

Abstract
Diabetic nephropathy is the major cause of end-stage renal disease worldwide. Despite its prevalence, identification of specific factors that cause or predict diabetic nephropathy has been delayed in part by lack of reliable animal models that mimic the disease in humans. The Animal Models of Diabetic Complications Consortium (AMDCC) was created 8 years ago by the National Institutes of Health to develop and characterize models of diabetic nephropathy and other complications. This interim report details the progress made toward that goal, specifically in the development and testing of murine models. Updates are provided on validation criteria for early and advanced diabetic nephropathy, phenotyping methods, the effect of background strain on nephropathy, current best models of diabetic nephropathy, negative models and views of future directions. AMDCC investigators and other investigators in the field have yet to validate a complete murine model of human diabetic kidney disease. Nonetheless, the critical analysis of existing murine models substantially enhances our understanding of this disease process.
Diabetes mellitus is the major cause of End-Stage Renal Disease (ESRD) in the United States and elsewhere, and its incidence has increased by more than 50% in the last 10 years.1 Despite this disturbing trend only a minority of individuals with either type 1 or type 2 diabetes develop nephropathy, indicating that specific environmental or genetic factors must contribute to its initiation and progression. While clear progress has been made in understanding the disease process, there has been limited success in identifying specific factors causing or even predicting human nephropathy and its progression. One of the reasons for the slow evolution in our understanding of diabetic nephropathy is the lack of reliable animal models that mimic human disease. Hence, eight years ago the Animal Models of Diabetic Complications Consortium (AMDCC) was initiated by the National Institutes of Health to develop and characterize such models to enhance the development and testing of effective therapies and preventative strategies for diabetic nephropathy and other complications. While the primary goal of the consortium—to develop murine models of diabetic micro and macro-vascular complications that completely replicate the human diseases—has not yet been attained, much progress has been made in model development, mouse phenotyping, strain analysis, and understanding of pathogenesis of diabetic complications.
Approximately 4 years ago the nephropathy subcommittee of the AMDCC published an account of phenotyping standards, validation criteria, and observations about currently available murine strains and models of diabetic nephropathy.2 This article set the standard for phenotyping methods of mouse models, and for diagnostic criteria of early and progressive diabetic nephropathy. These standards were valuable to the field and, as of June, 2009, this article had been cited 85 times in the biomedical literature.3 Since this article was published, the AMDCC investigators studying nephropathy have established more detailed validation criteria for phenotyping murine models of diabetic nephropathy and a number of exciting new models that replicate various features of the human disease. Conversely, several initially promising models develop little or no nephropathy. A number of new approaches to the study of diabetic nephropathy have emerged since the 2005 publication. The purpose of this article is to summarize these new observations and advances.
Validation criteria for diabetic nephropathy in the mouse
One of the first issues for the AMDCC was to define criteria for validating murine models of human diabetes and diabetic complications. These validation criteria are available on the AMDCC website (www.amdcc.org). The criteria for nephropathy have been modified recently and are listed in Table 1. AMDCC investigators conclude that, although an ideal model of diabetic nephropathy would display all of these criteria, no current model meets them all. The criteria should therefore be viewed as goals rather than requirements. In addition, validation of any model should include reasonable efforts to exclude other types of kidney disease or damage unrelated to that from diabetes.
TABLE 1.
Research Criteria for Validating a Progressive Mouse Model Of Diabetic Nephropathy
| A. Greater than 50% decline in GFR over the lifetime of the animal |
| B. Greater than 10-fold increase in albuminuria compared to controls for that strain at the same age and gender |
| C. Pathology of kidneys |
| a. Advanced mesangial matrix expansion +/- nodular sclerosis and mesangiolysis |
| b. Any degree of arteriolar hyalinosis |
| c. Glomerular basement membrane thickening by >50% over baseline |
| d. Tubulointerstitial fibrosis |
Phenotyping methods
Another early task of the AMDCC was to standardize phenotyping of murine models of diabetic nephropathy so that models could be compared between laboratories within and outside the consortium. These phenotyping protocols are recently updated and are available on the AMDCC website at www.amdcc.org.
Current basal phenotyping for diabetic nephropathy includes urinary albumin determination by ELISA and urinary creatinine testing by standard methods on both spot urines and 24 h urine collections. Unfortunately, the albuminuria data from 24-hour collections and those from spot samples are not always concordant, with one value sometimes suggesting a greater increase in albuminuria than the other.4 As discussed below, this discrepancy may be somewhat strain dependent4 and is due in part to variations in creatinine excretion. At present, it is not clear which value is the more accurate indicator of relative changes in albuminuria and AMDCC investigators recommend reporting both measurements.
Measurements of serum or plasma creatinine in mouse models need to be performed by High Performance Liquid Chromatography (HPLC), LC MS/MS,5, 6 or a similar detection system since the Jaffe alkaline picrate method overestimates creatinine levels by 3-5 fold in plasma of mice.7-9 Also it is important to recognize that the enzymatic assay of creatinine used in clinics, which converts creatinine into creatine, should not be used for mouse urine because mice excrete significant amounts of creatine in their urine.10 When measured by these more definitive assays, normal mouse serum creatinine values are in the range of 0.08-0.11mg/dl, as opposed to 0.4-0.6 when measured by the Jaffe method. Many facilities (including some MMPC sites) utilize an autoanalyzer enzymatic assay based on creatinase activity. In normal mice this method overestimates creatinine values approximately twice that of HPLC determined values (i.e. ~0.2mg/dl).11-13 Autoanalyzer values are also susceptible to underestimation of creatinine in the presence of hemolyzed blood.12 Nevertheless, if assessed together with the blood urea nitrogen, the enzymatic determination of creatinine may be suitable for high throughput screening of renal function. When the plasma creatinine is elevated, glomerular filtration rate (GFR) can be best determined by FITC-inulin clearance.14, 15 Estimation of GFR by creatinine clearance using HPLC values for creatinine in serum or plasma may not be reliable, as a preliminary report suggests that tubular secretion of creatinine may account for up to 50% of creatinine clearance in some strains of mice16 and the contribution of creatinine secretion to its clearance in diabetes and renal insufficiency is unclear.
Renal histopathology includes periodic acid Schiff (PAS) staining of kidney tissues for assessment of mesangial matrix expansion and arteriolopathy, tubulointerstitial disease of any type, and general glomerular morphology. However, PAS stained tissue does not clearly distinguish between extracellular matrix and cell cytoplasm and this can confound determinations of altered matrix accumulation. Silver methenamine is a more specific stain for extracellular matrix but is more technically difficult to reliably perform (Figure 1). For models with advanced lesions a more comprehensive assessment should take place, as noted in Table 2. Mesangiolysis (Figure 2) is a common feature of diabetic nephropathy in humans and should be assessed in mice. In addition, the presence of a concurrent glomerular immune complex deposition process should be excluded by immunofluorescence or immunohistochemical studies of renal tissue in all validated models of diabetic nephropathy.
Figure 1.
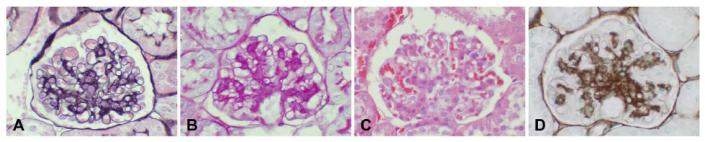
Comparable appearance of four histologic and/or immunohistochemical stains utilized to characterize the morphologic changes in a 24 week old BKS db/db mouse. A. Jones’ silver methenamine/H&E stain shows expansion of mesangial matrix (black stain) and clear depiction of cellularity (cell nuclei). This stain also clearly delineates basement membranes, and allows good definition of capillary loops versus mesangial structures. B. PAS stain. Like the Jones’ stain depicted in A, this stain shows good delineation of basement membranes and capillary loops as distinct from mesangial regions. Expanded mesangial regions are well detected by this stain. Compared with the silver methenamine stain some cellular detail is lost with PAS as the distinction between matrix and cell cytoplasm can be obscured due to staining of carbohydrate structures in both compartments (see figure 2). C. H&E stain. This stain is valuable for identifying overall cellularity, but is unsatisfactory for quantification of glomerular matrix changes, particularly if they involve modest expansion of mesangial regions. This stain does not highlight basement membranes well, and does not clearly distinguish cellular versus matrix components in expanded mesangial regions. D. Immunohistochemical staining for collagen IV, with methyl green counter stain. This stain clearly defines increased matrix components within the glomerular tuft, and also is a good marker of basement membranes in the interstitium. Overall, Jones’ silver methenamine, PAS, and collagen IV immunohistochemical staining can be reliably used for morphometric analysis of glomerular changes that involve matrix expansion. When these changes are pronounced, and there is greater distortion of glomerular tuft architecture than depicted in this example, the PAS stain may be less reliable than the silver methenamine stain in separating cellular versus matrix contributions to expanded glomerular compartments due to its greater recognition of some cellular components.
TABLE 2.
Additional Histological Phenotyping for Diabetic Nephropathy Models with Advanced Disease
| a. Quantitate mesangial matrix expansion, ideally with morphometric analyses, mesangiolysis, and microaneurysms with silver methenamine staining or with immunohistochemical stains that identify relevant matrix components. |
| c. Rule out immune complex disease with immunohistochemistry on frozen sections for IgG, IgM, IgA. |
| d. Determine the presence of either afferent or efferent arteriolar hyalinosis by silver methenamine staining or PAS (renin can give a false positive reading with PAS). |
| e. Demonstrate glomerular basement membrane thickening by electron microscopy. |
| f. Podocyte loss should be demonstrated by a reasonable morphometric system. |
| g. Additional optional analyses: |
| i) Macrophage detection by Mac-2 immunohistochemistry |
| ii) Lipid detection by oil Red O staining |
Figure 2.

Morphologically advanced murine diabetic nephropathy demonstrating prominant mesangiolysis. A. There are broad areas of lucency (arrows) within expanded mesangial regions. Mesangiolysis is most easily recognized with a Jones’ silver methenamine stain, in which the normally homogeneous and compact silver staining (black) matrix (shown in Figure 1A) is disrupted as indicated by areas of lucency and/or spongiform appearance. B. In contrast, a PAS stain shows equivalent mesangial expansion, but the demarcation of cellular and matrix components and lytic regions is less distinct with this stain as compared to the Jones’ stain.
Update on strain effects in diabetic nephropathy
The previous AMDCC article2 and two subsequent reports from AMDCC investigators4, 17 compare the effects of genetic background on the degree of diabetic nephropathy in a variety of inbred mouse strains. These findings will not be repeated here, except to note the relative resistance of the C57BL/6 (B6) mouse to nephropathy and the relative susceptibility of the DBA/2 strain to nephropathy (Figure 3). The inbred strains that develop the most profound diabetic nephropathy, use of outbred strains, and models negative for diabetic nephropathy are described in the online supplement. The Mouse Phenome Database is a useful source for comparative data of basal metabolic parameters distinguishing the more commonly used inbred strains (http://phenome.jax.org/pub-cgi/phenome/mpdcgi?rtn=docs/home). For example, the website contains albumin/creatinine ratio (ACR) data for males and females of 30 inbred strains http://phenome.jax.org/pub-cgi/phenome/mpdgrcgi?rtn=views/measplot& brieflook=29722&userhilite= .
Figure 3.

Phenotypic comparison of the Ins2Akita mutation in males of 3 stocks shows comparable hyperglycemia in Akita/+ males on all 3 backgrounds (A), but development of early signs of diabetic nephropathy [increased albumin/creatinine ratio (ACR)] was limited to the DBA/2 strain background (B) when necropsy was done at 6 months of age.
New mouse models of diabetic nephropathy
While some inbred mouse strains are more susceptible to nephropathy than others, no current murine strain of type 1 or type 2 diabetes reliably develops all the features of human diabetic nephropathy listed in Table 1. AMDCC investigators as well as others outside the consortium have endeavored to augment diabetic nephropathy by genetic breeding and other means in both susceptible and resistant strains of mice. For these efforts a number of recently described models develop features of human diabetic nephropathy appear more robust than those in standard models. Many but not all of these new models have been developed by members of the AMDCC. A number of models that AMDCC investigators believe are the most promising are discussed below.
eNOS deficiency (C57BL/6 and C57BLKS backgrounds)
Endothelial damage is a hallmark of diabetes and contributes to the development of ESRD.18 Endothelial cell-derived vasodilators such as nitric oxide are important modulators of permeability in the vasculature. Vascular endothelial nitric oxide synthase (eNOS) activity is altered in diabetes, and functionally significant polymorphisms in the NOS3 gene lead to lower production of nitric oxide,19-21 and are associated with the development of advanced nephropathy in patients with type 1 and type 2 diabetes.22-24 Recent studies indicate that targeting of Nos3, the gene encoding eNOS and denoted here as eNOS−/−, induce nephropathic changes in mouse models of both type 1 and type 2 diabetes that mimic many aspects of human disease.25-28
To generate a model of type I diabetes with deficient eNOS activity, two groups induced diabetes by streptozotocin (STZ) injection in eNOS−/− mice on the nephropathy-resistant B6 background (JAX #2684). Nakagawa et al.25 used high-dose STZ while Kanetsuta et al.26 used multiple low-dose injections. Compared to wild-type B6 mice, diabetic eNOS−/− mice develop a 10-fold increase in diabetic albuminuria with low-dose STZ and a 40-fold increase with high dose STZ. While mice do not develop significant reductions in GFR compared to B6 mice, their GFR was 40% lower than in wild-type low-dose STZ diabetic mice, because the wild-type mice develop significant diabetic hyperfiltration, which the eNOS−/− mice fail to develop. Finally, diabetic B6-eNOS−/− mice develop significant increases in mesangial expansion, mesangiolysis, and focal sclerosis. In the low-dose STZ diabetic B6-eNOS-/- mice, both glomerulosclerosis and tubulointerstitial injury developed gradually, while high-dose STZ B6-eNOS−/− mice exhibit earlier and more robust glomerular and tubulointerstitial pathology, even though the blood glucose levels are higher in low-dose STZ diabetic mice. The high-dose STZ diabetic mice also exhibit significant hypertension and mortality. It is likely that some of the pathologic changes observed in these high-dose STZ diabetic eNOS−/− mice reflect a toxic response to STZ rather than diabetic nephropathy per se, and the AMDCC recommends that low-dose STZ protocols be utilized for mouse models.
Preliminary studies by AMDCC investigators indicate that the B6-eNOS−/−Ins2Akita/+ mice die soon after weaning and therefore do not live long enough to develop diabetic nephropathy (Takahashi et al., unpublished observation). However, eNOS−/−Ins2Akita/+ diabetic mice that are F1 between B6 and 129SvEv develop diabetes with similar timing and severity as in B6-eNOS−/− Ins2Akita/+ mice, but survive as long as diabetic B6-eNOS+/+Ins2Akita/+ mice. The F1 mice develop diabetic nephropathy similar to B6-eNOS−/− males made diabetic by low-dose STZ, and so may also be a useful type 1 model. F1 mice between inbred strains are as genetically uniform as their parents, but are more robust because most strain-specific detrimental genes will no longer be homozygous.
To generate a model of type 2 diabetes with deficient eNOS activity, C57BLKS (BKS)-db/db mice were crossed with eNOS−/− mice also on the BKS background27, 28 (JAX# 8340). These mice exhibit mild-moderate hypertension as is generally true with eNOS−/− mice on the B6 background. They develop significant albuminuria, decreased GFR, mesangial expansion, glomerular basement membrane (GBM) thickening, arteriolar hyalinosis, mesangiolysis, nodular glomerulosclerosis and tubulointerstitial injury that is significantly greater than found in low-dose STZ diabetic B6-eNOS−/− mice (Figure 4). Hyperglycemia is first apparent at 6-8 weeks of age and mice exhibit full-blown functional and nephropathic changes by 16-20 weeks. In general, BKS-eNOS −/− db/db mice exhibit more profound diabetic nephropathy than the low-dose STZ diabetic B6-eNOS−/− mice. This divergence between strains may result from strain differences (nephropathy-resistant B6 versus nephropathy-sensitive BKS) or could reflect differences in pathologic responses to endothelial nitric oxide deficiency in type 1 versus type 2 diabetes. To address this question, AMDCC investigators are examining diabetic nephropathy in type 1 diabetic eNOS−/− mice crossed onto other nephropathic strains (DBA/2J and KK/HIJ, JAX #2106). Currently, eNOS−/− mice are available on B6 (JAX# 2684), BKS-db/db (JAX# 8340) and BALB/cBy (JAX# 7073) backgrounds from The Jackson Laboratory.
Figure 4.

Representative glomerular histology at 24 weeks of age from eNOS+/+m/m (control) mice, eNOS−/− m/m mice (eNOS−/−), eNOS+/+db/db (db/db) and eNOS−/−db/db (db/db eNOS−/−) mice. All mice are on the BKS background. Note the markedly increased mesangial expansion and glomerulosclerosis in the BKS eNOS−/−db/db mice.
Bradykinin B2 receptor deficiency (C57BL/6 and C57BLKS backgrounds)
Several groups have reported an association between the onset and progression of type 1 diabetic nephropathy in humans and the D allele of the Angiotensin Converting Enzyme (ACE) gene.29, 30 In addition, ACE inhibitors and angiotensin receptor blockers are mainstays of renal protection in human diabetic nephropathy. In mice, a 50% increase in ACE gene expression has minimal effects on blood pressure and angiotensin II levels but leads to substantial decreases in bradykinin, suggesting bradykinin rather than angiotensin II is more important in renal responses in diabetes.31, 32 Therefore, AMDCC investigators have studied the contribution of targeted deletion of the bradykinin 2 receptor (B2R) to the evolution of diabetic nephropathy in Ins2Akita/+ mice on a B6 background.33 By 6 months of age in this model, chronically diabetic Ins2Akita/+ mice with homozygous deletions of Bdkrb2 (the gene coding for B2R) develop a 4-fold increase in albuminuria and profound mesangial expansion that resembles the glomerular changes seen in human diabetic glomerulosclerosis. There were no changes in the glomerular endothelial cells or podocytes. The mice also develop mitochondrial DNA damage in the kidneys and other tissues suggestive of generalized senescence.34 Although the mechanisms of enhanced nephropathy in this model remain unclear, there is normally a high level of bradykinin 2 receptor expression in mesangial cells, and knockout of these receptors is associated with enhanced renal expression of several genes involved in progressive glomerulosclerosis, including TGFβ1, connective tissue growth factor (CTGF), and p53.34 The mice also have a substantial increase in expression of the bradykinin 1 receptor (B1R), but this does not protect them from developing nephropathy.
Other investigators have reported similar findings. For example, treatment of diabetic rats35 and BKS-db/db mice36 with a specific non-peptidic bradykinin 2 receptor antagonist shows reversal of almost all the salutary effects of ACE inhibitors on albuminuria, glomerular ERK and TGFβ signaling pathways, and changes in glomerular gene expression. The antagonist also results in enhanced oxidative stress in glomeruli from treated diabetic rats. One exception to the general principle that activation of B2R protects against nephropathy has been reported in mice on a mixed 129S6/SvEvTac and B6 background made diabetic with low-dose STZ.37 In this study, in contrast to the others, the B2R deletion is protective with respect to the mesangial expansion seen after 6 months of diabetes and with respect to total excretion of albumin over the 6 months. Nonetheless, by the end of the 6-month period the STZ diabetic mice lacking B2R excrete more albumin than STZ diabetic mice with intact receptor. Moreover, a marked increase in renal expression of B1R occurs in mice lacking B2R, and this could be sufficient to partially protect the low-dose STZ diabetic mice but not the more severely affected Ins2Akita/+ diabetic mice. Subsequent studies by AMDCC investigators (Kakoki et al., unpublished) demonstrate that Ins2Akita/+ diabetic mice lacking both B1R and B2R have more severe nephropathy than Ins2Akita/+ mice lacking only B2R. The general importance of bradykinin and the whole of the kallikrein/kinin system in maintaining renal function has recently been reviewed by AMDCC investigators.38 On balance, it appears that increased kinin levels following treatment with ACE inhibitors are protective with respect to diabetic nephropathy, while decreased levels (such as result from the D allele of the human ACE gene) are harmful.
Decorin deficiency (B6 background)
Decorin is a small leucine rich proteoglycan which is primarily secreted and stored in the extracellular matrix.39 In the extracellular milieu it can have multiple functions by virtue of its ability to bind growth factors.40 Decorin can inhibit TGFβ activation by binding to the active form of TGFβ as well as regulate PDGF and Epidermal Growth Factor (EGF) activity.41 As decorin is consistently stimulated in diabetic nephropathy,42-44 AMDCC investigators evaluated the decorin null mouse as a potential model of progressive diabetic nephropathy. The decorin null mouse on a B6 background develops enhanced features of nephropathy as characterized by a 50% increase in mesangial matrix expansion and a 23% decrease in renal function over wild-type diabetic mice despite having only a modest 50% increase in urine albumin excretion.45 The diabetic decorin null mouse exhibits increased TGFβ levels and activity in glomeruli, as well as increased inflammation, marked by macrophage infiltration and up-regulation of Nox4.45 However, the mice do not develop nodular sclerosis or tubulointerstitial lesions. Features of nephropathy in the diabetic decorin null mice diverge from those in diabetic wild-type B6 mice after 6 months of diabetes, again, suggesting that aging is an important aspect of progressive diabetic nephropathy.
In order to determine whether decorin deficiency in the setting of hyperlipidemia further accentuates diabetic injury, AMDCC investigators evaluated the degree of disease in a double null mouse for decorin and the LDL receptor. Despite a higher degree of LDL cholesterol, there is no evidence of worsening diabetic nephropathy based on albuminuria, mesangial matrix expansion, or plasma creatinine levels in the diabetic double null mice (Sharma, K, et al., unpublished data).
NONcNZO10/LtJ mice
NONcNZO10/LtJ (JAX # 4456) is an inbred congenic strain derived from a cross between the Nonobese Nondiabetic (NON/LtJ, JAX#2423) strain and the New Zealand Obese (NZO/HlLt, JAX# 2105) mouse, which provides a model of polygenic type 2 diabetes mellitus. NONcNZO10/LtJ male mice were developed at The Jackson Laboratory as a model of obesity-induced type 2 diabetes and metabolic syndrome resulting from polygenic interactions producing moderate obesity in contradistinction to the massive obesity elicited by mutations in the leptin/leptin receptor axis. In the NONcNZO10/LtJ congenic strain, the NZO/HlLt contributes segments from chromosomes 1, 4, 5, 11, 12, 15, and 18 onto the NON background.46, 47 Male NONcNZO10/LtJ mice weaned onto a chow diet containing 10-11% fat (by weight) develop visceral obesity, maturity-onset hyperglycemia, dyslipidemia, moderate liver steatosis, and pancreatic islet atrophy. Notably, after approximately eight months of age, these mice also develop significant and progressively increasing albuminuria, with urine ACRs greater than 1000 μg/mg after 1 year. Glomerular histopathology is impressively abnormal but, in addition to glomerulosclerosis, exhibits features that are atypical for diabetic nephropathy, including intraglomerular capillary thrombi and lipid deposition and evidence of immunoglobulin deposition. Moreover, several of these features including the intracapillary glomerular lipid deposits and periarteriolar lymphoid infiltrates have been observed in the nondiabetic NON parental strain48 and so appear to be unrelated to diabetic effects in this model.
FVB-OVE26 (FVB background) mice
The FVB-OVE26 mouse (JAX # 5564) is a transgenic model of early-onset type 1 diabetes, generated directly onto the FVB/N background in the laboratory of Paul Epstein.49 These mice develop diabetes within the first weeks of life due to beta cell toxicity in response to over-expression of an insulin promotor-driven calmodulin gene. However, like Ins2Akita/+ heterozygotes, a low level of beta cell survival allows OVE26 heterozygotes to live and maintain their body weight well over a year with no insulin treatment in the Epstein laboratory. In their studies of this model, Dr. Epstein and his colleagues report progressively increasing albuminuria that exceeds 15,000 μg/24 hr at 9 months of age in conjunction with hypoalbuminemia, high blood pressure, and decreasing GFR.49 These animals develop progressively enlarged glomeruli, enlarging mesangium with diffuse and nodular expansion of mesangial matrix, tubulointerstitial fibrosis, and thickening of the GBM. AMDCC investigators have not yet examined FVBOVE26 heterozygotes. Although re-derived stock is available from The Jackson Laboratory, high mortality was experienced both during shipment and by investigators shortly after receipt.
Renin overexpression (129S6/SvEvTac background)
Mice having a series of transgenes (RenTgARE, RenTgKC, and RenTgMK) on a 129S6/SvEvTac coisogenic background exhibit progressively greater plasma renin levels, ranging from near physiological to 8X normal, and develop graded kidney and cardiovascular disease.50 Urinary protein excretion in these mice ranges from normal to 10X normal, and renal pathology at 6 months ranges from normal to severe glomerular sclerosis with inflammatory cell infiltration and tubulointerstitial fibrosis together with proteinaceous casts. The cardiovascular phenotype is increased blood pressure (up to 150 mmHg) accompanied by age-dependent cardiac hypertrophy and vascular fibrosis. AMDCC members have made the RenTgMK mice diabetic with low-dose STZ or with the Ins2Akita/+ mutation, and find the proteinuria and glomerulosclerosis caused by the diabetes is dramatically increased by the greater renin levels (Gurley, S, et al., unpublished).
Negative Mouse Models
Although a number of genetic modifications and alternate strains have resulted in models of diabetic nephropathy that more closely resemble the human complication, there are a number of AMDCC models, which do not result in worsening nephropathy. Some of these models have been surprises or have not always agreed with previously published models. The data for some of these models are discussed in the online supplement for this article, and data for all models are available on the AMDCC website (www.amdcc.org). Some of the surprising negative models included diabetic mice with the following gene deficiencies, gene duplications or transgenes, when compared with control diabetic mice on the same genetic background lacking such changes: B6-ACE2-deficient mice; 129S6/SvEvTac mice with an AT1 angiotensin receptor isoform gene duplication; B6-Sod2 (superoxide dismutase 2) heterozygous mice; B6 mice with podocyte-specific deficiency in the Sod2 gene; CD2AP (CD2-associated protein) heterozygous mice on a B6 background, and; endothelial-specific RAGE monotransgenic mice on a mixed B6 and CD-1 background. More details about each of these models, and possible explanations for their lack of efficacy in producing accelerated nephropathy are contained in the online supplement.
Current understanding of diabetic nephropathy in mouse models
As noted above, AMDCC investigators and other investigators in the field have yet to validate a complete murine model of human diabetic kidney disease. Nonetheless, the critical analysis of extant murine models by the AMDCC and others has substantially enhanced our understanding of this nephropathy over the past few years. Perhaps the most obvious insight derived from this work is the confirmation that genetic background has a profound effect on the evolution of disease. While this has been clear from human studies,51-55 the degree of difference in glomerulopathy between inbred mouse strains is remarkable, even between those that share most genetic information (B6 and BKS mice). Of all genetic backgrounds tested or reported so far, the DBA/2 mouse is most susceptible to diabetic glomerulopathy while the B6 mouse appears to be the most resistant. Emerging studies regarding the 129S6/SvEvTac and KK/HIJ strains also support the potential utility of these strains in the study of diabetic nephropathy.
The role of environmental factors, including diet, caging, light cycle, handling, gut microbiome, and pathogen exposure has not been addressed comprehensively. Nonetheless, it is clear that environmental factors have a major impact on disease phenotypes. For example, AMDCC investigators (Brosius, FC, et al., unpublished) and others find that the fat content in the diet can have a marked impact on the degree of hyperglycemia found in B6-db/db mice (but not in BKS-db/db mice). B6-db/db mice on a standard chow that contains only 4% fat (weight/weight) exhibit only transient hyperglycemia and still become obese while those on as little as 8% fat maintain hyperglycemia as well as obesity. Whether differences in environmental conditions can lead to differences in the phenotype of diabetic nephropathy has not yet been reported.
Another key conclusion from findings from AMDCC and other investigators is that abnormalities arising in each type of glomerular cell (endothelial cell, podocyte, mesangial cell) as well as a number of extra-glomerular cells play important roles in the evolution of diabetic glomerulosclerosis. While historically most studies focus on regulation of mesangial cell extracellular matrix and the involvement of these cells in the pathogenesis of diabetic nephropathy,56-58 more recent reports indicate that isolated podocyte damage and loss59, 60 lead to glomerulosclerosis, and that podocyte loss is a requisite early event in diabetic nephropathy.61-64 Additionally, models of endothelial dysfunction result in glomerulosclerosis,25, 27, 65-69 suggesting, not surprisingly, a substantial crosstalk between endothelial and mesangial cells. Indeed, mice with a targeted mutation in the Nos3 gene encoding endothelial nitric oxide synthase25-28 is the most robust model of diabetic nephropathy on a number of genetic backgrounds. This nephropathy presumably results from endothelial cell abnormalities in the glomerulus and possibly the tubulointerstitium. This model is one of the very few to develop appreciable degrees of tubulointerstitial fibrosis, a critical lesion of progressive diabetic nephropathy. Finally, there is reason to believe that extra-glomerular cells, such as bone marrow-derived mesangial cell progenitors70 and macrophages71, 72 significantly contribute to diabetic glomerulopathy.
Perhaps one of the most significant findings of the AMDCC is that only a limited number of monogenic changes in gene expression (eNOS−/−, B2R−/−, and renin transgene) substantially worsen diabetic nephropathy, and none of those monogenic models fully recapitulate human disease. Thus, monogenic changes in gene expression are likely to be insufficient to induce full-fledged diabetic nephropathy, at least on the resistant B6 background. The section on negative models in the online supplement and the results in the archived model database on the AMDCC website (www.amdcc.org) document the relative lack of effect of reduction in antioxidant defenses, increased glucose transport, increased AGE receptors and other changes that resulted from the manipulation of expression of candidate genes.
A final repeated theme has been that aging is a critical feature of even the best mouse models of diabetic nephropathy. Aging of diabetic mice to 12 months of age or older may be required for consistent development of diabetic nephropathy in many of the models heretofore evaluated by AMDCC investigators and others. This theme appears to hold in the diabetic DBA/2 mouse, as well as the diabetic eNOS, B2R and decorin null mice. It is supported by the phenotyping of the Ins2Akita/+ diabetic mice on various backgrounds, where highly variable kidney histopathology suggests that lesions are just beginning to develop at 6 months of age. Effects of the sex of the animal seem to be much less clear-cut. While the severity of nephropathy in many spontaneously diabetic animal models (db/db and Ins2Akita/+) is generally less in females than in males this could be due to the lower blood glucose levels in females as well as to differences in body weight or food intake (Susztak K, et al., unpublished observation, and AMDCC archival database: www.amdcc.org). It is clear that further research is needed to define sex and gender differences in diabetic nephropathy in rodents and humans.
Future directions
While the AMDCC has made vital progress during the last 8 years toward developing better models of diabetic complications and has helped advance the understanding of human diabetic nephropathy, there remains much to accomplish. Although it is difficult to predict the directions of future research that are most likely to drive our understanding forward, there are several aspects of this enterprise about which it is tempting to speculate. First, it seems likely that a combination of sophisticated comparisons between murine models and humans with diabetic kidney disease may promote relatively rapid development of new ideas and models. The focus on candidate pathways, while informative, has for the moment clearly limited the ability to expand our understanding of the nephropathy. A more unbiased approach to identification of pathways and networks of abnormalities in diabetic nephropathy and other complications seems likely now to be well suited to expand our understanding. One recent example of an unbiased approach utilized a transcriptomic analysis of both humans with diabetic nephropathy and mouse models of the disease to uncover a signaling pathway that may be important in diabetic nephropathy.73 This AMDCC study found high levels of many JAK/STAT family members in humans with progressive diabetic nephropathy but not in STZ diabetic DBA/2 or BKS-db/db mice with the relatively mild kidney disease seen in these models. Activation of JAK/STAT signaling may be important in human diabetic nephropathy and induction of such signaling in mouse models might accelerate complications. Similar approaches are being taken by other AMDCC investigators to identify new pathways.
Importantly, it is likely to be difficult to generate a single mouse model that recapitulates all the features of human diabetic nephropathy. Perhaps rather than continuing the search for this holy grail we should settle for a suite of mouse models that recapitulate individually the findings of the human disease. Each of these models individually will teach us something new about the multiple molecular abnormalities that together produce this deadly diabetic complication. In the event that human genetic variants are identified that account for a major percentage of diabetic nephropathy, it will be possible to use a susceptible strain such as DBA/2 as a building block to “knock-in” the human diabetic nephropathy risk variant into the orthologous mouse gene.
Supplementary Material
Footnotes
Disclosures: This work was supported by National Institutes of Health Grants (AMDCC): U24DK076169 (RAM), U01DK061018 (RCH), U01DK076139 (FCB), U01DK076133 (KS), U01DK076136 (TMC), U01DK060995 (EPB), U01DK076134 (ML), U01DK076131 (OS); MMPC grant: U01DK076126 (CA). The authors wish to thank the AMDCC External Advisory Board (Drs. Thomas Hostetter, John Sedor, Juergen Naggert, Phil Tsao, and Doug Zochodne) as well as Dr. Helen Nickerson (Juvenile Diabetes Research Foundation International) for their advice and support. The authors also thank Peter Reifsnyder and Racheal Wallace (Type 1 Diabetes Resource/AMDCC Core at The Jackson Laboratory) for phenotyping data in Fig. 3. Finally, the authors would like to thank the heroically committed AMDCC Program Staff, Drs. Chris Ketchum (NIDDK), Kristin Abraham (NIDDK) and Cristina Rabadan-Diehl (NHLBI), for unwavering support and guidance.
References
- 1.U.S. Renal Data System . USRDS 2008 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; Bethesda, MD: 2008. [Google Scholar]
- 2.Breyer MD, Bottinger E, Brosius FC, 3rd, Coffman TM, Harris RC, Heilig CW, Sharma K. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2005;16(1):27–45. doi: 10.1681/ASN.2004080648. [DOI] [PubMed] [Google Scholar]
- 3.ISI Web of Knowledge. Thomson Reuters; 2008. [Google Scholar]
- 4.Qi Z, Fujita H, Jin J, Davis LS, Wang Y, Fogo AB, Breyer MD. Characterization of susceptibility of inbred mouse strains to diabetic nephropathy. Diabetes. 2005;54(9):2628–2637. doi: 10.2337/diabetes.54.9.2628. [DOI] [PubMed] [Google Scholar]
- 5.Takahashi N, Boysen G, Li F, Li Y, Swenberg JA. Tandem mass spectrometry measurements of creatinine in mouse plasma and urine for determining glomerular filtration rate. Kidney Int. 2007;71(3):266–271. doi: 10.1038/sj.ki.5002033. [DOI] [PubMed] [Google Scholar]
- 6.Takahashi N, Boysen G, Li F, Li Y, Swenberg JA. Tandem mass spectrometry measurements of creatinine in mouse plasma and urine for determining glomerular filtration rate. Kidney international. 2007;71:266–271. doi: 10.1038/sj.ki.5002033. [DOI] [PubMed] [Google Scholar]
- 7.Meyer MH, Meyer RA, Jr., Gray RW, Irwin RL. Picric acid methods greatly overestimate serum creatinine in mice: more accurate results with high-performance liquid chromatography. Anal Biochem. 1985;144(1):285–290. doi: 10.1016/0003-2697(85)90118-6. [DOI] [PubMed] [Google Scholar]
- 8.Dunn SR, Qi Z, Bottinger EP, Breyer MD, Sharma K. Utility of endogenous creatinine clearance as a measure of renal function in mice. Kidney international. 2004;65(5):1959–1967. doi: 10.1111/j.1523-1755.2004.00600.x. [DOI] [PubMed] [Google Scholar]
- 9.Yuen PS, Dunn SR, Miyaji T, Yasuda H, Sharma K, Star RA. A simplified method for HPLC determination of creatinine in mouse serum. American journal of physiology. 2004;286(6):F1116–1119. doi: 10.1152/ajprenal.00366.2003. [DOI] [PubMed] [Google Scholar]
- 10.Bernstein SE, Green E, editors. Biology of the laboratory mouse. Dover Publications, Inc.; New York: 1975. Physiological Characteristics. [Google Scholar]
- 11.Jung K, Wesslau C, Priem F, Schreiber G, Zubek A. Specific creatinine determination in laboratory animals using the new enzymatic test kit “Creatinine-PAP”. J Clin Chem Clin Biochem. 1987;25(6):357–361. doi: 10.1515/cclm.1987.25.6.357. [DOI] [PubMed] [Google Scholar]
- 12.Keppler A, Gretz N, Schmidt R, Kloetzer HM, Groene HJ, Lelongt B, Meyer M, Sadick M, Pill J. Plasma creatinine determination in mice and rats: an enzymatic method compares favorably with a high-performance liquid chromatography assay. Kidney Int. 2007;71(1):74–78. doi: 10.1038/sj.ki.5001988. [DOI] [PubMed] [Google Scholar]
- 13.Palm M, Lundblad A. Creatinine concentration in plasma from dog, rat, and mouse: a comparison of 3 different methods. Veterinary clinical pathology / American Society for Veterinary Clinical Pathology. 2005;34(3):232–236. doi: 10.1111/j.1939-165x.2005.tb00046.x. [DOI] [PubMed] [Google Scholar]
- 14.Qi ZWI, Mehta A, Jin J, Zhao M, Harris RC, Fogo AB, Breyer MD. Serial Determination of Glomerular Filtration Rate in Conscious Mice Using FITC-Inulin Clearance. Am J Physiol. 2004 doi: 10.1152/ajprenal.00324.2003. [DOI] [PubMed] [Google Scholar]
- 15.Lorenz JN, Gruenstein E. A simple, nonradioactive method for evaluating single-nephron filtration rate using FITC-inulin. The American journal of physiology. 1999;276(1 Pt 2):F172–177. doi: 10.1152/ajprenal.1999.276.1.F172. [DOI] [PubMed] [Google Scholar]
- 16.Eisner C, Huang Y, Wang Y, Levine M, Schnermann J. Creatinine Excretion in MIce: Implications for the Estimation of Renal Fucntion in Murine Models of Kidney Disease. J Am Soc Nephrol. 2007;18:109A. [Google Scholar]
- 17.Gurley SB, Clare SE, Snow KP, Hu A, Meyer TW, Coffman TM. Impact of genetic background on nephropathy in diabetic mice. American journal of physiology. 2006;290(1):F214–222. doi: 10.1152/ajprenal.00204.2005. [DOI] [PubMed] [Google Scholar]
- 18.Futrakul N, Butthep P, Vongthavarawat V, Futrakul P, Sirisalipoch S, Chaivatanarat T, Suwanwalaikorn S. Early detection of endothelial injury and dysfunction in conjunction with correction of hemodynamic maladjustment can effectively restore renal function in type 2 diabetic nephropathy. Clin Hemorheol Microcirc. 2006;34(3):373–381. [PubMed] [Google Scholar]
- 19.Tsukada T, Yokoyama K, Arai T, Takemoto F, Hara S, Yamada A, Kawaguchi Y, Hosoya T, Igari J. Evidence of association of the ecNOS gene polymorphism with plasma NO metabolite levels in humans. Biochemical and biophysical research communications. 1998;245(1):190–193. doi: 10.1006/bbrc.1998.8267. [DOI] [PubMed] [Google Scholar]
- 20.Veldman BA, Spiering W, Doevendans PA, Vervoort G, Kroon AA, de Leeuw PW, Smits P. The Glu298Asp polymorphism of the NOS 3 gene as a determinant of the baseline production of nitric oxide. Journal of hypertension. 2002;20(10):2023–2027. doi: 10.1097/00004872-200210000-00022. [DOI] [PubMed] [Google Scholar]
- 21.Nakayama M, Yasue H, Yoshimura M, Shimasaki Y, Kugiyama K, Ogawa H, Motoyama T, Saito Y, Ogawa Y, Miyamoto Y, Nakao K. T-786-->C mutation in the 5′-flanking region of the endothelial nitric oxide synthase gene is associated with coronary spasm. Circulation. 1999;99(22):2864–2870. doi: 10.1161/01.cir.99.22.2864. [DOI] [PubMed] [Google Scholar]
- 22.Ksiazek P, Wojewoda P, Muc K, Buraczynska M. Endothelial nitric oxide synthase gene intron 4 polymorphism in type 2 diabetes mellitus. Mol Diagn. 2003;7(2):119–123. doi: 10.1007/BF03260027. [DOI] [PubMed] [Google Scholar]
- 23.Ezzidi I, Mtiraoui N, Mohamed MB, Mahjoub T, Kacem M, Almawi WY. Association of endothelial nitric oxide synthase Glu298Asp, 4b/a, and -786T>C gene variants with diabetic nephropathy. Journal of diabetes and its complications. 2008 doi: 10.1016/j.jdiacomp.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 24.Zanchi A, Moczulski DK, Hanna LS, Wantman M, Warram JH, Krolewski AS. Risk of advanced diabetic nephropathy in type 1 diabetes is associated with endothelial nitric oxide synthase gene polymorphism. Kidney international. 2000;57(2):405–413. doi: 10.1046/j.1523-1755.2000.00860.x. [DOI] [PubMed] [Google Scholar]
- 25.Nakagawa T, Sato W, Glushakova O, Heinig M, Clarke T, Campbell-Thompson M, Yuzawa Y, Atkinson MA, Johnson RJ, Croker B. Diabetic endothelial nitric oxide synthase knockout mice develop advanced diabetic nephropathy. J Am Soc Nephrol. 2007;18(2):539–550. doi: 10.1681/ASN.2006050459. [DOI] [PubMed] [Google Scholar]
- 26.Kanetsuna Y, Takahashi K, Nagata M, Gannon MA, Breyer MD, Harris RC, Takahashi T. Deficiency of endothelial nitric-oxide synthase confers susceptibility to diabetic nephropathy in nephropathy-resistant inbred mice. The American journal of pathology. 2007;170(5):1473–1484. doi: 10.2353/ajpath.2007.060481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao HJ, Wang S, Cheng H, Zhang MZ, Takahashi T, Fogo AB, Breyer MD, Harris RC. Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J Am Soc Nephrol. 2006;17(10):2664–2669. doi: 10.1681/ASN.2006070798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mohan S, Reddick RL, Musi N, Horn DA, Yan B, Prihoda TJ, Natarajan M, Abboud-Werner SL. Diabetic eNOS knockout mice develop distinct macro- and microvascular complications. Laboratory investigation; a journal of technical methods and pathology. 2008;88(5):515–528. doi: 10.1038/labinvest.2008.23. [DOI] [PubMed] [Google Scholar]
- 29.Marre M, Bernadet P, Gallois Y, Savagner F, Guyene TT, Hallab M, Cambien F, Passa P, Alhenc-Gelas F. Relationships between angiotensin I converting enzyme gene polymorphism, plasma levels, and diabetic retinal and renal complications. Diabetes. 1994;43(3):384–388. doi: 10.2337/diab.43.3.384. [DOI] [PubMed] [Google Scholar]
- 30.Doria A, Warram JH, Krolewski AS. Genetic predisposition to diabetic nephropathy. Evidence for a role of the angiotensin I--converting enzyme gene. Diabetes. 1994;43(5):690–695. doi: 10.2337/diab.43.5.690. [DOI] [PubMed] [Google Scholar]
- 31.Huang W, Gallois Y, Bouby N, Bruneval P, Heudes D, Belair MF, Krege JH, Meneton P, Marre M, Smithies O, Alhenc-Gelas F. Genetically increased angiotensin I-converting enzyme level and renal complications in the diabetic mouse. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(23):13330–13334. doi: 10.1073/pnas.231476798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takahashi N, Hagaman JR, Kim HS, Smithies O. Minireview: computer simulations of blood pressure regulation by the renin-angiotensin system. Endocrinology. 2003;144(6):2184–2190. doi: 10.1210/en.2002-221045. [DOI] [PubMed] [Google Scholar]
- 33.Kakoki M, Takahashi N, Jennette JC, Smithies O. Diabetic nephropathy is markedly enhanced in mice lacking the bradykinin B2 receptor. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(36):13302–13305. doi: 10.1073/pnas.0405449101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kakoki M, Kizer CM, Yi X, Takahashi N, Kim HS, Bagnell CR, Edgell CJ, Maeda N, Jennette JC, Smithies O. Senescence-associated phenotypes in Akita diabetic mice are enhanced by absence of bradykinin B2 receptors. J Clin Invest. 2006;116(5):1302–1309. doi: 10.1172/JCI26958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Allard J, Buleon M, Cellier E, Renaud I, Pecher C, Praddaude F, Conti M, Tack I, Girolami JP. ACE inhibitor reduces growth factor receptor expression and signaling but also albuminuria through B2-kinin glomerular receptor activation in diabetic rats. American journal of physiology. 2007;293(4):F1083–1092. doi: 10.1152/ajprenal.00401.2006. [DOI] [PubMed] [Google Scholar]
- 36.Buleon M, Allard J, Jaafar A, Praddaude F, Dickson Z, Ranera MT, Pecher C, Girolami JP, Tack I. Pharmacological blockade of B2-kinin receptor reduces renal protective effect of angiotensin-converting enzyme inhibition in db/db mice model. American journal of physiology. 2008;294(5):F1249–1256. doi: 10.1152/ajprenal.00501.2007. [DOI] [PubMed] [Google Scholar]
- 37.Tan Y, Keum JS, Wang B, McHenry MB, Lipsitz SR, Jaffa AA. Targeted deletion of B2-kinin receptors protects against the development of diabetic nephropathy. American journal of physiology. 2007;293(4):F1026–1035. doi: 10.1152/ajprenal.00203.2007. [DOI] [PubMed] [Google Scholar]
- 38.Kakoki M, Smithies O. The kallikrein-kinin system in health and in diseases of the kidney. Kidney international. 2009;75(10):1019–1030. doi: 10.1038/ki.2008.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weber I, Harrison R, Iozzo R. Model structure of decorin and implications for collagen fibrillogenesis. The Journal of Biological Chemistry. 1996;271(50):31767–31770. doi: 10.1074/jbc.271.50.31767. [DOI] [PubMed] [Google Scholar]
- 40.Schonherr E, Sunderkotter C, Iozzo RV, Schaefer L. Decorin, a novel player in the insulin-like growth factor system. Journal of Biological Chemistry. 2005;280(16):15767–15772. doi: 10.1074/jbc.M500451200. [DOI] [PubMed] [Google Scholar]
- 41.Takeuchi Y, Kodama Y, Matsumato T. Bone matrix decorin binds transforming growth factor-β and enhances its bioactivity. J Biol Chem. 1994;269:32634–32638. [PubMed] [Google Scholar]
- 42.Wahab N, Parker S, Sraer J-D, Mason R. The decorin high glucose response element and mechanism of its activation in human mesangial cells. J Am Soc Nephrol. 2000;11:1607–1619. doi: 10.1681/ASN.V1191607. [DOI] [PubMed] [Google Scholar]
- 43.Schaefer L, Raslik I, Grone H-J, Schonherr E, Macakova K, Ugorcakova J, Budny S, Schaefer R, Kresse H. Small proteoglycans in human diabetic nephropathy: discrepancy between glomerular expression and protein accumulation of decorin, biglycan, lumican, and fibromodulin. The FASEB Journal. 2001;15:559–561. doi: 10.1096/fj.00-0493fje. [DOI] [PubMed] [Google Scholar]
- 44.Mogyorosi A, Ziyadeh FN. Increased decorin mRNA in diabetic mouse kidney and in mesangial and tubular cells cultured in high glucose. American Journal of Physiology-Renal Physiology. 1998;275:F827–F832. doi: 10.1152/ajprenal.1998.275.5.F827. [DOI] [PubMed] [Google Scholar]
- 45.Williams KJ, Qiu G, Usui HK, Dunn SR, McCue P, Bottinger E, Iozzo RV, Sharma K. Decorin deficiency enhances progressive nephropathy in diabetic mice. Am J Pathol. 2007;171(5):1441–1450. doi: 10.2353/ajpath.2007.070079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leiter EH, Reifsnyder PC. Differential levels of diabetogenic stress in two new mouse models of obesity and type 2 diabetes. Diabetes. 2004;53(Suppl 1):S4–11. doi: 10.2337/diabetes.53.2007.s4. [DOI] [PubMed] [Google Scholar]
- 47.Reifsnyder PC, Leiter EH. Deconstructing and reconstructing obesity-induced diabetes (diabesity) in mice. Diabetes. 2002;51(3):825–832. doi: 10.2337/diabetes.51.3.825. [DOI] [PubMed] [Google Scholar]
- 48.Watanabe Y, Itoh Y, Yoshida F, Koh N, Tamai H, Fukatsu A, Matsuo S, Hotta N, Sakamoto N. Unique glomerular lesion with spontaneous lipid deposition in glomerular capillary lumina in the NON strain of mice. Nephron. 1991;58(2):210–218. doi: 10.1159/000186417. [DOI] [PubMed] [Google Scholar]
- 49.Zheng S, Noonan WT, Metreveli NS, Coventry S, Kralik PM, Carlson EC, Epstein PN. Development of late-stage diabetic nephropathy in OVE26 diabetic mice. Diabetes. 2004;53(12):3248–3257. doi: 10.2337/diabetes.53.12.3248. [DOI] [PubMed] [Google Scholar]
- 50.Caron KM, James LR, Lee G, Kim HS, Smithies O. Lifelong genetic minipumps. Physiol Genomics. 2005;20(2):203–209. doi: 10.1152/physiolgenomics.00221.2004. [DOI] [PubMed] [Google Scholar]
- 51.Iyengar SK, Abboud HE, Goddard KA, Saad MF, Adler SG, Arar NH, Bowden DW, Duggirala R, Elston RC, Hanson RL, Ipp E, Kao WH, Kimmel PL, Klag MJ, Knowler WC, Meoni LA, Nelson RG, Nicholas SB, Pahl MV, Parekh RS, Quade SR, Rich SS, Rotter JI, Scavini M, Schelling JR, Sedor JR, Sehgal AR, Shah VO, Smith MW, Taylor KD, Winkler CA, Zager PG, Freedman BI. Genome-wide scans for diabetic nephropathy and albuminuria in multiethnic populations: the family investigation of nephropathy and diabetes (FIND) Diabetes. 2007;56(6):1577–1585. doi: 10.2337/db06-1154. [DOI] [PubMed] [Google Scholar]
- 52.Knowler WC, Coresh J, Elston RC, Freedman BI, Iyengar SK, Kimmel PL, Olson JM, Plaetke R, Sedor JR, Seldin MF. The Family Investigation of Nephropathy and Diabetes (FIND): design and methods. Journal of diabetes and its complications. 2005;19(1):1–9. doi: 10.1016/j.jdiacomp.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 53.Imperatore G, Hanson RL, Pettitt DJ, Kobes S, Bennett PH, Knowler WC. Sib-pair linkage analysis for susceptibility genes for microvascular complications among Pima Indians with type 2 diabetes. Pima Diabetes Genes Group. Diabetes. 1998;47(5):821–830. doi: 10.2337/diabetes.47.5.821. [DOI] [PubMed] [Google Scholar]
- 54.Bowden DW, Sale M, Howard TD, Qadri A, Spray BJ, Rothschild CB, Akots G, Rich SS, Freedman BI. Linkage of genetic markers on human chromosomes 20 and 12 to NIDDM in Caucasian sib pairs with a history of diabetic nephropathy. Diabetes. 1997;46(5):882–886. doi: 10.2337/diab.46.5.882. [DOI] [PubMed] [Google Scholar]
- 55.Seaquist ER, Goetz FC, Rich S, Barbosa J. Familial clustering of diabetic kidney disease. Evidence for genetic susceptibility to diabetic nephropathy. N Engl J Med. 1989;320(18):1161–1165. doi: 10.1056/NEJM198905043201801. [DOI] [PubMed] [Google Scholar]
- 56.Chen S, Jim B, Ziyadeh FN. Diabetic nephropathy and transforming growth factor-beta: transforming our view of glomerulosclerosis and fibrosis build-up. Semin Nephrol. 2003;23(6):532–543. doi: 10.1053/s0270-9295(03)00132-3. [DOI] [PubMed] [Google Scholar]
- 57.Heilig CW, Brosius FC, Cunningham C. Role for GLUT1 in diabetic glomerulosclerosis. Expert Rev Mol Med. 2006;8(4):1–18. doi: 10.1017/S1462399406010490. [DOI] [PubMed] [Google Scholar]
- 58.Kikkawa R, Koya D, Haneda M. Progression of diabetic nephropathy. Am J Kidney Dis. 2003;41(3 Suppl 1):S19–21. doi: 10.1053/ajkd.2003.50077. [DOI] [PubMed] [Google Scholar]
- 59.Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, Saunders TL, Dysko RC, Kohno K, Holzman LB, Wiggins RC. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol. 2005;16(10):2941–2952. doi: 10.1681/ASN.2005010055. [DOI] [PubMed] [Google Scholar]
- 60.Shankland SJ. The podocyte’s response to injury: role in proteinuria and glomerulosclerosis. Kidney international. 2006;69(12):2131–2147. doi: 10.1038/sj.ki.5000410. [DOI] [PubMed] [Google Scholar]
- 61.Pagtalunan ME, Miller PL, Jumping-Eagle S, Nelson RG, Myers BD, Rennke HG, Coplon NS, Sun L, Meyer TW. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest. 1997;99(2):342–348. doi: 10.1172/JCI119163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marshall SM. The podocyte: a major player in the development of diabetic nephropathy? Horm Metab Res. 2005;37(Suppl 1):9–16. doi: 10.1055/s-2005-861397. [DOI] [PubMed] [Google Scholar]
- 63.Siu B, Saha J, Smoyer WE, Sullivan KA, Brosius FC., 3rd Reduction in podocyte density as a pathologic feature in early diabetic nephropathy in rodents: prevention by lipoic acid treatment. BMC Nephrol. 2006;7:6. doi: 10.1186/1471-2369-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55(1):225–233. [PubMed] [Google Scholar]
- 65.Morcos M, Borcea V, Isermann B, Gehrke S, Ehret T, Henkels M, Schiekofer S, Hofmann M, Amiral J, Tritschler H, Ziegler R, Wahl P, Nawroth PP. Effect of alpha-lipoic acid on the progression of endothelial cell damage and albuminuria in patients with diabetes mellitus: an exploratory study. Diabetes Res Clin Pract. 2001;52(3):175–183. doi: 10.1016/s0168-8227(01)00223-6. [DOI] [PubMed] [Google Scholar]
- 66.Isermann B, Vinnikov IA, Madhusudhan T, Herzog S, Kashif M, Blautzik J, Corat MA, Zeier M, Blessing E, Oh J, Gerlitz B, Berg DT, Grinnell BW, Chavakis T, Esmon CT, Weiler H, Bierhaus A, Nawroth PP. Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat Med. 2007;13(11):1349–1358. doi: 10.1038/nm1667. [DOI] [PubMed] [Google Scholar]
- 67.Ichinose K, Maeshima Y, Yamamoto Y, Kitayama H, Takazawa Y, Hirokoshi K, Sugiyama H, Yamasaki Y, Eguchi K, Makino H. Antiangiogenic endostatin peptide ameliorates renal alterations in the early stage of a type 1 diabetic nephropathy model. Diabetes. 2005;54(10):2891–2903. doi: 10.2337/diabetes.54.10.2891. [DOI] [PubMed] [Google Scholar]
- 68.Quaggin SE, Coffman TM. Toward a mouse model of diabetic nephropathy: is endothelial nitric oxide synthase the missing link? J Am Soc Nephrol. 2007;18(2):364–366. doi: 10.1681/ASN.2006121396. [DOI] [PubMed] [Google Scholar]
- 69.Toyoda M, Najafian B, Kim Y, Caramori ML, Mauer M. Podocyte Detachment and Reduced Glomerular Capillary Endothelial Fenestration in Human Type 1 Diabetic Nephropathy. Diabetes. 2007 doi: 10.2337/db07-0019. [DOI] [PubMed] [Google Scholar]
- 70.Zheng F, Cornacchia F, Schulman I, Banerjee A, Cheng QL, Potier M, Plati AR, Berho M, Elliot SJ, Li J, Fornoni A, Zang YJ, Zisman A, Striker LJ, Striker GE. Development of albuminuria and glomerular lesions in normoglycemic B6 recipients of db/db mice bone marrow: the role of mesangial cell progenitors. Diabetes. 2004;53(9):2420–2427. doi: 10.2337/diabetes.53.9.2420. [DOI] [PubMed] [Google Scholar]
- 71.Shikata K, Makino H. Role of macrophages in the pathogenesis of diabetic nephropathy. Contrib Nephrol. 2001;(134):46–54. doi: 10.1159/000060147. [DOI] [PubMed] [Google Scholar]
- 72.Tesch GH. Role of macrophages in complications of type 2 diabetes. Clinical and experimental pharmacology & physiology. 2007;34(10):1016–1019. doi: 10.1111/j.1440-1681.2007.04729.x. [DOI] [PubMed] [Google Scholar]
- 73.Berthier CC, Zhang H, Schin M, Henger A, Nelson RG, Yee B, Boucherot A, Neusser MA, Cohen CD, Carter-Su C, Argetsinger LS, Rastaldi MP, Brosius FC, Kretzler M. Enhanced expression of Janus kinase-signal transducer and activator of transcription pathway members in human diabetic nephropathy. Diabetes. 2009;58(2):469–477. doi: 10.2337/db08-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.