

Abstract
The ubiquitin-proteasome system and autophagy were long viewed as independent, parallel degradation systems with no point of intersection. By now we know that these degradation pathways share certain substrates and regulatory molecules and show coordinated and compensatory function. Two ubiquitin-like protein conjugation pathways were discovered that are required for autophagosome biogenesis: the Atg12-Atg5-Atg16 and Atg8 systems. Autophagy has been considered to be essentially a nonselective process, but it turned out to be at least partially selective. Selective substrates of autophagy include damaged mitochondria, intracellular pathogens, and even a subset of cytosolic proteins with the help of ubiquitin-binding autophagic adaptors, such as p62/SQSTM1, NBR1, NDP52, and Optineurin. These proteins selectively recognize autophagic cargo and mediate its engulfment into autophagosomes by binding to the small ubiquitin-like modifiers that belong to the Atg8/LC3 family.
1. Introduction
Two major pathways accomplish regulated protein catabolism in eukaryotic cells: the ubiquitin-proteasome system (UPS) and the autophagy-lysosomal system. The UPS serves as the primary route of degradation for thousands of short-lived proteins and many regulatory proteins and contributes to the degradation of defective proteins [1]. Autophagy, by contrast, is primarily responsible for degrading long-lived proteins and maintaining amino acid pools during stress conditions, such as in chronic starvation [2]. The critical factors that direct a specific substrate to one degradation route or the other are incompletely understood. Protein degradations performed by the UPS and autophagy were regarded for a long time as complementary but separate mechanisms [3]. However, on the basis of recent studies, there are overlaps between them. The way of degradation of a misfolded, redundant, or unneeded protein may be often governed by the momentary activity or capacity of these systems or, in some cases, determined by strict regulation. Moreover, the two pathways use common adaptors capable of directing ubiquitinylated target proteins to both.
2. Ubiquitin-Proteasome System
The ubiquitin-proteasome pathway plays a crucial role in governing many basic cellular processes, such as normal protein turnover, protein quality control by degrading misfolded and damaged proteins, signal transduction, metabolism, cell death, immune responses, and cell cycle control [4]. Ubiquitin is a small, globular protein containing 76 amino acid residues (Figure 1). There are only three amino-acid changes from yeast to human, so ubiquitin is highly conserved within eukaryotes. Ubiquitinylation, the covalent conjugation of ubiquitin to other proteins, is a special posttranslational modification, which may either serve as an essential degradation signal for proteins or it may alter their localisation, function, or activity.
Figure 1.
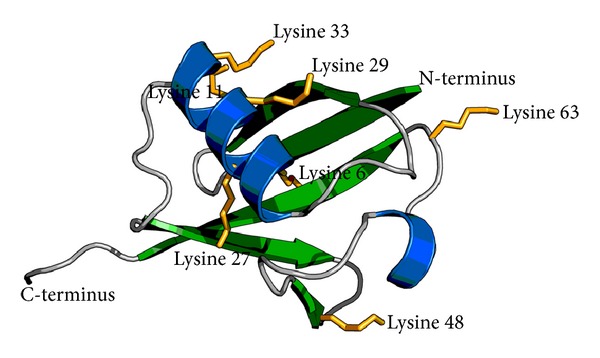
Ribbon model of ubiquitin exposing all the seven lysine side chains possibly involved in polyubiquitinylation reactions.
Before being covalently attached to other proteins, free ubiquitin is activated in an ATP-dependent manner with the formation of a thiolester linkage between a ubiquitin-activating enzyme (E1) and the carboxyl terminus of ubiquitin. Then, it is transferred to a ubiquitin-conjugating enzyme (E2). Finally, E2 associates with ubiquitin-ligases (E3s) which specifically bind the target substrate and attach ubiquitin through its carboxyl terminal glycine to the ε-amino group of a lysine residue in the target protein (Figure 2). The exact details of ubiquitinylation biochemistry are determined by the type of E3 enzyme involved. E3s can be grouped into two major classes: HECT (homologous to E6-AP carboxyl-terminus) domain E3s and RING-finger (really interesting new gene) domain E3s [5]. The identification of E6-AP as the E3 responsible for the human papilloma virus E6-dependent ubiquitinylation of p53 led to the discovery of the HECT domain enzymes [6]. HECT domain is a conserved C-terminus of the molecule, which is about 350 amino acids long. HECT domain E3s form thiolester intermediates with ubiquitin through a conserved cysteine residue, like in case of E1 and E2 enzymes. By contrast, RING-finger E3s do not generate a thiolester intermediate but just simply act as a scaffold to hold a ubiquitin-E2 intermediate close to a substrate and catalyze ubiquitin transfer [7] (Figure 2).
Figure 2.
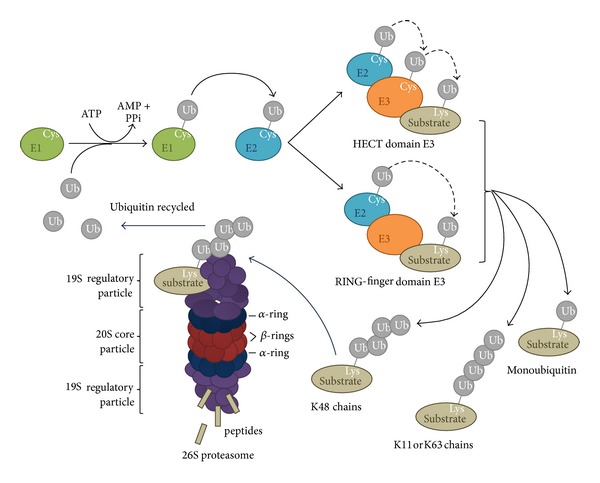
The ubiquitin-proteasome system. An enzyme cascade organizes the attachment of mono- or polyubiquitin to the substrates. Ubiquitin (Ub) is first activated in an ATP-consuming reaction by E1 (Ub-activating enzyme), to which it becomes attached by a high-energy thiolester bond. Then, the activated Ub is shifted to the active Cys residue of E2 (ubiquitin-conjugating enzyme). E2 catalyzes the transfer of ubiquitin to the substrate protein with the help of E3 (ubiquitin ligase). There are two major classes of E3 enzymes, characterized by the HECT domain or the RING-finger domain. In case of the HECT E3 enzymes, the activated Ub is transferred first to an active Cys residue in the HECT domain before it is finally moved to the substrate. RING-finger domain E3 enzymes bind to both the E2 enzyme and the substrate and catalyze the transfer of Ub directly from the E2 enzyme to the substrate. A polyubiquitin chain linked through Lys 48 is the signal for the proteasome to degrade the substrate. The 26S proteasome consists of the catalytic 20S core particle; a barrel of four stacked rings: two outer α-rings (blue) and two inner β-rings (red); and the 19S regulatory particle. The polyubiquitin chain is recognized by the regulatory particle, which then binds, unfolds, and translocates the polypeptide into the catalytic core. The substrate is hydrolyzed by the enzymatically active β-subunits inside the core particle producing short peptides. Ubiquitin is recycled in the process [102, 103].
The high specificity of the UPS system is tightly associated with the E3 enzymes, as they determine which substrate should be ubiquitinylated and hence usually degraded. Whether the attached ubiquitin is a modification signal or a sign for degradation depends on how it is linked to its substrates: conjugation of a single ubiquitin monomer (monoubiquitinylation) or sequential conjugation of several ubiquitin moieties (polyubiquitinylation) of variable length.
The ubiquitin chain could be lengthened by the E2 and E3, sometimes with the help of an accessory factor (E4). The carboxyl terminal glycine of the more distal ubiquitin molecule is bound to the previous ubiquitin molecule through an isopeptide bond with an ε-amino group of a lysine [8]. If the series of ubiquitin moieties is extended to at least four units, then it is sufficient to allow the ubiquitylated target protein to be recognized and degraded by the 26S proteasome [9].
The 26S proteasome is a 2.5 MDa multicatalytic multisubunit protease, which is made up of two subcomplexes: a barrel-shaped core particle (CP: also known as the 20S proteasome) and one or two 19S regulatory particle(s) (RP) on one or both ends of the core particle [10–12]. The 19S RP serves to recognize ubiquitinylated substrate proteins and plays a role in their unfolding and translocation into the interior of the 20S CP (Figure 2).
The 20S CP contains two outer α-rings and two inner β-rings, each of which is made up of seven structurally similar α and β subunits, respectively. The rings form an α 1–7 β 1–7 β 1–7 α 1–7 structure creating three continuous chambers inside the particle. Only three of the β-type subunits (β1, β2, and β5) in each inner ring are catalytically active. They have threonine residues at their N-termini and show N-terminal nucleophile hydrolase activity. Such a “self-compartmentalized” structure keeps the proteolytic active sites separated in the central chamber and allows regulated substrate degradation only. The proteasome is a multicatalytic protease because the β1, β2, and β5 subunits are associated with caspase-like, trypsin-like, and chymotrypsin-like activities, respectively, which are able to cleave amide bonds at the C-terminal side of acidic, basic, and hydrophobic amino-acid residues, respectively.
The ubiquitin chains are called K6, K11, K27, K29, K33, K48, or K63 chains depending on which of the seven lysine (K) residues is involved in linkage of monomers in the polyubiquitin polymer (Figures 1 and 2). K48 ubiquitin chain was first identified as the signal to target proteins for proteasomal degradation. In contrast, K11 or K63 chains or single ubiquitin moieties (monoubiquitinylation) were thought to signal mainly for nonproteolytic functions [13]. These chain types are involved in controlling several processes such as gene transcription, DNA repair, cell cycle progression, apoptosis, and receptor endocytosis [14]. However, recent reports have demonstrated that all types of ubiquitin chains as well as monoubiquitinylation can target substrates for degradation via autophagy [15].
3. Ubiquitin-Like Proteins
There are more and more ubiquitin-like proteins (Ubls) identified and characterized. They resemble ubiquitin, as for all Ubls whose covalent attachment to other biomolecules has been experimentally demonstrated, the C-terminal residue is a glycine, and the carboxyl group of this glycine is the site of attachment to substrates [16]. On substrate proteins lysine side chains are the target sites so the Ubl and substrate are connected with an amide (or isopeptide) bond. Ubls also share a similar structural motif, the β-grasp fold, which contains a β-sheet with four antiparallel β-strands and a helical segment (Figure 3).
Figure 3.

Structures of ubiquitin and the ubiquitin-like proteins (Ubls) Atg12 and LC3B, shown as ribbon diagrams generated by Jmol 13.0 [104] upon the structural data deposited in PDB. The characteristic Ubl β-grasp fold: a β-sheet with four antiparallel β-strands (yellow) and a helical segment (green) is well observable. Other helical structures are blue (Protein Data Bank (PDB) accession codes: 1UBQ [105], 4GDK [106], and 1UGM [107], resp.).
4. Autophagy
Autophagy is another degradative pathway that occurs in all eukaryotic cells. It is the main system for the degradation of bulk cytoplasmic components in the cell, and it is induced by nutrient starvation for example. Autophagy is crucial for homeostasis in the cell, as it recycles proteins and organelles. In addition, autophagy plays a critical role in cytoprotection by preventing the accumulation of toxic proteins and acting in various aspects of immunity, including the elimination of invading microbes and its participation in antigen presentation. Macroautophagy is the best characterized type of autophagy. In this case the cell forms a double-membrane sequestering compartment called the phagophore, which develops into an autophagosome. After fusion with lysosomes, the content of the resulting autolysosome is degraded and the newly generated monomers are released back into the cytosol for reuse [2, 17] (Figure 4).
Figure 4.
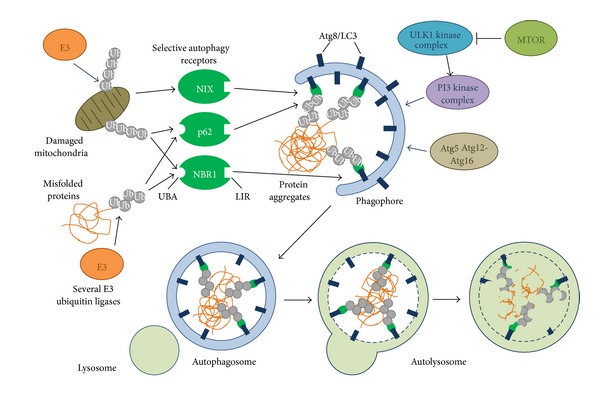
The process of autophagy. Initiation of autophagy is controlled by the ULK1 complex, followed by activation of the PI3-kinase complex leading to nucleation of the phagophore. Vesicle expansion is governed by two ubiquitin-like conjugation systems: the Atg5-Atg12- Atg16 and Atg8/LC3 pathways. Finally, autophagosomes fuse with lysosomes forming autolysosomes, where breakdown of the autophagic cargo takes place. Selective autophagy can distinguish and direct specific cargos to the lysosome. Autophagy receptors contain a short LIR (LC3-interacting region) sequence responsible for Atg8/LC3 binding. Recognition of ubiquitinylated proteins is mediated by interacting with ubiquitin noncovalently, via an ubiquitin-binding domain (UBA). NIX acts as a mitophagy receptor; it has a LIR motif but lacks an UBA domain and is localized within the mitochondrial outer membrane; this is why ubiquitinylation is not required for NIX-dependent delivery of damaged mitochondria to autophagosomes.
There are 38 known autophagy-related (Atg) genes regulating the steps of autophagosome formation and breakdown. These were identified in yeast genetic screens but they are evolutionarily well conserved also in plants and animals, including Drosophila and mammalian cells [18, 19]. Initiation of autophagy is controlled by the Atg1/ULK complex, consisting of Atg1, Atg13, Atg17, Atg29, and Atg31 in yeast and ULK1/2, mAtg13, FIP200, and Atg101 in mammals. The ULK1/2, mAtg13, and FIP200 proteins form a complex independently of nutrient supply. MTORC1 (mechanistic target of rapamycin complex 1) phosphorylates and inhibits ULK1/2 and mAtg13 in nutrient-rich conditions, disrupting the contact between ULK1 and AMPK, an energy sensor kinase with activating effect on ULK1. On the contrary, MTOR is released from its complex under starvation, resulting in activation of ULK1/2 (Figure 4), which, in turn, phosphorylates and activates mAtg13 and FIP200 [20].
The transmembrane protein Atg9 and regulators of its trafficking (Atg2 and Atg18) play a role in membrane delivery to the expanding phagophore after the assembly of the Atg1 complex at the single phagophore assembly site (PAS), which is marked by the selective cargo proaminopeptidase I aggregate in yeast. Nucleation of the phagophore at the PAS is controlled by the phosphatidylinositol-3-kinase (PI3 K) complex (Vps34/hVPS34, Vps15/hVPS15, Vps30/Atg6/Beclin 1, and Atg14/ATG14L). Finally, there are two Ubl conjugation systems: the Atg12 (Atg5, Atg7, Atg10, Atg12, and Atg16) and Atg8 (Atg3, Atg4, Atg7, and Atg8) pathways which are responsible for vesicle expansion [18, 21] (Figure 4).
Autophagosomes undergo a maturation process in animal cells, which involves the recruitment of the SNARE protein syntaxin 17 [22–24]. Interaction of syntaxin 17 with the HOPS (homotypic fusion and vacuole protein sorting) tethering complex promotes the fusion of autophagosomes with lysosomes, where breakdown of autophagic cargo takes place [25, 26] (Figure 4).
Macroautophagy has long been considered as a nonselective process responsible for bulk degradation of cytoplasmic components. The autophagy pathway appeared during evolution as an adaptation mechanism of the eukaryotic cell to starvation, allowing mobilization of nutrients in the cell by forfeit materials of the cytosol. Additionally, it became indispensable for specific degradation of unnecessary or toxic structures: proteins, organelles, and intracellular pathogens [27]. In contrast to the bulk autophagy, which ensures the more or less random sequestration of cytosol, selective autophagy operates under nutrient-rich conditions as well and is characterized by the presence of specialized autophagosomes. These autophagosomes lock up substrates in an exclusive way, which means that other parts of the cytoplasm are largely absent from them [18, 28, 29] (Figure 4).
4.1. Atg12 and Atg8
Autophagy requires the Ubls Atg12 and Atg8/LC3 (Figures 3 and 4). Atg12, which is 2.5 times larger than ubiquitin, was the first Ubl identified as a core autophagy protein [30]. It is synthesized in an active form that does not require proteolytic maturation. The C-terminal glycine of Atg12 is first activated by the E1 enzyme Atg7, and is then transferred to an E2 enzyme, Atg10, before finally forming a conjugate with Atg5 [30]. This Atg12-Atg5 conjugate is essential for autophagy. This system is well conserved in mammals; there is only one orthologue for each of the components of the Atg12 system in mice and humans [21].
Atg8, the other Ubl regulator of autophagy, is expressed with a C-terminal arginine residue in yeast, which is removed by the cysteine protease Atg4 leaving a glycine residue at the C-terminus [31]. Biochemical studies revealed the existence of another ubiquitinylation-like conjugation system [32]. The C-terminal glycine residue of Atg8 is activated by the same E1-like enzyme, Atg7, as in case of Atg12. Then Atg3, an E2-like enzyme, together with an Atg12-5-16 complex catalyzes the transfer of the activated Atg8 to phosphatidylethanolamine, the target lipid substrate. This way Atg8 becomes tightly membrane associated. Atg8 therefore can be utilized as a marker of the autophagosomal membrane and a key molecule during autophagosome formation (Figures 3 and 4). The conjugation of Atg8 to and its removal from phosphatidylethanolamine are essential for autophagy. There are three families of Atg8 homologues in mice and humans called LC3s, GABARAPs, and GABARAP-like proteins.
4.2. Selective Autophagy and Its Specific Adaptors
In the last decade, emerging evidence revealed that autophagy can distinguish and direct specific cargos to the lysosome. Different terms were coined to distinguish between different targets. The most investigated processes are mitophagy: the selective removal of defective or excess mitochondria [33]; aggrephagy: the disposal of aberrant, misfolded protein aggregates [34]; xenophagy: the selective autophagy of pathogenic intracellular bacteria, protozoa, or viruses [35, 36], and pexophagy: peroxisome autophagy first described in detail during peroxisome degradation in methylotrophic yeast species but also responsible for the destruction of 70–80% of the peroxisomal mass in mammalian cells [37]. The selective nature of autophagy is ensured mainly by specific adaptors, but direct interactions between the target molecule and the core autophagy machinery are also observed.
A molecule convenient to link a process with its substrate needs to carry at least two distinct functional domains: one that recognizes the target and another that transports it to the site of operation. How does it work in the case of selective autophagy? The best known mechanism to solve the problem of distinction between the different cytoplasmic components deemed for engulfment is to bring properly marked cargos to the inner surface of the growing phagophore. Accordingly, the precise delivery is generally ensured by interaction of the adaptor both with the membrane-anchored form of Atg8/LC3 and the main targets that are usually polyubiquitinylated (Figure 4).
The first clues for the role of protein ubiquitinylation as a signal for selective autophagy came from Atg knockout mice and some Drosophila experiments. They showed that the loss of basal autophagy in the brain resulted in large-scale accumulation of ubiquitinylated proteins [38–40].
Recognition of ubiquitinylated proteins during autophagy is mediated by ubiquitin receptors interacting with ubiquitin noncovalently, via their ubiquitin-binding domains. p62/SQSMT1 (hereafter p62), the first protein reported to have such an adaptor function [41], was originally discovered as a scaffold in signaling pathways regulating cell growth and proliferation; however, it was also detected in ubiquitinylated protein aggregates [42] (Figure 4). p62 possesses a C-terminal ubiquitin-binding domain (UBA) [43] and a short LIR (LC3-interacting region) sequence responsible for LC3 interaction [41]. In addition, it has a PB1 domain promoting self-aggregation and association with other adaptors such as NBR1, neighbour of BRCA1 gene 1 [15] (Figure 5). Knockout studies in mice and Drosophila revealed that p62 is required for the aggregation of ubiquitinylated proteins and thus plays essential roles for their autophagic clearance [44, 45]. The levels of p62 usually inversely correlate with autophagic degradation, as the loss of Atg genes or factors required for the fusion of autophagosomes with lysosomes all result in a marked increase of p62-positive aggregates [46, 47]. p62 can also deliver ubiquitinylated cargos to the proteasome, although they are mainly degraded by autophagy [48, 49].
Figure 5.
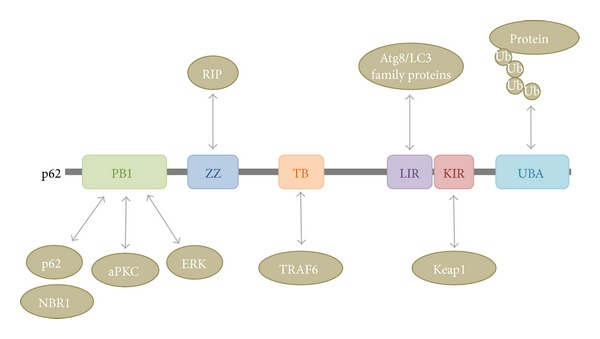
Domain structure of p62 and its interacting partners. There are six main domains/motifs in the p62 protein, necessary for its interaction with the autophagic machinery and with signaling pathways. The N-terminal Phox and Bem1 (PB1, 21-103 aa) domain is involved in the self-oligomerization of p62 or in heterodimerization with NBR1, a protein similar to p62. The PB1 domain is also responsible for the binding to atypical PKC (aPKC) or to ERK1. The central zinc finger ZZ domain (128-163 aa) and the TRAF6-binding domain (TB, 225-250 aa) interact with the RIP and TRAF6 proteins, respectively, to regulate the NF-κB pathway. Through the LC3-interacting region (LIR, 321-345 aa) and the C-terminal ubiquitin-associated domain (UBA, 386-440 aa), p62 links the autophagic machinery to ubiquitinylated protein substrates to promote the selective degradation of these molecules. Finally, the Keap-interacting region (KIR, 346-359 aa) binds Keap1 leading to stabilization and nuclear translocation of the transcription factor Nrf2, engaged in the control of ROS level.
Another adaptor used in selective autophagy is the above-mentioned NBR1, which, via its own PB1 domain, is able to interact with p62, and through its own UBA domain and LIR it can participate in the recruitment and autophagosomal degradation of ubiquitinylated proteins [50]. In plants, a functional hybrid homologue of p62 and NBR1 (NBR1 in Arabidopsis, Joka2 in tobacco) plays an important role in the disposal of polyubiquitinylated proteins accumulated under abiotic stress conditions [51, 52].
Optineurin and NDP52 have been recently described as xenophagy receptors, utilizing the autophagic machinery for restriction of ubiquitinylated intracellular pathogens [53]. Both of them also participate in the clearance of protein aggregates [54, 55] and are required for the regulation of NF-κB signaling [56, 57].
While these receptors all mediate degradation of ubiquitinylated cargos, there are other more specific adaptors acting on removal of damaged or surplus mitochondria (e.g., Atg32 in yeast and NIX in mammals) or peroxisomes (such as Atg30 and Atg36). They recognize particular binding partners on the surface of their target organelle and, through their LIR sequence, ensure their delivery to the maturing autophagosome [58, 59]. It is worth noting that additional autophagic adaptors may be identified by software prediction of LIR sequences in suspected protein candidates [60] (see a recent review for more details on the structural basis of how the Atg8/LC3 and Atg12 Ubls interact with specific autophagy adaptors [21]).
4.2.1. Role of p62 in Autophagosome Formation
As individual p62-ubiquitin interactions are rather weak, the starting point of the polyubiquitinylated aggregate formation is presumably the p62 self-oligomerization via its PB1 domain [61]. However, the original “simple” concept of delivery through bridging the polyubiquitin side chain on the cargo and the Atg8/LC3 decoration on the phagophore surface by p62 is now changing. In fact, these aggregates containing p62 and ubiquitinylated proteins may even serve as a nucleating scaffold for autophagosome biogenesis, potentially by binding multiple Atg proteins [61–63].
Moreover, it was recently reported that phagophores may preferentially form at p62 aggregates near lysosomes in Drosophila cells, which is very similar to the location of PAS near the vacuole/lysosome in yeast [64, 65]. It is worth noting that p62 also associates with MTORC1 [66]. MTORC1 is active when bound to lysosomes and promotes cell growth and inhibits autophagy by phosphorylating Atg1 (ULK1/2) [67–69]. These data suggest the direct assembly of early autophagic structures on the surface of protein aggregates, which may be mediated by interactions between p62 and upstream Atg proteins. Later on, Atg8/LC3 will be recruited to the forming phagophore, and the growing double membrane will enclose the p62-containing aggregate due to interactions between p62, Atg8/LC3, and other Atg proteins [70, 71].
4.2.2. p62 in Autophagy Regulation
The role of p62 in the regulation of autophagy is controversial. It was suggested to promote MTORC1 activation by contributing to its translocation to the lysosomal surface. Therefore, p62 reduction, similarly to MTORC1 inactivation, may activate autophagy [72]. However, in HEK293 and HeLa cells p62 was suggested to liberate Beclin1 (an Atg6 homologue) by disrupting the association of Bcl-2 and Beclin1, and thus p62 may positively regulate the induction of bulk autophagy [73]. In addition, p62 interacts with and regulates the deacetylase activity of HDAC6, a known modifier of F-actin network involved in selective autophagy [74]. In carcinoma cells, while p62 silencing suppressed cell proliferation and induced autophagy, abnormal autophagosomes were found and p62 inhibition finally resulted in autophagic cell death [75]. We have recently found that p62 is not required for proteasome inhibition-induced autophagy in Drosophila fat body cells [76]. Thus, the role of p62 in autophagy induction appears to be complex and probably context-dependent.
As p62 can shuttle between the nucleus and the cytoplasm (in the nucleus it is thought to recruit proteasomes to nuclear polyubiquitinylated protein aggregates), it can even export ubiquitinylated substrates from the nucleus into the cytosol, where autophagy offers a more robust degradative capacity [77].
4.2.3. Cytoplasmic p62 Level as an Autophagy Indicator
Since p62 itself is removed from the cytoplasm mainly by autophagy, its amount is generally considered to inversely correlate with autophagic activity [46, 47]. Accumulation of p62-positive inclusions during immunocytochemistry or elevated p62 levels on Western blots are frequently used as signs of autophagy impairment. In some cases, transgenic p62 reporter systems are also used to monitor the rate of autophagic degradation, although their use requires caution as overexpressed p62 tends to self-aggregate and may no longer indicate autophagy activity [78]. In addition, long term starvation may positively influence the amount of p62 in certain mammalian cell types, via both its transcriptional upregulation and promoting de novo p62 protein synthesis by providing autophagy-derived amino acids [49].
5. Interplay between p62 and Signaling Pathways
p62 was originally described as a scaffold protein ensuring the formation of signaling hubs, since, through different binding domains, it can establish interactions with many types of enzymes. As a consequence, it is able to integrate signaling routes involving particular kinases and ubiquitin-mediated pathways (Figure 5). This way, p62 regulates inflammatory processes in TNFα-activated cells. The complex including the RIP kinase, atypical PKCs and TRAF6, and a K63 ubiquitin ligase (interactions formed through the ZZ, PB1, and TB domain of p62, resp.) plays a critical role in the phosphorylation of IKKβ leading to activation of the NF-κB transcription factor [79]. Enhanced p62 level (under inflammatory conditions induced by impaired proteasomal degradation) was demonstrated to contribute to elevated IL-1β production: p62 was found to bind the JNK and ERK kinases, hence further increasing NF-κB activation and, as a consequence, pro-IL-1β expression. In addition, p62 accumulation was found to promote caspase-1 activation in inflammasomes, which is required for IL-1β proteolytic processing [80]. Interestingly, an opposite effect of p62 is suggested in Legionella-infected p62-deficient mice that showed more severe pulmonary inflammation than control animals, because the production and secretion of IL-1β was significantly enhanced due to elevated caspase-1 activity in their macrophages [81].
p62, likewise in association with TRAF6 and aPKCs, is needed for the NF-κB-mediated neuronal survival and differentiation in response to NGF [82] and also for osteoclastogenesis [83]. p62 mutations are among the genetic alterations that play a role in Paget disease of bone, where osteoclasts are overactive because of disturbed NF-κB signalization [84]. The p62-NF-κB connection has a role in tumorigenesis as well, since p62 is necessary to NF-κB-dependent survival in Ras-transformed cells [85].
The autophagy adaptor function of p62 also has an impact on the NF-κB signaling pathway. In human monocytes, high level of inflammation due to autophagy impairment is associated with p62 accumulation and the consequent overactivation of the NF-κB pathway [86]. In accordance with the positive role of p62 in caspase-1 activation [80], a previous study demonstrated that stimulated autophagy, by enhanced degradation of p62, also eliminates activated inflammasomes and reduces inflammation, while blocking autophagy has an opposite effect [87]. In addition, NF-κB signalization may be regulated directly by the rate of NF-κB removal. Targeted degradation of the p62-NF-κB p65 subunit complex by p62-mediated selective autophagy may play a key role in bone marrow derived macrophage differentiation [88].
The important role of p62 in innate immunity does not only rely on regulation of immune signaling responses. As an autophagy adaptor, p62 takes part in the elimination of ubiquitinylated intracellular pathogens; some infecting agents even target this step to escape from the defensive system of the cell. The coxsackievirus B3, through the activity of one of its proteases, cleaves p62 which results in impairment of selective autophagy and host defense [89]. Moreover, selective autophagy induced by pathogen-specific TLR4 activation requires transcriptional upregulation of p62 [90]. Interestingly, p62 also participates in the synthesis of neoantimicrobial peptides, by bringing inactive precursors such as Fau to autophagic degradation, where they are processed to active fragments [91].
p62 is also involved in the regulation of apoptosis. p62-mediated aggregation is needed for the activation of polyubiquitinated caspase-8 [92]. It was shown recently that caspase-8 colocalizes not only with p62, but also with Atg8/LC3 and Atg5, and its full self-processing requires the autophagosomal membrane as a platform for the assembly of the death-inducing signaling complex [93]. On the other hand, failure of autophagy may contribute to enhanced apoptosis because of impaired degradation of p62-complexed apoptosis proteins, as found in T-cells [94], while in autophagy-inhibited cancer cells, caspase-8 dependent cell death was mainly associated with the concomitantly elevated p62 level [95].
Another well-known signaling pathway influenced by p62 is the oxidative stress response, which is regulated by the Keap1-Nrf2 system. Through its KIR motif (Figure 5), p62 is able to bind to Keap1, a Cullin3-ubiquitin E3 ligase complex adaptor protein. In turn, Keap1-promoted polyubiquitinylation and subsequent proteasomal degradation of the transcription factor Nrf2 are inhibited. As a consequence, the expression of cytoprotective, antioxidant Nrf2 target genes is increased [96, 97]. Moreover, the p62 gene itself is a target for Nrf2; thus, the appropriate oxidative stress response is supported by a positive feedback regulation between p62 and Nrf2 [98]. Autophagy has a strong impact on Nrf2 activation, since p62 not only disrupts Keap1-Nrf2 interaction but also removes Keap1 from the cytosol via selective autophagy [99]. The well-known antioxidant effect of sestrins is, at least partly, due to their influence on the p62-dependent autophagic degradation of Keap1 [100]. In case of autophagy impairment, accumulation of p62 and the subsequent overactivation of Nrf2 may contribute to development of liver carcinomas [96]. Interestingly, in these cancer cells, phosphorylation of p62 by the MTORC1 complex increases its affinity for Keap1, so MTORC1 activity further enhances stabilization of Nrf2 and the transcription of its target genes [101].
6. Conclusions
Ubiquitin and ubiquitin-like proteins (Ubl) share functional similarity. The different Ubls are activated and conjugated to substrates by similar biochemical mechanisms.
Ubiquitinylation is frequently needed for substrate recognition and renders selectivity to autophagy in eukaryotes.
The connection between ubiquitinylation and autophagy is provided by autophagic adaptor proteins (or autophagy receptors), which bind both ubiquitin and autophagy specific Ubl modifiers like Atg8/LC3 family proteins.
Atg8/LC3 is required for the biogenesis of autophagosomal membrane and also mediates selective autophagy via the recruitment of LIR-containing autophagy receptors that recognize and select cargo.
Autophagy receptors such as p62 regulate the selective autophagosomal degradation of large protein aggregates, mitochondria, and bacterial pathogens.
p62 may play an important role also as a regulator of autophagy; moreover, it may even be involved in the formation of the autophagosome.
As a scaffold protein, p62 operates in signaling pathways which, through the link provided by p62, can also be regulated by selective autophagy.
Acknowledgments
The authors thank Vilmos Tóth for his excellent assistance in completing Figure 3. They apologize to the investigators whose works are not cited here.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Wolf DH. Proteasomes: a historical retrospective. In: Hilt W, Wolf DH, editors. Proteasomes: The World of Regulatory Proteolysis. Madame Curie Bioscience Database: Landes Bioscience; 2000. pp. 1–7. [Google Scholar]
- 2.Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Research. 2014;24(1):24–41. doi: 10.1038/cr.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Korolchuk VI, Menzies FM, Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Letters. 2010;584(7):1393–1398. doi: 10.1016/j.febslet.2009.12.047. [DOI] [PubMed] [Google Scholar]
- 4.Tai H-C, Schuman EM. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nature Reviews Neuroscience. 2008;9(11):826–838. doi: 10.1038/nrn2499. [DOI] [PubMed] [Google Scholar]
- 5.Huang L, Kinnucan E, Wang G, et al. Structure of an E6AP-UbcH7 complex: insights into ubiquitination by the E2-E3 enzyme cascade. Science. 1999;286(5443):1321–1326. doi: 10.1126/science.286.5443.1321. [DOI] [PubMed] [Google Scholar]
- 6.Huibregtse JM, Scheffner M, Beaudenon S, Howley PM. A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(7):2563–2567. doi: 10.1073/pnas.92.7.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nature Reviews Molecular Cell Biology. 2005;6(1):9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 8.Hochstrasser M. Lingering mysteries of ubiquitin-chain assembly. Cell. 2006;124(1):27–34. doi: 10.1016/j.cell.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 9.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. The EMBO Journal. 2000;19(1):94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Annual Review of Biochemistry. 1996;65:801–847. doi: 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- 11.Baumeister W, Walz J, Zühl F, Seemüller E. The proteasome: paradigm of a self-compartmentalizing protease. Cell. 1998;92(3):367–380. doi: 10.1016/s0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- 12.Murata S, Yashiroda H, Tanaka K. Molecular mechanisms of proteasome assembly. Nature Reviews Molecular Cell Biology. 2009;10(2):104–115. doi: 10.1038/nrm2630. [DOI] [PubMed] [Google Scholar]
- 13.Behrends C, Harper JW. Constructing and decoding unconventional ubiquitin chains. Nature Structural and Molecular Biology. 2011;18(5):520–528. doi: 10.1038/nsmb.2066. [DOI] [PubMed] [Google Scholar]
- 14.Welchman RL, Gordon C, Mayer RJ. Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nature Reviews Molecular Cell Biology. 2005;6(8):599–609. doi: 10.1038/nrm1700. [DOI] [PubMed] [Google Scholar]
- 15.Walinda E, Morimoto D, Sugase K, Konuma T, Tochio H, Shirakawa M. Solution atructure of the ubiquitin-associated (UBA) domain of human autophagy receptor NBR1 and its interaction with ubiquitin and polyubiquitin. Journal of Biological Chemistry. 2014;289(20):13890–13902. doi: 10.1074/jbc.M114.555441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hochstrasser M. Origin and function of ubiquitin-like proteins. Nature. 2009;458(7237):422–429. doi: 10.1038/nature07958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mulakkal N, Nagy P, Takáts S, Tusco R, Juhász G, Nezis IP. Autophagy in Drosophila: from historical studies to current knowledge. BioMed Research International. 2014;2014:24 pages. doi: 10.1155/2014/273473.273473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mizushima N, Yoshimori T, Ohsumi Y. The role of atg proteins in autophagosome formation. Annual Review of Cell and Developmental Biology. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 19.Érdi B, Nagy P, Zvara Á, et al. Loss of the starvation-induced gene Rack1 leads to glycogen deficiency and impaired autophagic responses in Drosophila . Autophagy. 2012;8(7):1124–1135. doi: 10.4161/auto.20069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim J, Kundu M, Viollet B, Guan K-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature Cell Biology. 2011;13(2):132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klionsky DJ, Schulman BA. Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nature Structural and Molecular Biology. 2014;21(4):336–345. doi: 10.1038/nsmb.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012;151(6):1256–1269. doi: 10.1016/j.cell.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 23.Hegedüs K, Takáts S, Kovács AL, Juhász G. Evolutionarily conserved role and physiological relevance of a STX17/Syx17 (syntaxin 17)-containing SNARE complex in autophagosome fusion with endosomes and lysosomes. Autophagy. 2013;9(10):1642–1646. doi: 10.4161/auto.25684. [DOI] [PubMed] [Google Scholar]
- 24.Takáts S, Nagy P, Varga Á, et al. Autophagosomal Syntaxin17-dependent lysosomal degradation maintains neuronal function in Drosophila . Journal of Cell Biology. 2013;201(4):531–539. doi: 10.1083/jcb.201211160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang P, Nishimura T, Sakamaki Y, et al. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Molecular Biology of the Cell. 2014;25(8):1327–1337. doi: 10.1091/mbc.E13-08-0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takats S, Pircs K, Nagy P, et al. Interaction of the HOPS complex with Syntaxin 17 mediates autophagosome clearance in Drosophila . Molecular Biology of the Cell. 2014;25(8):1338–1354. doi: 10.1091/mbc.E13-08-0449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Molecular Cell. 2009;34(3):259–269. doi: 10.1016/j.molcel.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 28.Johansen T, Lamark T. Selective autophagy goes exclusive. Nature Cell Biology. 2014;16(5):395–397. doi: 10.1038/ncb2961. [DOI] [PubMed] [Google Scholar]
- 29.Sawa-Makarska J, Abert C, Romanov J, Zens B, Ibiricu I, Martens S. Cargo binding to Atg19 unmasks additional Atg8 binding sites to mediate membrane-cargo apposition during selective autophagy. Nature Cell Biology. 2014;16(5):425–433. doi: 10.1038/ncb2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mizushima N, Noda T, Yoshimori T, et al. A protein conjugation system essential for autophagy. Nature. 1998;395(6700):395–398. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- 31.Kirisako T, Baba M, Ishihara N, et al. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. Journal of Cell Biology. 1999;147(2):435–446. doi: 10.1083/jcb.147.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ichimura Y, Kirisako T, Takao T, et al. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408(6811):488–492. doi: 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- 33.Dengjel J, Abeliovich H. Musical chairs during mitophagy. Autophagy. 2014;10(4):706–707. doi: 10.4161/auto.28150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lamark T, Johansen T. Aggrephagy: selective disposal of protein aggregates by macroautophagy. International Journal of Cell Biology. 2012;2012:21 pages. doi: 10.1155/2012/736905.736905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pareja ME, Colombo MI. Autophagic clearance of bacterial pathogens: molecular recognition of intracellular microorganisms. Frontiers in Cellular and Infection Microbiology. 2013;3, article 54 doi: 10.3389/fcimb.2013.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wileman T. Autophagy as a defence against intracellular pathogens. Essays in Biochemistry. 2013;55:153–163. doi: 10.1042/bse0550153. [DOI] [PubMed] [Google Scholar]
- 37.Till A, Lakhani R, Burnett SF, Subramani S. Pexophagy: the selective degradation of peroxisomes. International Journal of Cell Biology. 2012;2012:18 pages. doi: 10.1155/2012/512721.512721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441(7095):885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 39.Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441(7095):880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 40.Juhász G, Érdi B, Sass M, Neufeld TP. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila . Genes and Development. 2007;21(23):3061–3066. doi: 10.1101/gad.1600707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. Journal of Biological Chemistry. 2007;282(33):24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 42.Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137(6):1001–1004. doi: 10.1016/j.cell.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ciani B, Layfield R, Cavey JR, Sheppard PW, Searle MS. Structure of the ubiquitin-associated domain of p62 (SQSTM1) and implications for mutations that cause Paget’s disease of bone. Journal of Biological Chemistry. 2003;278(39):37409–37412. doi: 10.1074/jbc.M307416200. [DOI] [PubMed] [Google Scholar]
- 44.Komatsu M, Waguri S, Koike M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131(6):1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 45.Nezis IP, Simonsen A, Sagona AP, et al. Ref(2)P, the Drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain. Journal of Cell Biology. 2008;180(6):1065–1071. doi: 10.1083/jcb.200711108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bartlett BJ, Isakson P, Lewerenz J, et al. p62, Ref(2)P and ubiquitinated proteins are conserved markers of neuronal aging, aggregate formation and progressive autophagic defects. Autophagy. 2011;7(6):572–583. doi: 10.4161/auto.7.6.14943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy ,Autophagy. 2012;8(4):445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Myeku N, Figueiredo-Pereira ME. Dynamics of the degradation of ubiquitinated proteins by proteasomes and autophagy: association with sequestosome 1/p62. Journal of Biological Chemistry. 2011;286(25):22426–22440. doi: 10.1074/jbc.M110.149252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sahani MH, Itakura E, Mizushima N. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy. 2014;10(3) doi: 10.4161/auto.27344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lamark T, Perander M, Outzen H, et al. Interaction codes within the family of mammalian Phox and Bem1p domain-containing proteins. Journal of Biological Chemistry. 2003;278(36):34568–34581. doi: 10.1074/jbc.M303221200. [DOI] [PubMed] [Google Scholar]
- 51.Zhou J, Wang J, Cheng Y, et al. NBR1-mediated selective autophagy targets insoluble ubiquitinated protein aggregates in plant stress responses. PLoS Genetics. 2013;9(1) doi: 10.1371/journal.pgen.1003196.e1003196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zientara-Rytter K, Sirko A. Significant role of PB1 and UBA domains in multimerization of Joka2, a selective autophagy cargo receptor from tobacco. Frontiers in Plant Science. 2014;5, article 13 doi: 10.3389/fpls.2014.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boyle KB, Randow F. The role of “eat-me” signals and autophagy cargo receptors in innate immunity. Current Opinion in Microbiology. 2013;16(3):339–348. doi: 10.1016/j.mib.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 54.Korac J, Schaeffer V, Kovacevic I, et al. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. Journal of Cell Science. 2013;126(2):580–592. doi: 10.1242/jcs.114926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jo C, Gundemir S, Pritchard S, Jin YN, Rahman I, Johnson GV. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nature Communications. 2014;5, article 3496 doi: 10.1038/ncomms4496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhu G, Wu C-J, Zhao Y, Ashwell JD. Optineurin negatively regulates TNFα-induced NF-κB activation by competing with NEMO for ubiquitinated RIP. Current Biology. 2007;17(16):1438–1443. doi: 10.1016/j.cub.2007.07.041. [DOI] [PubMed] [Google Scholar]
- 57.Inomata M, Niida S, Shibata K-I, Into T. Regulation of toll-like receptor signaling by NDP52-mediated selective autophagy is normally inactivated by A20. Cellular and Molecular Life Sciences. 2012;69(6):963–979. doi: 10.1007/s00018-011-0819-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Novak I. Mitophagy: a complex mechanism of mitochondrial removal. Antioxidants and Redox Signaling. 2012;17(5):794–802. doi: 10.1089/ars.2011.4407. [DOI] [PubMed] [Google Scholar]
- 59.Farré J-C, Burkenroad A, Burnett SF, Subramani S. Phosphorylation of mitophagy and pexophagy receptors coordinates their interaction with Atg8 and Atg11. EMBO Reports. 2013;14(5):441–449. doi: 10.1038/embor.2013.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kalvari I, Tsompanis S, Mulakkal NC, et al. iLIR: a web resource for prediction of Atg8-family interacting proteins. Autophagy. 2014;10(5):913–925. doi: 10.4161/auto.28260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rogov V, Dotsch V, Johansen T, Kirkin V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Molecular Cell. 2014;53(2):167–178. doi: 10.1016/j.molcel.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 62.Fujita N, Morita E, Itoh T, et al. Recruitment of the autophagic machinery to endosomes during infection is mediated by ubiquitin. Journal of Cell Biology. 2013;203(1):115–128. doi: 10.1083/jcb.201304188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nagy P, Hegedus K, Pircs K, Varga A, Juhasz G. Different effects of Atg2 and Atg18 mutations on Atg8a and Atg9 trafficking during starvation in Drosophila . FEBS Letters. 2014;588(3):408–413. doi: 10.1016/j.febslet.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Scott SV, Nice DC, III, Nau JJ, et al. Apg13p and Vac8p are part of a complex of phosphoproteins that are required for Cytoplasm to vacuole targeting. Journal of Biological Chemistry. 2000;275(33):25840–25849. doi: 10.1074/jbc.M002813200. [DOI] [PubMed] [Google Scholar]
- 65.Nagy P, Karpati M, Varga A, et al. Atg17/FIP200 localizes to perilysosomal Ref(2)P aggregates and promotes autophagy by activation of Atg1 in Drosophila . Autophagy. 2014;10(3):453–467. doi: 10.4161/auto.27442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moscat J, Diaz-Meco MT. P62: a versatile multitasker takes on cancer. Trends in Biochemical Sciences. 2012;37(6):230–236. doi: 10.1016/j.tibs.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hosokawa N, Hara T, Kaizuka T, et al. Nutrient-dependent mTORCl association with the ULK1-Atg13-FIP200 complex required for autophagy. Molecular Biology of the Cell. 2009;20(7):1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141(2):290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Juhász G. Interpretation of bafilomycin, pH neutralizing or protease inhibitor treatments in autophagic flux experiments: novel considerations. Autophagy. 2012;8(12):1875–1876. doi: 10.4161/auto.21544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Alemu EA, Lamark T, Torgersen KM, et al. ATG8 family proteins act as scaffolds for assembly of the ULK complex: sequence requirements for LC3-interacting region (LIR) motifs. Journal of Biological Chemistry. 2012;287(47):39275–39290. doi: 10.1074/jbc.M112.378109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hegedüs K, Nagy P, Gáspári Z, Juhász G. The putative HORMA domain protein Atg101 dimerizes and is required for starvation-induced and selective autophagy in Drosophila . BioMed Research International. 2014;2014:13 pages. doi: 10.1155/2014/470482.470482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Duran A, Amanchy R, Linares JF, et al. P62 is a key regulator of nutrient sensing in the mTORC1 pathway. Molecular Cell. 2011;44(1):134–146. doi: 10.1016/j.molcel.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou L, Wang H-F, Ren H-G, et al. Bcl-2-dependent upregulation of autophagy by sequestosome 1/p62 in vitro. Acta Pharmacologica Sinica. 2013;34(5):651–656. doi: 10.1038/aps.2013.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yan J, Seibenhener ML, Calderilla-Barbosa L, et al. SQSTM1/p62 interacts with HDAC6 and regulates deacetylase activity. PLoS ONE. 2013;8(9) doi: 10.1371/journal.pone.0076016.e76016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nihira K, Miki Y, Ono K, Suzuki T, Sasano H. An inhibition of p62/SQSTM1 caused autophagic cell death of several human carcinoma cells. Cancer Science. 2014;105(5):568–575. doi: 10.1111/cas.12396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Low P, Varga Á, Pircs K, et al. Impaired proteasomal degradation enhances autophagy via hypoxia signaling in Drosophila . BMC Cell Biology. 2013;14(1, article 29) doi: 10.1186/1471-2121-14-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pankiv S, Lamark T, Bruun J-A, Øvervatn A, Bjørkøy G, Johansen T. Nucleocytoplasmic shuttling of p62/SQSTM1 and its role in recruitment of nuclear polyubiquitinated proteins to promyelocytic leukemia bodies. Journal of Biological Chemistry. 2010;285(8):5941–5953. doi: 10.1074/jbc.M109.039925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pircs K, Nagy P, Varga A, et al. Advantages and limitations of different p62-based assays for estimating autophagic activity in Drosophila . PLoS ONE. 2012;7(8) doi: 10.1371/journal.pone.0044214.e44214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sanz L, Sanchez P, Lallena M-J, Diaz-Meco MT, Moscat J. The interaction of p62 with RIP links the atypical PKCs to NF-κB activation. The EMBO Journal. 1999;18(11):3044–3053. doi: 10.1093/emboj/18.11.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Choe JY, Jung HY, Park KY, Kim SK. Enhanced p62 expression through impaired proteasomal degradation is involved in caspase-1 activation in monosodium urate crystal-induced interleukin-1beta expression. Rheumatology. 2014;53(6):1043–1053. doi: 10.1093/rheumatology/ket474. [DOI] [PubMed] [Google Scholar]
- 81.Ohtsuka S, Ishii Y, Matsuyama M, et al. SQSTM1/p62/A170 regulates the severity of Legionella pneumophila pneumonia by modulating inflammasome activity. European Journal of Immunology. 2014;44(4):1084–1092. doi: 10.1002/eji.201344091. [DOI] [PubMed] [Google Scholar]
- 82.Wooten MW, Seibenhener ML, Mamidipudi V, Diaz-Meco MT, Barker PA, Moscat J. The atypical protein kinase C-interacting protein p62 is a Scaffold for NF-κB activation by nerve growth factor. Journal of Biological Chemistry. 2001;276(11):7709–7712. doi: 10.1074/jbc.C000869200. [DOI] [PubMed] [Google Scholar]
- 83.Durán A, Serrano M, Leitges M, et al. The atypical PKC-interacting protein p62 is an important mediator of RANK-activated osteoclastogenesis. Developmental Cell. 2004;6(2):303–309. doi: 10.1016/s1534-5807(03)00403-9. [DOI] [PubMed] [Google Scholar]
- 84.Laurin N, Brown JP, Morissette J, Raymond V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in paget disease of bone. The American Journal of Human Genetics. 2002;70(6):1582–1588. doi: 10.1086/340731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Duran A, Linares JF, Galvez AS, et al. The signaling adaptor p62 is an important NF-κB mediator in tumorigenesis. Cancer Cell. 2008;13(4):343–354. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 86.Takeda-Watanabe A, Kitada M, Kanasaki K, Koya D. SIRT1 inactivation induces inflammation through the dysregulation of autophagy in human THP-1 cells. Biochemical and Biophysical Research Communications. 2012;427(1):191–196. doi: 10.1016/j.bbrc.2012.09.042. [DOI] [PubMed] [Google Scholar]
- 87.Shi C-S, Shenderov K, Huang N-N, et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nature Immunology. 2012;13(3):255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chang C-P, Su Y-C, Hu C-W, Lei H-Y. TLR2-dependent selective autophagy regulates NF-κB lysosomal degradation in hepatoma-derived M2 macrophage differentiation. Cell Death and Differentiation. 2013;20(3):515–523. doi: 10.1038/cdd.2012.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shi J, Wong J, Piesik P, et al. Cleavage of sequestosome 1/p62 by an enteroviral protease results in disrupted selective autophagy and impaired NFKB signaling. Autophagy. 2013;9(10):1591–1603. doi: 10.4161/auto.26059. [DOI] [PubMed] [Google Scholar]
- 90.Fujita K-I, Srinivasula SM. TLR4-mediated autophagy in macrophages is a p62-dependent type of selective autophagy of aggresome-like induced structures (ALIS) Autophagy. 2011;7(5):552–554. doi: 10.4161/auto.7.5.15101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ponpuak M, Davis AS, Roberts EA, et al. Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity. 2010;32(3):329–341. doi: 10.1016/j.immuni.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jin Z, Li Y, Pitti R, et al. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell. 2009;137(4):721–735. doi: 10.1016/j.cell.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 93.Young MM, Takahashi Y, Khan O, et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. Journal of Biological Chemistry. 2012;287(15):12455–12468. doi: 10.1074/jbc.M111.309104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kovacs JR, Li C, Yang Q, et al. Autophagy promotes T-cell survival through degradation of proteins of the cell death machinery. Cell Death and Differentiation. 2012;19(1):144–152. doi: 10.1038/cdd.2011.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Huang S, Okamoto K, Yu C, Sinicrope FA. p62/sequestosome-1 up-regulation promotes ABT-263-induced caspase-8 aggregation/activation on the autophagosome. Journal of Biological Chemistry. 2013;288(47):33654–33666. doi: 10.1074/jbc.M113.518134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Komatsu M, Kurokawa H, Waguri S, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nature Cell Biology. 2010;12(3):213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 97.Jain A, Lamark T, Sjøttem E, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. Journal of Biological Chemistry. 2010;285(29):22576–22591. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ishii T, Itoh K, Takahashi S, et al. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. Journal of Biological Chemistry. 2000;275(21):16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- 99.Taguchi K, Fujikawa N, Komatsu M, et al. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(34):13561–13566. doi: 10.1073/pnas.1121572109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bae SH, Sung SH, Oh SY, et al. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of keap1 and prevent oxidative liver damage. Cell Metabolism. 2013;17(1):73–84. doi: 10.1016/j.cmet.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 101.Ichimura Y, Waguri S, Sou Y-S, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Molecular Cell. 2013;51(5):618–631. doi: 10.1016/j.molcel.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 102.Nalepa G, Rolfe M, Harper JW. Drug discovery in the ubiquitin: proteasome system. Nature Reviews Drug Discovery. 2006;5(7):596–613. doi: 10.1038/nrd2056. [DOI] [PubMed] [Google Scholar]
- 103.Ravid T, Hochstrasser M. Diversity of degradation signals in the ubiquitin-proteasome system. Nature Reviews Molecular Cell Biology. 2008;9(9):679–689. doi: 10.1038/nrm2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tully SP, Stitt TM, Caldwell RD, Hardock BJ, Hanson RM, Maslak P. Interactive web-based pointillist visualization of hydrogenic orbitals using jmol. Journal of Chemical Education. 2013;90(1):129–131. [Google Scholar]
- 105.Vijay-kumar S, Bugg CE, Cook WJ. Structure of ubiquitin refined at 1.8 Å resolution. Journal of Molecular Biology. 1987;194(3):531–544. doi: 10.1016/0022-2836(87)90679-6. [DOI] [PubMed] [Google Scholar]
- 106.Otomo C, Metlagel Z, Takaesu G, Otomo T. Structure of the human ATG12∼ATG5 conjugate required for LC3 lipidation in autophagy. Nature Structural and Molecular Biology. 2013;20(1):59–66. doi: 10.1038/nsmb.2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sugawara K, Suzuki NN, Fujioka Y, Mizushima N, Ohsumi Y, Inagaki F. The crystal structure of microtubule-associated protein light chain 3, a mammalian homologue of Saccharomyces cerevisiae Atg8. Genes to Cells. 2004;9(7):611–618. doi: 10.1111/j.1356-9597.2004.00750.x. [DOI] [PubMed] [Google Scholar]