

Abstract
Parkinson’s disease (PD) is a progressive neurodegenerative disease that causes a debilitating movement disorder. While most cases of PD appear to be sporadic, rare Mendelian forms have provided tremendous insight into disease pathogenesis. Accumulating evidence suggests that impaired mitochondria underpin PD pathology. In support of this theory, data from multiple PD models has linked PINK1 and parkin, two recessive PD genes, in a common pathway impacting mitochondrial health, prompting a flurry of research to identify their mitochondrial targets. Recent work has focused on the role of PINK1 and parkin in mediating mitochondrial autophagy (mitophagy), however, emerging evidence casts parkin and PINK1 as key players in multiple domains of mitochondrial health and quality control.
Parkinson’s Disease is a Mitochondrial Disease of Aging
Parkinson’s disease (PD) is a common neurodegenerative disease of complex etiology marked by the insidious onset of a constellation of characteristic movement symptoms, including resting tremor, bradykinesia, rigidity and difficulty initiating movement. These movement symptoms are attributed to the relatively selective loss of dopamine producing neurons in the substantia nigra pars compacta (SNc). Over the past 30 years, basic and clinical research points to mitochondrial compromise as a central or contributing factor in PD pathogenesis [1–3].
While 90% of PD cases are considered sporadic, a handful of Mendelian forms of PD discovered over the past 15 years are now thought to make up at least 10% of disease burden [4, 5]. Mutations in genes coding for LRRK2 (Leucine rich repeat kinase 2), α-synuclein, parkin, PINK1 (PTEN Induced Putative Kinase 1), and DJ-1 among others lead, with varying penetrance, to the development of PD. Their discovery has brought substantial genetic tools to bear on the problem of PD pathogenesis. Years of studying pathogenic variants of these genes has yielded evidence tying each to mitochondrial health [2], but perhaps none more so than the autosomal-recessive PD (AR-PD) linked PINK1 and parkin [1–3, 6]. Here we review the evolving body of evidence characterizing PINK1 and parkin’s role as key regulators of mitochondrial quality control.
PINK1 and Parkin in PD
Mutations in the parkin gene are the most common cause of autosomal recessive PD [4, 5, 7]. The gene codes for a 465 amino acid E3 ubiquitin ligase capable of mediating mono or polyubiquitination using different ubiquitin linkages via lysine 29, 48 and 63 of ubiquitin. To date, more than 100 pathogenic parkin mutations disrupt the protein’s E3 ligase activity, either directly or by altering the solubility or stability of the protein, leading to dopaminergic cell death [4, 5, 8]. Moreover, recent evidence from post-mortem PD brain samples and mouse models suggest that parkin is inactivated by post-translational modifications. These include oxidation, nitrosylation, addition of dopamine and phosphorylation by c-Abl, an important stress-activated non-receptor tyrosine kinase that is activated in sporadic PD brains and in animal models of PD (reviewed in [9, 10]). These findings imply that parkin dysfunction may play a broader role in sporadic PD [9]. Parkin seems to mediate its effects in multiple cellular compartments including the cytosol, synaptic terminals, mitochondria and nucleus. Crystallization of parkin suggests that it exists in an auto-inhibited state with the requirement that it undergoes structural changes to be fully active [11, 12]. How parkin is activated is not known, but translocation to the mitochondria or association with its substrate, E2 conjugating enzyme or CHIP are likely to be key activators [12, 13].
The PINK1 (or PARK6) gene was first connected to PD by linkage analysis of consanguineous families with early-onset autosomal recessive PD [4, 14]. Loss of PINK1 is the second most common cause of autosomal recessive PD. The gene encodes a mitochondrial targeted serine/threonine kinase and loss of this kinase function is associated with the development of PD. Recent studies show that mitochondrial localized PINK1 is mainly situated in the outer mitochondrial membrane (OMM) with its C-terminal and kinase domain facing the cytosol [15], suggesting that disease-relevant PINK1 substrates may be found in the cytosol and/or on the OMM. However, alternatively processed forms may also be found in the inner mitochondrial membrane (IMM) and in the cytosol [15–18]. While there is ample evidence tying each of these genes to mitochondrial health recent studies suggest that parkin operates together with PINK1 in a common genetic pathway with the loss of either leading to altered mitochondrial dynamics and impaired mitochondrial function [19, 20].
Mitochondrial Quality Control
Mitochondrial quality control is a term used to describe the coordination of mitochondrial dynamics, mitophagy and biogenesis so as to maintain a healthy pool of mitochondria. Mitochondria are highly dynamic organelles that form complex networks. Their very morphology is constantly being modified by fission and fusion events, all while they are being shuttled throughout the cell, phenomena collectively known as mitochondrial dynamics. Controlling mitochondrial dynamics is an efficient method for tailoring mitochondria to meet the needs of different cellular compartments and for reorganizing working pieces into well-functioning machines. However, worn out parts often need to be removed altogether. This is accomplished through both proteasomal degradation and mitophagy [21]. Complementary to these removal processes, mitochondrial biogenesis leads to the synthesis of new mitochondrial proteins either to replenish depleted parts or to meet increased cellular demands. The coordination of these two important quality control pillars results in a balanced turnover of mitochondrial proteins. Adjusting the balance between biogenesis and mitophagy allows the cell to adjust bioenergetic efficiency, meeting energy demands while minimizing the accumulation of damaged mitochondrial proteins. Moreover, the nature of the dynamic equilibrium maintained by these processes necessitates that deficits in one pillar lead to alterations in another sometimes obscuring the primary insult [2].
Deficits in PINK1 and Parkin Alter Mitochondrial Fission and Fusion
A number of genetic and biochemical studies link the PINK1/parkin pathway to mitochondrial fission and fusion (Figure 1). Loss of parkin or PINK1 in Drosophila results in swollen mitochondria in the indirect flight muscles and in dopaminergic neurons, suggesting that the balance of fission to fusion is pushed towards fusion in these mutants [19, 20, 22]. This is supported by the fact that decreasing fission or increasing fusion exacerbates this phenotype, while increasing fission or decreasing fusion suppresses it [23–25]. In contrast to what was shown in Drosophila, most studies in mammalian cells demonstrate that the PINK1/parkin pathway is pro-fusion as overexpression of PINK1 leads to elongated, interconnected mitochondria, whereas knockdown of PINK1 yields fragmented mitochondria [26, 27]. However, not all studies are in agreement as to how PINK1 and parkin affect fission and fusion in mammalian cells [28]. It is possible that parkin and PINK1 may affect fission and fusion differently in diverse cell types, as distinct homologs or splice variants of the GTPases involved in fission and fusion are differentially expressed in different cell types [29], which may be related to cell-specific bioenergetic demands. In addition, the effects of the loss of PINK1 and parkin on fission and fusion may not be direct, but instead downstream of other mitochondrial defects. Indeed, the cell may compensate for mitochondrial injury by modifying the balance of mitochondrial fission to fusion. At the molecular level, one protein that may link PINK1 and parkin to fission and fusion is mitofusin (Mfn), a GTPase important for mitochondrial fusion. Mfn has been shown to be a substrate for both PINK1 and parkin and its ubiquitination appears to accelerate its proteasomal degradation [30].
Figure 1. Mitochondrial Fission and Fusion.

PINK1 and Parkins’ role in regulating the balance of mitochondrial fission to fusion. (a) PINK1 localized to healthy mitochondria is cleaved and exported from mitochondria to be rapidly degraded by the proteasome. Drp1 accumulates on mitochondria targeted for fission and allows mitochondria to divide. Conversely, in mitochondria destined to fuse, Mfn molecules from separate mitochondria tether mitochondria together and allow fusion of the OMM to occur. Subsequently, fusion of the IMM of the two mitochondria ensues via Opa1. PINK1 localized on damaged mitochondria is stabilized on the OMM. PINK1 phosphorylates Mfn, priming it for degradation by parkin. This results in a decreased rate of mitochondrial fusion. It is unknown whether parkin or PINK1 directly affect any of the proteins involved in mitochondrial fission. (b) In PD, the loss of PINK1 or parkin prevents cells from responding to mitochondrial damage by altering the balance of fission to fusion. Abbreviations: Ub, ubiquitin; Drp1, Dynamin related protein 1, Mfn, mitofusin; Opa1, optic atrophy one; E2, ubiquitin conjugating enzyme
Though the evidence points to the PINK1/parkin pathway affecting fission and fusion, it is not known how this could cause dopaminergic neuron degeneration, nor even whether it could do so. One hypothesis is that fission and fusion allow damaged mitochondria to complement each other. For instance, mtDNA can routinely accumulate mutations amongst other damage caused by exposure to oxidative stress. As this mtDNA encodes indispensable components of the ETC, mutations can have devastating consequences on mitochondrial function. The fusion of two damaged mitochondria that harbour mutations in different genes can allow functional complementation to occur by the diffusion of RNA and protein components across the newly formed mitochondria, rescuing mitochondrial function. This is believed to be an important prosurvival process under stressful conditions such as nutritional deprivation [31]. Therefore, the loss of this ability to rescue mitochondrial function by complementation may render cells more vulnerable to mitochondrial deficits. Conversely, mitochondrial fission allows mitochondria to break into smaller pieces, facilitating transport as well as the autophagic degradation of damaged mitochondria. Interestingly, fission often leads to the segregation of damaged components to one of the resulting mitochondria, producing one highly functional mitochondrion and another damaged mitochondrion with reduced membrane potential [32]. Therefore, impaired fission could reduce the ability of cells to degrade dysfunctional mitochondria by mitophagy and even to transport them to and from neurites, which is essential in long and energetically demanding cells such as neurons.
PINK1 and parkin regulate the transport of mitochondria
Impairment in the fission fusion balance is often accompanied by perinuclear clustering of mitochondria. Fission and fusion can directly impact mitochondrial transport as small mitochondria are preferentially trafficked (reviewed in [33]). Parkin and PINK1 may directly affect transport by interacting with the transport machinery (Figure 2). The OMM protein Miro participates directly in the axonal and dendritic trafficking of mitochondria through its recruitment of milton, an adapter protein that facilitates binding of mitochondria to kinesin-1 heavy chain. PINK1 phosphorylates Miro and parkin ubiquitinates Miro, targeting this protein for proteasomal degradation and arresting mitochondrial motility [34, 35]. As PINK1 is selectively stabilized on unhealthy mitochondria, damaged mitochondria could selectively be immobilized through this mechanism, potentially decreasing their probability of fusing with health mitochondria until they can be degraded. Intriguingly, the OMM GTPase mitofusin (Mfn) interacts with Miro and is required for mitochondrial transport even independently of its role in fusion [36]. This suggests that PINK1 and parkin may also modulate transport through their effects on steady state levels of Mfn.
Figure 2. Mitochondrial Transport.

PINK1 and Parkin halt the transport of damaged mitochondria. (a) Mitochondria are transported along microtubules throughout the cell by interacting with kinesin motor proteins. The OMM protein Miro tethers mitochondria to kinesins via its interaction with Milton. PINK1 localized to healthy mitochondria is cleaved and exported from mitochondria to be rapidly degraded by the proteasome, allowing mitochondria to be transported throughout the cell. Conversely, PINK1 localized to severely damaged mitochondria is stabilized on the OMM where it can phosphorylate Miro. This phosphorylation primes Miro for ubiquitination by Parkin and degradation by the 26S proteasome. As a result, mitochondria dissociate from kinesins and their transport is halted. (b) In PD, the loss of PINK1 or parkin prevents cells from halting transport as a response to mitochondrial damage. Abbreviations: Ub, ubiquitin; ROS, reactive oxygen species; RNS, reactive nitric oxide species.
The thought that the dysregulation of mitochondrial transport may be implicated in PD is especially interesting because of the increased requirements of neurons for transport given how far neurites extend from the cell soma. PINK1 and or parkin loss could eliminate this sorting function allowing suboptimal mitochondria to be trafficked to axon terminals where they could undermine local energy production
PINK1 and parkin initiate the removal of mitochondrial proteins
Many recent studies peg initiation of mitophagy at damaged mitochondria as one of the functions of PINK1-parkin cooperation (Figure 3). In mammalian cell line models [37] and in human fibroblasts [38], over-expressed parkin or in some situations endogenous parkin is recruited to mitochondria in a PINK1-dependant manner after widespread mitochondrial depolarization using mitochondrial uncouplers such as carbonyl cyanide m-chlorophenyl hydrazone (CCCP). Mitochondrial mass is subsequently reduced and mitochondrial proteins co-localize with autophagosomal and lysosomal markers.
Figure 3. Mitochondrial Autophagy.
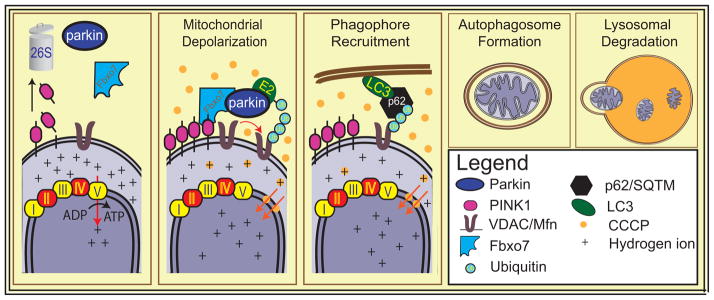
Parkin mediates removal of damaged mitochondria. After widespread mitochondrial depolarization by CCCP, PINK1 is stabilized on the OMM, enabling parkin to translocate to mitochondria and ubiquitinate OMM proteins including mitofusins and VDAC. Some of these ubiquitin moieties may serve as signals to recruit autophagic machinery including HDAC6, or p62. Mitochondria are engulfed by the autophagosome, which eventually fuses with the lysosome, leading to the degradation of the dysfunctional organelles. Abbreviations: Ub, ubiquitin; Mfn, mitofusin; VDAC, valtage dependent anion channel; Fbxo7, F-box protein 7; p62/SQTM, p62 sequestome; CCCP, Carbonyl cyanide m-chlorophenyl hydrazine; LC3, light chain 3
What accounts for the translocation of parkin from the cytosol to the mitochondria after widespread mitochondrial depolarization is not known. Mitochondrial localized PINK1, which is normally rapidly degraded is stabilized on the OMM when mitochondria are depolarized [37]. Since PINK1 kinase activity is required for the translocation of parkin and for mitophagy, this suggests that there is an as of yet unexplained kinase-dependent process/cascade that is required for parkin translocation to the mitochondria and subsequent mitophagy. Some studies suggest that the phosphorylation of substrates by PINK1 primes them to interact with parkin, thus sequestering cytosolic parkin to the mitochondria or alternatively are ubiquitinated by parkin [34]. One candidate substrate for this role is Mfn2. PINK1 phosphorylates Mfn2, which results in Mfn2 acting as a receptor to dock parkin at the mitochondria [39]. Alternatively voltage-dependent anion channels (VDACs) or Miro may act as parkin receptors at the mitochondria. However, when PINK1 is overexpressed and artificially targeted to peroxisomes or lysosomes, parkin can be recruited to these organelles, suggesting that a direct interaction of PINK1 and parkin might mediate parkin translocation [40]. Some evidence suggests that direct phosphorylation of parkin by PINK1 may also be required for parkin’s translocation or activation [41]. However, to date, the evidence for this is debated. Other players may be required for parkin recruitment. Fbxo7, an AR-PD linked protein encoded by PARK15, participates in mitochondrial maintenance through a direct interaction with PINK1 and parkin. Fbxo7 harbours a mitochondrial translocation sequence (MTS) and, after mitochondrial depolarization, Fbxo7 appears to form complexes with parkin and either full-length or N-terminal cleaved PINK1 and is found to be enriched in mitochondrial fractions [42]. Another recent study also indicates that hexokinase activity is required for parkin mitochondrial recruitment [43]. In addition, in the setting of parkin overexpression, a number of enhancers and suppressors of recruitment to the mitochondria have been described [44].
Once recruited to mitochondria, overexpressed parkin ubiquitinates a number of OMM proteins including Mfn 1 and 2, VDAC1 and Miro among others [45]. These proteins do not seem to be required for mitochondrial degradation since fibroblasts from VDAC1/3 or Mfn1/2 knockout mice still undergo mitophagy [2, 6]. What triggers the autophagy of mitochondria is not known. While some of these proteins are targeted for proteasomal degradation, parkin’s mediation of K63 and K27 linked monoubiquitylation of other proteins, including VDAC1, suggests a possible signalling function. After mitochondrial uncoupling with CCCP treatment, HDAC6 and p62/SQTM accumulate on OMMs. p62 and HDAC6 interact with ubiquitin moieties as well as components of the autophagosome such as Ambra1 (activating molecule in Beclin-regulated autophagy) and Beclin and could thus serve as linker proteins that recruit the autophagosome to mitochondria [46, 47]. HDAC6 is necessary for CCCP mediated mitophagy whereas a recent study in p62 knockout mice suggests that p62 may mediate perinuclear clustering, but not mitophagy [48]. The PINK1/parkin-dependent mitophagy pathway uses, at least in part, the canonical autophagy machinery as well as the ubiquitin-proteasome system [49].
While the parkin translocation phenotype is robust in multiple mammalian cell lines that overexpress parkin, questions remain as to whether these observations are relevant in neurons or in PD pathogenesis [49]. A major concern is that most of these observations are in cells manipulated to overexpress parkin raising the concern that endogenous parkin may not mediate mitophagy [50]. Furthermore, in neurons mitochondrial translocation of overexpressed parkin is modest and sporadic and seems to only occur after prolonged CCCP exposure or in the setting of co-administration of apoptosis inhibitors or lysosomal inhibitors [51] or special culture conditions [49]. There are many differences between neurons and transformed cell lines that may explain why they respond differently to CCCP treatment. Probably most important is that most cell lines are glycolytic, whereas neurons depend heavily on oxidative phosphorylation for ATP production. Some observations suggest that forcing transformed cells to rely more heavily on oxidative phosphorylation substantially mitigates CCCP induced mitophagy [52]. Thus, the bioenergetic crisis engendered by uncouplers particularly may not elicit endogenous parkin translocation in neurons due to the unique energy requirements of these cells.
Another important concern with this model is that CCCP treatment induces a rapid and almost complete loss of mitochondrial membrane potential in all mitochondria, a harsh insult that probably never occurs in PD. Using light activated generation of reactive oxygen species, it was shown that parkin recruitment and mitophagy can be mediated in subsets of the mitochondrial pool after mitochondrial depolarization stemming from overwhelming oxidative damage [53]. However other mitochondrial stressors that may be more physiologic fail to induce parkin translocation in vivo. For instance, mitoPARK mice, which harbour the deletion of the mitochondrial transcription factor A (TFAM) selectively in DA neurons, show the formation of large mitochondrial aggregates, but no evidence of parkin recruitment even when parkin is overexpressed [54]. Knocking out parkin in these mice had no observable effect on the accumulation of mitochondrial aggregates or mitophagy. Moreover, cytosolic dynamin-like GTPase, Drp1, knockout neurons, which accumulate damaged mitochondria, showed markers of mitophagy such as mitochondrial ubiquitination that were present even when parkin was knocked out [55]. Whether the mitoPARK neurons or the Drp1 knockout neurons have sufficient mitochondrial depolarization to stabilize PINK1 on mitochondria and initiate parkin translocation is not known. Furthermore, the role of PINK1 and parkin in mitophagy has not been described in patients with mutations in these genes and there is no accumulation of abnormal damaged mitochondria in PINK1 or parkin knockout mice [56, 57]. Thus, it remains to be seen whether there are mechanisms that can trigger PINK1/parkin-dependent mitophagy in neurons of higher organisms and what compensatory mechanisms might be at play in vivo that can substitute for PINK1/parkin mediated mitophagic pathways. Alternatively, Parkin and PINK1 may exert more subtle control of mitochondrial turnover by responding to mild oxidative stress to generate mitochondrial-derived vesicles (MDV) that are transported to lysosomes for degradation, thus favouring repair of mitochondria over replacement [58].
Parkin deficits impair mitochondrial biogenesis
While the recent expansion of knowledge regarding the role of PINK1 and parkin in mitochondrial dynamics, transport and autophagy is potentially important, there have also been advances in our understanding of the role of mitochondrial biogenesis in PD. Over the years, a number of observations have suggested that impaired mitochondrial biogenesis may contribute to PD. For instance, reduced levels of Complex I found in PD patients and in models of PD may be attributed to defects in biogenesis. There is decreased Complex I activity in patients with parkin mutations [59] and in parkin and PINK1 knockout mice [57, 60]. Notably, in PINK1 mutant flies, Complex I deficiency impairs ATP production, and supplementation of the PINK1 deficient flies with Complex 1 or with electron carrier Vitamin K2 or ubiquinone corrects deficits in ATP production and rescues flight muscle [61]. Moreover, in both PINK1 and parkin null flies there is an overall defect with protein turnover that is more pronounced in ETC proteins, consistent with the possibility that PINK1 and parkin could regulate mitochondrial content via mechanisms independent from mitophagy, such as by affecting biogenesis [62]. There is also evidence that parkin may mediate some of its protective effects by maintaining the integrity of the mitochondrial genome. Parkin overexpression appears to increase mitochondrial transcription and suppress mitochondrial DNA damage through a poorly characterized interaction with TFAM in both proliferating cells and differentiated SH-SY5Y cells [63, 64].
While parkin may have a hand in maintaining mtDNA integrity, new evidence suggests that the degeneration of DA neurons that occurs upon the loss of parkin may result from the dysregulation of 2 key proteins, AIMP2 (aminoacyl tRNA synthetase complex-interacting multifunctional protein 2) and PARIS (parkin Interacting Substrate) that accumulate in the absence of parkin’s E3 ligase activity [65, 66]. Adult conditional knockout of parkin recapitulates key features of PD pathogenesis, most notably progressive degeneration of SNc DA neurons. Both AIMP2 and PARIS accumulate in these mice, as well as in patients with parkin mutations and sporadic PD [9]. PARIS is responsible for the degeneration of DA neurons that occurs following the adult inactivation of parkin since knockdown of PARIS attenuates this loss of DA neurons [66]. Interestingly, PARIS directly impacts the nuclear transcription of mitochondrial proteins.
PARIS is a zinc-finger protein, first identified by a yeast two-hybrid screen, which mediates neurodegeneration via transcriptional repression of peroxisome proliferator-activated receptor gamma (PPARγ) coactivator-1α (PGC-1α), a master regulator of mitochondrial biogenesis that is required to match mitochondrial output to energetic demands (Figure 5). By serving as a transcriptional cofactor for numerous mitochondrial proteins, PGC-1α is able to fine tune mitochondrial function to meet the needs of the cell and mediate protection from oxidative injury (reviewed in [67]). Consistent with the notion that PGC-1α defects play a role in PD are the observations that PGC-1α levels are decreased in the SNc of PD patients [66] and mice lacking PGC-1α are more susceptible to MPTP, a neurotoxin associated with PD [68]. PGC-1α overexpression prevents the loss of DA neurons when PARIS is overexpressed suggesting that it may be the key target linking PARIS to DA degeneration. In addition, overexpression of PGC-1α protects against MPTP, rotenone, oxidative stress and α-synuclein induced neurodegeneration [67]. PGC-1α polymorphisms are associated with age of onset and risk of PD [69] and PGC-1α is dysfunctional in patients with parkin-associated PD [70]. Moreover, reductions in PGC-1α levels may be an important cause of PD pathogenesis since PGC-1α-responsive genes are downregulated in micro-dissected dopaminergic neurons [71]. Thus accumulating evidence strongly supports the notion that defects in PGC-1α signalling contribute to the pathogenesis of PD. PARIS and its regulation by parkin via the ubiquitin proteasome system may provide the underlying molecular mechanism linking PGC-1α to PD. Although exciting and potentially important, the parkin, PARIS, PGC-1α pathway of neurodegeneration in PD still awaits further independent validation. Interestingly, PPARγ activation also rescues mitochondrial function due to loss of PINK1 through increased mitochondrial biogenesis suggesting that PINK1 may be part of PARIS and PGC-1α pathway [72]. Moreover, enhancing nucleotide metabolism, which can promote mitochondrial biogenesis, protects against mitochondrial dysfunction and neurodegeneration in PINK1 models of PD. This provides further evidence that PINK1 may regulate biogenesis [73]. It will be important to determine what role if any PINK1 plays in PARIS regulation and how it regulates mitochondrial biogenesis.
Figure 5. Mitochondrial Quality Control.

Parkin and PINK1 regulate mitochondrial quality control. PINK1 and parkin modulate mitochondrial dynamics both by tipping the fission-fusion balance towards fission and by stalling the transport of damaged mitochondria. PINK1 and parkin may regulate the turnover of mitochondria by recruiting the autophagic machinery to damaged mitochondria and allowing mitophagy to occur, while also playing an important role in the production of novel mitochondrial components by biogenesis. Abbreviations: PGC-1α, Peroxisome proliferator-activator receptor gamma co-activator 1α; ROS, reactive oxygen species; RNS, reactive nitric oxide species; E2, ubiquitin conjugating enzyme; Ub, ubiquitin; OMM, outer mitochondrial membrane.
Further investigation is needed to characterize the nature and extent of mitochondrial dysfunction downstream of adult parkin loss. Moreover, since parkin seems to have other pathogenic substrates, such as AIMP2 [65], it will be important to determine how PARIS and AIMP2 and potentially other substrates interact in the pathogenesis of PD due to parkin inactivation. Do they intersect in a common pathway or are they part of separate pathways? Since parkin inactivation impairs mitochondrial quality control at multiple levels, does the accumulation of AIMP2 play a role in mitochondrial QC or does it contribute to DA degeneration by affecting upstream or downstream pathways?
Concluding Remarks
There is evidence implicating parkin and PINK1 in each axis of mitochondrial quality control including fission and fusion, mitophagy, transport and biogenesis (Figure 5). However, it remains unclear whether insult to these different mitochondria control axis is sufficient to cause PD. For instance, altering the balance of fission to fusion in PINK1 or parkin mutant Drosophila significantly corrects the phenotype seen in these models, but even this is not sufficient to establish that a defect in fission is responsible for this phenotype rather than a downstream consequence of another mitochondrial insult. In support of the latter, correction of mitochondrial dynamics does not correct Complex I dysfunction, hinting that the bioenergetic compromise may be an upstream mediator of fission-fusion defects in flies [61, 74]. Also, mutations in Mfn2 and OPA1 do lead to cell death of selective neuronal populations as they can respectively cause Charcot Marie Tooth type 2a [75] and optic atrophy [76, 77]. But the dopaminergic cell death that would be expected if an imbalance of fission and fusion were at the heart of PD pathogenesis has not been reported in these diseases. Although, the loss of PINK1 or of parkin affect the transport of damaged mitochondria, it is unknown whether correcting this has any effect on PD pathogenesis in any existing model. As for mitophagy, it is not clear whether PINK1 and parkin regulate mitophagy in neurons. One confounding observation is that, while the Drosophila genetic studies show that parkin overexpression can rescue defects in PINK1 mutants, overexpressed parkin cannot be recruited to mitochondria to initiate mitophagy in the absence of PINK1. This suggests that the loss of parkin/PINK1 mediated mitophagy may not be responsible for the degeneration seen in Drosophila and possibly not in PD patients either. Finally, parkin plays a role in mitochondrial biogenesis by both safeguarding mtDNA and by regulating PGC-1α-levels via its ubiquitination of PARIS. This is promising both because of the compelling evidence tying injury to mtDNA to PD pathogenesis and the emerging role of PGC-1α in PD. Under normal conditions, reductions in mitochondria via the PINK1/parkin-dependent mitophagy pathway could be counter-balanced by the parkin/PARIS mitochondrial biogenesis pathway as part of a homeostatic mechanism to maintain mitochondrial mass to meet the energy demands of a cell. It will be of interest to determine whether PINK1 also plays a role in mitochondrial biogenesis and also to determine whether other genetic and chemical models of PD converge upon mitochondrial biogenesis pathways. Additional questions regarding the role of parkin and PINK1 in mitochondrial function and quality control remain (Box I). Determining whether defects in any of these processes are sufficient to cause PD in multiple different models will be central to furthering our understanding of PD pathogenesis.
BOX I. Remaining Questions.
What is the primary deficit caused by PINK1 or Parkin loss and how does it drive mitochondrial compromise and dopaminergic cell death?
Are impaired mitochondrial dynamics a cause or consequence of impaired mitochondrial bioenergetics?
If deficits in PINK1 dependant mitophagy underlie pink1 deficiency phenotypes, how are these phenotypes rescued by Parkin overexpression?
How does PINK1 play a role in mediating mitochondrial biogenesis and is it linked to PARIS and parkin in PD pathogenesis?
Why are DA neurons primarily affected if Parkin and PINK1 are expressed in most neurons?
Figure 4. Mitochondrial Biogenesis.

Mitochondrial biogenesis involves the transcription, translation and assembly of new mitochondrial proteins and the replication of mitochondrial DNA. A common link in the coordination of cellular responses to metabolic demands and oxidative stress is the Peroxisome proliferator-activator receptor gamma co-activator (PGC) family of proteins and in particular PGC-1α. PGC-1α is often called the master regulator of mitochondrial biogenesis. The gene for PGC-1α is inducible and responsive to many cellular events leading to AMPK activation and CREB dependent transcription. PGC-1α increases expression of nuclear respiratory factor 1 (NRF-1) and estrogen response receptor (ERR) α and also catalyzes the transcriptional activity of NRF-1/2, ERR and Peroxisome proliferator-activated receptor (PPAR) families of transcription factors. Converging evidence from sporadic PD and adult-onset parkin KO mice implicate transcriptional repression of PGC-1α by PARIS as an important pathogenic mechanism in dopamine neuron degeneration downstream of parkin loss [66]. Abbreviations: Tfam, Mitochondrial transcription factor A; CREB, cAMP responsive element binding protein, mtDNA, mitochondrial DNA; Enz, enzyme; ETC, electron transport subunits; β-Ox, β-oxidation; TCA Cyc, tricarboxylic acid cycle; MnSOD, manganese superoxide dismutase; UCP, mitochondrial uncoupling proteins; NADH, Nicotinamide adenine dinucleotide; FA-βO; fatty acid β-oxidation.
Highlights.
Parkinson’s disease is a mitochondrial disease of aging
PINK1 and parkin are key players in multiple domains of mitochondrial health
PINK1 and parkin are key regulators of mitochondrial quality control
Acknowledgments
This work was supported by grants from the NIH/NINDS NS38377, the Cure Parkinson’s Trust and the JPB Foundation. The authors acknowledge the joint participation by the Adrienne Helis Malvin Medical Research Foundation through their direct engagement in the continuous active conduct of medical research in conjunction with The Johns Hopkins Hospital and the Johns Hopkins University School of Medicine and the Foundation’s Parkinson’s Disease Program. L.A.S. is the recipient of a Canadian Institutes of Health Research Doctoral Foreign Study Award. T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson’s disease. Science. 2003;302:819–822. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- 2.Exner N, et al. Mitochondrial dysfunction in Parkinson’s disease: molecular mechanisms and pathophysiological consequences. The EMBO journal. 2012;31:3038–3062. doi: 10.1038/emboj.2012.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schon EA, Przedborski S. Mitochondria: the next (neurode)generation. Neuron. 2011;70:1033–1053. doi: 10.1016/j.neuron.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corti O, et al. What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol Rev. 2011;91:1161–1218. doi: 10.1152/physrev.00022.2010. [DOI] [PubMed] [Google Scholar]
- 5.Martin I, et al. Recent advances in the genetics of Parkinson’s disease. Annu Rev Genomics Hum Genet. 2011;12:301–325. doi: 10.1146/annurev-genom-082410-101440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Corti O, Brice A. Mitochondrial quality control turns out to be the principal suspect in parkin and PINK1-related autosomal recessive Parkinson’s disease. Curr Opin Neurobiol. 2013;23:100–108. doi: 10.1016/j.conb.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 7.Kitada T, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 8.Houlden H, Singleton AB. The genetics and neuropathology of Parkinson’s disease. Acta Neuropathol. 2012;124:325–338. doi: 10.1007/s00401-012-1013-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dawson TM, Dawson VL. Parkin Plays a Role in Sporadic Parkinson’s Disease. Neurodegener Dis. 2013 doi: 10.1159/000354307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walden H, Martinez-Torres RJ. Regulation of Parkin E3 ubiquitin ligase activity. Cell Mol Life Sci. 2012;69:3053–3067. doi: 10.1007/s00018-012-0978-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dove KK, Klevit RE. Structural Biology: Parkin’s Serpentine Shape Revealed in the Year of the Snake. Curr Biol. 2013;23:R691–693. doi: 10.1016/j.cub.2013.07.039. [DOI] [PubMed] [Google Scholar]
- 12.Byrd RA, Weissman AM. Compact Parkin only: insights into the structure of an autoinhibited ubiquitin ligase. The EMBO journal. 2013;32:2087–2089. doi: 10.1038/emboj.2013.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Imai Y, et al. CHIP is associated with Parkin, a gene responsible for familial Parkinson’s disease, and enhances its ubiquitin ligase activity. Mol Cell. 2002;10:55–67. doi: 10.1016/s1097-2765(02)00583-x. [DOI] [PubMed] [Google Scholar]
- 14.Valente EM, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 15.Zhou C, et al. The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc Natl Acad Sci U S A. 2008;105:12022–12027. doi: 10.1073/pnas.0802814105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Becker D, et al. Pink1 kinase and its membrane potential (Deltapsi)-dependent cleavage product both localize to outer mitochondrial membrane by unique targeting mode. The Journal of biological chemistry. 2012;287:22969–22987. doi: 10.1074/jbc.M112.365700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beilina A, et al. Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc Natl Acad Sci U S A. 2005;102:5703–5708. doi: 10.1073/pnas.0500617102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin SM, et al. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. The Journal of cell biology. 2010;191:933–942. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clark IE, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- 20.Park J, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 21.Fischer F, et al. Mitochondrial quality control: an integrated network of pathways. Trends in biochemical sciences. 2012;37:284–292. doi: 10.1016/j.tibs.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 22.Yang Y, et al. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci U S A. 2006;103:10793–10798. doi: 10.1073/pnas.0602493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deng H, et al. The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci U S A. 2008;105:14503–14508. doi: 10.1073/pnas.0803998105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poole AC, et al. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Y, et al. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci U S A. 2008;105:7070–7075. doi: 10.1073/pnas.0711845105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dagda RK, et al. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. The Journal of biological chemistry. 2009;284:13843–13855. doi: 10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lutz AK, et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. The Journal of biological chemistry. 2009;284:22938–22951. doi: 10.1074/jbc.M109.035774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu W, et al. The PINK1/Parkin pathway regulates mitochondrial dynamics and function in mammalian hippocampal and dopaminergic neurons. Hum Mol Genet. 2011;20:3227–3240. doi: 10.1093/hmg/ddr235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akepati VR, et al. Characterization of OPA1 isoforms isolated from mouse tissues. Journal of neurochemistry. 2008;106:372–383. doi: 10.1111/j.1471-4159.2008.05401.x. [DOI] [PubMed] [Google Scholar]
- 30.Ziviani E, et al. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc Natl Acad Sci U S A. 2010;107:5018–5023. doi: 10.1073/pnas.0913485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tondera D, et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. The EMBO journal. 2009;28:1589–1600. doi: 10.1038/emboj.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Itoh K, et al. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 2013;23:64–71. doi: 10.1016/j.tcb.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X, et al. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893–906. doi: 10.1016/j.cell.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu S, et al. Parkinson’s disease-associated kinase PINK1 regulates Miro protein level and axonal transport of mitochondria. PLoS Genet. 2012;8:e1002537. doi: 10.1371/journal.pgen.1002537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Misko A, et al. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci. 2010;30:4232–4240. doi: 10.1523/JNEUROSCI.6248-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Narendra D, et al. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. The Journal of cell biology. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rakovic A, et al. Mutations in PINK1 and Parkin impair ubiquitination of Mitofusins in human fibroblasts. PLoS One. 2011;6:e16746. doi: 10.1371/journal.pone.0016746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Y, Dorn GW., 2nd PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340:471–475. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lazarou M, et al. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Developmental cell. 2012;22:320–333. doi: 10.1016/j.devcel.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iguchi M, et al. Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. The Journal of biological chemistry. 2013;288:22019–22032. doi: 10.1074/jbc.M113.467530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burchell VS, et al. The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat Neurosci. 2013;16:1257–1265. doi: 10.1038/nn.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McCoy MK, et al. Hexokinase activity is required for recruitment of parkin to depolarized mitochondria. Hum Mol Genet. 2013 doi: 10.1093/hmg/ddt407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hasson SA, et al. High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature. 2013;504:291–295. doi: 10.1038/nature12748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sarraf SA, et al. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496:372–376. doi: 10.1038/nature12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Michiorri S, et al. The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy. Cell Death Differ. 2010;17:962–974. doi: 10.1038/cdd.2009.200. [DOI] [PubMed] [Google Scholar]
- 47.Van Humbeeck C, et al. Parkin interacts with Ambra1 to induce mitophagy. J Neurosci. 2011;31:10249–10261. doi: 10.1523/JNEUROSCI.1917-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Narendra DP, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grenier K, et al. Parkin- and PINK1-Dependent Mitophagy in Neurons: Will the Real Pathway Please Stand Up? Front Neurol. 2013;4:100. doi: 10.3389/fneur.2013.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rakovic A, et al. Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. The Journal of biological chemistry. 2013;288:2223–2237. doi: 10.1074/jbc.M112.391680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cai Q, et al. Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr Biol. 2012;22:545–552. doi: 10.1016/j.cub.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Van Laar VS, et al. Bioenergetics of neurons inhibit the translocation response of Parkin following rapid mitochondrial depolarization. Hum Mol Genet. 2011;20:927–940. doi: 10.1093/hmg/ddq531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Y, et al. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy. 2012;8:1462–1476. doi: 10.4161/auto.21211. [DOI] [PubMed] [Google Scholar]
- 54.Sterky FH, et al. Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc Natl Acad Sci U S A. 2011;108:12937–12942. doi: 10.1073/pnas.1103295108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kageyama Y, et al. Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. The Journal of cell biology. 2012;197:535–551. doi: 10.1083/jcb.201110034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Palacino JJ, et al. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. The Journal of biological chemistry. 2004;279:18614–18622. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- 57.Gautier CA, et al. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A. 2008;105:11364–11369. doi: 10.1073/pnas.0802076105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McLelland GL, et al. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. The EMBO journal. 2014;33:282–295. doi: 10.1002/embj.201385902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Muftuoglu M, et al. Mitochondrial complex I and IV activities in leukocytes from patients with parkin mutations. Mov Disord. 2004;19:544–548. doi: 10.1002/mds.10695. [DOI] [PubMed] [Google Scholar]
- 60.Morais VA, et al. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol Med. 2009;1:99–111. doi: 10.1002/emmm.200900006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vos M, et al. Vitamin K2 is a mitochondrial electron carrier that rescues pink1 deficiency. Science. 2012;336:1306–1310. doi: 10.1126/science.1218632. [DOI] [PubMed] [Google Scholar]
- 62.Vincow ES, et al. The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc Natl Acad Sci U S A. 2013;110:6400–6405. doi: 10.1073/pnas.1221132110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kuroda Y, et al. Parkin enhances mitochondrial biogenesis in proliferating cells. Hum Mol Genet. 2006;15:883–895. doi: 10.1093/hmg/ddl006. [DOI] [PubMed] [Google Scholar]
- 64.Rothfuss O, et al. Parkin protects mitochondrial genome integrity and supports mitochondrial DNA repair. Hum Mol Genet. 2009;18:3832–3850. doi: 10.1093/hmg/ddp327. [DOI] [PubMed] [Google Scholar]
- 65.Lee Y, et al. Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat Neurosci. 2013 doi: 10.1038/nn.3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shin JH, et al. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell. 2011;144:689–702. doi: 10.1016/j.cell.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhu J, et al. After the banquet: Mitochondrial biogenesis, mitophagy and cell survival. Autophagy. 2013;9 doi: 10.4161/auto.24135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.St-Pierre J, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 69.Clark J, et al. Association of PGC-1alpha polymorphisms with age of onset and risk of Parkinson’s disease. BMC Med Genet. 2011;12:69. doi: 10.1186/1471-2350-12-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pacelli C, et al. Mitochondrial defect and PGC-1alpha dysfunction in parkin-associated familial Parkinson’s disease. Biochim Biophys Acta. 2011;1812:1041–1053. doi: 10.1016/j.bbadis.2010.12.022. [DOI] [PubMed] [Google Scholar]
- 71.Zheng B, et al. PGC-1alpha, a potential therapeutic target for early intervention in Parkinson’s disease. Sci Transl Med. 2010;2:52ra73. doi: 10.1126/scitranslmed.3001059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Corona JC, et al. PPARgamma activation rescues mitochondrial function from inhibition of complex I and loss of PINK1. Experimental neurology. 2014;253:16–27. doi: 10.1016/j.expneurol.2013.12.012. [DOI] [PubMed] [Google Scholar]
- 73.Tufi R, et al. Enhancing nucleotide metabolism protects against mitochondrial dysfunction and neurodegeneration in a PINK1 model of Parkinson’s disease. Nature cell biology. 2014;16:157–166. doi: 10.1038/ncb2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vilain S, et al. The yeast complex I equivalent NADH dehydrogenase rescues pink1 mutants. PLoS Genet. 2012;8:e1002456. doi: 10.1371/journal.pgen.1002456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zuchner S, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- 76.Alexander C, et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000;26:211–215. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- 77.Delettre C, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207–210. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]