of 3–5 independently processed mice/group and are representative of 3 independent assays which gave similar results. Significance was analyzed using the non-parametric Mann- Whitney U test on pooled data from replicate experiments. *P<0.0115; **P<0.0374; ***P<0.0001; NS= Not significant.
When DCs were taken from naïve mice, (a) pulsed with MSP119 or (b) un-pulsed, and transferred to naïve or MSP119-vaccinated mice, respectively, very low numbers of ASC were generated (Figure 1B). In contrast, if DCs were taken from naïve mice, pulsed with MSP119 and transferred to MSP119- vaccinated mice, >660% more MSP119-specific ASC were generated in the recipient mice (c, Figure 1B). Thus the transfer of vaccine-pulsed DCs had an effect similar to a vaccine boost. Significantly, when MSP119-vaccinated mice were given MSP119-pulsed DCs from (d) lethal or (e) non-lethal P. yoelii -infected mice, significantly fewer MSP119-specific ASCs were found respectively (69% and 34%) compared with those given MSP119-pulsed DCs from naïve mice (Figure 1B). These experiments showed that DCs from malaria-infected mice were less efficient at boosting MSP119-specific MBC responses than DCs from naïve mice either by a direct failure to provide MBCs with activation signals or indirect effect such as failure to activate helper T cells.
BAFF expression is reduced on DCs during malaria
As BAFF expression on DCs has been shown to be required for survival of ASCs that had differentiated from antigen-activated MBCs [12, 19, 20], we next examined BAFF-expression on DCs during P. yoelii infections (Figure 2). Groups of mice were infected with P. yoelii YM and DCs were analyzed by flow cytometry at 3 and 7 days post-infection and compared with DC from naive mice (Figure 2A–F). We examined BAFF expression on two major populations of DCs: B220+CD11c+ plasmacytoid DCs (pDC) and B220-CD11c+ cDC (Figure 2A). We found that <0.5% of pDCs (Figure 2C) and >6% of cDCs (Figure 2D) from naïve mice expressed BAFF, translating to approximately 7x103 pDCs and 3.1x105 cDCs per spleen respectively (Figure 2G). After 3 and 7 days post-infection, the percentage of pDCs that were BAFF+ were relatively unchanged from day 0 (not shown), although total BAFF+ pDC increased slightly in line with the increase in total pDC per spleen (Figures 2G and H). In contrast, the percentage of cDCs expressing BAFF dropped to 4% on day 3 (Figure 2E) and 0.5% on day 7 (Figure 2F) following infection with P. yoelii YM. Consequently, the total number of BAFF+ cDCs in the spleen was reduced significantly by approximately 40% and 83% on days 3 and 7 post-infection respectively (Figure 2G) even though total numbers of DCs increased significantly (Figure 2H). In addition, the level of BAFF expressed on individual cDCs also clearly declined with infection, as shown by the change in mean fluorescence intensity within the BAFF+ population (MFI- Figures 2D–F).
Figure 2. Loss of BAFF-expressing DCs in the spleen during malaria.

Groups of mice were infected with lethal P. yoelii YM or non-lethal P. yoelii 17XNL and DCs in spleens were examined at days 3 and/or 7 post-infection (p.i) and compared with splenic DCs from naïve mice. (A) Spleen cells from naïve mice, with CD19+ B cells gated out, contain both CD11c+B220− cDCs and CD11c+B220+ pDCs. (B) Isotype control antibody delineates BAFF expression on DCs. (C–F) BAFF expression on (C) pDCs from naïve mice, (D) cDCs from naïve mice, (E) cDCs on day 3 p.i. and (F) cDCs on day 7 p.i. are shown. (D–F) Box gate shows the MFI of CD11c+BAFF+ cells. (G) The absolute numbers of BAFF+ DC sub-populations per spleen in naïve and P. yoelii YM-infected mice are shown. (H) The absolute numbers of DC sub-populations in naïve and P. yoelii YM- infected mice are shown. (I) The absolute numbers of BAFF+ DC sub-populations per spleen in naïve and P yoelii 17XNL-infected mice at day 7 p.i. are shown. (A–I) Data are shown as mean ± SEM of 3–6 independently processed mice/group and are representative of 2–4 independent assays which gave similar results. (J) BAFF mRNA was compared with an average of 3 housekeeping genes by real time PCR in total spleen DCs from day 7 p.i. with P. yoelii YM and P yoelii 17XNL. Data are shown as the mean and range of mRNA levels obtained using RNA prepared in two independent experiments. Significance was analyzed using the non-parametric Mann-Whitney U test on pooled data from replicate experiments. *P=0.0173; **P=0.0238; ***P=0.0286; ****P=0.0136.
We next examined BAFF-expression on DCs from non-lethal P. yoelii 17XNL infections on day 7, to explain their significantly higher support for MSP119-specific ASCs compared with DCs from P. yoelii YM infections (Figure 1B). The numbers of cDCs expressing BAFF protein, decreased by nearly 80% within 7 days of infection (Figure 2I), similar to the lethal infection. However, real time PCR studies found that the levels of BAFF mRNA expression on day 7, in CD11c+ DCs from non- lethal P. yoelii 17XNL infections was significantly higher than DCs from lethal P. yoelii YM–infected mice (Figure 2J). Furthermore, compared with DCs from naïve mice, P. yoelii YM–infected mice had >75% reduction in BAFF mRNA in their DCs (Figure 2J).
Finally, an examination of overall BAFF expression in spleen cells showed that approximately 20% of CD11c-negative cells (i.e., non-DC cell populations) expressed BAFF in naïve mice (Figure 3A and B), and this increased to 29.5% within 3 days of a P. yoelii YM infection (Figure 3C), but then dropped to 10.6% by day 7 (Figure 3D). This was not however, an absolute loss of BAFF+ non DC- cells, as total spleen cell numbers increase several-fold within 7 days of infection (data not shown), thus adjusting absolute numbers of non-DC expressing BAFF to pre-infection levels (Figure 3E).
Figure 3. Expression of BAFF on CD11c− cells (non DCs) during lethal P. yoelii YM malaria.

Groups of mice were infected with P. yoelii YM and spleen cells from these and naïve mice were examined at days 3 and 7 post-infection (p.i). (A) Isotype control antibody delineates BAFF and CD11c expression on total spleen cells from naïve mice. (B) BAFF and CD11c expression on spleen cells from naïve mice. (C) BAFF and CD11c expression on spleen cells day 3 p.i. (D) BAFF and CD11c expression on spleen cells day 7 p.i. (A–D) Gates highlight the percentage of CD11c− cells (non-DCs) which expressed BAFF. (E) Absolute numbers of BAFF+ non-DCs per spleen. Data are shown as mean ± SEM of 5–6 independently processed mice/group and are representative of replicate experiments.
Transgenic BAFF mice can support MSP119-specific ASC survival during malaria
To assess the overall contribution of BAFF to survival of ASCs during malaria, we compared the activation of MSP119-specific MBCs in BAFF-Transgenic (BAFF-Tg) and WT mice, by P. yoelii YM malaria. BAFF-Tg mice have abundant oligomeric (60-mer) soluble BAFF (sBAFF) in their serum [28]. For testing, pooled spleen cells (containing MBCs and memory T cells) were taken from MSP119-vaccinated mice (after resting the mice >12 weeks) and transferred into sub-lethally irradiated WT or BAFF-Tg mice. Thus, both cohorts of recipient mice had the same starting population of cells and any differences would be related to survival of ASC that had differentiated from antigen-activated MBCs, a process normally dependent on BAFF expression on DCs [12, 19, 20]. These recipient mice were then either, infected with P. yoelii YM, given MSP119-boost, or given no stimulus, and MSP119-specific ASCs were assessed after 4 days by ELISPOT (Figure 4A).
Figure 4. Transgenic BAFF can support MSP119-specific ASCs survival during malaria.

(A) A representation of the protocol used to show that soluble BAFF in transgenic mice improves MSP119-specific ASC survival during malaria is shown. (B) Bar chart shows a comparison of the numbers of IgG MSP119-specific ASCs/spleen in BAFF-Tg and WT mice during P. yoelii YM-malaria or following MSP119-boost. (C) The percentage parasitemia over 150 days in naive WT and BAFF-Tg mice infected with P. yoelii YM are shown. (B, C) Data are shown as mean ± SEM of 5–6 independently processed mice/group and are representative of 2–3 independent assays which gave similar results. Significance was analyzed using the non-parametric Mann-Whitney U test on pooled data from replicate experiments. *P<0.0403; **P<0.0014; ***P<0.0007; ****P=<.0.0004.
WT mice given MSP119-boosts had on average 315 ASCs per spleen and this number decreased significantly (~72%) following P. yoelii YM infections (Figure 4B). In contrast, BAFF-Tg mice had on average 860 ASCs per spleen following MSP119-boosts and >200% more ASCs per spleen (~1828 ASCs per spleen) following P. yoelii YM-infections (Figure 4B). MSP119-specific ASCs were not detected in recipient mice without stimulus, indicating that all MSP119-specific ASC seen were induced by in vivo re-stimulation either by a MSP119-boost or malaria infection. Overall, these studies found 21-fold more ASCs during P. yoelii YM malaria in the presence of excess transgenic sBAFF compared with WT mice (1828 versus 88 ASCs per spleen). By contrast, following MSP119-boosts, excess sBAFF in transgenic mice only improved ASCs-survival compared with that of WT mice by 3-fold (860 versus 315 ASCs per spleen). Studies on survival mediated by the vaccine were not undertaken, as BAFF-Tg mice survived lethal P. yoelii YM malaria independent of immunization (Figure 4c).
BAFF+ DCs from infected mice can activate vaccine-specific MBCs
We then investigated whether the residual BAFF+ DCs from mice infected with P. yoelii YM malaria were competent to activate MBCs. For this, total DC populations were isolated from spleens of naive or P. yoelii YM-infected mice, and BAFF+ DCs were isolated from infected mice (Figure 5A). These DC populations were then pulsed with MSP119 and transferred into mice vaccinated with MSP119, 14 to17 weeks earlier. After 5 days, the numbers of IgG-secreting ASCs in the spleens of recipient mice were quantified using an MSP119-specific-ELISPOT assay (Figure 5B). Similar to data in Figure 1, MSP119-vaccinated mice given DCs from infected mice had significantly (67%) fewer ASCs compared with those given DCs from naïve mice (Figure 5B). However, MSP119- vaccinated mice given BAFF+ DCs from infected mice had significantly (280%) more ASCs compared with the total DC populations from naive mice (Figure 5B). Thus, the few DCs in infected mice that expressed BAFF were competent for the activation of MSP119-specific MBCs.
Figure 5. BAFF signals from DCs can support survival of MSP119 and Diptheria toxoid (DT) vaccine–specific ASCs.

BAFF+CD11c+ and total CD11c+ DCs were isolated from P. yoelii YM- infected mice, and along with total CD11c+ DCs from naïve mice were pulsed with MSP119 or DT and transferred to groups of MSP119 or DT vaccinated mice respectively. (A) A representative flow cytometry profiles of BAFF+CD11c+ DCs isolated from spleens of P. yoelii YM-infected mice is shown. (B) The numbers of IgG MSP119-specific ASCs/spleen in MSP119-vaccinated mice following transfer of MSP119-pulsed total CD11c+ DCs from naïve or infected mice or MSP119- pulsed BAFF+ CD11c+ DCs from infected mice are shown. (C) The numbers of IgG DT-specific ASCs/spleen in DT-vaccinated mice following transfer of DT-pulsed DCs from naïve or infected mice or DT-pulsed BAFF+CD11c+ DCs from infected mice are shown. (B, C) Data are shown as mean ± SEM of 5–6 independently processed mice/group and are representative of 5 independent experiments which gave similar results. Significance was analyzed using the non-parametric Mann- Whitney U test on pooled data from replicate experiments. *P=0.0140; **P=0.0043; ***P=0.0022; P=0.0238.
To determine whether the functional defect of malaria-induced DCs extended to other MBC responses, experiments were repeated in mice vaccinated with DT. For this, mice were vaccinated with DT, rested for 20 weeks and then given DCs from naïve or P. yoelii YM-infected mice pulsed with DT. Mice vaccinated with DT and given DT-pulsed DCs from infected mice had significantly (78%) fewer DT-specific ASCs compared with those given DT-pulsed DCs from naïve mice (Figure 5C). However, BAFF+ DCs generated similar numbers of DT-specific ASCs as DCs from naïve mice but 850% more than DT-pulsed total DCs from infected mice (Figure 5C). This suggests that the loss of BAFF expression on DCs during P. yoelii YM malaria significantly affected the survival of all vaccine-derived ASCs and was not specific to only the MSP119-vaccine.
MBCs require direct BAFF signals
To determine if the loss of DCs capable of expressing BAFF directly affected B cells, we used B cell-restricted tumor necrosis factor receptor-associated factors 3-knockout mice (TRAF3KO) where B cells can function independent of BAFF but other cells still depend on this signal. The inactivation of TRAF3 expression was achieved specifically within B cells by crossing mice with the Traf3lox allele with the Cd19-CRE line [29]. Normally, BAFF signals to B cells leads to TRAF2-dependent depletion of TRAF3, which promotes survival [29]. Thus, B cells without TRAF2 and/or TRAF3 develop and survive independently of obligatory BAFF. However, other cells (e.g. T cells) in these mice are still dependent on BAFF. Groups of these TRAF3KO or their WT littermate controls were vaccinated with MSP119 and rested 12 or 17 weeks. One half of the mice in each group were then infected with P. yoelii YM and the other half given soluble MSP119. After 5 days, the spleens were taken to quantify numbers of MSP119-specific ASCs (Figure 6).
Figure 6. MSP119-specific ASCs require direct BAFF signals for their survival during malaria.
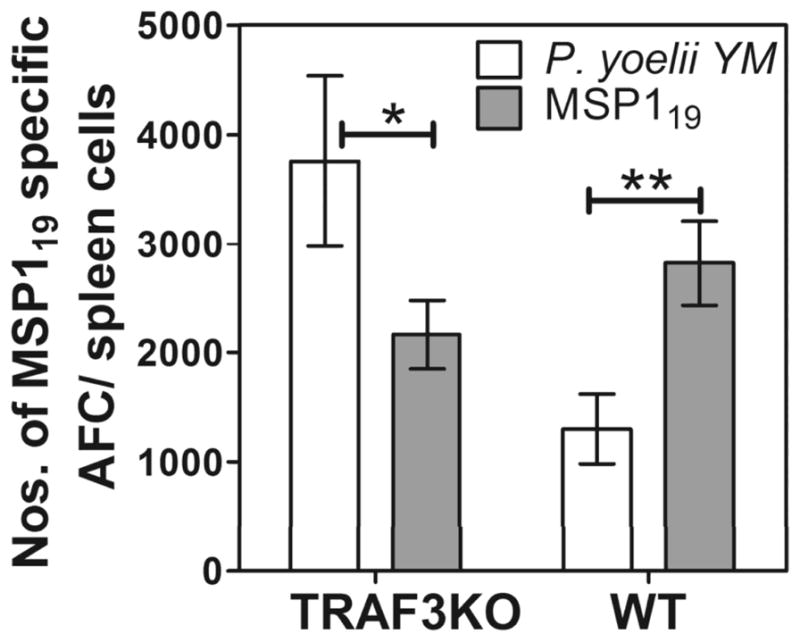
TRAF3-knockout mice (TRAF3KO), where B cells survive independently of obligatory BAFF, and their WT littermate controls (WT) were vaccinated with MSP119, rested for 14 weeks and either given 10 μg MSP119 or infected with P. yoelii YM. MSP119-specific ASCs in the spleen were enumerated after 5 days by MSP119-specific ELISPOT assay. The numbers of MSP119-specific ASCs/spleen in TRAF3-knockout or WT mice, following either MSP119-boost or P. yoelii YM infection are shown. Data are shown as mean ± SEM of 5 independently processed mice/group and are representative of 2 independent experiments that gave similar results. Significance was analyzed using the non-parametric Mann-Whitney U test on pooled data from replicate experiments. *P<0.0161; **P<0.0066.
Similar to the observations presented in Figure 4, WT mice had significantly reduced ASC survival during P. yoelii YM-malaria compared to WT mice boosted with MSP119 (Figure 6). However, the reverse was true in the TRAF3KO mice, where infection stimulated significantly better ASC survival in the infected mice than in those that received the MSP119-boost. Thus, when MBC- derived ASCs are no longer dependent on BAFF signals for survival, as in the TRAF3KO mice, the infection stimulated better responses than the vaccine. This suggests that during malaria, BAFF is a key signal for survival of ASCs following their differentiation from activated MBCs.
DISCUSSION
This study found that despite the generation of MBC responses to MSP119, a leading anti-malarial vaccine candidate, their function was ablated due to loss of BAFF expression on cDCs during malaria. Conventional DCs are not the only cell type to express BAFF in the spleen, but they were the cells with the most profound loss of BAFF during the course of infection. This, together with the observations that BAFF-Tg mice or transfer of BAFF+ DCs into vaccinated mice were able to elicit effective MBC responses, suggests that the overall loss of BAFF on cDCs mediated poor support for activation of memory responses. Further, the significantly higher numbers of MSP119-specific ASCs in TRAF3KO mice compared with those of WT mice during malarial infections is consistent with suboptimal BAFF signaling for survival of MBCs activated by the infection. Finally, the critical role of BAFF in protection during malaria was seen when BAFF-Tg mice survived lethal malaria. Together, these studies indicate that BAFF, a key signal required for maintenance of B-cell function, is compromised during malarial infection, limiting the effectiveness of MBC responses elicited by malaria vaccines.
BAFF is a member of the TNF family of ligands, expressed on DCs, macrophages, neutrophils and stromal cells [30]. BAFF supports B-cell proliferation [15] and plasma cell generation [19, 20, 31]. In contrast, the maintenance of MBCs in mice prior to activation is independent of BAFF [31]. This was noted when the administration of soluble BAFF receptors (BAFF-R or TACI) to vaccinated mice, to block BAFF activity, did not affect numbers of MBCs subsequently activated by antigen when BAFF activity was available [31]. However, BAFF was required for the survival of ASCs which differentiated from antigen-activated human MBCs [19, 20] via TACI signals [28]. Finally, TACI can only respond to oligomeric BAFF 60-mer but not the BAFF 3-mer, which is the main form of BAFF found in circulation [28]. We thus focused on the role of BAFF in survival of antigen-activated MBCs. In our studies, in addition to the significant role of BAFF in supporting MBC responses, we also noted that BAFF-Tg mice which have soluble BAFF 60-mers in their serum [28], survived lethal malaria independent of vaccination. This finding indicates that BAFF also has a role in primary immunity against malaria. Furthermore, the significantly better survival of ASCs in malaria-infected TRAF3KO mice compared with that of WT mice shows that the effect of BAFF is not on T-cell help, as only B cells were free from the requirement of obligatory BAFF in TRAF3KO mice.
A recent study suggested that BAFF was an indicator of disease severity during and after acute malaria in children, as it was found that serum BAFF levels increased during acute malarial infection which correlated with the severity of the disease [32]. Further, numbers of MBCs expressing TACI, the receptor for BAFF on MBCs increased following acute infection [32]. This observation appears to contradict our study. Another study reported that the soluble schizont fraction of P. falciparum antigens and hemozoin (HZ) could activate monocytes to express BAFF on their surface and release soluble BAFF into the supernatant within 72 h of stimulation [33]. However, studies have shown that TACI, the receptor for BAFF on MBCs, only mediates survival via oligomeric (60-mer) BAFF signals [28]. Given that the main form of soluble BAFF found in serum is only a 3-mer [28], we propose that the soluble BAFF found in children with acute malaria may not activate MBCs. In contrast, we found oligomeric BAFF, which occurs in the serum of BAFF-Tg mice [28] did support ASCs. The 3-mer sBAFF may even prevent oligomeric BAFF from mediating survival of ASC. These findings could explain why parasite-specific ASCs may not survive in children with acute malaria even though they appear to have soluble BAFF in their serum. Although we utilised the BAFF-Tg mice to show boosted ASC survival, we propose that the most relevant form of BAFF normally mediating ASC-survival is on the surface of the cDC.
Malaria is not the only infectious disease to be associated with a dysregulation of BAFF. In particular, high serum levels of BAFF at the onset of acute hepatitis C infections can predict both its evolution into a chronic infection [34]. Similarly, high expression levels of BAFF by DCs correlate with higher HIV-related B-cell disease progression in humans [35] while the differential expression of BAFF receptors BMCA and BAFF-R increased the propensity of B-cell death during HIV infections [36]. Further, BAFF also contributes to the loss of various B-cell populations during Trypanosoma brucei infections in mice [37]. The role of dysregulated BAFF in mediating the severity of infectious diseases is emerging and needs consideration where vaccine development has been ineffective.
Only one malaria vaccine candidate out of a multitude has entered Phase IIIb clinical trials [38], highlighting our incomplete understanding of malaria. However, new data strongly supports further consideration of MBCs in the field [3]. Our current studies, in rodent models, provide a mechanism that might explain why individuals in endemic areas fail to maintain parasite-specific MBCs [3]. We also suggest that MBC responses against malaria-vaccines cannot be boosted in endemic areas [4, 39] because the malaria parasite infects pDCs [40] which then are unable to support BAFF expression by cDCs (Figure 2), necessary to sustain ASC survival [12, 19, 20]. Finally, this study highlights a novel mechanism by which the malaria parasite prevents long-term immunity, and results suggest this may also compromise B, cell responses to co-infections with malaria.
Methods and Materials
Animals
All animal studies were reviewed and approved by the QIMR Animal Ethics Committee (A02-626M) under National Health and Medical Research Council guidelines. Specific-pathogen- free 6–8 week old, female C57BL/6J mice were obtained from the Animal Resources Centre (Murdoch, Western Australia). The CD19+ conditional TRAF3KO mice were kindly provided by Dr. Robert Brink (Garvan Institute, Sydney, Australia) and BAFF-transgenic mice by Dr. Susan Kalled (Biogen Idec, Cambridge, MA, US), both on a C57BL/6 background.
Infection of mice
Groups of 3–15 C57BL/6J mice were infected intravenously with 104 lethal P. yoelii YM or 105 P. yoelii 17XNL infected RBC (pRBC), and spleens taken after 3, 7 or 10 days.
Immunization protocol for protection studies
Mice were vaccinated subcutaneously with 50 μg MSP119 in Freund’s complete adjuvant per mouse, followed by 3 additional booster doses of 20 μg MSP119 in Freund’s incomplete adjuvant given at 3-week intervals. The first booster dose was given subcutaneously followed by three doses given intra-peritoneally. This protocol was shown previously to induce complete protection against a lethal infection given within 2–3 weeks of vaccination [8]. The mice were rested after the last immunization for 12–17 weeks to allow vaccine- specific primary T and B cells to subside and when only memory cells survive.
Isolation of spleen DCs for cell transfer studies
CD11c+ DCs were obtained from spleens of naive mice or mice infected with either 104 P. yoelii YM (lethal) or 105 P. yoelii 17XNL (non-lethal) pRBC. On day 4 of the lethal infection or day 7 of non-lethal infections, when parasitemia levels were ~5–10%, mice were treated with 250 μg Pyrimethamine (i.p) daily for 3 days, to clear the infection. The P. yoelii YM infections were started 3 days after the P. yoelii 17XNL infections so that both groups of mice could be cured and tested at the same time. Infected mice were treated with Pyrimethamine, to clear the infection prior to isolation and transfer of DCs, to prevent transfer of parasites within DCs [40]. Previous studies showed that DC taken from Pyrimethamine -treated P. yoelii 17XNL-infected mice remained functional [21].
Spleens from naive or infected mice were digested as previously described in [11] and in the supplementary section of [40]. RBCs were lysed with Gey’s solution and the digested spleen cell suspension was then incubated with 100 μg/ml Rat IgG to block FcR binding of labeling antibodies. CD11c+ DCs were isolated using Dynal and MACS microbeads as described in the main and supplementary section of [40].
BAFF+ DCs
To isolate BAFF+ DCs, total DC populations were enriched as described using Dynal beads [40], then labeled with anti-BAFF-FITC antibody and then with anti-FITC MACS microbeads for isolation on multiple MACS columns. BAFF+ CD11c+ DCs were then sorted on a MoFlo or ARIA cell sorter to obtain a purity of >98%.
DC transfer studies
Several groups of mice were vaccinated with MSP119 or GMP grade Diptheria Toxoid (DT) to generate vaccine-specific MBCs and then rested for 12–17 weeks. The immunization protocol included 50 μg MSP119 or DT in Freund’s complete adjuvant per mouse as for the protection studies. A second group of mice which served as donors of DCs were either naïve or infected with P. yoelii YM (lethal parasite) or P. yoelii 17XNL (non-lethal parasite) and then drug cured. Spleen DCs were isolated from these mice as described above, pulsed with 100 μg/ml MSP119 or DT, in Iscove’s media with 2% FCS at 37°C for 1 hour. Cells were then washed in x50 volume of ice cold PBS with 20mM EDTA, spun, washed in cold PBS and 106 DCs were administered per mouse intravenously in a 200 μL volume to each mouse vaccinated with MSP119 or DT, 12–17 weeks earlier (Fig. 1). The 20 mM EDTA was added to the wash buffer as it was previously shown to clear surface antigen, prior to transfer, so that the only source of the vaccine would be from within transferred DCs [11]. After a further 4 or 5 days, the number of MBCs which had differentiated into IgG-secreting ASCs in the spleen were quantified using an MSP119 or DT- specific-ELISPOT assay (Fig. 1A).
Flow cytometry of DCs
To assess expression of BAFF on DC and stroma, spleens from P. yoelii YM and P. yoelii 17XNL-infected mice on days 3 and 7 post-infection and from naïve mice were digested to release these cells. Cells were then labeled and examined by flow cytometry as described in the supplementary section of [40].
ELISPOT assay
Multiscreen-HA plates were coated with 10 μg/ml MSP119 or DT in an alkaline carbonate buffer, overnight at 4°C. Isolated spleen cells were directly tested for IgG-secreting ASCs using the published method [8] on these coated plates. The use of purified MSP119 or DT on the ELISPOT plates allowed measurement of vaccine-specific ASC but not responses to the adjuvant.
BAFF-Transgenic (BAFF-Tg) mice and cell transfer studies
To compare activation of MBCs by infection versus vaccine boosts, spleen cells containing vaccine-specific MBCs and T cells were transferred to naïve mice before activation. For this, lymphocytes were enriched from spleens of vaccinated mice (after >10 weeks resting), depleted of stromal and antigen presenting cells which adhere to plastic at 37°C, within 30 min. The pooled lymphocytes were then transfused intravenously into sub-lethally irradiated (550 cGy), naive, C57BL/6 (WT) or BAFF-Tg (C57BL/6 background) mice. Recipient mice were sub-lethally irradiated, so that their immune cells could not influence the responses of donor cells while their DC functions were maintained. The ratio of cells transferred was one spleen from a vaccinated mouse (with approximately 5x107-108 cells) to one irradiated recipient mouse.
In vivo activation of memory responses in BAFF-Tg and TRAFKO mice
To measure MBC activation in vivo, vaccinated and rested TRAF-3KO mice (with memory B and T cells) or recipient BAFF-Tg mice with transfused spleen cells (with memory B and T cells) were boosted intravenously with 10μg MSP119 or infected with 104 P. yoelii YM pRBC. The 10μg MSP119 dose of vaccine without adjuvant is sufficient to activate MBCs but not initiate new IgG responses, especially within 5 days. The memory IgG, anti-MSP119 responses were measured after 4 or 5 days by an MSP119-specific ELISPOT assay of recipients’ total spleen cells. Of particular note, since BAFF-Tg and WT mice received similar numbers of total spleen cells from vaccinated mice, the numbers of memory B and T cells would also be the similar in both groups. Similarly, since TRAF3 deletion in TRAF3-KO mice was restricted to CD19+ B cells, T cell function was similar in both WT and knockout mice. This also means that while B cells were free from the requirement for obligatory BAFF in knockout mice, T cells were still dependent on BAFF. TRAF3-KO mice were used to show that BAFF directly signaled B cells, and that the effect of BAFF was not on T cells.
Real Time PCR
Groups of 3–5 C57BL/6 mice were infected with 104 P. yoelli YM and 105 P. yoelii 17XNL. Spleens were harvested after 7 days and CD11c+ cells isolated as above. The cells were passed through the several MACS columns until purity was >99% as assessed by flow cytometry. Cells were lysed and RNA prepared by RNAeasy kit (Qiagen, Hilden, Germany). cDNA was synthesized using oligo(dT) priming, and levels of gene expression quantified by real-time PCR using Sybr Green and the CT method on an ABI 7900 machine (Applied Biosystems). Results for expression of BAFF were corrected for the amount of cDNA by using the average CT of three housekeeping genes, TATA binding protein (TBP), CXXC1, and mRpl13a. The sequences of primers used were: BAFF - CACGCCGACTATACGAAAAGGA, BAFFr - TTGCCTCACCACTATTTTGTTCTCT, CXXC1f – CAGACGTCTTTTGGGTCCA, CXXC1r – AGACCTCATCAGCTGGCAC, TBPf - GACCTAAAGACCATTGCACTTCGT, TBPr - GCAGTTGTCCGTGGCTCTCT, RPL13af -GAGGTCGGGTGGAAGTACCA, RPL13ar - TGCATCTTGGCCTTTTCCTT.
Statistics
Except where noted, data shown are mean ± Standard Error of Mean (SEM). Statistical differences between groups were determined using the non-parametric Mann-Whitney U test on pooled data from replicate experiments, using GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego, CA) with p<0.05 considered statistically significant.
Acknowledgments
We sincerely thank Dr Susan Kalled for kindly providing the BAFF transgenic mice and Dr Robert Brink for providing TRAF3-knockout mice. We also thank Drs, Michael Good, Danielle Stanisic, Robin Anders, Christian Engwerda, Denise Doolan, Sharon Johnatty, Geoffrey Hill and Kenneth Shortman for their critical reading and suggestions for the manuscript. We especially thank Ms Grace Chojnowski and Paula Hall for their excellent cell sorting skills. This work was supported by National Health and Medical Research Council (Australia) grant to MW and SP, in part by the Intramural Research Program of NIAID, NIH, and Queensland Smart State Fellowship to MW and an Australian Research Council Future Fellowship to KS.
Abbreviations
- MBC
Memory B cell
- BAFF/BLyS
B cell activating factor
- MSP119
Merozoite surface protein 1-19kD subunit
- LLPC
Long -lived Plasma cell
- ASC
Antibody secreting cell
- AMA-1
Apical Membrane-1 antigen
- DC
Dendritic Cell
Footnotes
Conflict of interest
The authors have no financial or commercial conflicts of interest.
Contributor Information
Liu Q. Xue, Email: xue.liu@griffith.edu.au.
Katryn J. Stacey, Email: katryn.stacey@uq.edu.au.
Joshua M. Horne-Debets, Email: Joshua.Horne-Debets@qimr.edu.au.
Jasmyn A. Cridland, Email: j.dunn1@uq.edu.au.
Katja Fischer, Email: Katja.Fischer@qimr.edu.au.
David Narum, Email: dnarum@niaid.nih.gov.
Fabienne Mackay, Email: Fabienne.Mackay@med.monash.edu.au.
Susan K. Pierce, Email: SPierce@niaid.nih.gov.
Michelle N. Wykes, Email: Michelle.Wykes@qimr.edu.au.
References
- 1.Cohen S, McGregor I, Carrington S. Gamma-globulin and acquired immunity to human malaria. Nature. 1961;192:733–737. doi: 10.1038/192733a0. [DOI] [PubMed] [Google Scholar]
- 2.Crompton PD, Kayala MA, Traore B, Kayentao K, Ongoiba A, Weiss GE, Molina DM, et al. A prospective analysis of the Ab response to Plasmodium falciparum before and after a malaria season by protein microarray. Proc Natl Acad Sci U S A. 2010;107:6958–6963. doi: 10.1073/pnas.1001323107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weiss GE, Traore B, Kayentao K, Ongoiba A, Doumbo S, Doumtabe D, Kone Y, et al. The Plasmodium falciparum-specific human memory B cell compartment expands gradually with repeated malaria infections. PLoS Pathog. 2010;6:e1000912. doi: 10.1371/journal.ppat.1000912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dicko A, Diemert DJ, Sagara I, Sogoba M, Niambele MB, Assadou MH, Guindo O, et al. Impact of a Plasmodium falciparum AMA1 vaccine on antibody responses in adult Malians. PLoS ONE. 2007;2:e1045. doi: 10.1371/journal.pone.0001045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Migot F, Chougnet C, Henzel D, Dubois B, Jambou R, Fievet N, Deloron P. Anti-malaria antibody-producing B cell frequencies in adults after a Plasmodium falciparum outbreak in Madagascar. Clin Exp Immunol. 1995;102:529–534. doi: 10.1111/j.1365-2249.1995.tb03848.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wipasa J, Suphavilai C, Okell LC, Cook J, Corran PH, Thaikla K, Liewsaree W, et al. Long-lived antibody and B Cell memory responses to the human malaria parasites, Plasmodium falciparum and Plasmodium vivax. PLoS Pathog. 2010;6:e1000770. doi: 10.1371/journal.ppat.1000770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ogutu BR, Apollo OJ, McKinney D, Okoth W, Siangla J, Dubovsky F, Tucker K, et al. Blood stage malaria vaccine eliciting high antigen-specific antibody concentrations confers no protection to young children in Western Kenya. PLoS ONE. 2009;4:e4708. doi: 10.1371/journal.pone.0004708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wykes MN, Zhou YH, Liu XQ, Good MF. Plasmodium yoelii can ablate vaccine-induced long-term protection in mice. J Immunol. 2005;175:2510–2516. doi: 10.4049/jimmunol.175.4.2510. [DOI] [PubMed] [Google Scholar]
- 9.Batista FD, Harwood NE. The who, how and where of antigen presentation to B cells. Nat Rev Immunol. 2009;9:15–27. doi: 10.1038/nri2454. [DOI] [PubMed] [Google Scholar]
- 10.Qi H, Egen JG, Huang AY, Germain RN. Extrafollicular activation of lymph node B cells by antigen-bearing dendritic cells. Science. 2006;312:1672–1676. doi: 10.1126/science.1125703. [DOI] [PubMed] [Google Scholar]
- 11.Wykes M, Pombo A, Jenkins C, MacPherson GG. Dendritic cells interact directly with naive B lymphocytes to transfer antigen and initiate class switching in a primary T-dependent response. J Immunol. 1998a;161:1313–1319. [PubMed] [Google Scholar]
- 12.Balazs M, Martin F, Zhou T, Kearney J. Blood dendritic cells interact with splenic marginal zone B cells to initiate T-independent immune responses. Immunity. 2002;17:341–352. doi: 10.1016/s1074-7613(02)00389-8. [DOI] [PubMed] [Google Scholar]
- 13.Wykes M, MacPherson G. Dendritic cell-B-cell interaction: dendritic cells provide B cells with CD40-independent proliferation signals and CD40-dependent survival signals. Immunology. 2000;100:1–3. doi: 10.1046/j.1365-2567.2000.00044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dubois B, Vanbervliet B, Fayette J, Massacrier C, Van Kooten C, Briere F, Banchereau J, et al. Dendritic cells enhance growth and differentiation of CD40-activated B lymphocytes [see comments] J Exp Med. 1997;185:941–951. doi: 10.1084/jem.185.5.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Craxton A, Magaletti D, Ryan EJ, Clark EA. Macrophage- and dendritic cell-- dependent regulation of human B-cell proliferation requires the TNF family ligand BAFF. Blood. 2003;101:4464–4471. doi: 10.1182/blood-2002-10-3123. [DOI] [PubMed] [Google Scholar]
- 16.Sapoznikov A, Pewzner-Jung Y, Kalchenko V, Krauthgamer R, Shachar I, Jung S. Perivascular clusters of dendritic cells provide critical survival signals to B cells in bone marrow niches. Nat Immunol. 2008;9:388–395. doi: 10.1038/ni1571. [DOI] [PubMed] [Google Scholar]
- 17.Garcia De Vinuesa C, Gulbranson-Judge A, Khan M, O’Leary P, Cascalho M, Wabl M, Klaus GG, et al. Dendritic cells associated with plasmablast survival. Eur J Immunol. 1999;29:3712–3721. doi: 10.1002/(SICI)1521-4141(199911)29:11<3712::AID-IMMU3712>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 18.Mackay F, Schneider P. Cracking the BAFF code. Nat Rev Immunol. 2009;9:491–502. doi: 10.1038/nri2572. [DOI] [PubMed] [Google Scholar]
- 19.Avery DT, Kalled SL, Ellyard JI, Ambrose C, Bixler SA, Thien M, Brink R, et al. BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J Clin Invest. 2003;112:286–297. doi: 10.1172/JCI18025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ettinger R, Sims GP, Robbins R, Withers D, Fischer RT, Grammer AC, Kuchen S, et al. IL-21 and BAFF/BLyS Synergize in Stimulating Plasma Cell Differentiation from a Unique Population of Human Splenic Memory B Cells. J Immunol. 2007;178:2872–2882. doi: 10.4049/jimmunol.178.5.2872. [DOI] [PubMed] [Google Scholar]
- 21.Wykes MN, Liu XQ, Beattie L, Stanisic DI, Stacey KJ, Smyth MJ, Thomas R, et al. Plasmodium Strain Determines Dendritic Cell Function Essential for Survival from Malaria. PLoS Pathog. 2007;3:e96. doi: 10.1371/journal.ppat.0030096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ocana-Morgner C, Mota MM, Rodriguez A. Malaria blood stage suppression of liver stage immunity by dendritic cells. J Exp Med. 2003;197:143–151. doi: 10.1084/jem.20021072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilson NS, Behrens GM, Lundie RJ, Smith CM, Waithman J, Young L, Forehan SP, et al. Systemic activation of dendritic cells by Toll-like receptor ligands or malaria infection impairs cross-presentation and antiviral immunity. Nat Immunol. 2006;7:165–172. doi: 10.1038/ni1300. [DOI] [PubMed] [Google Scholar]
- 24.Wykes MN, Liu XQ, Jiang S, Hirunpetcharat C, Good MF. Systemic tumor necrosis factor generated during lethal Plasmodium infections impairs dendritic cell function. J Immunol. 2007;179:3982–3987. doi: 10.4049/jimmunol.179.6.3982. [DOI] [PubMed] [Google Scholar]
- 25.Sponaas AM, Cadman ET, Voisine C, Harrison V, Boonstra A, O’Garra A, Langhorne J. Malaria infection changes the ability of splenic dendritic cell populations to stimulate antigen-specific T cells. J Exp Med. 2006;203:1427–1433. doi: 10.1084/jem.20052450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Urban BC, Ferguson DJ, Pain A, Willcox N, Plebanski M, Austyn JM, Roberts DJ. Plasmodium falciparum-infected erythrocytes modulate the maturation of dendritic cells [see comments] Nature. 1999;400:73–77. doi: 10.1038/21900. [DOI] [PubMed] [Google Scholar]
- 27.van Essen D, Kikutani H, Gray D. CD40 ligand-transduced co-stimulation of T cells in the development of helper function. Nature. 1995;378:620–623. doi: 10.1038/378620a0. [DOI] [PubMed] [Google Scholar]
- 28.Bossen C, Cachero TG, Tardivel A, Ingold K, Willen L, Dobles M, Scott ML, et al. TACI, unlike BAFF-R, is solely activated by oligomeric BAFF and APRIL to support survival of activated B cells and plasmablasts. Blood. 2008;111:1004–1012. doi: 10.1182/blood-2007-09-110874. [DOI] [PubMed] [Google Scholar]
- 29.Gardam S, Sierro F, Basten A, Mackay F, Brink R. TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to B cells by the BAFF receptor. Immunity. 2008;28:391–401. doi: 10.1016/j.immuni.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 30.Gorelik L, Gilbride K, Dobles M, Kalled SL, Zandman D, Scott ML. Normal B cell homeostasis requires B cell activation factor production by radiation-resistant cells. J Exp Med. 2003;198:937–945. doi: 10.1084/jem.20030789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benson MJ, Dillon SR, Castigli E, Geha RS, Xu S, Lam KP, Noelle RJ. Cutting edge: the dependence of plasma cells and independence of memory B cells on BAFF and APRIL. J Immunol. 2008;180:3655–3659. doi: 10.4049/jimmunol.180.6.3655. [DOI] [PubMed] [Google Scholar]
- 32.Nduati E, Gwela A, Karanja H, Mugyenyi C, Langhorne J, Marsh K, Urban BC. The plasma concentration of the B cell activating factor is increased in children with acute malaria. J Infect Dis. 2011;204:962–970. doi: 10.1093/infdis/jir438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumsiri R, Potup P, Chotivanich K, Petmitr S, Kalambaheti T, Maneerat Y. Blood stage Plasmodium falciparum antigens induce T cell independent immunoglobulin production via B cell activation factor of the TNF family (BAFF) pathway. Acta Trop. 2010 doi: 10.1016/j.actatropica.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 34.Tarantino G, Marco VD, Petta S, Almasio PL, Barbaria F, Licata A, Bosco GL, et al. Serum BLyS/BAFF predicts the outcome of acute hepatitis C virus infection. J Viral Hepat. 2009;16:397–405. doi: 10.1111/j.1365-2893.2009.01093.x. [DOI] [PubMed] [Google Scholar]
- 35.Poudrier J, Roger M. Dendritic Cell Status Modulates the Outcome of HIV-Related B Cell Disease Progression. PLoS Pathog. 2011;7:e1002154. doi: 10.1371/journal.ppat.1002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moir S, Malaspina A, Pickeral OK, Donoghue ET, Vasquez J, Miller NJ, Krishnan SR, et al. Decreased survival of B cells of HIV-viremic patients mediated by altered expression of receptors of the TNF superfamily. J Exp Med. 2004;200:587–599. [PubMed] [Google Scholar]
- 37.Radwanska M, Guirnalda P, De Trez C, Ryffel B, Black S, Magez S. Trypanosomiasis-induced B cell apoptosis results in loss of protective anti-parasite antibody responses and abolishment of vaccine-induced memory responses. PLoS Pathog. 2008;4:e1000078. doi: 10.1371/journal.ppat.1000078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bejon P, Lusingu J, Olotu A, Leach A, Lievens M, Vekemans J, Mshamu S, et al. Efficacy of RTS,S/AS01E vaccine against malaria in children 5 to 17 months of age. N Engl J Med. 2008;359:2521–2532. doi: 10.1056/NEJMoa0807381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bojang KA, Milligan PJ, Pinder M, Vigneron L, Alloueche A, Kester KE, Ballou WR, et al. Efficacy of RTS,S/AS02 malaria vaccine against Plasmodium falciparum infection in semi-immune adult men in The Gambia: a randomised trial. Lancet. 2001;358:1927–1934. doi: 10.1016/S0140-6736(01)06957-4. [DOI] [PubMed] [Google Scholar]
- 40.Wykes MN, Kay JG, Manderson A, Liu XQ, Brown DL, Richard DJ, Wipasa J, et al. Rodent blood-stage Plasmodium survive in dendritic cells that infect naive mice. Proc Natl Acad Sci U S A. 2011;108:11205–11210. doi: 10.1073/pnas.1108579108. [DOI] [PMC free article] [PubMed] [Google Scholar]