

Abstract
Purpose
In order to investigate Poly (lactic-co-glycolic acid) (PLGA) nanoparticles (NP) as potential vehicles for efficient tumor antigen (TA) delivery to dendritic cells (DC), this study aimed to optimize encapsulation/release kinetics before determining immunogenicity of antigen-containing NP.
Methods
Various techniques were used to liberate TA from cell lines. Single (gp100) and multiple (B16-tumor lysate containing gp100) antigens were encapsulated within differing molecular weight PLGA co-polymers. Differences in morphology, encapsulation/release and biologic potency were studied. Findings were adopted to encapsulate fresh tumor lysate from patients with advanced tumors and compare stimulation of tumor infiltrating lymphocytes (TIL) against that achieved by soluble lysate.
Results
Four cycles of freeze-thaw + 15 s sonication resulted in antigen-rich lysates without the need for toxic detergents or protease inhibitors. The 80KDa polymer resulted in maximal release of payload and favorable production of immunostimulatory IL-2 and IFN-γ. NP-mediated antigen delivery led to increased IFN-γ and decreased immunoinhibitory IL-10 synthesis when compared to soluble lysate.
Conclusions
Four cycles of freeze-thaw followed by 15 s sonication is the ideal technique to obtain complex TA for encapsulation. The 80KDa polymer has the most promising combination of release kinetics and biologic potency. Encapsulated antigens are immunogenic and evoke favorable TIL-mediated anti-tumor responses.
Keywords: antigen delivery, dendritic cell, immunotherapy, molecular weight, nanoparticles
INTRODUCTION
Efficient delivery of tumor antigens (TA) is a major challenge facing dendritic cell (DC)-based immunotherapy for many solid organ malignancies like malignant melanoma and head and neck squamous cell carcinoma (HNSCC). One option to achieve this is by using the inherent advantages of micro- or nano-particles (MP/NP) made from poly (lactic-co-glycolic acid) (PLGA) (1–3). By selecting the appropriate polymer composition with a known rate of degradation, MP/NP can be exploited to produce a delivery system that releases an active agent at a predetermined rate (4,5). Release rate depends on particle properties such as PLGA composition (lactide: glycolide ratio), loading, size, porosity and ultrastructure (6,7). An additional factor of importance is the polymer molecular weight (MW). This is crucial because it alters hydrophilicity, diffusion and degradation via non-enzymatic autocatalysis. Moreover it affects the viscosity of polymer solution and sol–gel phase inversion induced by precipitation following solvent removal, both of which have a bearing on release rates of encapsulated payload (8,9). It is therefore important to evaluate and define the influence of polymer MW on delivery of either single, or multiple TA. While there have been isolated reports of encapsulation of cellular material (10,11), only one study has so far investigated the role of polymer MW on encapsulation and in vitro release of complex mixture of TA (12). However it did not evaluate the impact of MW on extent of DC-mediated antigen cross-presentation to cytolytic T cells (CTL), something to consider when using PLGA polymers as antigen delivery vehicles.
Stability of encapsulated material is key to the quality and magnitude of immune responses generated by particulate-based vaccine delivery systems (13,14). When fabricating NP/MP using the double emulsion and solvent evaporation technique, TA are exposed to a range of chemical and physical insults like excess heat (15), high speed homogenization-induced shearing forces and organic solvents (16). These can result in denaturation, aggregation, adsorption, hydrolysis, oxidation, β-elimination, racemization or deamidation (17,18), each of which could adversely affect their potency. Concerns about antigen stability have led to modifications in polymer particle fabrication like using poly-D,L-lactide-poly (ethylene glycol) (PELA) instead of PLGA, incorporating protein stabilizers to shield antigens from organic solvents at the water/oil interface (19) and coupling cationic amphiphile molecules with encapsulation targets (20). It is therefore important to confirm the stability and immunogenicity of encapsulated material when considering this technology in anti-tumor immunotherapy. While there have been earlier attempts at evaluating single TA containing-NP (15,16), encapsulation and release of mixture of TA have only infrequently been studied. In an earlier study, our group reported that encapsulated melanoma antigens loaded onto C57BL/6 mice derived- bone marrow DC (BMDC) resulted in greater T helper (Th)-1-type responses when compared to soluble lysates (12). But the delivery of patient-derived TA has not been fully evaluated underscoring the importance of obtaining additional information regarding polymer composition and stability.
As part of ongoing efforts to develop a PLGA-based anti-tumor DC vaccination protocol, we herein report our experience with in vitro evaluation of encapsulated TA derived from a variety of sources. We hypothesized that: a) PLGA MW influenced downstream biological activity of NP and b) by using optimal techniques to generate and then encapsulate whole tumor lysates, TA could efficiently be delivered to DC for cross-presentation to CTL. The aims of the study were to: (i) improve antigen stability and potency during encapsulation by attempting to increase antigen concentration within tumor lysates, (ii) identify the PLGA formulation that resulted in favorable encapsulation and release of both single and multiple TA and (iii) compare immunostimulatory capacity of various formulations utilizing bonafide murine and human T-cell readouts. A number of techniques were initially used to solubilize HNSCC lines. The intention was to enhance antigens within the lysate and adopt the clinically compatible technique consistently resulting in maximal release of TA from a finite amount of cellular material. TA were then encapsulated within differing MW of 50:50 PLGA polymers using a double emulsion solvent evaporation method. The physico-chemical characteristics, in vitro controlled release profile and differences in biological activity of these formulations were evaluated. Using naïve tumor-specific CD8+ T cells derived from transgenic mice, we measured NP potency through DC-based cross-presentation of encapsulated antigen (s). Our observations suggest that PLGA MW could influence this phenomenon. Fresh samples from patients with advanced tumors were then employed in similar experiments. Cytokine profile of autologous tumor infiltrating lymphocytes (TIL) stimulated by DC loaded with TA contained within either PLGA NP or soluble tumor lysates were compared. The findings indicate the potency of encapsulated TA and represent a critical step forward in the development of PLGA-based reagents for tumor immunotherapy.
MATERIALS AND METHODS
Antigen Encapsulation Procedures
Individual Antigen
gp100 conjugated to myelin basic protein (MBP) (MBP-gp100; a kind gift of the Rothberg Institute for Childhood Diseases, Guilford, CT, USA) was concentrated on YM-10 Ultracel YM membrane Microcon filtration units (Millipore Corporation, Billerica, MA, USA) by centrifugation at 12000 rpm for 30 min at 4°C to attain a final concentration of 10 μg/μl. Recombinant gp100 protein was also spiked (5% w/w) into B16 murine melanoma cell lysates prepared from freshly excised tumors as described below.
Cell Lines
Cells from FaDu and FAT7 (HNSCC) cell lines [American Type Culture Collection (ATCC), Manassas, VA, USA] were grown to more than 90% confluence in monolayers in ATCC recommended media in a humidified atmosphere containing 5% CO2. FaDu was grown in 1% sodium pyruvate (Sigma-Aldrich, St. Louis, MO, USA) + Eagle’s Minimal Essential Medium + 10% fetal bovine serum + 1% non-essential amino acids + 1.5 mg/ml sodium bicarbonate + 1% penicillin/streptomycin (all reagents from Invitrogen, Carlsbad, CA, USA). FAT7 cells were grown using Ham’s F12K medium + 10% fetal bovine serum (non-heat inactivated) + 1% penicillin/streptomycin (all reagents from Invitrogen, Carls-bad, CA, USA) + 0.1% insulin-HEPES + 0.5% hydrocortisone (both from Sigma-Aldrich, St. Louis, MO, USA) and 0.5% Transferrin (Calbiochem, San Diego, CA, USA). These served as sources of TA for the initial set of experiments aimed at enhancing stability of antigens.
Determination of Optimal Lysate Generation Technique
Confluent cells from FAT 7 and FaDu lines were re-suspended in ice cold PBS at concentrations ranging from 2–4×108/ml. In order to maximize release of protein antigens from cells and thus increase concentration of the lysate used for encapsulation, cell suspensions were converted into lysates using different techniques and compared. To alleviate the need for additional (cytotoxic) protease inhibitors, cell solutions were maintained on ice prior to and during lysate preparation. Methods screened initially included: Freeze-thaw (four cycles of alternating liquid nitrogen and 37°C water bath treatment) (F/T) alone, sonication (Ultrasonic Processor, Tekmar, Cincinnati, OH, USA) (S) for 60 s on ice at 38% amplitude, F/T + S for 15 s and F/T + Dounce homogenization (DH). DH involves disruption of cultured cells by forcing the cell suspension through a narrow space causing shearing of cell membranes. This is done using a Dounce homogenizer which consists of a round glass pestle that is manually driven into a glass tube. In all instances, cell death and lysis were confirmed by trypan blue exclusion. Lysates were initially spun at 12000 rpm for 20 min at 4°C to remove cellular debris and supernatants collected. These were either used immediately or stored at −20°C for future assays. Protein content of lysate preparations was measured by the standard test tube procedure using the Bicinchonic acid (BCA) protein assay kit (Thermo Scientific, Rockford, IL, USA) as per the manufacturer’s protocol. Absorbance at 562 nm was measured using a spectrophotometer and protein concentrations determined by comparing absorbance readings to a standard calibration curve of bovine serum albumin (BSA) in the concentration range between 5 μg/ml and 250 μg/ml.
Differences in protein content of selected lysates were also evaluated by Western Blot (WB) analysis and Coomassie blue staining. Samples were run on a sodium dodecyl sulfate poly-acrylamide gel electrophoresis (SDS-PAGE) mini gel system (Pre-cast 8–16% Tris–HCl 10-well, Jule Inc, CT, USA) @ 30–35 mA per gel before being transferred to nitrocellulose membranes (0.2 μm, Invitrogen, Carlsbad, CA, USA). Rainbow™ coloured protein molecular weight (MW) marker (Amersham Life Science, Amersham, Buckinghamshire, England) was used to determine the relative molecular weights of the proteins. WB was performed to detect HNSCC-associated protein p53. This was done using a combination of polyclonal goat anti-p53 antibody (AF1355, R&D Systems, Minneapolis, MN, USA) and horse radish peroxidase-conjugated donkey anti-goat immunoglobulin (Ig) (HAF 109, R&D Systems, Minneapolis, MN, USA) before detection by the enhanced chemiluminescence method (GE Healthcare, Amersham, Buckinghamshire, U.K.). Differences in intensity of bands served as indirect indicators of differences in antigen content of lysates. The spectrum and intensity of proteins released by various techniques were further evaluated by Coomassie blue staining. Membranes were placed in a staining jar containing Coomasie stain solution (0.2% Brilliant Blue R250 in 20% methanol) for 60 min. The stain solution was discarded before briefly rinsing the membranes in Milli-Q water followed by destaining over a further 60 min using 30% methanol. Based on the results of these experiments, the method favoring optimal protein release was used to make antigen rich-lysates for purposes of assays or nanoparticle fabrication.
Whole Tumor Isolates
The above results were adopted when generating lysates from freshly isolated murine tumor material. Melanoma tumors were initially induced by subcutaneous injection of cells from B16.F10 (murine melanoma; ATCC, Manassas, VA, USA) line into the flanks of female syngeneic C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME, USA). Institutional Animal Care and Use Committee (IACUC) granted approval for rodent usage. Animals were housed and cared for as per national guidelines specified in the Guide for the Care and Use of Laboratory Animals, Institute for Laboratory Animal Research (National Academies Press). Tumors were surgically resected and dissected under sterile conditions prior to admixing with a tumor digest solution (collagenase, dispase and hyaluronidase) for 2 h in an incubator at 37°C. Following removal of the undigested tumor material by filtering through a cell strainer (BD Falcon™ 70 μm filter; BD, Franklin Lakes, NJ, USA), cell solution was washed twice in ATCC-recommended B16.F10 medium, counted and re-suspended in sterile ice cold PBS.
Nanoparticle Fabrication
PLGA NP were fabricated employing the solvent evaporation method from a water/oil/water (W2/O/W1) emulsion as described in an earlier publication from our group (12). Encapsulated material included MBP-gp100 @10 μg/μl or typically lysates of 80×106 cells (@ 1×107 per 30 μL of sterile PBS) from either cultured FaDu or FAT7 lines or B16.F10 murine melanoma explants described above. To evaluate the ideal MW of PLGA co-polymers (50% lactide: 50% glycolide), differing preparations with inherent viscosity (i.v.) of 0.39 dL/g (45 KDa),0.59dL/g (80 KDa) or 0.67dL/g (105 KDa, carboxylated end group) (DURECT Corp., Pelham, AL, USA) were dissolved in methylene chloride (Fisher Scientific, Pittsburgh, PA) prior to being used for encapsulation with 0.5% Polyvinyl alcohol (PVA) (Sigma-Aldrich, St. Louis, MO, USA) acting as the stabilizer. Control NP (blanks) were synthesized by identical techniques utilizing sterile PBS instead of aqueous protein solutions.
Nanoparticle Characterization
Assessment of Morphology
Scanning Electron Microscopy
Scanning electron microscopy (SEM) was used to characterize individual NP formulations. A thin film of test sample was smeared onto a metal stub with double-sided adhesive carbon tape (Nisshin EM. Co. Ltd, Tokyo, Japan) and coated with gold (Cressington Sputter Coater 108 Auto, Redding, CA, USA). Imaging was completed using a XL-30 Environmental Scanning Electron Microscope-FEG (FEI, Hillsboro, OR, USA) with an acceleration voltage of 10 kV in a high vacuum mode to achieve magnifications between 9000x and 22,000x. Diameters from representative images of test samples (n=150 NP per image) were analyzed using Image J (image analysis software developed by National Institutes of Health, USA).
Dynamic Light Scattering
Dynamic light scattering (DLS) and zeta potential measurements were performed on a Malvern Zetasizer NanoZS system (Worcestershire, UK) with illumination from a 633 nm He-Ne laser. Samples were introduced in a clear folded capillary cell at a concentration of 100 μg per ml of phosphate buffer saline (PBS). Zeta potential was determined from the electrophoretic mobility using the Smoluchowski approximation.
Assessment of Protein Encapsulation
Extent of protein encapsulation was calculated based on the amount of protein extracted from a fixed mass of NP. Five mg of NP were degraded in 1 mL of 100 mM NaOH + 0.05% SDS (Ricca Chemicals, Arlington, TX, USA) by incubation at 37°C and continuous agitation in an orbital shaker. Samples were centrifuged at 16,100 × g for 5 min at room temperature (RT). Supernatants were analyzed for total protein as described above. Encapsulation efficiency was calculated using the formula:
Measurement of Protein Release Rate
The rate of release of encapsulated proteins by nanoparticle degradation was measured during incubation under controlled conditions. Five mg of NP were suspended in 1 ml of PBS and incubated at 37°C with continuous agitation in an orbital shaker. At designated time points, the suspension was centrifuged at 16,100 × g for 5 min at RT. Supernatants were collected and stored at −20°C for future studies. The particles were re-suspended in 1 ml of fresh PBS in the original tube for further incubation. To increase the extent of re-suspension and reduce clumping that could affect the release profile, a 1 ml pipette was used to pipette the samples as they were being vortexed. On occasions, NP were also briefly sonicated (≤ 15 s; Tekmar sonicator) to ensure re-suspension. Supernatant samples were analyzed for total protein as described above. Cumulative release was studied for at least eight days (the approximate functional lifespan of Ag-loaded DC), to reflect the amount of antigen available to DC for cross-presentation to CD8+ T cells. Percentage cumulative release was calculated using the formula:
Cytometric Bead Array
Influence of PLGA MW on extent of BMDC-mediated cross-presentation to antigen-specific CD8+ T cells was determined utilizing a recombinant version of the melanoma-associated protein gp100 and naïve transgenic T cells specific for a gp100-derived epitope. Syngeneic H-2Kd-restricted BMDC were obtained from C57BL6 mice as previously described (21) and plated in 6-well polystyrene flat-bottom cell culture plates (BD, NJ, USA). Day 5 DC were loaded with differing MW PLGA NP (0.5 mg of NP per 1×106 DC, a ratio previously determined to minimize the presence of unincorporated NP following co-culture) containing recombinant gp100 protein on its own or spiked into B16 tumor lysates. Four to six hours later, Lipopolysaccharide (LPS) (Sigma-Aldrich, St. Louis, MO, USA) at 10 ng/ml was added to trigger DC maturation. Following overnight incubation cells were harvested, washed twice, re-suspended in complete RPMI and plated in 96-well polystyrene U-bottom cell culture plates. These were admixed with gp100-specific CD8+ T cells isolated from the spleens of pmel transgenic mice [TCR transgenic mice with 95% gp100 (25–33)-specific, H-2Kd-restricted splenic CD8+ T cells (22); experiments were done in triplicate. After a 24-hour co-culture, the supernatants were removed and analyzed for production of cytokines associated with DC/T cell stimulation [such as Interleukin (IL)-2, Interferon (IFN)-γ and Tumor Necrosis Factor (TNF)-α]. T cell stimulation was quantified using a mouse Th1/Th2 cytometric bead array (CBA) cytokine kit (BD Biosciences, San Diego, CA, USA) as previously described by our group (12). Blank NP served as a control to rule out the possibility of non-specific stimulation by molecules such as endotoxins or the particles themselves.
Evaluation of Immunogenicity of Encapsulated TA from Patient Tumor-Derived Material
The results of the above experiments were adopted for the assessment of the immunogenicity of human tumor-derived material as TA in parallel experiments utilizing autologous TIL as T cell stimulation targets. Ethical permission from the Yale School of Medicine Human Investigation Committee was obtained to use freshly excised tumor and blood from patients with advanced tumors. Patients signed an informed consent and procedures followed were in accordance with institutional guidelines and the tenets of the Declaration of Helsinki promulgated in 1964. Tumor tissue served as source of TA as well as TIL while peripheral blood (drawn on the same day) was used to obtain DC from blood monocyte precursors.
Generation of Tumor Lysate and Isolation of TIL
Fresh tumor isolates were dissociated to generate both a soluble lysate as well as to isolate tumor-derived TIL. Tumor tissue (collected from surgical specimens with minimum delay, directly from surgical suites) was washed three times in modified FaDu medium (Eagle’s Minimal Essential Medium + 10% fetal bovine serum + 1% sodium pyruvate + 1% non-essential amino acids + 1.5 mg/ml sodium bicarbonate + 10% penicillin/streptomycin), dissected under sterile conditions prior to admixing with tumor digest solution as described above. Single cell suspensions were washed twice in modified FaDu medium and a portion, re-suspended in sterile ice cold PBS for lysate-NP production (as mentioned below). Cell solutions were brought up in minimum volumes of PBS (depending on original tumor volume and number of cells isolated) to ensure that tumor lysate concentrations were as high as possible. Protein content was determined using the BCA assay as described above. Remainder of the single cell suspension referred to above was suspended in complete TIL medium [(DMEM + 5% FBS + 1%L-Glutamine + 0.2% Gentamicin) (all Invitrogen, Carlsbad, CA, USA) + 100 IU/ml IL-2 (Peprotech, Rocky Hill, NJ, USA)]. TIL were isolated and expanded using CD3/CD28 Dynabeads (Dynal Prod No: 111.31) as per manufacturer’s instructions.
Nanoparticle Fabrication and Characterization
A portion of the single cell suspension obtained from tumor tisssue (mentioned above), was encapsulated within 50:50 PLGA copolymers by employing the solvent evaporation method (12). Rate of tumor antigen release was evaluated by nanoparticle degradation during incubation under controlled conditions.
Generation of DC from Peripheral Blood Monocyte (PBMC) Precursors
With a goal of generating DC from PBMC for use in co-culture with TIL, whole blood was collected at the time of surgery in heparin-containing 50 ml conical tubes (BD Falcon™). This was diluted 1:1 in sterile 1x PBS, layered over Isolymph gradient solution (CTL Scientific Supply Corp, Deer Park, NY, USA) and centrifuged at 1500 × g for 30 min at RT. The interface was collected and washed twice with sterile Hanks’ BSS (Invitrogen, Carlsbad, CA, USA). Cells were counted and viability checked by Trypan blue exclusion prior to suspension in complete RPMI (RPMI 1640 plus 10% FBS, 2% Pen/Strep and 25 mM HEPES) @ 2×106 cells/ml. Cells were plated in 5 ml aliquots in 6-well polystyrene flat-bottom cell culture plates and monocytes were allowed to adhere for 1 h at 37°C with 5% CO2. Adherent cells were incubated with complete RPMI + 0.25 ng/ml of IL-4 and 0.8 ng/ml of granulocyte-macrophage colony-stimulating factor (GM-CSF; Peprotech, Rocky Hill, NJ, USA) with media changes on days 1, 3 and 5. On day 6, the cells were harvested by gentle scraping, washed and counted. A portion of the DC was used immediately in antigen presentation assays. The remainder was re-suspended in 90% medium (complete RPMI)/10% Dimethyl sulphoxide (DMSO) prior to storage in cryovials at -80°C for subsequent use.
Assessing Immunogenicity Using DC/TIL Co-Culture
Stability and immunogenicity of encapsulated TA from human tumor samples were assessed utilizing TIL stimulation by antigen-loaded DC. Cultured DC were plated in 6-well plates and incubated with equimolar quantities of antigen in either of two forms –soluble lysate (conventional antigen delivery) or NP lysate (novel antigen delivery) as described above. TIL (@ 0.5×106 cells per ml of TIL medium) were added to the wells containing DC in a ratio between 1:3 and 2:3. The cell medium was changed on days 3 and 5. On day 6, autologous frozen DC were thawed, counted and re-plated as above for a second round of TIL stimulation. In order to compare the immunogenicity of encapsulated material as well as to confirm the increased effectiveness of NP as antigen delivery vehicles, for this round of stimulation, DC were loaded with a 5-fold molar excess of soluble lysate for comparison to NP-mediated Ag delivery. DC and pre-stimulated TIL were co-incubated in IL-2-free medium at a ratio of 1:2; experiments were done in triplicate. After 24 h, the supernatants were harvested and used immediately or stored at -20°C for future assays. Cytokine profiling of DC/T cell stimulation was performed on supernatants using a human Th1/Th2 CBA kit (BD Biosciences, San Diego, CA, USA) as described above.
Statistical Analysis
Statistical analysis was done using Statistical Package for the Social Sciences (SPSS) version 17.0 (SPSS, Surrey, UK). Differences were assessed using the independent samples T-test. Pearson’s coefficient of correlation was used to assess relationship between MW and extent of encapsulation. Significance was accepted at the 5% level.
RESULTS
Optimal Method for Lysate Preparation
A series of preliminary experiments were performed to determine the ideal technique for generating cellular lysate from cultured tumor cell lines. Different methodologies were evaluated in order to maximize protein (antigen) liberation. An initial screen showed that F/T (freeze/thaw) and F/T + S (sonication) yielded relatively high concentrations of lysates as measured by the BCA assay (Table I-column 1). In addition, Trypan Blue exclusion study revealed maximum cell lysis and fragmentation of cellular membranes following F/T + S (Table I- columns 2 and 3). During repeated treatments, F/T + S consistently yielded greater protein than F/T alone (mean ± SD=47± 21.5 versus 35.8±22.1; p>0.05) Additional characterization of lysates made by these two techniques through SDS/PAGE and western blot confirmed that F/T + S resulted in the greatest overall release of a mixture of TA as well as the specific release of p53. This was noted as increased intensity on Coomassie Blue stained gels (Fig. 1a) as well as a significantly larger p53-associated band by western blot (Fig. 1b). Taken together, the results indicated that F/T + S was an effective, reproducible and non-toxic/chemical-free methodology for the generation of tumor lysates for NP fabrication.
Table I.
Profile of Tumor Antigen Lysates made by different Techniques. Protein Content was measured using the Bicinchonic acid assay while Cell Death and Extent of Cellular Fragmentation were Assessed by Light Microscopy. Despite Starting with Similar Amounts of Cellular Material, there were Identifiable differences in the Final Lysate Content and Viability of Cells. Freeze-thaw Plus Sonication for 15 s proved to be Highly effective in Triggering Cellular Fragmentation and release of Antigens from Suspensions, making it the Preferred Method to Liberate Antigens from whole Tumor Cells. Data represent mean and Standard Deviation of Pooled Observations from repeat Experiments
| Lysate preparation technique | Protein (antigen) (mg/ml) | Trypan Blue positivity (%) | Cellular fragmentation (%) |
|---|---|---|---|
| F/T + S | 50±28.3 | 98.3±1.2100 | 98±1.9100 |
| F/T alone | 35±23 | 93.7±5.890 | 93±5.870 |
| F/T + DH | 10.5±7.3 | 93±2.995 | 45±750 |
| Sonication for 60 s alone | 26±19.3 | 91.7±2.895 | 93±5.8100 |
F/T + S, Freeze-thaw + sonication for 15 s; F/T, Freeze-thaw alone; F/T + DH, Freeze-thaw + Dounce homogenization
Fig. 1.
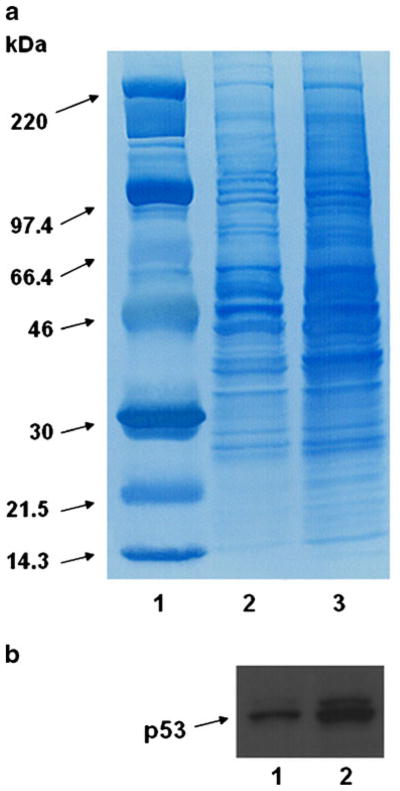
Differences in antigen release from tumor cells by freeze thaw (F/T) alone and freeze-thaw + 15 sonication (F/T + S). In order to determine differences in protein content, lysate samples from identical numbers of starting cells were electrophoresed on a SDS-PAGE mini-gel system and transferred onto nitrocellulose membranes. After an initial screen, two techniques (F/Tand F/T + S) resulting in relatively greater protein liberation from cellular lysates were compared against one another. Greater intensity of staining in the lanes loaded with lysate subject to F/T + S implied greater antigen release by this method, an observation confirmed by increased release of the defined tumor Ag (p53). (a) Coomassie Blue staining: Lane 1: Molecular weight ladder; lane 2: F/Talone; lane 3: F/T + S. (b) Western blot utilizing an anti-p53 antibody shows increased release of this protein: Lane 1: F/Talone; lane 2: F/T + S.
Effect of Differing PLGA MW
NP Morphology
Tissue culture lysates were encapsulated within PLGA copolymers of differing MW (45KDa, 80KDa and 105KDa), using the double emulsion procedure. SEM of resultant NP demonstrated a broad distribution in size and moderate differences in shape. They were largely spherical, non-porous in appearance and within the size range favoring efficient phagocytosis by DC (≥ 1 μm) (data not shown). Mean diameters (nm) ± S.D for 45KDa, 80KDa and 105KDa NP were 204±17, 252±22 and 337±18 respectively. NP made using 105KDa were larger than 80KDa (p=0.007) which in turn was larger than 45KDa particles (p=0.044), indicating that polymer had a proportional effect on final NP size (longer polymer = larger NP). DLS analysis showed a similar pattern (Fig. 2) where an increase in NP size was associated with increase in PLGA MW. Mean diamters (nm) were 342, 439 and 477 for the 45 KDa, 80KDa and 105 KDa formulations (p <0.05 for 80 and 105 KDa vs. 45 KDa formulations), with a large size polydispersion observed (Fig. 2b, c). The proportionally increased NP size determinations by DLS vs SEM may have resulted from DLS sizes representing a hydrodynamic radius. In addition, small differences in zeta potential were observed for the three MW formulations (Fig. 2a).
Fig. 2.
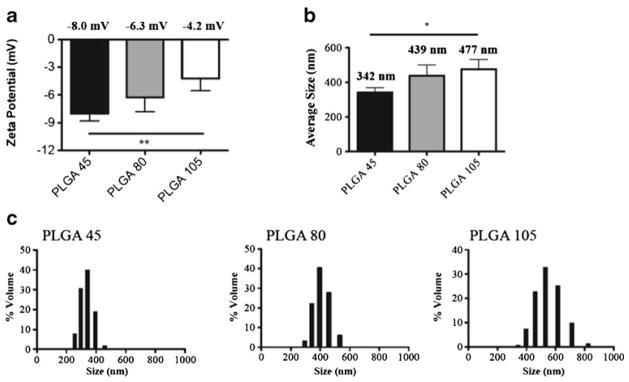
DLS and charge measurements of nanoparticles synthesized using different molecular weights of PLGA. (a) Average zeta potential values for PLGA 45, 80 and 105. (b) Average particle diameters for PLGA 45, 80 and 105 as measured by DLS. (c) Size distributions for PLGA 45, 80 and 105 as measured by DLS. Data are represented as mean +/− SD. In all experiments, statistical analysis was performed using student t-test where (*) represents p<0.05, (**) represents p<0.01.
Encapsulation Profile
To determine the overall effects of polymer MW on protein encapsulation, TA were encapsulated within the three different PLGA MW polymer formulations. NP were rapidly degraded and total protein content quantified. In a reproducibly observed pattern, total protein encapsulated and efficiency of encapsulation increased with higher polymer MW (Table II). Irrespective of protein (antigen) concentration of lysate used for encapsulation, Pearson’s coefficient of correlation was ≥0.9 suggesting a strong linear relationship between MW and extent of encapsulation.
Table II.
Effect of PLGA MW on Encapsulation Profile. Extent and Efficiency of Encapsulation Increased with Increasing MW. Rotein Content in Lysate (μg)/Unit of Polymer (mg). Encapsulation Efficiency was Calculated as: (mass protein encapsulated ÷ mass protein used in encapsulation) × 100% formula: Mass Protein Encapsulated/mass Protein loaded) × 100%. Data represent mean and Standard Deviation of Pooled Observations from repeat Experiments and Values from a Representative Experiment
| PLGA MW (KDa) | Protein content in lysate (μg) | Encapsulated protein (μg/mg of NP) | Encapsulation Efficiency (%) |
|---|---|---|---|
| 45 | 3600±20181950 | 22±1512 | 24±9.530 |
| 80 | 3600±20181950 | 26±1715.6 | 33±15.540 |
| 105 | 3600±20181950 | 34±19.718 | 42±1746.2 |
Release Kinetics
To determine which PLGA formulation was most likely to release TA in a clinically-relevant eight day period (the approximate function lifespan of an Ag-loaded DC), cumulative release was estimated for this duration. There was a biphasic pattern of protein release: a “burst release”- within the first 24 h and a “plateau phase” – a more sustained release over the subsequent days. Initial burst (μg/5 mg of NP; mean ± SD) for 45KDa, 80KDa and 105KDa were: 8.8±0.8, 44.5±4 and 27.4±4.4%, respectively. This was greatest with 80KDa PLGA (p=0.008 versus 105KDa and p=0.003 versus 45KDa). Total release (μg/5 mg of NP) was 8.85, 61.4 and 35.3%, respectively. This was highest with 80KDa MW polymer (p= 0.002 versus 105KDa and p=0.001 versus 45 KDa) (Fig. 3). Percentage cumulative release for 45KDa, 80KDa and 105KDa were 11, 39 and 25%, respectively. Similar results were observed in repeat experiments.
Fig. 3.

Effect of PLGA molecular weight (MW) on release of encapsulated tumor material. Proteins were released under controlled conditions from 5 mg of the three PLGA formulations; samples taken at designated time points and analyzed for protein content by the BCA assay. The initial burst of protein release was increased in association with the 80KDa PLGA (open square); p=0.008 versus 105KDa (open circle) and p=0.003 versus 45KDa (open triangle). Cumulative protein release over 8 days was maximal with 80KDa MW polymer; p =0.002 versus 105KDa and p =0.001 versus 45KDa. Data refers to mean ± SD of triplicates from a representative experiment.
Biological Activity of Differing Formulations of TA-Loaded NP
To determine whether a particular MW formulation was associated with superior TA delivery and cross-presentation by DC, melanoma-associated antigen gp100 (either in isolation or contained within a murine melanoma B16.F10 tumor lysate) was evaluated in a CD8+-based antigen-specific stimulation assay. Equimolar amounts of each PLGA formulation were loaded onto syngeneic murine BMDC and differences in stimulation of naïve gp100-specific CD8+ T cells assessed by characterizing profiles of anti-tumor cytokines. As shown in Fig. 4, delivery of gp100 (in isolation) by 80KDa polymer led to greatest immunostimulatory IL-2 production (p<0.001 versus 45 KDa and p >0.05 versus 105KDa) by antigen-specific T cells. Also, 105KDa MW PLGA resulted in greater IL-2 production when compared to 45KDa (p =0.001) (Fig. 4a). Similarly, 80KDa polymer resulted in maximal immunostimulatory IFN-γ production (p <0.001 versus 45KDa and p =0.014 versus 105KDa). Once again, the 105KDa polymer associated with increased IFN-γ production as compared to 45KDa (p<0.001) (Fig. 4b). When the gp100-specific stimulation was followed with the target Ag admixed with a large group of cellular proteins (i.e. B16 lysate), similar but not identical results were observed. Delivery of gp 100 within B16 tumor lysate utilizing the 45KDa polymer formulation led to greatest IL-2 production (p=0.002 versus 80KDa and p=0.003 versus 105KDa) (Fig. 4c). There were no significant differences when comparing 80KDa and 105 KDa polymers. However, IFN-γ synthesis was once again maximal with 80KDa (p=0.001 versus 105KDa and p>0.05 versus 45KDa); there were no significant differences when comparing 45KDa and 105KDa polymers (Fig. 4d). Taken together, this data suggest that the 80KDa polymer would be the best overall choice for delivery of encapsulated antigen to DC.
Fig. 4.

Effects of differing PLGA formulations on DC-mediated antigen cross-presentation to gp100-specific CD8+ T cells. Experiments were performed with recombinant gp100 protein encapsulated alone (a and b) or after co-encapsulation with murine B16 tumor lysate (c and d). Differences in T cell stimulation was assessed by measuring production of the immunostimulatory cytokines IL-2 and IFN-γ. DC loaded with blank NP (diagonal bar) were used as negative controls for assays. Single Ag delivery: (a) 80KDa polymer (gray bar) led to greatest IL-2 production; *p<0.001 versus 45KDa (black bar) and p> 0.05 versus 105KDa (open bar). Also, 105KDa MW PLGA was associated with increased IL-2 production when compared to 45KDa (**p=0.001). (b) 80 K polymer resulted in significantly higher IFN-γ production; *p<0.001 versus 45KDa and #p=0.014 versus 105KDa. Again, 105KDa polymer caused greater IFN-γ synthesis as opposed to 45KDa (**p<0.001). Ag in complex lysate: (c) IL-2 production was greatest with 45KDa; *p=0.002 versus 80KDa; #p= 0.003 versus 105KDa. There were no significant differences when comparing 80KDa and 105KDa polymers. (d) IFN-γ synthesis was highest in association with 80KDa polymer; *p=0.001 versus 105KDa and p>0.05 versus 45KDa. There were no significant differences when comparing 45KDa and 105KDa polymers.
Evaluation of Immunogenicity of Tumor Lysate-NP
To confirm whether the results observed with the model TA (gp100) were broadly applicable to lysates and T cell responses found in cancer patients, the above observations were extended to patient-derived whole tumor lysate containing NP. Since the 80KDa polymer appeared to have the most favorable overall release profile and immunostimulatory cytokine production, it was the polymer of choice for the experiments. NP were generated and loaded onto monocyte-derived autologous DC mobilized from patient PBMC. TA-containing DC were used as APC in stimulation assays which targeted tumor-derived TIL. Tumor material dissociated from fresh surgical samples acted as source of tumor NP (from pure tumor) as well as TIL (cultured from tumor interstitium and expanded by CD3/CD28 bead stimulation) (Fig. 5a). Autologous DC were loaded with either soluble tumor lysate (in 5-fold molar excess) or lysate NP (Fig. 5b). Stimulation of autologous TIL was monitored from DC/TIL co-cultures by phase contrast microscopy (Fig. 5c) and quantification of two cytokines known to be critical to anti-tumor immune responses. These were IFN-γ (emblimatic of an active Th1 or inflammatory anti-tumor response) and IL-10 (associated with non-productive tumor-tolerized T cells). As shown in Fig. 6, NP-mediated antigen delivery led to increased immunostimulatory IFN-γ (p=0.002 versus lysate and p=0.035 versus blank-NP) (Fig. 6a). Critically, NP delivery was associated with decreased immunoinhibitory IL-10 production (p=0.002 versus lysate and p= 0.015 versus blank-NP) when compared to soluble lysate (Fig. 6b).
Fig. 5.

Phase contrast microscopy of components of DC/TIL co-culture: (a) TIL were isolated and expanded using CD3/CD28 Dynabeads®; black arrow indicating conglomeration of expanded TIL. (b) PBMC-derived DC were loaded with TA containing-80KDa PLGA NP; cells demonstrating characteristic dendrites and visible NP associated with the cell surface and surface invaginations. (c) Co-culture of freshly obtained autologous antigen loaded-DC with TIL; white arrow depicting representative DC/TIL complex.
Fig. 6.
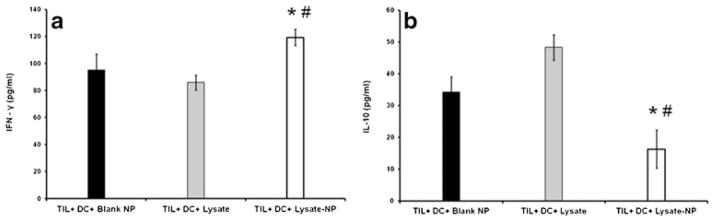
Enhanced stimulation of TIL after cross-presentation of NP encapsulated tumor antigens by autologous DC. Antigen loaded-DC and TIL were co-incubated overnight and supernatants collected and analyzed for Tcell production of IFN-γ and IL-10. NP-mediated antigen delivery led to: (a) increased immunostimulatory IFN-γ (*p=0.002 versus lysate and #p=0.035 versus blank-NP) and (b) decreased immunoinhibitory IL-10 (*p=0.002 versus lysate and #p=0.015 versus blank-NP).
DISCUSSION
Superior bioavailability and CTL responses associated with the PLGA system can help overcome many of the limitations of current DC-based adoptive immunotherapies (23,24). Key components that need to be optimized prior to employing this novel vaccine strategy in human clinical trials include evaluation of antigen loading and release kinetics, assessment of preclinical safety and estimation of biological potency (25,26). An ideal TA-NP formulation should satisfy three main criteria: high encapsulation efficiency, sustained antigen delivery over 7–8 days (the approximate period of functional antigen presentation by APC) and heightened stimulation of cell-mediated immunity. Although polymer MW can influence encapsulation and release (27), its role in TA presentation has not been clearly elucidated to date (28). Additionally, it is important to establish that the encapsulated substrate retains its structural integrity and remains antigenic. This is a concern because of the well-documented degradation of payload both during NP synthesis and release (29,30). The scope of the present study was to determine the best methodology to generate a celluar lysate, the PLGA MW formulation that was associated with efficient TA encapsulation anddelivery to DC, and to assess whether such TA-containing NP could ultimately evoke superior anti-tumor cytolytic CD8+ T cell responses.
Different methods were compared when trying to maximize the spectrum and content of tumor antigens in the pre-encapsulation lysate. Since there have been reports of up to 20% antigen instability (31,32), we hypothesized that increasing the lysate concentration would improve encapsulation, provide a buffering effect and eventually increase the amount of stable antigen within PLGA particles. Increasing antigen content of the loading lysate is purported to reduce the extent of antigen adsorbed and denatured at the water-oil interface during particle fabrication. This is due to irreversible adsorbtion of proteins at the interface. At higher concentrations these adsorbed proteins act as “self-protectants” preventing further degradation of remaining proteins present in the lysate and causing enhanced encapsulation (33). In a previous study, increasing the theoretical drug content from 20 to 50%, resulted in an increase in the mean amount of drug entrapped from 19.67±3.21 to 51.53±3.15 (34). Factors influencing the choice of a suitable protein isolation technique include safety (for clinical use), reproducibility and efficacy in liberating TA. It was observed that four cycles of freeze-thaw plus 15-second sonication (Fig. 1 and Table I) satisfied these criteria. In the only other report pertaining to encapsulation of a complex mixture of tumor antigens, our group employed four cycles of freeze-thaw and octyl β-glucoside (a dialyzable detergent) to generate lysates (12). The present study suggests that such detergents are not necessary for significant protein liberation. Our initial observations suggest that liberation of critical target proteins such as gp100 (17) and p53 (Fig. 1) by F/T + sonication adequately solubilizes critical target proteins.
The effects of PLGA polymer MW on the efficiency of encapsulation of both single and complex mixtures of TA within tumor lysate mixtures were provocative. While keeping the concentration of protein in the internal aqueous phase of the double emulsion constant, we observed that increase in polymer molecular weight resulted in concomitant increase in encapsulation rate and efficiency (Table II). This was in keeping with the previous report by our group (12). Increase in molecular mass of polymer increases viscosity of the polymer which in turn improves stability of the primary w/o emulsion. This results in enhanced encapsulation of hydrophilic molecules (proteins and peptides) present in aqueous droplets (35). Our observation is important when attempting to encapsulate tumor tissue as part of NP-based vaccine therapy. Should only meagre amounts of surgical tissue be available (as is often the case), the PLGA MW preparation associated with highest encapsulation should be used in an effort to maximize entrapment of TA. As will be discussed below, this should be considered in conjunction with other factors such as the rate of release and biological potency.
There was an increase in particle diameter associated with increasing polymer molecular weight as determined by both on SEM and DLS (Fig. 2). Similar observations were recorded previously; particle size increased from 98.3 nm to 155.4 nm with increase in polymer MW from 14.5 KDa to 213 KDa, during the encapsulation of estradiol (27). A possible explanation for this is that as polymer molecular weight increases, there is an increase in the viscosity of the emulsion leading to lower stirring efficiency at the same agitation speed. This makes it difficult to generate smaller organic droplets and in turn small sized spheres (27). Our findings confirmed that the even the largest NP were well within the size range that could be efficiently internalized by DC, something of importance when considering NP as antigen delivery vehicles in a DC-based anti-tumor immunotherapy (36).
As reported previously (12), we saw that the extent of release of protein encapsulates varied with alterations in the molecular weight of PLGA co-polymer. Total release over 8 days was greatest from NP made using 80KDa (Fig. 3). Percentage cumulative release for 45KDa, 80KDa and 105KDa were 11, 39 and 25%, respectively. These findings underline the fact that polymer molecular weight plays a role in rate of degradation and release kinetics of payload. Lower molecular weight polymers tend to have shorter chain lengths causing them to be less lipophilic. This favors rapid water hydration causing a drop in the glass transition temperature (Tg) (called plasticizing effect of the water) resulting in greater mobility of polymer chain segments in turn leading to increased degradation (9). Maximal release with the mid-range 80KDa polymer, could imply that additional factors like polymer shape and ionic interaction of encapsulated material with PLGA terminal end-groups influence release (37). Nevertheless, the biological effect of this phenomenon has only infrequently been described. For instance, testosterone suppression in Sprague–Dawley rats following Leuprolide acetate delivered using differing PLGA (50:50) formulations indicated that 8.6 KDa demonstrated quicker but less lasting effect than 28.3 K (38). The present study reports differences in immunological potency attributable to alteration of polymer MW. In all but one instance (Fig. 4c), 80 KDa polymer was associated with greatest production of anti-tumor inflammatory/Th1 cytokines (Fig. 4a, b and d) implying superior antigen cross-presentation. The overall antigen encapsulation,/release and T cell stimulation profile suggest that 80 KDa PLGA could be the formulation of choice for encapsulating fresh tumor material from patients. This could enhance CD8+ T cell stimulation and reduce chances of tumor immune escape by targeting multiple TA in a “personalized” vaccination strategy.
As part of a stepwise approach to generating important pre-clinical data prior to adoption of NP reagents in clinical trials, we conducted further ex vivo experiments aimed at assessing the immunogenicity of encapsulated complex TA derived from freshly isolated tumor. These involved samples from patients with loco-regionally advanced malignancies because this sub-group would most likely be the targets for early (Phase I/II) clinical trials testing novel therapies like NP-mediated DC-based immunotherapy. The experimental design paralleled a potential vaccination protocol; the two rounds of TIL stimulation by antigen loaded-DC mimicked a primary and a booster dose of antigen-loaded DC vaccine. To approximate the trial setting, fresh human tumor samples and TIL rather than established tumor or epitope-specific T cell lines were used. The results suggest that encapsulated material is not only immunogenic but also more efficient at T cell stimulation when compared to conventional lysate. NP-mediated antigen delivery led to significantly increased immunostimulatory IFN-γ (Fig. 6a) and decreased immunoinhibitory IL-10 (Fig. 6b). These phenomena could be critical in overcoming immune resistance that has become the hallmark of solid organ malignancies (39). The data reported in the present study suggests that whole cell lysates (a staple in DC-based immunotherapy outside of melanoma) may be a new and powerful source of antigenic reagents, provided they are delivered to DC through a NP intermediate, for potentially overcoming real-world tumor tolerance. This work is in agreement with earlier reports of favorable T cell stimulation evoked by single antigen-containing NP (40,41). But it extends these observations to encapsulation of multiple (unidentified) TA present within many solid tumors.
This paper reports novel data from encapsulation, release and biological activity of complex mixture of antigens derived from cell lines and whole tumor lysate. More specifically, various techniques were evaluated with a view to finding out the best method to maximize release of mixture of tumor antigens and ensure production of an antigen rich lysate. Enhancing the release of tumour antigens would in turn ensure that relatively large amounts of antigens are available for encapsulation into and release from PLGA particles. This would then lead to greater delivery of antigens to dendritic cells and potentially cause broader and more intense stimulation of anti-tumor T cells. We present data on the differences in extent of DC-mediated cross-presentation of complex panoply of whole tumor antigens to cytolytic T cells brought about by varying PLGA molecular wieght. This is a crucial factor to consider when using PLGA polymers as antigen delivery vehicles. Our results were derived following experiments involving naïve, tumor-specific CD8+ T cells derived from gp100-specific transgenic mice in a highly specific T cell assay. These observations were subsequently adopted in encapsulating patient derived tumor antigens and then delivering them via DC for stimulation of freshly isolated autologous T cells. The experimental setup closely mimicked a vaccination protocol that could be used in a clinical trial of NP-DC vaccine, and represents an incremental step forward in the development of PLGA-based reagents for tumor immunotherapy.
It is noteworthy that the NP reagents described here were fabricated without the inclusion of additional maturation stimuli (for example pathogen associated molecular patterns) co-encapsulated or decorating the NP surface. This was a conscious decision aimed at safely using these reagents clinically in the near-term. But our group is actively investigating the use of additional NP formulations utilizing DC-targeting pathogen associated molecules or direct DC ligands on the NP surface. We expect to use these reagents in the future as a stepwise approach to NP-based vaccination strategies leading to overall improvements in vaccine potency.
CONCLUSIONS
Four cycles of freeze-thaw followed by 15 s sonication of tumor cell suspension is an effective, reproducible and non-toxic methodology for the generation of rich tumor Ag containing-lysates for NP fabrication. Irrespective of polymer MW, resultant particles fabricated from a variety of PLGA MWs are within the size range favoring easy phagocytosis by antigen presenting cells. While extent of encapsulation increases with rising PLGA MW, the 80KDa PLGA formulation appears to have the most promising combination of release kinetics and biologic potency for TA delivery. This indicates that polymer MW may directly influence efficient access to antigen cross-presentation compartments found specifically within DC. Encapsulated antigens within such a formulation are immunogenic and evoke favorable TIL-mediated anti-tumor responses when compared to conventional soluble lysate. A favorable cytokine profile in the form of increased immunostimulatory IFN-γ and reduced immunoinhibitory IL-10 could surmount the immune tolerance millieu prevalent in loco-regional advanced or recurrent solid tumours like HNSCC, thus suppporting their use in novel DC-based adoptive solid tumor immunotherapy in the future.
Acknowledgments
The authors thank Prof Mark Saltzman and Dr Camille Solbrig in Biomedical Engineering for assistance and supply of reagents associated with NP encapsulation, Dr Michael Girardi and Kacie Carlson in Dermatology for their help in animal handling, and members of the Girardi and Cresswell laboratory for helpful discussions. We also thank Prof Sasaki and Shelley Jolie in Otolaryngology Head and Neck Surgery, Dr Diane Kowalski and Lori Patruno in Surgical Pathology, and members of the team of anaesthetists involved in care of patients recruited to this study. This work was partially funded by intra mural grant available to the Department of Dermatology, Yale University School of Medicine and partially by a NCI/NTRAC grant managed by Cancer Research UK.
ABBREVIATIONS
- Ags
antigens
- APC
antigen presenting cell
- ATCC
American Type Culture Collection
- BCA
bicinchonic acid
- BMDC
bone marrow dendritic cells
- BSA
bovine serum albumin
- CBA
cytometric bead array
- CD
cluster of differentiation
- CTL
cytotoxic T cells
- DC
dendritic cell
- FT + S
freeze-thaw + sonication
- GM-CSF
granulocyte macrophage-colony stimulating factor
- HNSCC
head and neck squamous cell carcinoma
- IFN-γ
interferon-γ
- IL
interleukin
- LPS
lipopolysaccharide
- MP
microparticles
- MW
molecular weight
- NP
nanoparticles
- PBS
phosphate buffered saline
- PLGA
poly (lactic-co-glycolic acid)
- PVA
polyvinyl alcohol
- RT
room temperature
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SEM
scanning electron microscopy
- SPSS
Statistical Package for the Social Sciences
- TA
tumor associated antigen
- Th
T-helper
- TIL
tumor infiltrating lymphocytes
- TNF-α
tumor necrosis factor-α
- WB
Western blot
Footnotes
DISCLOSURES
There is no perceived, potential or real conflict of interest.
Contributor Information
Shashi Prasad, Department of Dermatology, Yale University, New Haven, Connecticut 06520-8260, USA.
Virginia Cody, Department of Dermatology, Yale University, New Haven, Connecticut 06520-8260, USA.
Jennifer K. Saucier-Sawyer, Department of Biomedical Engineering, Yale University, New Haven, Connecticut 06520-8260, USA
Tarek R. Fadel, Department of Biomedical Engineering, Yale University, New Haven, Connecticut 06520-8260, USA
Richard L. Edelson, Department of Dermatology, Yale University, New Haven, Connecticut 06520-8260, USA
Martin A. Birchall, UCL Center for Stem Cells & Regenerative Medicine & UCL Ear Institute, Royal National Throat Nose & Ear Hospital, London WC1X 8DA, UK
Douglas J. Hanlon, Email: douglas.hanlon@yale.edu, Department of Dermatology, Yale University, New Haven, Connecticut 06520-8260, USA. Yale University, P.O. Box 208059, New Haven, Connecticut 06520-8059, USA
References
- 1.Cruz LJ, Tacken PJ, Fokkink R, Joosten B, Stuart MC, Albericio F, et al. Targeted PLGA nano-but not microparticles specifically deliver antigen to human dendritic cells via DC-SIGN in vitro. J Control Rel. 2010:118–126. doi: 10.1016/j.jconrel.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Thomas C, Gupta V, Ahsan F. Influence of surface charge of PLGA particles of recombinant hepatitis B surface antigen in enhancing systemic and mucosal immune responses. Int J Pharm. 2009;379:41–50. doi: 10.1016/j.ijpharm.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Tel J, Lambeck AJA, Cruz LJ, Tacken PJ, de Vries IJM, Figdor CG. Human Plasmacytoid Dendritic Cells Phagocytose, Process, and Present Exogenous Particulate Antigen. J Immunol. 2010;184:4276–83. doi: 10.4049/jimmunol.0903286. [DOI] [PubMed] [Google Scholar]
- 4.Tuncay M, Calis S, Kas HS, Ercan MT, Peksoy I, Hincal AA. Diclofenac sodium incorporated PLGA (50:50) microspheres: formulation considerations and in vitro/in vivo evaluation. Int J Pharm. 2000;195:179–88. doi: 10.1016/s0378-5173(99)00394-4. [DOI] [PubMed] [Google Scholar]
- 5.Paolicelli P, Prego C, Sanchez A, Alonso MJ. Surface-modified PLGA-based nanoparticles that can efficiently associate and deliver virus-like particles. Nanomed. 2010;5:843–53. doi: 10.2217/nnm.10.69. [DOI] [PubMed] [Google Scholar]
- 6.Rajapaksa TE, Lo DD. Microencapsulation of vaccine antigens and adjuvants for mucosal targeting. Curr Immunol Rev. 2010;6:29–37. [Google Scholar]
- 7.Duan Y, Sun X, Gong T, Wang Q, Zhang Z. Preparation of DHAQ-loaded mPEG-PLGA-mPEG nanoparticles and evaluation of drug release behaviors in vitro/in vivo. J Mat Sci Mat Med. 2006;17:509–16. doi: 10.1007/s10856-006-8933-3. [DOI] [PubMed] [Google Scholar]
- 8.Astaneh R, Erfan M, Moghimi H, Mobedi H. Changes in morphology of in situ forming PLGA implant prepared by different polymer molecular weight and its effect on release behavior. J Pharm Sci. 2009;98:135–45. doi: 10.1002/jps.21415. [DOI] [PubMed] [Google Scholar]
- 9.Fu X, Ping Q, Gao Y. Effects of formulation factors on encapsulation efficiency and release behaviour in vitro of huperzine A-PLGA microspheres. J Microencapsul. 2005;22:57–66. doi: 10.1080/02652040400026509. [DOI] [PubMed] [Google Scholar]
- 10.Murillo M, Grillo MJ, Rene J, Marin CM, Barberan M, Blasco JM, et al. A Brucella ovis antigenic complex bearing (poly-e-caprolactone) microparticles confer protection against experimental brucellosis in mice. Vaccine. 2001;19:4099–106. doi: 10.1016/s0264-410x(01)00177-3. [DOI] [PubMed] [Google Scholar]
- 11.Ren JM, Zou QM, Wang FK, He Q, Chen W, Zen WK. PELA microspheres loaded H Pylori lysates and their mucosal immune response. World J Gastroenterol. 2002;8:1098–102. doi: 10.3748/wjg.v8.i6.1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Solbrig CM, Saucier-Sawyer JK, Cody V, Saltzman WM, Hanlon DJ. Polymer nanoparticles for immunotherapy from encapsulated tumor associated antigens and whole tumor cells. Mol Pharm. 2007;4:47–57. doi: 10.1021/mp060107e. [DOI] [PubMed] [Google Scholar]
- 13.Jiang W, Gupta RK, Deshpande MC, Schwendeman SP. Biodegradable poly(lactic-co-glycolic acid) microparticles for injectable delivery of vaccine antigens. Adv Drug Deliv Rev. 2005;57:391–410. doi: 10.1016/j.addr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 14.Li XY, Kong XY, Shi S, Zheng XL, Guo G, Wei YQ, et al. Preparation of alginate coated chitosan microparticles for vaccine delivery. BMC Biotechnol. 2008;8:89. doi: 10.1186/1472-6750-8-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marazuela EG, Prado N, Moro E, Fernandez-Garcia H, Villalba M, Rodriguez R. Intranasal vaccination with poly (lactide-co-glycolide) microparticles containing a peptide of T of Ole 1 prevents mice against sensitization. Clin Exp Allerg. 2008;38:520–8. doi: 10.1111/j.1365-2222.2007.02922.x. [DOI] [PubMed] [Google Scholar]
- 16.Zhou S, Liao X, Liang Z, Li X, Deng X, Li H. Preparation and characterization of biodegradable microspheres containing Hepatitis B surface antigen. Macromol Biosci. 2004;4:47–52. doi: 10.1002/mabi.200300044. [DOI] [PubMed] [Google Scholar]
- 17.Costantino HR, Langer R, Kilbanov KM. Solid-phase aggregation of proteins under pharmaceutically relevant conditions. J Pharm Sci. 1994;83:1662–9. doi: 10.1002/jps.2600831205. [DOI] [PubMed] [Google Scholar]
- 18.Fu K, Kilbanov KM, Langer R. Protein instability in controlled-release systems. Nat Biotechnol. 2000;18:24–5. doi: 10.1038/71875. [DOI] [PubMed] [Google Scholar]
- 19.Jaganathan KS, Singh P, Prabhakaran D, Mishra V, Vyas SP. Development of a single-dose stabilized poly (D, L lactic-co-glycolic acid) microsphere-based vaccine against Hepatitis B. J Pharm Pharmacol. 2004;56:1243–50. doi: 10.1211/0022357044418. [DOI] [PubMed] [Google Scholar]
- 20.Ganguli MJK, Maiti S. Nanoparticles from cationic coploymers and DNA that are soluble and stable in common organic solvents. J Am Chem Soc. 2004;126:26–7. doi: 10.1021/ja037534v. [DOI] [PubMed] [Google Scholar]
- 21.Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–80. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johansen P, Gomez JMM, Gander B. Development of synthetic biodegradable microparticulate vaccines: a roller coaster story. Exp rev vaccines. 2007;6:471–4. doi: 10.1586/14760584.6.4.471. [DOI] [PubMed] [Google Scholar]
- 24.Nestle FO, Farkas A, Conrad C. Dendritic-cell-based therapeutic vaccination against cancer. Curr Opinion Immunol. 2005;17:163–9. doi: 10.1016/j.coi.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 25.Johansen P, Men Y, Merkle HP, Gander B. Revisiting PLA/PLGA microspheres: an analysis of their potential in parenteral vaccination. Eur J Pharm and Biopharm. 2000;50:129–46. doi: 10.1016/s0939-6411(00)00079-5. [DOI] [PubMed] [Google Scholar]
- 26.Sesardic D, Dobbelaer R. European union regulatory developments for new vaccine adjuvants and delivery systems. Vaccine. 2004;22:2452–6. doi: 10.1016/j.vaccine.2003.11.071. [DOI] [PubMed] [Google Scholar]
- 27.Mittal G, Sahana DK, Bhardwaj V, Ravi MNV. Estradiol loaded PLGA nanoparticles for oral administration: effect of polymer molecular weight and copolymer composition on release behavior in vitro and in vivo. J Control Release. 2007;119:77–85. doi: 10.1016/j.jconrel.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 28.Mundargi RC, Babu VR, Rangaswamy V, Patel P, Aminabhavi TM. Nano/micro technologies for delivering macromolecular therapeutics using poly (d, l-lactide-co-glycolide) and its derivatives. J Control Release. 2008;125:193–209. doi: 10.1016/j.jconrel.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 29.Houchin M, Topp EM. Chemical degradation of peptides and proteins in PLGA: a review of reactions and mechanisms. J Pharm Sci. 2007;97:2395–404. doi: 10.1002/jps.21176. [DOI] [PubMed] [Google Scholar]
- 30.Schwendeman SP. Recent advances in the stabilization of proteins encapsulated in injectable PLGA delivery systems. Crit Rev Ther Drug Carr Syst. 2002;19:73–98. doi: 10.1615/critrevtherdrugcarriersyst.v19.i1.20. [DOI] [PubMed] [Google Scholar]
- 31.Schwendeman SP, Costantino HR, Gupta RK, Tobio M, Chang AC, Alonso MJ, et al. Strategies for stabilizing tetanus toxoid toward the development of a single-does tetanus vaccine. Dev Biol Stand. 1996;87:293–306. [PubMed] [Google Scholar]
- 32.Kersten G, Donders D, Akkermans A, Beuvery EC. Single shot with tetanus toxoid in biodegradable microspheres protects mice despite acid-induced denaturation of the antigen. Vaccine. 1996;14:1627–32. doi: 10.1016/s0264-410x(96)00145-4. [DOI] [PubMed] [Google Scholar]
- 33.Tamber H, Johansen P, Merkle HP, Gander B. Formulation as pects of biodegradable polymeric microspheres for antigen delivery. Adv Drug Deliv Rev. 2005;57:357–76. doi: 10.1016/j.addr.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 34.Jelvehgari M, Valizadeh H, Rezapour M, Nokhodchi A. Control of encapsulation efficiency in polymeric microparticle system of tolmetin. Pharm Dev Technol. 2010;15:71–9. doi: 10.3109/10837450903002173. [DOI] [PubMed] [Google Scholar]
- 35.Wang D, Robinson D, Kwona GS, Samuel J. Encapsulation of plasmid DNA in biodegradable poly(lactic-co-glycolic acid) micro-spheres as a novel approach for immunogene delivery. J Control Release. 1999;57:9–18. doi: 10.1016/s0168-3659(98)00099-6. [DOI] [PubMed] [Google Scholar]
- 36.Fogeda C, Brodina B, Frokjaera S, Sundblad A. Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int J Pharm. 2005;298:315–22. doi: 10.1016/j.ijpharm.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 37.Jaraswekin S, Prakongpan S, Bodmeier R. Effect of poly(lactide-co-glycolide) molecular weight on the release of dexamethasone sodium phosphate from microparticles. J Microencapsul. 2007;24:117–28. doi: 10.1080/02652040701233655. [DOI] [PubMed] [Google Scholar]
- 38.Ravivarapu HB, Burton K, De Luca PP. Polymer and micro-sphere blending to alter the release of a peptide from PLGA microspheres. Eur J Pharm Biopharm. 2000;50:263–70. doi: 10.1016/s0939-6411(00)00099-0. [DOI] [PubMed] [Google Scholar]
- 39.Mocellin S, Mandruzzato S, Zanovello P, Bronte V. Cancer rejection by the immune system: forcing the check-points of tumor immune escape. Drug Discov Today: Dis Mech. 2005;2:191–7. [Google Scholar]
- 40.Bharali DJ, Mousa SA, Thanavala Y. Micro- and nanoparticle-based vaccines for hepatitis B. Adv Exp Med Biol. 2007;601:415–21. doi: 10.1007/978-0-387-72005-0_44. [DOI] [PubMed] [Google Scholar]
- 41.Elamanchili P, Lutsiak CME, Hamdy S, Diwan M, Samuel J. Pathogen-mimicking nanoparticles for vaccine delivery to dendritic cells. J Immunother. 2007;30:378–95. doi: 10.1097/CJI.0b013e31802cf3e3. [DOI] [PubMed] [Google Scholar]