

Abstract
Human endometrium undergoes major gene expression changes, resulting in altered cellular functions in response to cyclic variations in circulating estradiol and progesterone, largely mediated by transcription factors and nuclear receptors. In addition to classic modulators, epigenetic mechanisms regulate gene expression during development in response to environmental factors and in some diseases and have roles in steroid hormone action. Herein, we tested the hypothesis that DNA methylation plays a role in gene expression regulation in human endometrium in different hormonal milieux. High throughput, genome-wide DNA methylation profiling of endometrial samples in proliferative, early secretory, and midsecretory phases revealed dynamic DNA methylation patterns with segregation of proliferative from secretory phase samples by unsupervised cluster analysis of differentially methylated genes. Changes involved different frequencies of gain and loss of methylation within or outside CpG islands. Comparison of changes in transcriptomes and corresponding DNA methylomes from the same samples revealed association of DNA methylation and gene expression in a number of loci, some important in endometrial biology. Human endometrial stromal fibroblasts treated in vitro with estradiol and progesterone exhibited DNA methylation changes in several genes observed in proliferative and secretory phase tissues, respectively. Taken together, the data support the observation that epigenetic mechanisms are involved in gene expression regulation in human endometrium in different hormonal milieux, adding endometrium to a small number of normal adult tissues exhibiting dynamic DNA methylation. The data also raise the possibility that the interplay between steroid hormone and methylome dynamics regulates normal endometrial functions and, if abnormal, may result in endometrial dysfunction and associated disorders.
Human endometrium, the obligate tissue for blastocyst implantation and embryonic development (1), undergoes complex molecular, cellular, and functional changes on a cyclic basis in response to the ovarian steroid hormones, estradiol (E2) and progesterone (P4) (2–4). In the proliferative phase, E2 induces cellular proliferation and angiogenesis (2), and after ovulation in the secretory phase, P4 induces secretory transformation of the epithelium and differentiation (decidualization) of the stromal fibroblasts (2–4). Extensive changes in the transcriptome and proteome are required for these morphologic and functional changes, which involve well-orchestrated gene expression regulation by well-characterized transcription factors, nuclear receptors, and coregulators (5–10). In addition to these well-characterized regulatory mechanisms, there is increasing evidence in steroid hormone–responsive tissues (11, 12), including endometrium (13), of changes in gene activity due to epigenetic modifications, although the involvement of epigenetic mechanisms such as DNA methylation is less well understood in cycling endometrium.
Methylation of the carbon-5 position of cytosine, mostly in the context of CpG dinucleotides, is the main epigenetic modification of DNA (14–17) and is essential for a properly functioning genome, including maintenance of chromosome stability and transcriptional repression, and is a rate-limiting step for many cellular transitions (eg, embryonic and germ cell development and nuclear reprogramming) (14, 18–20). DNA methylation at the promoter-associated CpG islands of genes is usually associated with gene silencing (21), whereas promoter CpG island hypomethylation (22, 23) and gene body CpG dinucleotide hypermethylation are associated with gene activation (24, 25). About 10% to 20% of genes display DNA methylation patterns in a tissue-specific manner, usually associated with tissue-specific patterns of gene expression (26). In human endometrium, DNA methyltransferase (DNMT1, DNMT3A, and DNMT3B) expression is cycle dependent (8, 27) and in endometrial explants DNMTs are regulated by P4 (27). Furthermore, the DNA demethylating agent 5-aza-2′-deoxycytidine increases ESR2 (estrogen receptor-β) expression in human endometrial stromal fibroblasts (eSFs) (28), underscoring the importance of epigenetic modifications in the endometrial cellular steroid hormone response. Whether the human endometrial DNA methylome is cycle dependent has not been established and is the subject of this article.
Herein, using a bisulfite-based quantitative assay that measures DNA methylation at 27 578 CpG sites, we investigated the DNA methylome of human endometrium to better understand the involvement of epigenetic mechanisms in hormonally regulated cyclic endometrial activity. Our results show dynamic, cycle phase–dependent DNA methylation correlating with gene expression changes in the endometrium at specific loci, underscoring an important role of this epigenetic modification in the steroid hormone response of this tissue.
Materials and Methods
Sample collection and processing
This study was approved by the Committee on Human Research of the University of California, San Francisco (UCSF). Eighteen eutopic endometrial tissue samples, n = 6 in each of the proliferative (PE), early secretory (ESE), and midsecretory (MSE) phases, were obtained after informed consent from subjects undergoing endometrial biopsy, hysterectomy, or gynecologic surgery for a benign condition (Table 1). All subjects were documented as not having endometriosis and not being pregnant and had no hormonal treatments within 3 months before sample acquisition. Tissue acquisition, processing, and storage were conducted by the UCSF National Institutes of Health Specialized Cooperative Centers Program in Reproductive and Infertility Research Human Endometrial Tissue and DNA Bank, as described previously (29). All subjects were nonsmokers with one exception (Table 1). Menstrual cycle phase was determined by histologic evaluation by 3 independent readers according to the Noyes criteria (30), by blood levels of estradiol and progesterone, and/or by unsupervised principal component analysis and independent hierarchical clustering analysis of microarray gene expression data (31). The mean ages of participants were 42.5 ± 1.44 years for the PE phase, 40.5 ± 2.72 years for the ESE phase, and 46.3 ± 1.44 years for the MSE phase (P > .05).
Table 1.
Sample Characteristics.
| Sample No. | Age, y | Weight, kg | BMI, kg/m2 | Ethnicity | Histology Reading | Microarray Phasing |
|---|---|---|---|---|---|---|
| Z19 | 39 | 82.6 | 25.5 | White | PE | PE |
| Z20 | 42 | NA | NA | White | PE | PE |
| Z21 | 44 | 59.0 | 23.9 | White | PE | PE |
| Z22 | 43 | 88.0 | 32.5 | Black | PE | PE |
| Z23 | 40 | 67.1 | 25.3 | White | PE | PE |
| Z24 | 47 | 111.6 | NA | White | PE | PE |
| Z25 | 44 | 65.0 | 24.5 | White | ESE | ESE |
| Z26 | 46 | 82.3 | 30.2 | White | ESE | ESE |
| Z27 | 40 | 83.4 | 31.4 | White | ESE | ESE |
| Z28 | 34 | 95.7 | 35.2 | White | ESE | ESE |
| Z29 | 45 | 65.8 | NA | Black | ESE | ESE |
| Z30 | 34 | 96.6 | 36.4 | White | ESE | ESE |
| Z31 | 49 | 64.5 | 25.2 | White | MSE | MSE |
| Z32 | 50 | 60.8 | 21.5 | White | MSE | MSE |
| Z33 | 42 | 61.3 | 21.2 | White | MSE | MSE |
| Z34 | 45 | 56.3 | 20.7 | White | MSE | MSE |
| Z35 | 46 | 79.0 | 28.0 | White | MSE | MSE |
| Z36 | 46 | 61.9 | 23.3 | White | MSE | MSE |
All subjects were nonsmokers (except Z21, PE, a past smoker), were not exposed to hormonal medications within 3 months before tissue sampling, and were confirmed as being not pregnant. Cycle phase was determined by histologic evaluation according to Noyes criteria and also by sample clustering in unsupervised principal component analysis of microarray data. Dating by both methods was consistent for all subjects.
DNA extraction
Genomic DNA was extracted from endometrial tissue samples using a NucleoSpin Tissue Kit (Macherey-Nagel Inc), according to the manufacturer's protocol, and was stored at −20°C until use.
DNA methylation analysis
Endometrial sample genomic DNA was bisulfite-converted using the EZ-96 DNA Methylation Kit (Zymo Research), according to the manufacturer's protocol, at the University of Southern California (USC) Epigenome Center. Quality controls for the amount and completeness of bisulfite conversion were conducted by a panel of MethyLight reactions, as described previously (32). All samples passed the initial quality control test and were further assayed by the Illumina Infinium HumanMethylation27 DNA methylation platform at the USC Epigenome Center, based on Illumina's specifications, as described previously (33). This Illumina platform interrogates DNA methylation levels of 110 microRNA-coding and 14 475 protein-coding genes at 27 578 CpG sites. Illumina platform probes are selected based on their differentially methylated status in cancers and during development.
DNA methylation values were scored as β values, calculated for each probe as the ratio of the methylated signal over the total fluorescent signal. β values range from 0 to 1, with lower levels of DNA methylation close to 0 and higher levels of DNA methylation close to 1 (33). DNA methylation measurement quality was assessed by the detection P value of each probe in each sample. Detection P values were calculated based on the difference in signal intensity of each probe compared with that of a set of 16 negative control probes. Probes with detection values of P < .05 were considered to have statistically significant differences from background and were retained for further data analysis. Probes with a detection value of P > .05 were marked with NA (not acceptable) and were excluded from analysis as nonsignificant DNA methylation detection values. By stringent technical quality criteria, 1 MSE sample with the highest probe dropout rate (1.8%) and an overall deviant DNA methylation distribution pattern on density plot compared with the rest of the samples was eliminated from subsequent analysis (Supplemental Figure 1).
The Illumina Infinium platform has been used in several studies (34, 35), and data obtained with this platform have been validated by other assays such as bisulfite genomic sequencing (36). The reproducibility of these data has been shown previously (33, 37), and we observed similar reproducibility with technical replicates of a subset of our samples as well (2 PE, 2 ESE, and 2 MSE; data not shown).
DNA methylation clustering and visualization
To investigate DNA methylation profiling across all samples, we selected the probes with the highest variation across all samples. The SD of each probe across all samples was calculated to identify the most variable probes, and 10% of all probes (2758) with the most variable DNA methylation levels across all samples were selected for cluster analysis. Probes with more than 1 NA dropout data point were eliminated. Two-way average-linkage unsupervised hierarchical clustering analysis was performed on the selected probes for both CpG sites and samples, using JMP software (38).
Global DNA Methylation Profile and Patterns of Endometrium
To better understand the global pattern of endometrial methylome, we identified loci that were highly methylated (β >0.8), were highly unmethylated (β <0.2), or had intermediate methylation levels (0.2 ≤ β ≤ 0.8) in each phase. We removed 25 probes with missing data in >1 sample from further analysis. We identified 2881 CpG sites to be highly methylated and 16 730 CpG sites to be highly unmethylated in each phase. We next identified 828 CpG sites that were highly methylated and 569 CpG sites that were highly unmethylated in 1 or 2 but not all 3 phases. The remaining 6746 CpG sites had intermediate levels of DNA methylation, with either similar or variable β values across the phases. Next, we determined what fractions of CpG sites within each group were located within or outside CpG islands.
Phase-specific changes in DNA methylation
CpG sites showing differential DNA methylation between cycle phases were identified using median β value differences as described previously (39). In brief, the median β value was calculated for each probe in each phase of the cycle excluding from the analysis probes with >2 missing values across all samples. Median β value differences between cycle phases were derived by subtracting the median β value of each probe in 1 phase from the corresponding value in another phase for the following comparisons: ESE vs PE, MSE vs ESE, and MSE vs PE. In all comparisons analysis included only differentially methylated probes with a detectable difference of 95% confidence (Δβ of ≥0.136), as described previously (33).
DNA methylation of human eSFs treated with hormones in vitro
Human endometrial tissue was obtained from normal controls through the UCSF Human Endometrial Tissue and DNA Bank, after written informed consent (as mentioned above). eSFs were treated with 10 nM E2, 10 nM E2 + 1 μM P4, or vehicle for 15 days in culture, as described previously (40). IGFBP1, a decidualization marker, was determined by ELISA (Alpha Diagnostic International). Cells from n = 4 subjects exhibiting robust decidualization after 15 days and parallel treatment groups and controls were selected for further analysis. DNA was extracted, bisulfite converted, and assayed on an Illumina Infinium 450K methylation platform at USC as described above. Genes with differential DNA methylation in E2 + P4 (corresponding to secretory phase) vs E2 (corresponding to proliferative phase) were selected and compared with genes observed in PE vs ESE and PE vs MSE from eutopic endometrium.
Gene expression microarray
RNA isolation, cDNA conversion, and microarray
Portions of the same tissue samples used for DNA methylation analysis were processed using TRIzol reagent (Invitrogen), and total RNA was isolated with DNase treatment using an RNeasy Plus Kit (QIAGEN), according to the manufacturer's protocol. RNA quality was assessed using the Bioanalyzer 2100 (Agilent Technologies). RNA samples were prepared for microarray analysis according to Affymetrix specifications. For each sample, 5 μg of total RNA was reversed transcribed to cDNA, followed by second-stand DNA generation using DNA polymerase and cRNA generation by in vitro transcription. Sample quality was assessed using the Agilent Bioanalyzer, and hybridization to an Affymetrix HU133 Plus 2.0 gene array interrogating >38 500 genes (Affymetrix, Inc) was conducted at the UCSF Genome Core facility as described previously (41).
Comparison of DNA methylation and gene expression data
The raw .cel expression data were normalized by the GCRMA method using GeneSpring GX 12.0 software (Agilent Technologies). Normalized expression and DNA methylation data were imported into R, and the corresponding probes from the 2 platforms were matched using the transcription unit identifier. The Spearman correlation was used to investigate the relationship between DNA methylation and gene expression values for each locus. In this analysis every DNA methylation probe for a given locus was compared individually with all transcripts of the corresponding locus to discern the effect of the localization of methylation changes on the corresponding gene expression.
Pathway analysis
Differentially methylated loci that were associated with gene expression changes (either positively or negatively) in each phase of the endometrium were examined using the DAVID (Database for Annotation, Visualization and Integrated Discovery) database (42) to elucidate functionally important genes and/or pathways.
Results
DNA methylation profiles of endometrium during the menstrual cycle
Cluster analysis
Genome-scale DNA methylation profiles of 27 578 CpG sites, associated with 14 475 genes, were assessed in 17 samples from human endometrium. Two-dimensional unsupervised hierarchical cluster analysis of all samples and the top 10% (2758) most variable probes revealed 2 main branches (Figure 1). The first branch included only secretory phase samples (4 of the 6 ESE and 4 of the 5 MSE); whereas the second branch included all of the PE samples and 3 secretory phase samples. It should be noted that for gene transcription changes, the greatest differences are seen in PE vs secretory endometrium and that ESE clusters with PE and not with MSE (31). The observation that cluster 1 included only secretory endometrium but not any PE samples indicates that the secretory phase has a methylation profile different from that of the proliferative phase, suggesting that DNA methylation profiles change during the menstrual cycle.
Figure 1.
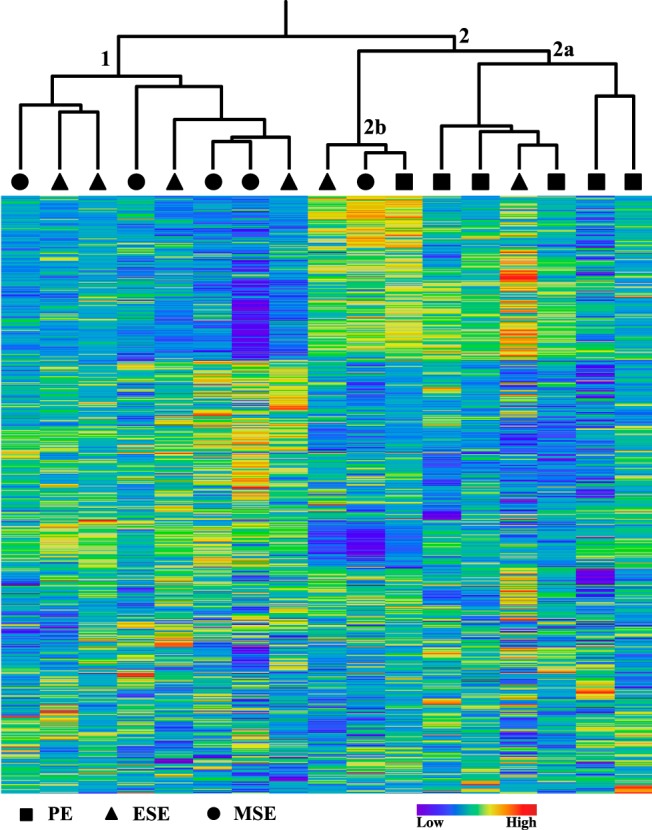
Two-dimensional unsupervised hierarchical cluster analysis of 17 samples and the top 10% (2578) of probes with the most variable DNA methylation levels. Cluster 1 includes only secretory samples, whereas cluster 2 includes all PE samples and 1 ESE sample in 2a and 1 PE, 1 ESE, and 1 MSE sample in 2b. Diamonds, PE; triangles, ESE; circles, MSE. The heat map represents less methylated (dark blue) to more methylated (dark red).
DNA methylation profile of the endometrium
The following analyses examined the changes in profiles and in patterns of DNA methylation in the endometrium and across the cycle by assessing the frequency of differentially methylated loci across the phases, whether changes involved gain or loss of methylation, chromosomal distribution, and genetic location of these changes, and whether differentially methylated loci were located within or outside a CpG island.
We compared the methylation level of every CpG site across all 3 phases to assess the overall methylation pattern and profile of endometrium (see Materials and Methods). We observed (Figure 2A) that more than 60% of CpG sites are highly unmethylated (β value <0.2), whereas only 10% of the CpG sites are highly methylated (β value >0.8) in all 3 phases. Only 4% of the CpG sites were highly methylated (2%) or highly unmethylated (2%) in 1 or 2 but not all 3 phases. The remaining 25% of the CpG sites showed moderate levels of methylation (0.2 ≤ β value ≤ 0.8), with either similar or variable levels across the 3 phases (Figure 2A).
Figure 2.
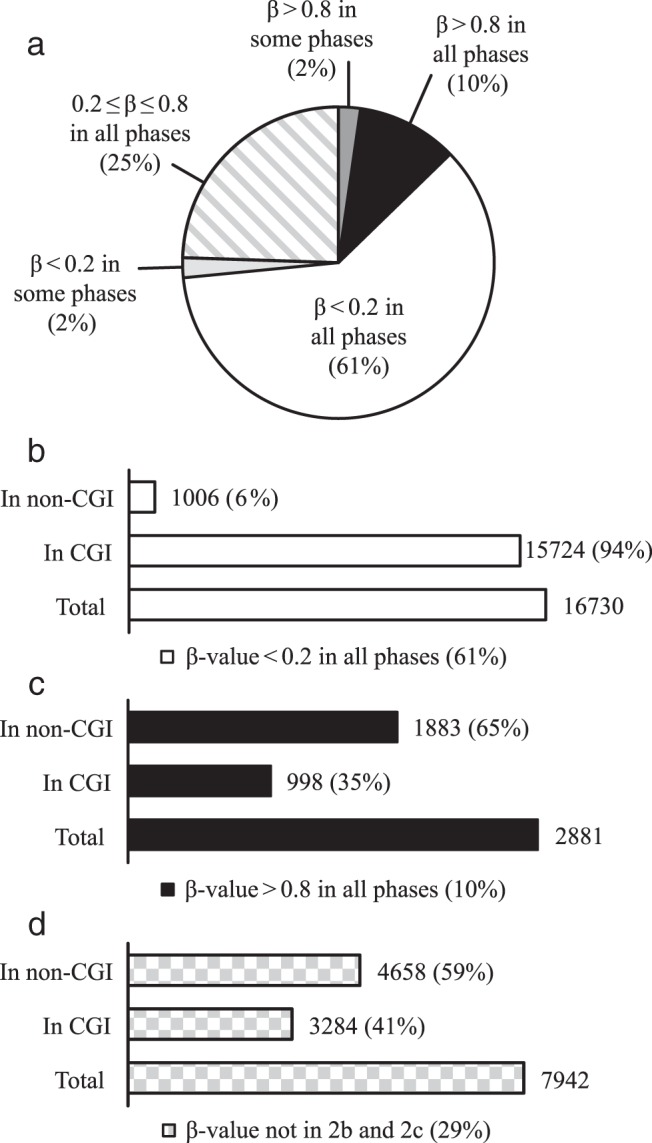
Global DNA methylation pattern of human endometrium. A, percentage of CpG sites that are highly methylated or highly unmethylated in all 3 phases, CpG sites that are either highly methylated or highly unmethylated in some but not all phases, and CpG sites with intermediate levels of methylation. B and C, distribution of highly unmethylated or highly methylated sites in all phases of the cycle based on their location within or outside CpG islands, respectively. D, distribution within or outside CpG islands of the highly methylated and highly unmethylated CpG sites in some (but not all) phases plus the CpG sites with intermediate DNA methylation levels. β, β value; CGI, CpG island; In CGI, within CpG island; non-CGI, outside CpG island.
We further investigated whether the methylation state of these CpG sites is associated with their location within or outside CpG islands. In the highly unmethylated group, 94% of CpG sites are located within a CpG island (Figure 2B). The highly unmethylated CpG islands also represent 79% of total CpG islands in the platform (Figure 3). Furthermore, of the highly methylated group, only 35% are within a CpG island (Figure 2C), representing a mere 5% of total CpG islands in the platform (Figure 3). On the other hand, the majority of highly methylated CpG sites are not associated with a CpG island (Figure 2C). This observation indicates that only a small fraction of the CpG islands is highly methylated in the endometrium, whereas most CpG islands are unmethylated. The data also indicate that hypermethylated CpG sites are mostly located outside CpG islands, in line with previous reports in somatic tissues that a small portion of CpG islands are methylated, whereas the majority remain unmethylated (19, 26) and most hypermethylated CpG sites are located outside CpG islands (19, 26) (see Supplemental Figure 2 for the overall distribution of DNA methylation levels in relation to location within or outside CpG islands).
Figure 3.
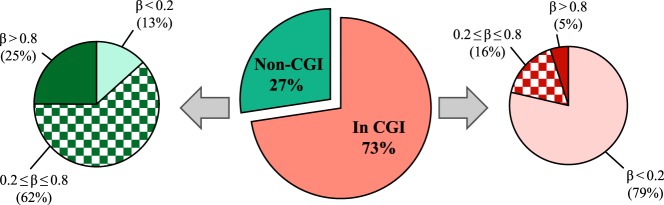
Distribution of CpG sites based on their location within or outside CpG islands. The middle pie chart represents the distribution of probes within or outside CpG islands on the 27K platform. Smaller pie charts depict the frequency of hypermethylation, hypomethylation, or intermediate methylation within each category. Light green and light red represent hypomethylated sites, dark green and dark red represent hypermethylated sites, and checkered green and checkered red represent intermediate methylated sites. β, β value; CGI, CpG island; In CGI, within CpG island; non-CGI, outside CpG island.
The remaining 29% of the CpG sites with variable and/or intermediate DNA methylation levels (Figure 2D) were subsequently analyzed to find differentially methylated CpG sites between the 3 cycle phases. In this group, 41% of the CpG sites are located within and 59% located outside CpG islands (Figure 2D), which corresponds to 16% of total CpG islands and 62% of all non-CpG islands, respectively (Figure 3).
Differentially methylated loci in different phases
We compared DNA methylation profiles and patterns in ESE vs PE, MSE vs ESE, and MSE vs PE to better understand epigenetic differences corresponding to distinct hormonal environments across the cycle, ie, the E2-dominant proliferative phase and the P4-dominant secretory phase.
We observed the most differences between MSE vs PE (66 CpG sites), followed by ESE vs PE (27 CpG sites), and the fewest changes between MSE vs ESE (22 CpG sites) (Figure 4). This is consistent with the unsupervised cluster analysis (Figure 1). Changes involved both gain and loss of methylation in a manner that roughly half of differentially methylated loci displayed gain of methylation and the other half loss of methylation in each comparison (Figure 4 and Supplemental Table 1). These changes were distributed across the genome, and we did not observe concentration of DNA methylation changes on specific chromosomes or on specific chromosomal hot spots in any of the comparisons (Supplemental Table 1). Of note are genes with known importance in endometrial biology in different phases of the cycle (Figure 4).
Figure 4.

Heat map of differentially methylated CpG sites between cycle phases. A, Differentially methylated CpG sites between ESE and PE. B, Differentially methylated CpG sites between MSE and ESE. C, Differentially methylated CpG sites between MSE and PE. Data are mean-centered and arranged top to bottom from low to high median β value in PE in panel A, in ESE in panel B, and in PE in panel C. Blue represents less methylation, and yellow represents more methylation compared with the mean. The location of CpG sites within or outside CpG islands is shown on the right. Black represents within and white represents outside CpG islands; gray represents NA. CGI, CpG island.
Assessment of the location of differentially methylated loci within or outside a CpG island revealed that methylation changes involved both CpG islands and non-CpG islands (Figures 4 and 5). Nearly 60% of the probes differentially methylated in ESE vs PE and in MSE vs ESE and 47% of the probes differentially methylated in MSE vs PE were located within a CpG island (Figure 5 and Supplemental Table 1).
Figure 5.
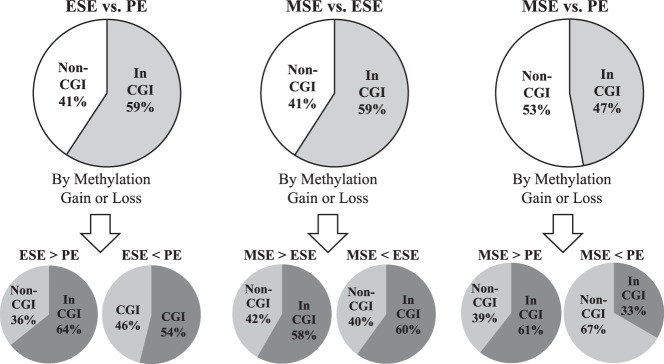
Distribution of differentially methylated loci between phases based on location within or outside a CpG island. The top panel shows the frequency of location within or outside a CGI for all differentially methylated loci between each comparison regardless of gain or loss of methylation, which is further divided into 2 pie charts based on gain or loss of methylation shown in the bottom panel. CGI, CpG island; Non-CGI, outside CpG island; In CGI, within CpG island.
In MSE vs PE, although the differentially methylated CpG sites are located almost equally within or outside CpG islands, we observe a different pattern when assessing the direction of the methylation changes. CpG sites that show gain of methylation in MSE compared with PE are mostly located within a CpG island (Figure 5), whereas CpG sites that show loss of methylation in MSE compared with MSE compared with PE are mostly located outside CpG islands (Figure 5). This is different from what is observed in MSE vs ESE or ESE vs PE (Figure 5); however, the number of differentially methylated CpG sites is smaller in these comparisons making conclusions less robust.
Differentially methylated genes between secretory and proliferative endometrium
Differentially methylated loci were surveyed to identify genes that differed in methylation status in the secretory phases (ESE and MSE) compared with those in the proliferative phase. As shown in Table 2, some loci including COL11A2, ID2, NFAM1, RUNX3, and ZNF57 were more methylated in MSE and ESE vs PE (Figure 6, panel I), whereas other loci including ADORA1, C21orf128, SMCR7, and TRPV3 were less methylated in MSE and ESE vs PE (Figure 6, panel II). Methylation changes of most of these loci were associated with gene expression changes (see below).
Table 2.
Selection of Differentially Methylated Probes Between Cycle Phases
| Illumina Probe ID | Gene Symbol | Product | CpG Island | Median Difference (Absolute Value) |
|---|---|---|---|---|
| PE < ESE | ||||
| cg03958979 | NR2E1 | Nuclear receptor subfamily 2; group E; member 1 | Y | 0.14 |
| cg16823701 | IFNA1 | Interferon; α 1 | N | 0.16 |
| cg12626411 | PRG4 | Proteoglycan 4 | N | 0.16 |
| cg17901463 | GSTM1 | Glutathione S-transferase M1 isoform 1 | Y | 0.18 |
| cg17568996 | NFAM1 | NFAT activation molecule 1 precursor | N | 0.18 |
| cg03171924 | RUNX3 | Runt-related transcription factor 3 isoform 2 | Y | 0.14 |
| cg10143146 | COL11A2 | Collagen; type XI; α 2 isoform 3 preproprotein | Y | 0.18 |
| cg13055278 | ID2 | Inhibitor of DNA binding 2 | Y | 0.17 |
| cg22074666 | C21orf7 | Chromosome 21 open reading frame 7 | N | 0.16 |
| PE > ESE | ||||
| cg11719784 | ADORA1 | Adenosine A1 receptor | N | 0.16 |
| cg18085517 | TRPM1 | Transient receptor potential cation channel; subfamily M; member 1 | N | 0.16 |
| cg12728032 | ELF5 | E74-like factor 5 ESE-2b | Y | 0.14 |
| cg16706631 | HIST1H4E | H4 histone family; member J | Y | 0.14 |
| cg19766460 | C21orf128 | Hypothetical protein LOC150147 | N | 0.27 |
| cg26771272 | SMCR7 | Smith-Magenis syndrome chromosome region; candidate 7 | Y | 0.15 |
| cg05790038 | TRPV3 | Transient receptor potential cation channel; subfamily V; member 3 | N | 0.15 |
| cg08158289 | KIAA0141 | Hypothetical protein LOC9812 | Y | 0.16 |
| cg00852964 | VNN1 | Vanin 1 precursor | N | 0.18 |
| cg00204262 | ECH1 | Peroxisomal enoyl-coenzyme A hydratase-like protein | Y | 0.21 |
| ESE < MSE | ||||
| cg20022541 | FAM181A | Hypothetical protein LOC90050 | N | 0.26 |
| cg08085267 | C17orf57 | Hypothetical protein LOC124989 | Y | 0.19 |
| cg20098118 | UXT | Ubiquitously-expressed transcript isoform 2 | Y | 0.19 |
| cg06074920 | KRT34 | Type I hair keratin 4 | N | 0.17 |
| cg16242770 | KRTAP17-1 | Keratin associated protein 17-1 | N | 0.15 |
| cg15928398 | ST6GAL1 | Sialyltransferase 1 isoform a | Y | 0.15 |
| cg00662775 | TCEAL4 | Transcription elongation factor A (SII)-like 4 | Y | 0.14 |
| ESE > MSE | ||||
| cg27202708 | C1orf65 | Hypothetical protein LOC164127 | Y | 0.16 |
| cg05333568 | C1orf65 | Hypothetical protein LOC164127 | Y | 0.16 |
| cg27371741 | TDGF1 | Teratocarcinoma-derived growth factor 1 | Y | 0.17 |
| cg26511075 | RANBP3 liter | Hypothetical protein LOC202151 | N | 0.17 |
| cg19195724 | SNORD109A | N | 0.18 | |
| cg03882305 | TRIM74 | Hypothetical protein LOC378108 | Y | 0.22 |
| cg13181019 | MPP7 | Palmitoylated membrane protein 7 | N | 0.23 |
| cg01120761 | CLEC4C | C-type lectin domain family 4; member C isoform 1 | N | 0.29 |
| PE < MSE | ||||
| cg16242770 | KRTAP17-1 | Keratin associated protein 17-1 | N | 0.21 |
| cg04797323 | SOCS2 | Suppressor of cytokine signaling-2 | Y | 0.18 |
| cg15928398 | ST6GAL1 | Sialyltransferase 1 isoform a | Y | 0.21 |
| cg10143146 | COL11A2 | Collagen; type XI; α 2 isoform 3 preproprotein | Y | 0.14 |
| cg03171924 | RUNX3 | Runt-related transcription factor 3 isoform 2 | Y | 0.14 |
| cg00662775 | TCEAL4 | Transcription elongation factor A (SII)-like 4 | Y | 0.14 |
| cg08209724 | ZNF207 | Zinc finger protein 207 isoform b | Y | 0.19 |
| cg15308737 | ARSG | Arylsulfatase G | N | 0.16 |
| cg12111714 | ATP8A2 | ATPase; aminophospholipid transporter-like; class I; type 8A; member 2 | Y | 0.14 |
| cg25569462 | TRIML2 | Hypothetical protein LOC205860 | N | 0.18 |
| cg20022541 | FAM181A | Hypothetical protein LOC90050 | N | 0.26 |
| cg08634464 | ZNF57 | Hypothetical protein LOC126295 | Y | 0.14 |
| cg20098118 | UXT | Ubiquitously-expressed transcript isoform 2 | Y | 0.18 |
| cg06074920 | KRT34 | Type I hair keratin 4 | N | 0.15 |
| cg17568996 | NFAM1 | NFAT activation molecule 1 precursor | N | 0.15 |
| cg13055278 | ID2 | Inhibitor of DNA binding 2 | Y | 0.14 |
| PE > MSE | ||||
| cg11719784 | ADORA1 | Adenosine A1 receptor | N | 0.14 |
| cg21296602 | TAF1D | Hypothetical protein MGC5306 | N | 0.16 |
| cg25141490 | IL17B | Interleukin 17B precursor | N | 0.19 |
| cg24512973 | MUC1 | MUC1 mucin isoform 1 precursor | Y | 0.14 |
| cg12743398 | SULT1A2 | Sulfotransferase family; cytosolic; 1A; phenol-preferring; member 2 | N | 0.17 |
| cg12493906 | MMP26 | Matrix metalloproteinase 26 preproprotein | N | 0.14 |
| cg26771272 | SMCR7 | Smith-Magenis syndrome chromosome region; candidate 7 | Y | 0.17 |
| cg19766460 | C21orf128 | Hypothetical protein LOC150147 | N | 0.24 |
| cg15928132 | CCKAR | Cholecystokinin A receptor | N | 0.14 |
| cg03870862 | ZC3H3 | Zinc finger CCCH-type domain containing 3 | Y | 0/14 |
| cg03270204 | DDR1 | Discoidin domain receptor family; member 1 isoform b | N | 0.15 |
| cg04576021 | HLA-DOB | Major histocompatibility complex; class II; DO β precursor | N | 0.15 |
| cg20993403 | EPB41L1 | Erythrocyte membrane protein band 4.1-like 1 isoform a | N | 0.14 |
| cg00035347 | NT5C2 | 5′-Nucleotidase; cytosolic II | N | 0.14 |
| cg05790038 | TRPV3 | Transient receptor potential cation channel; subfamily V; member 3 | N | 0.15 |
| cg03665457 | HSD17B7P2 | 17-β-Hydroxysteroid dehydrogenase type VII-like isoform 2 | Y | 0.17 |
| cg25527547 | PLOD3 | Procollagen-lysine; 2-oxoglutarate 5-dioxygenase 3 precursor | N | 0.18 |
| cg14862827 | SUSD1 | Sushi domain containing 1 | Y | 0.19 |
| cg16173109 | FLJ38379 | Y | 0.20 | |
| cg13181019 | MPP7 | Palmitoylated membrane protein 7 | N | 0.30 |
| cg01120761 | CLEC4C | C-type lectin domain family 4; member C isoform 1 | N | 0.24 |
| cg27202708 | C1orf65 | Hypothetical protein LOC164127 | Y | 0.14 |
Figure 6.

Heat map of a selected number of loci differentially methylated between phases. I, Genes more methylated in MSE and ESE compared to PE. II, Genes less methylated in MSE and ESE than in PE. III, Genes more methylated in MSE than in PE and ESE. IV, Genes less methylated in MSE than in PE and ESE. Blue represents less methylation, and yellow represents more methylation compared with the mean.
An additional search identified genes whose methylation status remained unchanged throughout the proliferative (peak E2) and early secretory (low P4) phases but differed in the MSE (peak P4) phase (ie, MSE vs ESE and in MSE vs PE). Loci with more methylation in MSE were FAM181A, KRT34, KRTAP17–1, ST6GAL1, TCEAL4, and UXT; loci with less methylation in MSE compared with that in the earlier phases of the cycle included C1orf65, CLEC4C, and MPP7 (Table 2 and Figure 6, panels III and IV; see Supplemental Table 1 for the full list of phase-dependent differentially methylated genes).
Relationship between DNA methylation status and gene expression
To investigate the relationship between DNA methylation status and gene expression in normal cycling endometrium, DNA methylation data from the Infinium platform were compared with the corresponding gene transcript levels on the Affymetrix U133 Human Plus 2.0 array (Tables 3 and 4), using the same (paired) endometrial samples. Although it is customary to merge the data of the different probes for individual loci, this practice precludes discriminating localization of methylation changes within loci and the potential effect on gene regulation. Therefore, in this analysis, data from each probe for every locus on the Illumina platform were matched to all the transcripts of the corresponding locus in the Affymetrix platform (see Materials and Methods).
Table 3.
Gene Expression and DNA Methylation Association Based on Location Within or Outside CpG Island
| ESE vs PE |
MSE vs PE |
MSE vs ESE |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Mean ρ | Total Probe | % | Mean ρ | Total Probe | % | Mean ρ | Total Probe | % | |
| All negative association | −0.34 | 34 | 63 | −0.32 | 82 | 58 | −0.26 | 12 | 30 |
| All positive association | 0.25 | 20 | 37 | 0.29 | 60 | 42 | 0.26 | 28 | 70 |
| Within CpG island (negative association) | −0.35 | 14 | 54 | −0.24 | 33 | 51 | −0.26 | 6 | 30 |
| Within CpG island (positive association) | 0.28 | 12 | 46 | 0.29 | 32 | 49 | 0.27 | 14 | 70 |
| Outside CpG island (negative association) | −0.34 | 20 | 71 | −0.37 | 49 | 64 | −0.26 | 6 | 30 |
| Outside CpG island (positive association) | 0.22 | 8 | 29 | 0.30 | 28 | 36 | 0.25 | 14 | 70 |
Table 4.
Selected Loci With Strong and Moderate Correlation of Gene Expression and DNA Methylation
| Illumina Probe ID | Affymetrix Identification | Gene Symbol | Gene Name | CpG Island | Spearman ρ |
|---|---|---|---|---|---|
| ESE vs PE | |||||
| cg11719784 | 216220_s_at | ADORA1 | Adenosine A1 receptor | No | −0.433 |
| cg12728032 | 220625_s_at | ELF5 | E74-like factor 5 | Yes | 0.300 |
| cg17901463 | 215333_x_at | GSTM1 | Glutathione S-transferase μ 1 | Yes | −0.367 |
| cg09879869 | 236004_at | MUDENG | Adaptor-related protein complex 5, μ 1 subunit | No | −0.700 |
| cg17568996 | 230322_at | NFAM1 | NFAT activating protein with ITAM motif 1 | No | −0.533 |
| cg03958979 | 207443_at | NR2E1 | Nuclear receptor subfamily 2, group E, member 1 | Yes | −0.317 |
| cg12626411 | 206007_at | PRG4 | Proteoglycan 4 | No | −0.483 |
| cg03171924 | 234928_x_at | RUNX3 | Runt-related transcription factor 3 | Yes | −0.417 |
| cg01009664 | 206622_at | TRH | Thyrotropin-releasing hormone | Yes | −0.400 |
| cg05790038 | 1555291_at | TRPV3 | Transient receptor potential cation channel, subfamily V, member 3 | No | −0.500 |
| cg00186701 | 213122_at | TSPYL5 | TSPY-like 5 | Yes | −0.667 |
| MSE vs PE | |||||
| cg11719784 | 205481_at | ADORA1 | Adenosine A1 receptor | No | 0.627 |
| cg00629217 | 223611_s_at | LNX1 | Ligand of numb-protein × 1, E3 ubiquitin protein ligase | No | 0.500 |
| cg08477744 | 203417_at | MFAP2 | Microfibrillar-associated protein 2 | No | −0.700 |
| cg12493906 | 220541_at | MMP26 | Matrix metallopeptidase 26 | No | −0.745 |
| cg24512973 | 207847_s_at | MUC1 | Mucin 1, cell surface associated | Yes | −0.655 |
| cg17568996 | 243099_at | NFAM1 | NFAT activating protein with ITAM motif 1 | No | −0.527 |
| cg00035347 | 236703_at | NT5C2 | 5′-nucleotidase, cytosolic II | No | −0.791 |
| cg03171924 | 234928_x_at | RUNX3 | Runt-related transcription factor 3 | Yes | −0.455 |
| cg21296602 | 222728_s_at | TAF1D | TATA box binding protein (TBP)-associated factor, RNA polymerase I, D | No | −0.545 |
| cg06194808 | 235817_at | TMEM184A | Transmembrane protein 184A | No | −0.818 |
| cg25569462 | 1552580_at | TRIML2 | Tripartite motif family-like 2 | No | −0.609 |
| cg05790038 | 1555291_at | TRPV3 | Transient receptor potential cation channel, subfamily V, member 3 | No | −0.327 |
| MSE vs ESE | |||||
| cg25598083 | 202982_s_at | ACOT2 | Acyl-CoA thioesterase 2 | No | −0.714 |
| cg05130485 | 207686_s_at | CASP8 | Caspase 8, apoptosis-related cysteine peptidase | No | 0.500 |
| cg13181019 | 238778_at | MPP7 | Membrane protein, palmitoylated7 (MAGUK p55 subfamily member 7) | No | −0.333 |
| cg27371741 | 206286_s_at | TDGF1 | Teratocarcinoma-derived growth factor 1 | Yes | −0.452 |
Changes in DNA methylation show positive or negative correlation with changes in gene expression
Because the relationship between DNA methylation and gene expression is not linear (16), the Spearman correlation was used to examine this relationship. Hypermethylation of CpG dinucleotides located in a CpG island at the 5′ region of a gene has been shown to be associated with gene silencing, whereas hypermethylation in the body of the gene is suggested to be associated with gene activation (19) (see Discussion). Therefore, the relationship between changes in DNA methylation and changes in gene expression was assessed for both positive and negative associations as well as the location of the interrogated CpG in the context of CpG islands (Tables 3 and 4).
We compared the frequency of loci that showed a negative association vs those with a positive association. In ESE vs PE, the percentage of loci with negative association (63%) was higher than that with positive association (37%), and negative association was also stronger (mean ρ = −0.341 vs mean ρ = 0.252, respectively) (Table 3). Similar results were observed in the MSE vs PE comparison (Table 3). However, in MSE vs ESE, the percentage of loci showing a negative association was less than that of those with a positive association (30% vs 70%, respectively); however, associations in each direction were of similar strength (ρ = −0.262 vs ρ = 0.255) (Table 3).
Association of DNA methylation with gene expression based on location within or outside a CpG island
The association of DNA methylation with gene expression was further analyzed by dividing differentially methylated CpG sites into 2 groups: within a CpG island or outside a CpG island (Table 3).
For ESE vs PE and MSE vs PE, equal numbers of differentially methylated loci showed positive or negative association with gene expression, when located within a CpG island. However, when outside a CpG island, most differentially methylated loci showed a negative association with gene expression (Table 3). In MSE vs ESE, most differentially methylated loci showed a positive association with gene expression, either within or outside a CpG island (Table 3).
In summary, overall, in the proliferative vs each of the secretory subphases, equal numbers of differentially methylated loci located within a CpG island had positive or negative correlation with gene expression, whereas a majority of the differentially methylated sites located outside a CpG island had a negative association. This result is in line with current understandings that changes in CpG island methylation, sometimes, but not always, result in negative association with gene expression, whereas changes in DNA methylation in non-CpG island CpG sites, mostly at the 5′ positions of the genes are usually negatively associated with gene expression (19, 26).
Loci with high correlation between DNA methylation and gene expression
In response to E2 and P4, human endometrium undergoes global transcriptomic changes involving genes in various signaling pathways, accounting for many of the dynamic histologic and functional events observed across the cycle (31, 43). A goal herein was to investigate the biological importance of genes whose DNA methylation changes show a correlation with changes in gene expression. Table 4 shows a partial list of the genes with strong and moderate correlation of the changes in DNA methylation and gene expression in the different phase comparisons (see Supplemental Table 2 for the full gene list).
To better understand the biological relevance of these genes, we interrogated their involvement in molecular pathways and their potential functions within endometrium. Pathway analysis using the DAVID database (42) revealed principal pathways represented by genes with correlation between DNA methylation and gene expression changes in ESE vs PE and included regulation of transcription (ID2, ELF5, NFAM1, IFI16, NR2E1, and RUNX3), regulation of apoptosis (IFI16, ADORA1, NR2E1, and RUNX3), and cell proliferation and regulation of RNA metabolic process (ID2, ELF5, IFI16, NR2E1, and RUNX3). Main pathways associated with changes in MSE vs PE are transmembrane (MUC1, DDR1, ST6GAL1, DRD5, TRPV3, CLIC6, TMEM184A, NFAM1, ADORA1, HLA-DOB, and ST6GALNAC1) and extracellular matrix and secreted proteins (MUC1, DDR1, ST6GAL1, MMP26, MFAP2, and COL11A2). Main pathways related to changes in MSE vs ESE include blood vessel morphogenesis (TDGF1, CASP8, and WT1). Several of these genes are also known to be important either in endometrial cyclicity, preparing for receptivity to embryonic implantation, and apoptosis or are endometrial cell specific (see Discussion), including RUNX3, important in TGF-β signaling; GSTM1, important in the response to oxidative stress; members of the matrix metalloproteinase family (MMP26); nuclear receptor subfamily 2 (NR2E1); and others (see Table 4 for a selected list by cycle phase and Supplemental Table 2 for the full gene list).
DNA methylation changes in eSFs in different hormonal milieux
To gain insight beyond global endometrial tissue changes in DNA methylation in different hormonal milieux, we investigated the DNA methylation status of human eSFs treated with E2 + P4 vs E2 in vitro (mimicking secretory vs proliferative phases, respectively, in vivo). Our preliminary analysis revealed DNA methylation changes for a number of genes, corresponding to either gain or loss of methylation similar to the observed changes in the endometrial tissue across the phases (Supplemental Table 3). Several of these genes also showed association of DNA methylation with changes in gene expression in the endometrium (Supplemental Table 2). These data support our whole-tissue observations of changes in DNA methylation across the cycle in different hormonal milieux.
Discussion
Dynamic DNA methylation and association with cyclic ovarian hormones
Endometrial DNA methylation changes during the menstrual cycle
The finding of dynamic changes in DNA methylation in human endometrium in different hormonal milieux is unique. Although extensive epigenetic reprogramming occurs in the preimplantation embryo (18, 20, 44) and during gametogenesis in primordial germ cells (45, 46), dynamic genome-wide changes in DNA methylation in normal adult tissues have been reported only in brain (47–49) and hematopoietic stem cell differentiation (50) and now in cycling human endometrium. This is in addition to DNA methylation changes that occur in cancer (17, 51, 52), infertility (39), aging (53, 54) and in response to environmental factors (55). How the DNA methylation changes reported here occur within the relatively short time span of a human menstrual cycle is not clear and warrants further investigation in longitudinally collected samples.
Epigenetic mechanisms and modifications in human endometrium
Various enzymes important in epigenetic processes have been reported in human endometrium. These include constitutive expression of histone deacetylase inhibitor 3, modest elevation of histone deacetylase 2 in the secretory phase (56) and varying expression of DNA methyltransferases (DNMT1, DNMT3A, and DNMT3B) during the cycle (8, 27) and with E2 and P4 treatment of endometrial explants (57). In addition, levels of immunoreactive 5-methylcytosine vary in glandular epithelial and stromal cells across the cycle (58). Herein, we found that the DNA methylation profiles of human endometrium differ in different phases of the cycle, resulting in the segregation of proliferative from secretory phase samples by unsupervised cluster analysis of the differentially methylated loci. Furthermore, the greatest DNA methylation differences are between the proliferative (peak E2) and midsecretory (peak P4) phases, consistent with periods of greatest changes in gene expression and suggesting a relationship between DNA methylation and endometrial gene expression across the cycle. In addition, our preliminary in vitro experiments corresponding to hormonal milieux of peak E2 and peak P4 show DNA methylation changes of several of the same genes. The fact that these epigenetic modifications occur in human endometrium during its hormonally regulated cyclic activity further suggests a relationship between steroid hormone action and endometrial DNA methylation dynamics (see below).
Steroid hormones and epigenetic changes in human endometrium
A major characteristic of human endometrium is that it undergoes cellular proliferation, differentiation, and regeneration on a cyclic basis. The question arises as to whether changes observed in the endometrial methylome occur in response to ovarian hormones or whether the DNA methylation changes facilitate endometrial responsiveness to these hormones, or perhaps it is a combination of both.
A growing body of literature supports that the epigenome can be affected by hormones or their disruption, such as by estrogen agonists. For example, bisphenol A (BPA), a weak estrogen agonist (59) induces hypomethylation and increased expression of the Agouti gene (60) in mice exposed prenatally. Mouse models of prenatal diethylstilbestrol exposure and genital tract neoplasia show hypomethylation and overexpression of EGF and SRF, estrogen-sensitive LTF (61), and HOXA10 hypermethylation, resulting in altered expression in the developing mouse uterus (62). Diethylstilbestrol, an estrogen agonist, causes vaginal clear-cell adenocarcinoma in adult women exposed in utero (63) and has been associated with epigenetic modifications in uterine tissues. Methoxychlor, another estrogenic endocrine disruptor, affects DNA methylation and alters reproductive senescence and estrous cyclicity in mice exposed prenatally (64).
In contrast to steroid hormones affecting the epigenome as part of their mechanism of action, it is possible that epigenetic changes could modulate steroid hormone action per se. For example, during early mouse postnatal development, Esr1 mRNA expression decreases within the cortex of the brain, coinciding with progressive gain of methylation in several Esr1 gene promoters (65). In addition, the methyl-DNA binding protein, MeCP2, is associated with the Esr1 promoter as it becomes methylated, suggesting a role for DNA methylation in Esr1 mRNA suppression in the postnatal brain (65). The preoptic area and hypothalamus are organized by early estradiol exposure (48) and methylation of Esr1 is altered by estradiol (49). Moreover, the brain can undergo epigenetic changes during development (47) and across the lifespan in response to varying hormonal milieu (47, 48). In addition, hypomethylation of an ESR2 promoter CpG island was shown to result in higher mRNA and protein levels in endometriosis (28).
Taken together, these findings suggest a dynamic interplay between DNA methylation and a changing hormonal milieu. We postulate that, in human endometrium, hormonal changes affect DNA methylation and changes in DNA methylation patterns play a crucial role in endometrial responsiveness to hormonal changes. This hypothesis is of great interest in our laboratory and awaits further experimental testing and validation.
DNA methylation and gene expression in human endometrium
Global methylation profile and pattern in the endometrium
Besides the small fraction of CpG sites with dynamic DNA methylation levels, we observed that across all phases, most CpG islands are unmethylated and only a very small percentage are hypermethylated. In addition, most of the hypermethylated loci are in non-CpG islands. This observation is in line with somatic tissue global DNA methylation patterns of hypermethylation of non-CpG island CpG sites and a small fraction of CpG islands, with most CpG islands remaining unmethylated (19, 66). Furthermore, there are tissue-specific CpG island methylation and tissue-specific unmethylated regions (26). Understanding which fractions of these highly methylated CpG islands or the unmethylated regions in our data are shared with other somatic tissues and which are endometrium specific requires further investigation and comparison with other somatic tissue methylomes and remains to be elucidated.
In addition to tissue-specific CpG islands or unmethylated regions, partially methylated domains (PMDs) (67) usually covering long genomic regions that could mark transcriptionally repressed regions, including genes with methylation levels of 0.2 ≤ β ≤0.8, are also potentially tissue specific (68). Although there is evidence suggesting they may be found in fibroblasts (69, 70), they may not be present in all adult tissues (71). Whether some of the repressed genes observed in endometrium with intermediate methylation are in fact associated with PMDs specific to endometrium is yet to be determined, using in-depth sequencing methods that investigate large regions of the genome. It is possible that the localization of the genes within the PMDs may contribute to their down-regulation during the menstrual cycle.
Mechanisms of de novo methylation and demethylation in endometrium
De novo methylation.
We observed specific de novo methylation in different cycle phases. In other somatic cells, 2 main mechanisms are involved in de novo methylation of targeted CpG islands: (1) the CpG islands must contain signals to direct the methylation, eg, mediated by histone methyltransferases that recruit DNA methylases (72–74); or (2) removal of transcription factors and/or nucleosomes containing histone 3 lysine 4 trimethylation (H3K4me3) in response to hormonal changes that would otherwise protect against de novo methylation. The latter is based on data suggesting that many unmethylated CpG islands near transcription start sites contain transcription factor binding sites (66) and nucleosomes containing H3K4me3 (75, 76) that may be inhibiting DNA methylation. Methylation of non-CpG island regions can have a direct impact on binding to their recognition sequences of transcription factors that play crucial roles during the menstrual cycle.
Demethylation.
Besides de novo methylation of CpG islands, we also observed demethylation. In the case of tissue-specific demethylation, it has been suggested that tissue-specific transacting factors (77, 78) and transcription factor binding may play a role (79, 80). Demethylation of genes is usually associated with gene activation, as was observed in our analysis, although its causal role is not clear. In the endometrium, phase-specific transcription factors may be involved in this process. Demethylation with regard to hormonal changes has been reported in enhancers, resulting in their subsequent activation (79, 81). Enhancers, located at variable distances to transcription start sites (TSSs), are generally CpG-poor with variable levels of methylation (82, 83) and play crucial roles during development. The relationship between methylation, transcription factor binding, and enhancer activity is complex and not well understood. Further investigation is anticipated to understand better how ovarian hormones affect transcription factor binding and enhancers/insulators related to changing levels of DNA methylation at various locations of a transcriptional unit in human endometrium.
Relationship of DNA methylation with gene expression
To better understand the role of DNA methylation in cycling endometrium, we investigated its relationship to gene expression changes in paired samples and found that for some genes, increased DNA methylation correlated with decreased levels of transcription, but as observed before (84), for a number of other loci there was a positive correlation between methylation and gene expression. DNA methylation of CpG islands at the promoter is generally, but not always, assumed to be negatively associated with gene expression. We also observed that methylation at CpG islands showed negative, positive, or no association with gene expression. Besides changes in CpG islands, we observed DNA methylation changes at non-CpG island regions. Unlike CpG islands at the 5′ end of genes, substantial fluctuations occur in these regions; however, usually an inverse relationship between methylation of the non-CpG island loci and gene expression exists (19). Again, we observed that most of these changes are negatively associated with gene expression, particularly when changes in the proliferative vs secretory phases are compared.
The precise role of DNA methylation in gene expression control is not fully understood. Although strong evidence suggests that there is no transcription initiation when CpG islands at the TSS are methylated (85), it is not clear which comes first, methylation or silencing. It has been suggested that DNA methylation serves to lock the silent state (19), but there is evidence suggesting it may also be involved in initiation of silencing (50). This is important in the interpretation of our results. Given the relatively short time period between the phases of the cycle and the higher frequency of gain of methylation at the CpG islands than of non-CpG islands in our study, it is possible that these changes may play a more instructive role in gene silencing than just serving as a locking tool for already repressed transcription induced by other mechanisms.
Biologically important genes in cycling endometrium
Differentially methylated genes with functional importance
We observed herein that DNA methylation changes affecting gene expression involve genes in signaling pathways including regulation of transcription and proliferation and transmembrane proteins. Several of these genes, such as RUNX3, DDR1 (NEP), MMP26, and MUC1, have been shown by many groups to play significant roles in endometrial biology, function, and embryonic implantation. Others such as NR2E1 and GSTM1 are also known to be important in biological processes with potential significance in endometrium, which remain to be elucidated. Several of the genes also show changes in DNA methylation when eSF cells are treated with E2 + P4 vs E2 (Supplemental Table 3). These genes are discussed below.
RUNX3.
RUNX3 (runt-related transcription factor 3) is involved in TGF-β signaling (86) important in cycling endometrium (87). Knockout of Runx3 in mice resulted in atrophic uteri compared with those in the wild type with less developed endometrial layer, uterine glands, thinner stromal layer, and no E2-induced epithelial proliferation (88). The lower mRNA levels of TGF β1 and TGFβ3 and lack of E2-induced EGF expression in stromal cells may be associated with the suppressed E2-dependent epithelial proliferation and suggests a regulatory role of Runx3 in the E2-induced uterine growth (87). In wild-type mice, Runx3 showed differential expression during early pregnancy with low expression on days 1 to 4 and increased expression in the stromal cells surrounding the implanting blastocyst on day 5, followed by high expression in the decidual cells from days 6 to 8. Similar results were observed in artificial decidualization and activated implantation in delayed uterus, suggesting an important role for Runx3 during mouse implantation and decidualization (89). Multiple transcripts of this gene exist, for some of which the CpG island is located before the TSS, whereas for others it is located within the gene body (analysis on the UCSC genome database). Herein, we observed that RUNX3 is hypermethylated in the secretory phase in both ESE and MSE. Of the 3 transcripts represented on the Affymetrix platform, the CpG island hypermethylation was associated with decreased gene expression for 1 transcript and showed no association with the others. We also observed increased methylation of RUNX3 in eSFs treated with E2 + P4 (decidualized) vs E2, confirming our initial observation. Hypermethylation and the subsequent decrease in transcription of RUNX3 may be involved in the suppression of endometrial epithelial/stromal cell proliferation in secretory phase endometrium.
DDR1.
DDR1 (discoidin domain receptor discoidin domain receptor tyrosine kinase 1; NEP) is suggested to play an important role in regulating endometrial cellular proliferation. It cleaves and inactivates ET-1 (endothelin-1), which stimulates phosphorylation of Akt and DNA synthesis in endometrial stromal cells via the ET(A) receptor and phosphatidylinositol-3 kinase signaling pathways (90). It is expressed in decidualized eSFs but not in endometrial epithelium or nondecidualized stromal fibroblasts (90). Our data show that it is more methylated in whole endometrial tissue in PE and becomes less methylated in MSE, resulting in increased expression in MSE. Furthermore, our in vitro results show higher methylation when endometrial stromal fibroblasts are treated with E2 and lower methylation with E2 + P4 treatment, consistent with the in vivo data.
MMP26.
MMP26 (matrix metalloproteinase-26) activates MMP9, which exhibits proteolytic activity on a large number of extracellular and basement membrane proteins (91, 92). Its expression pattern begins in the proliferative phase, peaks at midcycle, and then decreases to nondetectable levels in the late secretory and menstrual phases (93). In vitro expression of MMP26 in endometrial explants requires both E2 and P4. In endometrium of pregnant rhesus monkeys (94) the average endometrial levels of MMP26 mRNA (and protein) are high on day 12 of pregnancy but decrease significantly on days 18 and 26 with intense localization in the glandular epithelium on day 12 and in the walls of spiral arterioles adjacent to the implantation site on day 26. The spatiotemporal expression in early pregnancy suggests an important role of MMP26 in tissue remodeling regulation of glandular epithelium and spiral arteries. The MMP26 CpG site is located at a non-CpG island promoter region and is demethylated in MSE vs PE, with a strong negative correlation with its gene expression. Our in vitro eSF data confirm these in vivo results. This is an example of a biologically important gene that shows that demethylation of a non-CpG island promoter is highly associated with increased gene expression, as discussed above.
MPP7.
MPP7 (membrane protein, palmitoylated 7) is a member of the membrane-associated guanylate kinases, important in cell-cell contact. MPP7 plays a role in epithelial cell polarity and tight junction formation in endometrium and other tissues (95). The MPP7 interrogated CpG site is not located in a CpG island and becomes less methylated in MSE vs PE, and this change is negatively associated with its gene expression.
MUC1.
MUC1 (mucin 1) is a glycoprotein in human endometrium that interacts with ESR1, stabilizing it and stimulating ESR1-mediated transcription (96). Suppression of MUC1 inhibits expression of EGFR (97). In vivo and in vitro studies have shown that P4 increases MUC1 expression in humans (98). The importance of MUC1 expressed in endometrial epithelium has been extensively studied and shown to be important in endometrial receptivity and embryo attachment (99) with abnormalities in infertile women with polycystic ovary syndrome or endometriosis (100). Herein, the MUC1 CpG island is less methylated in MSE vs PE, which is strongly correlated with an increase in gene expression. We did not detect changes in DNA methylation in our in vitro eSF study, suggesting that the changes observed in whole endometrial tissue reflect changes in the epithelial compartment, consistent with the known expression of MUC1 in endometrial epithelium.
NR2E1 and GSTM1.
NR2E1 (nuclear receptor subfamily 2, group E, member 1) and GSTM1 (glutathione S-transferase mu 1) are examples of genes with known biological importance, but their exact function in the endometrium remains to be elucidated. NR2E1 affects many downstream genes such as zinc finger transcription factor BCL11A (CTIP1/Evi9) (101), and it recruits lysine-specific demethylase 1 (LSD1), an important transcriptional coregulator that modulates histone methylation (102, 103). The CpG island located before the TSS of NR2E1 is hypermethylated in ESE vs PE and is negatively associated with its gene expression. We also observed higher methylation in eSF treated with E2 + P4 vs E2 confirming the in vivo results.
GSTM1 functions in detoxifying carcinogens and products of oxidative stress. Interestingly, glutathione S-transferases are cycle dependent in human endometrium (31). Alterations in GSTM1 methylation result in changes in gene expression and have been associated with various cancers (104, 105). Our data showed that the GSTM1 CpG island located at the TSS is more methylated in the secretory vs the proliferative phase, and this was correlated with decreased gene expression in the secretory phase. Because there are multiple GSTMs in human endometrium, the physiologic significance of GSTM1 in the context of its detoxification function in proliferative vs secretory phase tissue remains to be determined.
Important endometrial genes reported in other transcriptomic studies
Many highly differentially expressed genes across the cycle in our previous transcriptome studies (31, 40, 106) were not detected as differentially methylated in the current study. There are several potential explanations for this observation. First, many genes with differential gene expression are the direct or downstream targets of steroid hormones, which may not involve DNA methylation. Second, it is reported that the higher the level of expression, the less likely is a gene's promoter CpG island to become de novo methylated and vice versa (107, 108). Third, cellular heterogeneity may have a greater impact on DNA methylation data than it has on gene expression data. This occurs because a given CpG dinucleotide may either be completely methylated or unmethylated in any given cell, varying widely among its own population, as well as across different cell types, which leads to detection of less robust methylation changes in the whole tissue. Last, in addition to DNA methylation, various other mechanisms are known to regulate gene expression through different pathways and other epigenetic mechanisms. Therefore, we would not necessarily expect to find DNA methylation changes in the same genes reported in previous transcriptomic studies.
Limitations of this study
There are limitations and caveats to our study, including the small sample size, cross-sectional study design, cellular heterogeneity of tissue samples, and Illumina platform probe bias with higher representation of (1) loci reported to be differentially methylated in cancer and during development and (2) CpG islands but not loci with variable partial methylation. Despite these shortcomings, the observed DNA methylation changes pave the way to expand our analysis using an unbiased comprehensive genome-wide DNA methylation platform in a larger number of samples collected longitudinally and of isolated cell types to better understand the extent and role of epigenetic changes in cycling human endometrium and the role of steroid hormones in these changes in normal endometrial physiology and in endometrium-related disorders.
In summary, the data presented herein provide, to our knowledge, the first report of dynamic DNA methylation changes in human endometrium in different hormonal milieux. These include both gain and loss of methylation either within or outside CpG islands, with proportions of DNA methylation within vs outside a CpG island varying across cycle phases. Several genes, such as RUNX3, DDR1, MUC1, MMP26, and MPP7, with predicted biological relevance in endometrial biology, show DNA methylation changes associated with gene expression changes, underscoring the importance of epigenetic regulation of gene expression in different hormonal milieux in human endometrium. In addition to advancing our understanding of molecular changes in cycling endometrium, the current data are interesting from a broader biological perspective, as the cyclic changes in DNA methylation in normal adult endometrium described herein add to the limited number of methylome changes documented in normal adult human tissues. The extent and level of these changes in the different cell types within human endometrium remain to be elucidated and are under investigation in our laboratory. The data raise the possibility that DNA methylome modifications by exogenous hormonal exposures may contribute to altered endometrial cellular function and associated endometrial disorders.
Additional material
Supplementary data supplied by authors.
Acknowledgments
The authors would like to thank Kim Chi Vo at the VCSF Human Endometrial Tissue and DNA Bank for her extensive efforts in providing the endometrial tissue samples.
This work was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health through cooperative agreement 1U54HD 055764–04/05 as part of the Specialized Cooperative Centers Program in Reproduction and Infertility Research (L.C.G.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- DNMT
- DNA methyltransferase
- E2
- estradiol
- ESE
- early secretory
- eSF
- endometrial stromal fibroblast
- H3K4me3
- histone H3 lusine 4 trimethylation
- MSE
- midsecretory
- NA
- not acceptable
- P4
- progesterone
- PE
- proliferative
- PMD
- partially methylated domain
- TSS
- transcription start site
- UCSF
- University of California, San Francisco
- USC
- University of Southern California.
References
- 1. Aghajanova L, Hamilton AE, Giudice LC. Uterine receptivity to human embryonic implantation: histology, biomarkers, and transcriptomics. Semin Cell Dev Biol. 2008;19:204–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hess A NN, Giudice L. Oviduct and Endometrium: Cyclic Changes in Primate Oviduct and Endometrium. San Diego, CA: Academic Press; 2005. [Google Scholar]
- 3. Irwin JC, Giudice LG. The Decidua. San Diego, CA: Academic Press; 1998. [Google Scholar]
- 4. Gargett CE, Masuda H. Adult stem cells in the endometrium. Mol Hum Reprod. 2010;16:818–834. [DOI] [PubMed] [Google Scholar]
- 5. Velarde MC, Aghajanova L, Nezhat CR, Giudice LC. Increased mitogen-activated protein kinase kinase/extracellularly regulated kinase activity in human endometrial stromal fibroblasts of women with endometriosis reduces 3′,5′-cyclic adenosine 5′-monophosphate inhibition of cyclin D1. Endocrinology. 2009;150:4701–4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jeong JW, Lee HS, Lee KY, et al. Mig-6 modulates uterine steroid hormone responsiveness and exhibits altered expression in endometrial disease. Proc Natl Acad Sci USA. 2009;106:8677–8682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burney RO, Hamilton AE, Aghajanova L, et al. MicroRNA expression profiling of eutopic secretory endometrium in women with versus without endometriosis. Mol Hum Reprod. 2009;15:625–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zelenko Z, Aghajanova L, Irwin JC, Giudice LC. Nuclear receptor, coregulator signaling, and chromatin remodeling pathways suggest involvement of the epigenome in the steroid hormone response of endometrium and abnormalities in endometriosis. Reprod Sci. 2012;19:152–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Matsumoto H, Zhao X, Das SK, Hogan BL, Dey SK. Indian hedgehog as a progesterone-responsive factor mediating epithelial-mesenchymal interactions in the mouse uterus. Dev Biol. 2002;245:280–290. [DOI] [PubMed] [Google Scholar]
- 10. Simon L, Spiewak KA, Ekman GC, et al. Stromal progesterone receptors mediate induction of Indian hedgehog (IHH) in uterine epithelium and its downstream targets in uterine stroma. Endocrinology. 2009;150:3871–3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nugent BM, Tobet SA, Lara HE, et al. Hormonal programming across the lifespan. Horm Metab Res. 2012;44:577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang X, Ho SM. Epigenetics meets endocrinology. J Mol Endocrinol. 2011;46:R11–R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Munro SK, Farquhar CM, Mitchell MD, Ponnampalam AP. Epigenetic regulation of endometrium during the menstrual cycle. Mol Hum Reprod. 2010;16:297–310. [DOI] [PubMed] [Google Scholar]
- 14. Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(suppl):245–254. [DOI] [PubMed] [Google Scholar]
- 15. Bird A. Perceptions of epigenetics. Nature. 2007;447:396–398. [DOI] [PubMed] [Google Scholar]
- 16. Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet. 2010;11:191–203. [DOI] [PubMed] [Google Scholar]
- 17. Taby R, Issa JP. Cancer epigenetics. CA Cancer J Clin. 2010;60:376–392. [DOI] [PubMed] [Google Scholar]
- 18. Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet. 2002;3:662–673. [DOI] [PubMed] [Google Scholar]
- 19. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492. [DOI] [PubMed] [Google Scholar]
- 20. Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–432. [DOI] [PubMed] [Google Scholar]
- 21. Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nat Rev Genet. 2009;10:805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ooi SK, Bestor TH. The colorful history of active DNA demethylation. Cell. 2008;133:1145–1148. [DOI] [PubMed] [Google Scholar]
- 23. Inoue A, Zhang Y. Replication-dependent loss of 5-hydroxymethylcytosine in mouse preimplantation embryos. Science. 2011;334:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science. 2007;315:1141–1143. [DOI] [PubMed] [Google Scholar]
- 25. Larsen F, Solheim J, Prydz H. A methylated CpG island 3′ in the apolipoprotein-E gene does not repress its transcription. Hum Mol Genet. 1993;2:775–780. [DOI] [PubMed] [Google Scholar]
- 26. Bergman Y, Cedar H. DNA methylation dynamics in health and disease. Nat Struct Mol Biol. 2013;20:1236. [DOI] [PubMed] [Google Scholar]
- 27. Vincent ZL, Farquhar CM, Mitchell MD, Ponnampalam AP. Expression and regulation of DNA methyltransferases in human endometrium. Fertil Steril. 2011;95:1522–1525.e1. [DOI] [PubMed] [Google Scholar]
- 28. Xue Q, Lin Z, Cheng YH, et al. Promoter methylation regulates estrogen receptor 2 in human endometrium and endometriosis. Biol Reprod. 2007;77:681–687. [DOI] [PubMed] [Google Scholar]
- 29. Sheldon E, Vo KC, McIntire RA, et al. Biobanking human endometrial tissue and blood specimens: standard operating procedure and importance to reproductive biology research and diagnostic development. Fertil Steril. 2011;95:2120–2122, 2122.e1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Noyes RW, Hertig AT, Rock J. Dating the endometrial biopsy. Am J Obstet Gynecol. 1975;122:262–263. [DOI] [PubMed] [Google Scholar]
- 31. Talbi S, Hamilton AE, Vo KC, et al. Molecular phenotyping of human endometrium distinguishes menstrual cycle phases and underlying biological processes in normo-ovulatory women. Endocrinology. 2006;147:1097–1121. [DOI] [PubMed] [Google Scholar]
- 32. Campan M, Weisenberger DJ, Trinh B, Laird PW. MethyLight. Methods Mol Biol. 2009;507:325–337. [DOI] [PubMed] [Google Scholar]
- 33. Bibikova M, Le J, Barnes B, et al. Genome-wide DNA methylation profiling using Infinium assay. Epigenomics. 2009;1:177–200. [DOI] [PubMed] [Google Scholar]
- 34. Fackler MJ, Umbricht CB, Williams D, et al. Genome-wide methylation analysis identifies genes specific to breast cancer hormone receptor status and risk of recurrence. Cancer Res. 2011;71:6195–6207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Campan M, Moffitt M, Houshdaran S, et al. Genome-scale screen for DNA methylation-based detection markers for ovarian cancer. PLoS One. 2011;6:e28141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bibikova M, Lin Z, Zhou L, et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006;16:383–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dedeurwaerder S, Defrance M, Calonne E, Denis H, Sotiriou C, Fuks F. Evaluation of the Infinium methylation 450K technology. Epigenomics. 2011;3:771–784. [DOI] [PubMed] [Google Scholar]
- 38. JMP. JMP Statistical Software. www.jmp.com.
- 39. Houshdaran S, Cortessis VK, Siegmund K, Yang A, Laird PW, Sokol RZ. Widespread epigenetic abnormalities suggest a broad DNA methylation erasure defect in abnormal human sperm. PLoS One. 2007;2:e1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Aghajanova L, Tatsumi K, Horcajadas JA, et al. Unique transcriptome, pathways, and networks in the human endometrial fibroblast response to progesterone in endometriosis. Biol Reprod. 2011;84:801–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aghajanova L, Giudice LC. Molecular evidence for differences in endometrium in severe versus mild endometriosis. Reprod Sci. 2011;18:229–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. [DOI] [PubMed] [Google Scholar]
- 43. Ponnampalam AP, Weston GC, Susil B, Rogers PA. Molecular profiling of human endometrium during the menstrual cycle. Aust NZ J Obstet Gynaecol. 2006;46:154–158. [DOI] [PubMed] [Google Scholar]
- 44. Shi L, Wu J. Epigenetic regulation in mammalian preimplantation embryo development. Reprod Biol Endocrinol. 2009;7:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Seisenberger S, Peat JR, Reik W. Conceptual links between DNA methylation reprogramming in the early embryo and primordial germ cells. Curr Opin Cell Biol. 2013;25:281–288. [DOI] [PubMed] [Google Scholar]
- 46. Kobayashi H, Sakurai T, Miura F, et al. High-resolution DNA methylome analysis of primordial germ cells identifies gender-specific reprogramming in mice. Genome Res. 2013;23:616–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lister R, Mukamel EA, Nery JR, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McCarthy MM. How it's made: organisational effects of hormones on the developing brain. J Neuroendocrinol. 2010;22:736–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McCarthy MM, Auger AP, Bale TL, et al. The epigenetics of sex differences in the brain. J Neurosci. 2009;29:12815–12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Challen GA, Sun D, Jeong M, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2012;44:23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jones PA. Overview of cancer epigenetics. Semin Hematol. 2005;42:S3–S8. [DOI] [PubMed] [Google Scholar]
- 52. Laird PW. Cancer epigenetics. Hum Mol Genet 14(spec no 1):R65–R76. [DOI] [PubMed] [Google Scholar]
- 53. Issa JP, Ottaviano YL, Celano P, Hamilton SR, Davidson NE, Baylin SB. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet. 1994;7:536–540. [DOI] [PubMed] [Google Scholar]
- 54. Bjornsson HT, Sigurdsson MI, Fallin MD, et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA. 2008;299:2877–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Christensen BC, Houseman EA, Marsit CJ, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009;5:e1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Krusche CA, Vloet AJ, Classen-Linke I, von Rango U, Beier HM, Alfer J. Class I histone deacetylase expression in the human cyclic endometrium and endometrial adenocarcinomas. Hum Reprod. 2007;22:2956–2966. [DOI] [PubMed] [Google Scholar]
- 57. van Kaam KJ, Delvoux B, Romano A, D'Hooghe T, Dunselman GA, Groothuis PG. Deoxyribonucleic acid methyltransferases and methyl-CpG-binding domain proteins in human endometrium and endometriosis. Fertil Steril. 2011;95:1421–1427. [DOI] [PubMed] [Google Scholar]
- 58. Ghabreau L, Roux JP, Niveleau A, et al. Correlation between the DNA global methylation status and progesterone receptor expression in normal endometrium, endometrioid adenocarcinoma and precursors. Virchows Arch. 2004;445:129–134. [DOI] [PubMed] [Google Scholar]
- 59. Le HH, Carlson EM, Chua JP, Belcher SM. Bisphenol A is released from polycarbonate drinking bottles and mimics the neurotoxic actions of estrogen in developing cerebellar neurons. Toxicol Lett. 2008;176:149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dolinoy DC. The agouti mouse model: an epigenetic biosensor for nutritional and environmental alterations on the fetal epigenome. Nutr Rev 2008;66(suppl 1):S7–S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fleisch AF, Wright RO, Baccarelli AA. Environmental epigenetics: a role in endocrine disease? J Mol Endocrinol. 2012;49:R61–R67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bromer JG, Wu J, Zhou Y, Taylor HS. Hypermethylation of homeobox A10 by in utero diethylstilbestrol exposure: an epigenetic mechanism for altered developmental programming. Endocrinology. 2009;150:3376–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Robboy SJ, Scully RE, Welch WR, Herbst AL. Intrauterine diethylstilbestrol exposure and its consequences: pathologic characteristics of vaginal adenosis, clear cell adenocarcinoma, and related lesions. Arch Pathol Lab Med. 1977;101:1–5. [PubMed] [Google Scholar]
- 64. Gore AC, Walker DM, Zama AM, Armenti AE, Uzumcu M. Early life exposure to endocrine-disrupting chemicals causes lifelong molecular reprogramming of the hypothalamus and premature reproductive aging. Mol Endocrinol. 2011;25:2157–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Westberry JM, Trout AL, Wilson ME. Epigenetic regulation of estrogen receptor α gene expression in the mouse cortex during early postnatal development. Endocrinology. 2010;151:731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Straussman R, Nejman D, Roberts D, et al. Developmental programming of CpG island methylation profiles in the human genome. Nat Struct Mol Biol. 2009;16:564–571. [DOI] [PubMed] [Google Scholar]
- 67. Lister R, Pelizzola M, Dowen RH, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schroeder DI, Blair JD, Lott P, et al. The human placenta methylome. Proc Natl Acad Sci USA. 2013;110:6037–6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Aran D, Toperoff G, Rosenberg M, Hellman A. Replication timing-related and gene body-specific methylation of active human genes. Hum Mol Genet. 2011;20:670–680. [DOI] [PubMed] [Google Scholar]
- 70. Ball MP, Li JB, Gao Y, et al. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol. 2009;27:361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Schroeder DI, Lott P, Korf I, LaSalle JM. Large-scale methylation domains mark a functional subset of neuronally expressed genes. Genome Res. 2011;21:1583–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10:295–304. [DOI] [PubMed] [Google Scholar]
- 73. Epsztejn-Litman S, Feldman N, Abu-Remaileh M, et al. De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat Struct Mol Biol. 2008;15:1176–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Schlesinger Y, Straussman R, Keshet I, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007;39:232–236. [DOI] [PubMed] [Google Scholar]
- 75. Meissner A, Mikkelsen TS, Gu H, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Weber M, Hellmann I, Stadler MB, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39:457–466. [DOI] [PubMed] [Google Scholar]
- 77. Lichtenstein M, Keini G, Cedar H, Bergman Y. B cell-specific demethylation: a novel role for the intronic kappa chain enhancer sequence. Cell. 1994;76:913–923. [DOI] [PubMed] [Google Scholar]
- 78. Paroush Z, Keshet I, Yisraeli J, Cedar H. Dynamics of demethylation and activation of the alpha-actin gene in myoblasts. Cell. 1990;63:1229–1237. [DOI] [PubMed] [Google Scholar]
- 79. Stadler MB, Murr R, Burger L, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480:490–495. [DOI] [PubMed] [Google Scholar]
- 80. ENCODE Project Consortium, Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Saluz HP, Jiricny J, Jost JP. Genomic sequencing reveals a positive correlation between the kinetics of strand-specific DNA demethylation of the overlapping estradiol/glucocorticoid-receptor binding sites and the rate of avian vitellogenin mRNA synthesis. Proc Natl Acad Sci USA. 1986;83:7167–7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Stroud H, Feng S, Morey Kinney S, Pradhan S, Jacobsen SE. 5-Hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol. 2011;12:R54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Branco MR, Ficz G, Reik W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat Rev Genet. 2012;13:7–13. [DOI] [PubMed] [Google Scholar]
- 84. Houshdaran S, Hawley S, Palmer C, et al. DNA methylation profiles of ovarian epithelial carcinoma tumors and cell lines. PLoS One. 2010;5:e9359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Venolia L, Gartler SM. Comparison of transformation efficiency of human active and inactive X-chromosomal DNA. Nature. 1983;302:82–83. [DOI] [PubMed] [Google Scholar]
- 86. Hasegawa K, Yazumi S, Wada M, et al. Restoration of RUNX3 enhances transforming growth factor-β-dependent p21 expression in a biliary tract cancer cell line. Cancer Sci. 2007;98:838–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Tsuchiya Y, Saito Y, Taniuchi S, et al. Runx3 expression and its roles in mouse endometrial cells. J Reprod Dev. 2012;58:592–598. [DOI] [PubMed] [Google Scholar]
- 88. Sakuma A, Fukamachi H, Ito K, Ito Y, Takeuchi S, Takahashi S. Loss of Runx3 affects ovulation and estrogen-induced endometrial cell proliferation in female mice. Mol. Reprod. Dev. 2008;75:1653–1661. [DOI] [PubMed] [Google Scholar]
- 89. Bai ZK, Guo B, Tian XC, et al. Expression and regulation of Runx3 in mouse uterus during the peri-implantation period. J Mol Histol. 2013;44:519–526. [DOI] [PubMed] [Google Scholar]
- 90. Iwase A, Ando H, Nagasaka T, et al. Neutral endopeptidase expressed by decidualized stromal cells suppresses akt phosphorylation and deoxyribonucleic acid synthesis induced by endothelin-1 in human endometrium. Endocrinology. 2006;147:5153–5159. [DOI] [PubMed] [Google Scholar]
- 91. Marchenko GN, Ratnikov BI, Rozanov DV, Godzik A, Deryugina EI, Strongin AY. Characterization of matrix metalloproteinase-26, a novel metalloproteinase widely expressed in cancer cells of epithelial origin. Biochem J. 2001;356:705–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zhao YG, Xiao AZ, Newcomer RG, et al. Activation of pro-gelatinase B by endometase/matrilysin-2 promotes invasion of human prostate cancer cells. J Biol Chem. 2003;278:15056–15064. [DOI] [PubMed] [Google Scholar]
- 93. Pilka R, Noskova V, Domanski H, Andersson C, Hansson S, Casslén B. Endometrial TIMP-4 mRNA is expressed in the stroma, while TIMP-4 protein accumulates in the epithelium and is released to the uterine fluid. Mol Hum Reprod. 2006;12:497–503. [DOI] [PubMed] [Google Scholar]
- 94. Li Q, Wang H, Zhao Y, Lin H, Sang QA, Zhu C. Identification and specific expression of matrix metalloproteinase-26 in rhesus monkey endometrium during early pregnancy. Mol Hum Reprod. 2002;8:934–940. [DOI] [PubMed] [Google Scholar]
- 95. Stucke VM, Timmerman E, Vandekerckhove J, Gevaert K, Hall A. The MAGUK protein MPP7 binds to the polarity protein hDlg1 and facilitates epithelial tight junction formation. Mol Biol Cell. 2007;18:1744–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wei X, Xu H, Kufe D. MUC1 oncoprotein stabilizes and activates estrogen receptor α. Mol. Cell. 2006;21:295–305. [DOI] [PubMed] [Google Scholar]
- 97. Li X, Wang L, Nunes DP, Troxler RF, Offner GD. Suppression of MUC1 synthesis downregulates expression of the epidermal growth factor receptor. Cancer Biol Ther. 2005;4:968–973. [DOI] [PubMed] [Google Scholar]
- 98. Meseguer M, Aplin JD, Caballero-Campo P, et al. Human endometrial mucin MUC1 is up-regulated by progesterone and down-regulated in vitro by the human blastocyst. Biol Reprod. 2001;64:590–601. [DOI] [PubMed] [Google Scholar]
- 99. Horne AW, Lalani EN, Margara RA, White JO. The effects of sex steroid hormones and interleukin-1-β on MUC1 expression in endometrial epithelial cell lines. Reproduction. 2006;131:733–742. [DOI] [PubMed] [Google Scholar]
- 100. Margarit L, Taylor A, Roberts MH, et al. MUC1 as a discriminator between endometrium from fertile and infertile patients with PCOS and endometriosis. J Clin Endocrinol Metab. 2010;95:5320–5329. [DOI] [PubMed] [Google Scholar]
- 101. Estruch SB, Buzon V, Carbo LR, Schorova L, Luders J, Estebanez-Perpina E. The oncoprotein BCL11A binds to orphan nuclear receptor TLX and potentiates its transrepressive function. PLoS One. 2012;7:e37963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Sun G, Ye P, Murai K, et al. miR-137 forms a regulatory loop with nuclear receptor TLX and LSD1 in neural stem cells. Nat Commun. 2011;2:529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Yokoyama A, Takezawa S, Schüle R, Kitagawa H, Kato S. Transrepressive function of TLX requires the histone demethylase LSD1. Mol Cell Biol. 2008;28:3995–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Sharma R, Panda NK, Khullar M. Hypermethylation of carcinogen metabolism genes, CYP1A1, CYP2A13, and GSTM1 genes in head and neck cancer. Oral Dis. 2010;16:668–673. [DOI] [PubMed] [Google Scholar]
- 105. Sabatino MA, Geroni C, Ganzinelli M, Ceruti R, Broggini M. Zebularine partially reverses GST methylation in prostate cancer cells and restores sensitivity to the DNA minor groove binder brostallicin. Epigenetics. 2013;8:656–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Aghajanova L, Hamilton A, Kwintkiewicz J, Vo KC, Giudice LC. Steroidogenic enzyme and key decidualization marker dysregulation in endometrial stromal cells from women with versus without endometriosis. Biol Reprod. 2009;80:105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Macleod D, Charlton J, Mullins J, Bird AP. Sp1 sites in the mouse aprt gene promoter are required to prevent methylation of the CpG island. Genes Dev. 1994;8:2282–2292. [DOI] [PubMed] [Google Scholar]
- 108. Matys V, Kel-Margoulis OV, Fricke E, et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:D108–D110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.