

Abstract
Lung inflammation and alterations in endothelial cell (EC) micro- and macro-vascular permeability are key events to development of acute lung injury (ALI). Using ECs derived from human pulmonary artery (HPAECs) and lung microvasculature (HLMVECs), we investigated interplay between p38 stress MAPK and Rho GTPase signaling in the inflammatory and hyperpermeability response. Both cell types were treated with Staphylococcus aureus-derived peptidoglycan (PepG) and lipoteichoic acid (LTA) with or without pretreatment with p38 MAPK or Rho kinase inhibitors. LTA and PepG markedly increased permeability in both pulmonary macrovascular and microvascular EC. Agonist-induced hyper-permeability was accompanied by cytoskeletal remodeling, disruption of cell-cell contacts, formation of paracellular gaps, and activation of p38 MAPK, NFκB, and Rho/Rho kinase signaling. In macrovascular ECs, pharmacological inhibition of Rho kinase with Y27632 significantly suppressed p38 MAP kinase cascade activation, while inhibition of p38 MAPK with SB203580 had no effect on Rho activation. In contrast, inhibition of p38 MAPK in microvascular ECs suppressed LTA/PepG-induced activation of Rho, while Rho inhibitor suppressed activation of p38 MAPK. Inhibition of either p38 MAPK or Rho kinase substantially attenuated activation of NFκB signaling. These results demonstrate cell type-specific differences in signaling induced by Staphylococcus aureus-derived pathogens in pulmonary endothelium. Thus, although Gram-positive bacterial compounds caused barrier dysfunction in both EC types, it was induced by different pattern of crosstalk between Rho, p38 MAPK, and NFκB signaling. These observations may have important implications in defining microvasculature-specific therapeutic strategies aimed at the treatment of sepsis and acute lung injury induced by Gram-positive bacterial pathogens.
Keywords: LTA, PepG, inflammation, cytoskeleton, pulmonary endothelium, vascular leak
INTRODUCTION
Sepsis is the 10th leading cause of death in the United States (1) and a major contributor to development of Acute Respiratory Dystress Syndrome (ARDS). Reported mortality rates resulting from ARDS reach 35–40% (2). About 50% of all cases of sepsis are caused by gram-positive bacteria (3). Specifically, presence of Staphylococcus aureus has been associated with the acquisition of late-onset ventilator-associated pneumonia (VAP) in critically ill patients and represented 21% of recovered organisms in the bronchoalveolar lavage (4). Furthermore, development of methicillin resistant strains detected in 65.8% cases of Staphylococcus aureus infections was defined as a major risk factor for VAP (5) that can lead to the development of lung injury (6, 7).
Peptidoglycan (PepG) and lipoteichoic acid (LTA) are two major cell wall components in gram-positive bacteria. Both pathogens stimulate inflammatory responses via activation of toll-like receptors (TLRs) (8, 9). In the lungs, LTA and PepG induced dose-dependent acute pulmonary inflammation characterized by neutrophilic influx and IL-6 production detected in the bronchoalveolar lavage fluid (10). Importantly, synergistic relations between LTA and PepG have been reported which caused shock and multiple systems failure (11).
Components of both Gram-positive and Gram-negative bacteria are recognized by a family of TLRs. Of the ten TLRs known, only TLR2 has been clearly demonstrated to be involved in the host defense against gram-positive bacteria, although it also recognizes lipoproteins from other bacterial species. On the other hand, the crosstalk between TLR2 and TLR4 has been also suggested (12, 13). TIR domain-containing adaptors, such as MyD88, TIRAP, and TRIF, modulate TLR signaling pathways. MyD88 is essential for the induction of inflammatory cytokines triggered by all TLRs and TIRAP is specifically involved in the MyD88-dependent pathway via TLR2 and TLR4 (13). Recruitment of TIRAP/MyD88 by TLR2 leads to activation of mitogen-activated protein kinases (MAPK) p42/p44, JNK1/2, p38, as well as nuclear factor kappa-B (NFκB)-dependent gene transcritption (8, 14, 15). In addition, TLR activation induces phosphatidyl inositol 3-kinase-dependent signaling, which appear to be important component of NFκB activity following its translocation to the nucleus (16). NFκB in the cytoplasm is inactive as it is bound by the inhibitory IκB proteins. Activation of inflammatory signaling leads to IκB phosphorylation by IκB kinase and its subsequent degradation by the proteasome. As a result, activated NFκB translocates to the nucleus, where it triggers the transcription of multiple genes and production of pro-inflammatory cytokines TNFα, IL-1b, IL-6, and IL-8 (17). In turn, activation of p38 MAPK signaling contributes to both, the inflammatory gene expression and cytoskeletal remodeling leading to increased endothelial permeability (18–20).
Small Rho GTPases have been recently suggested as additional effectors of TLR signaling (21). A role of Rho signaling in endothelial permeability caused by bacterial pathogens including TLR2 receptor ligands has been described by our group (15, 22, 23) and others (24–27). Rho kinase mediated myosin light chain (MLC) phosphorylation via inactivation of myosin light chain phosphatase (MYPT1) by its phosphorylation at Thr695, Ser894, and Thr850 (28–30) leads to actomyosin-driven cell contraction and EC barrier compromise. In addition, Rho activity has been implicated in the loss of adherens and tight junctions and decreased intercellular gap formation observed in endothelial cells challenged with Staphylococcus aureus (31). In addition to direct effects on EC permeability, Rho activation by endotoxin has been shown to stimulate transcription of pro-inflammatory genes, while inhibition of Rho signaling reduced expression of TNFα, CXC chemokines, leukocyte infiltration, and endotoxin-induced lung edema (32, 33).
We have recently demonstrated that attenuation of Rho and p38 MAPK activities by atrial natriuretic peptide in pulmonary endothelium significantly reduced endothelial barrier dysfunction and attenuated lung inflammation (15, 22, 23). However, interrelationships between LTA/PepG-induced stress kinase (p38 MAPK), RhoA and NFκB signaling are not understood, and potential differences between lung macro- and micro-vascular endothelial cell responses to LTA/PepG await further investigation. This study investigated whether endothelial cells from different pulmonary vascular beds display different patterns of signaling cascades activation and barrier dysfunction in response to Gram-positive bacterial compounds LTA and PepG.
MATERIALS AND METHODS
Reagents and cell culture
Human pulmonary artery (HPAECs) and human lung microvascular (HLMVECs) endothelial cells were obtained from Lonza (Allendale, NJ). Cells were maintained in a complete culture medium according to the manufacturer’s recommendations and used for experiments at passages 5–8. Phospho-Hsp27, phospho-p38 MAPK, di-phospho-MLC, and IκBα antibodies were obtained from Cell Signaling (Beverly, MA); phospho-MYPT antibodies were purchased from Millipore (Billerica, MA). Reagents for immunofluorescence were purchased from Molecular Probes (Eugene, OR). SB203580 and Y27632 were purchased from EDM (La Jolla, CA). Unless specified, biochemical reagents including LTA and PepG were obtained from Sigma (St. Louis, MO).
Measurement of transendothelial electrical resistance across confluent HPAEC monolayers was performed using an electrical cell-substrate impedance sensing system (Applied Biophysics, Troy, NY), as previously described (34, 35). Experiments were conducted only on wells that achieved >1,000 Ω (10 microelectrodes/well) of steadystate resistance. Resistance was expressed by the in-phase voltage (proportional to the resistance), which was normalized to the initial voltage and expressed as a fraction of the normalized resistance value.
Immunofluorescence labeling
After agonist treatment endothelial cells grown on glass coverslips were fixed in PBS containing 3.7% formaldehyde, and F-actin was visualized by immunofluorescence staining of cell monolayers with Texas Red conjugated phalloidin as previously described (35, 36). Image analysis of gap formation in control and LTA/PepG-challenged EC monolayers with and without pretreatment with inhibitors was performed using an algorithm described in our previous studies (36, 37)
Western blot analysis
Protein extracts from mice lungs or ECs were separated by SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membranes which were then incubated with primary antibodies to proteins of interest, as described elsewhere (38). Immunoreactive proteins were detected using the enhanced chemiluminescent detection system according to the manufacturer’s protocol (Amersham, Little Chalfont, UK). Equal protein loading was verified by reprobing membranes with antibody to β-tubulin or specific protein of interest. The relative intensities of immunoreactive protein bands (RDU, relative density units) were analyzed and quantified by scanning densitometry using Image Quant software (Molecular Dynamics, Sunnyvale, CA).
In vivo model of acute lung injury
Adult male C57BL/6J mice, 8–10 week old, with average weight 20–25 grams (Jackson Laboratories, Bar Harbor, ME) were anesthetized with an intraperitoneal injection of ketamine (75 mg/kg) and acepromazine (1.5 mg/kg). A mixture of LTA (2.5 mg/kg) and PepG (2.5 mg/kg) or sterile saline solution were injected intratracheally in a small volume (20–30μl) using a 20 gauge catheter. The doses of Rho kinase inhibitor Y-27632 and p38 MAP kinase inhibitor SB203580 causing maximal inhibitory effects had been determined in previous studies by our (39) and other groups (32, 40). Mice were randomized to concurrently receive sterile saline solution, Y27632 (2 mg/kg) or SB203580 (10 mg/kg) by intravenous injection in the external jugular vein to yield the experimental groups: control, (LTA + PepG), (LTA + PepG) + Y27632, and (LTA + PepG) + SB203580. At 24 hours, animals were sacrificed by exsanguination under anesthesia. Tracheotomy was performed, and the trachea was canulated with a 20 gauge intravenous catheter, which was tied into place. Measurements of cell count and protein concentration in bronchoalveolar lavage fluid (BALF) were performed as previously described (41, 42). For histopathologic lung analysis, left lung was harvested from each treated mouse and immediately fixed with 10% buffered formalin overnight, followed by embedding in paraffin. Lung microsections were used for histological evaluation by H&E staining. Cellular infiltration in control and treated lungs was evaluated by light microscopy. All experimental protocols involving the use of animals were approved by the University of Chicago Institutional Animal Care & Use Committee for the humane treatment of experimental animals. The animals were housed in pathogen-free conditions in the University of Chicago Animal Care Facilities where they were cared for in accordance with the institutional and the National Institutes of Health (NIH) guidelines.
Statistical analysis
Results are expressed as means ±SD of three to eight independent experiments. Stimulated samples were compared with controls by using unpaired Student’s t-test. For multiple-group comparisons, a one-way variance analysis (ANOVA) and post hoc multiple comparisons tests were used, and results with P <0.05 were considered statistically significant.
RESULTS
Effects of Rho kinase and p38 MAPK inhibitors on lung injury induced by combined treatment with LTA and PepG
Concurrent intratracheal instillation of LTA (2.5 mg/kg) and PepG (2.5 mg/kg) induced significant lung injury manifested by 25-fold increase in the total cell count in BAL fluid and 6-fold elevation of protein content (Figure 1A). A single concurrent intravenous injection of Rho kinase inhibitor Y27632 (2 mg/kg) or p38 MAPK inhibitor SB203580 (10 mg/kg) significantly attenuated BALF cell counts and protein content in the animals treated with combination of LTA and PepG (0.314 ± 0.072 mg/ml protein in Y27632-pretreated group and 0.420 ± 0.061 mg/ml protein in SB203580-pretreated group vs. 0.616 ± 0.065 mg/ml protein LPS treatment alone; p<0.05). Of note, both Y27632 and SB203580 had a similar effect on the reduction of the total and neutrophil cell count after LTA/PepG treatment (Figure 1BC), whereas Y27632 was more effective against the LTA/PepG-induced increase in BALF protein concentration (p<0.05) (Figure 1A). LTA/PepG induced inflammatory cell infiltration (predominantly neutrophils and mononuclear cells) in both lung interstitium and alveolar compartments (Figure 1D). Pretreatment with Y27632 or SB203580 attenuated LTA/PepG-induced inflammatory cell infiltration in lung tissue.
Figure 1. Effects of Rho kinase and p38 MAPK inhibitors on the development of lung inflammation induced by LTA and PepG.
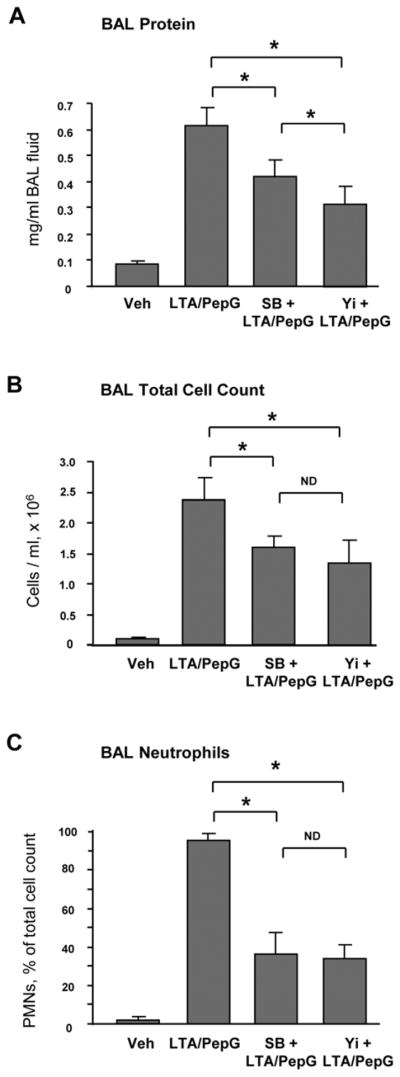

C57BL/6J mice were treated i.t. with a mixture of LTA (2.5 mg/kg) and PepG (2.5 mg/kg), with or without concurrent i.v. treatment with Y27632 (2 mg/kg), SB203580 (10 mg/kg) for 24 hours. Control animals were treated with sterile saline solution. A - Protein concentration; B - Total cell count; and C - Neutrophil count were measured in bronchoalveolar lavage fluid taken from control and experimental animals, n=6 per condition; *p<0.05. D – Histological analysis of lung tissue (×40 magnification). Whole lungs (4 to 6 animals from each experimental group) were agarose-inflated in situ, fixed with 10% formalin, and used for hematoxylin and eosin staining and histological evaluation.
Analysis of LTA/PepG-induced activation of inflammatory signaling in human lung microvascular and macrovascular ECs
All following in vitro experiments were performed using combined LTA and PepG co-treatment (50 ng/ml and 100 ng/ml, respectively). Activation of intracellular signaling by LTA/PepG was evaluated by increases in phosphorylation of signaling proteins involved in stress and inflammatory cascades. LTA/PepG treatment caused sustained phosphorylation of MYPT1 at the Rho kinase-specific site and increased myosin light chain phosphorylation in both EC types (Figure 2AB, upper panels). These parameters reflect activation of EC actomyosin machinery associated with activated Rho pathway (29, 38). LTA/PepG also activated stress signaling reflected by time-dependent phosphorylation of stress-activated p38 MAPK and its downstream target Hsp27 (Figure 2AB, middle panels), and led to activation of inflammatory signaling as detected by degradation of IκBα, an inhibitory subunit of the NFκB complex, which causes activation of NFκB-dependent transcription (Figure 2AB, lower panels).
Figure 2. Effects of combined LTA and PepG treatment on inflammatory cascade activation in pulmonary macro- and micro-vascular endothelium.


HPAEC (A) or HLMVEC (B) were treated with a combination of LTA (50 ng/ml) and PepG (100 ng/ml) for indicated periods of time. Phosphorylation of p38 MAPK, Hsp27, MYPT1, and MLC was determined by western blot with corresponding phospho-specific antibodies. Degradation of IκBα was detected using pan IκBα antibodies. Phosphorylation of the protein of interest was evaluated by quantitative densitometry and normalized to the total content of corresponding protein in cell lysates. Rate of IκBα protein degradation was normalized to β-tubulin content in cell lysates. Bar graphs represent results of three to six independent experiments; *p<0.01 vs vehicle control.
Rho kinase and p38 MAPK inhibitors attenuate LTA/PepG-induced actin cytoskeleton remodeling and barrier disruption in human lung microvascular and macrovascular endothelial monolayers
Morphological analysis of agonist-treated EC monolayers revealed that co-treatment with LTA/PepG induced formation of actin stress fibers and cell retraction accompanied by formation of paracellular gaps in both microvascular and macrovascular endothelium (Figure 3AB, marked by arrows), reflecting compromised EC monolayer integrity. Pretreatment with SB203580 (20 μM, 30 min) or Y27632 (2 μM, 30 min) attenuated LTA/PepG-induced stress fiber and paracellular gap formation in HPAECs and HLMVECs. However, in HPAECs, SB203580 was less efficient in inhibiting LTA/PepG-induced EC barrier dysfunction (Figure 3A) than in HLMVECs (Figure 3B), as detected by increased number of paracellular gaps after SB203580-LTA/PepG treatment (Figure 3C).
Figure 3. Effects of p38 MAPK and Rho pathway inhibition on LTA/PepG-induced cytoskeletal remodeling.

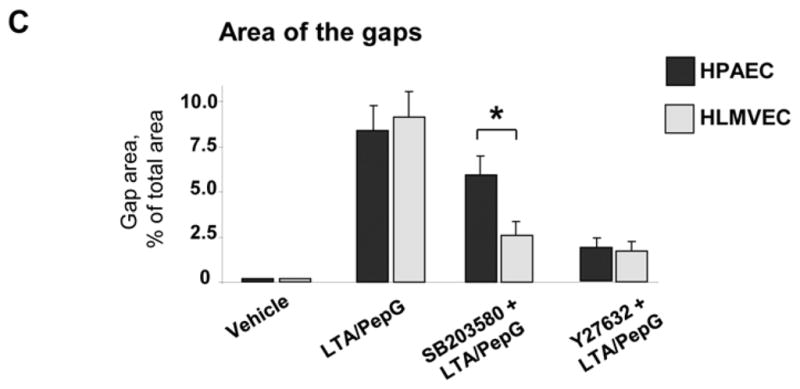
A - HPAEC or B - HLMVEC were challenged with a combination of LTA (50 ng/ml) and PepG (100 ng/ml) with or without pretreatment with p38 MAPK inhibitor SB203580 (20 μM) or Rho kinase inhibitor Y27632 (2 μM) for 30 min. Actin rearrangement was assessed after 6 hours by immunofluorescence staining for F-actin with Texas Red phalloidin. Paracellular gaps are marked by arrows. C - Quantitative image analysis of gap formation in control and stimulated endothelial monolayers was performed as described in Methods. (p<0.01; n=3 independent experiments; 10 microscopic fields per condition).
Inhibition of p38 MAPK and Rho kinase attenuates LTA/PepG-induced hyper-permeability in macrovascular and microvascular endothelial cells
The role of Rho and p38 MAPK signaling in LTA/PepG-induced barrier dysfunction in macrovascular and microvascular ECs was further investigated in functional assay by monitoring changes in transendothelial electrical resistance (TER) as a readout of EC permeability. Basal levels of electrical resistance were 1109+/−236 Ohm for HPAEC and 1749+/−312 for HLMVEC at the beginning of the experiments, and 1164+/−261 Ohm for HPAEC and 1801+/−337 for HLMVEC at the end of the experiments, for vehicle-treated cells. The magnitude of LTA/PepG-induced TER decline was similar between HPAECs and HLMVECs and reached maximal levels by 15 hrs of LTA/PepG stimulation (Figure 4AB). This time point was used for analysis of effects of Rho kinase and p38 MAPK inhibitors on LTA-PepG induced EC barrier dysfunction. Inhibition of Rho kinase caused more significant attenuation of LTA/PepG-induced permeability in HPAECs than inhibition of p38 MAPK (Figure 4C). In contrast, both p38 MAPK and Rho kinase inhibitors caused similar barrier protective effects in LTA/PepG-challenged lung microvascular ECs (Figure 4C). These results indicate potential differences in p38 MAPK - Rho signaling interrelationships in control of microvascular and macrovascular EC permeability responses to LTA/PepG. These differences were further investigated in biochemical studies.
Figure 4. Effects of p38 MAPK and Rho pathway inhibition on LTA/PepG-induced hyper-permeability in lung macrovascular and microvascular endothelium.

HPAEC or HLMVEC grown on microelectrodes to confluence were pretreated with 20 μM SB203580 (A) or 2 μM Y27632 (B) for 30 min followed by challenge with a combination of LTA (50 ng/ml) and PepG (100 ng/ml) and used for measurements of TER. C - Permeability changes were registered at the time of maximal permeability response (15 hrs post LTA/PepG treatment). Maximal TER decline reflecting endothelial barrier dysfunction induced by LTA/PepG was taken as 100%. Results are shown as mean ± SD, * p<0.05; n=8 experiments.
Analysis of interactions between Rho kinase and p38 MAPK pathways in lung microvascular and macrovascular EC stimulated with LTA/PepG
Effects of Rho kinase and p38 MAPK inhibitors on cross-regulation of p38 MAPK, Rho, and NFκB pathways were evaluated by western blot analysis (Figure 5AB). Quantitative analysis of western blot data showed that inhibition of p38 MAPK abolished LTA/PepG-induced Hsp27 phosphorylation in both cell types, but did not affect the Rho kinase-dependent phosphorylation of MYPT1 and MLC in macrovascular ECs (Figure 5C). In contrast, inhibition of p38 MAPK significantly decreased LTA/PepG induced MYPT1 and MLC phosphorylation in microvascular cells. These results suggest that LTA/PepG-induced activation of Rho kinase can be modulated by p38 MAPK in HLMVECs, but not in HPAECs. In turn, inhibition of Rho kinase abolished LTA/PepG-induced MYPT1 and MLC phosphorylation and significantly attenuated phosphorylation of p38 MAPK and Hsp27 in both cell types. Interestingly, inhibition of Rho kinase and p38 MAPK activities also attenuated LTA/PepG induced IκBα subunit degradation in both cell types, while the effect of p38 MAPK inhibition on IκBα preservation in HLMVECs was more pronounced. These results suggest unique patterns of p38 MAPK – Rho – NFκB signaling balance in lung macrovascular and microvascular endothelium.
Figure 5. Effects of p38 MAPK and Rho pathway inhibition on LTA/PepG-induced inflammatory signaling.


HPAEC (A) or HLMVEC (B) were pretreated with SB203580 (20 μM) or Y27632 (2 μM) for 30 min followed by LTA/PepG stimulation for 4 hours. Activation of Rho cascade, p38 MAPK, and NFκB cascades was examined by analysis of specific protein phosphorylation and IκBα degradation. Equal protein loading was confirmed by probing of membranes with β-tubulin antibodies. C – Quantitative analysis of western blot data. Results of densitometry are shown as mean ± SD, * p<0.05; n=6 experiments.
DISCUSSION
The main finding of this study is a distinct relationship between Rho and p38 stress kinase signaling in macrovascular and microvascular endothelial cells, which leads to increased EC permeability and lung barrier dysfunction. Despite the common location in the lung and shared roles in control of pulmonary circulation, endothelial cells from different lung vascular compartments may exhibit different permeability responses to pathologic stimuli or pharmacological treatments (43). For example, direct activation of store-operated channel Ca2+ entry increased extra-alveolar, but not alveolar, EC permeability (44). These examples illustrate differential effects of same stimulation on endothelial micro- and macrovascular permeability, which reflect phenotypic differences of endothelium from these beds. More detailed characterization of endothelial heterogeneity in the lung is important for more complete understanding of the mechanisms of inflammation and permeability control specific for microvascular and macrovascular endothelium. This knowledge is also critical for development of targeted therapeutic treatments of lung vasculature as potential new approach to confront ALI/ARDS.
The present study shows that inhibition of p38 MAPK was less effective in suppressing the LTA/PepG-induced permeability in macrovascular EC, while effect of Rho kinase inhibitor was equal in both microvascular and macrovascular ECs. These results can be explained by different degrees of interaction between LTA/PepG-induced Rho and p38 MAPK signaling in macrovascular and microvascular endothelium. While Rho kinase inhibitor abolished phosphorylation of Rho kinase targets and partially attenuated p38 MAPK signaling in both macro microvascular and macrovascular ECs, the inhibition of p38 MAPK significantly attenuated LTA/PepG-induced activation of Rho signaling in microvascular ECs, but not in macrovascular EC. These data suggest bidirectional Rho - p38 MAPK crosstalk in LTA/PepG-challenged microvascular ECs, and uni-directional Rho/Rho kinase → p38 MAPK interactions in macrovascular ECs. Requirement for Rho in TLR-dependent NFκB activation has been described in lung epithelial cells and brain microvascular endothelium, although specific mechanism of such activation remains unclear (25, 26). Inhibition of both Rho kinase and p38 MAPK signaling significantly attenuated LTA/PepG-induced activation of the NFκB cascade as monitored by the levels of inhibitory IκBα subunit degradation. Of note, while maximal protection against LTA/PepG-induced IκBα degradation was achieved by Rho-kinase inhibitor in both macrovascular and microvascular ECs, the protective effect of p38 MAPK inhibition was stronger in microvascular cells. Figure 6 summarizes the observed differences between Rho and p38 MAPK crosstalk mechanisms induced by LTA/PepG challenge of human lung microvascular and macrovascular ECs. Taken together, these results demonstrate a possibility of more site-specific control of microvascular and macrovascular permeability in the lung by cell type-specific targeting of Rho/Rho kinase and p38 MAPK activities.
Figure 6. Schematic presentation of relations between Rho and p38 MAPK signaling leading to barrier dysfunction in human lung microvascular and macrovascular endothelium.
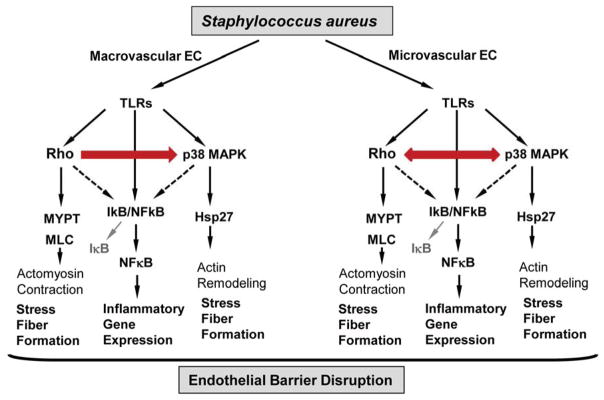
Stimulation of both, microvascular and macrovascular EC, with LTA/PepG triggers Rho, p38 MAPK and NFκB signaling leading to cytoskeletal remodeling, stress fiber formation and endothelial barrier dysfunction. In HPAEC, LTA/PepG-induced p38 MAPK activation is attenuated by inhibition of Rho kinase, while activation of Rho kinase is not affected by inhibition of p38 MAPK. In contrast, the Rho - p38 MAPK crosstalk in microvascular EC is bi-directional, as inhibition of either pathway affected its counterpart. Finally, both Rho and p38 MAPK pathways modulate LTA/PepG-induced activation of NFκB pathway.
The mechanisms which determine the differences between LTA/PepG-activated signaling in micro- and macro-vascular endothelium require further investigation. One potential explanation of cell type-specific differences is a multi-step mechanism of NFκB, p38 MAPK and Rho activation by TLR receptors, which involves intermediate signaling events. Activation of NFκB is dependent on NFκB-inducing kinase (NIK) and TAK1; p38MAPK and NFκB is also activated by MEKK3 (17, 45), while Rho activation is dependent on TAK1 and other yet-to-be-identified molecules such as recently described guanine nucleotide exchange factor AKAP13 activated by TLR2 ligation (26, 46). Thus, differences in the spectrum and magnitude of signaling pathways activated in pulmonary microvascular and macrovascular endothelium by LTA/PepG may be a result of complex interactions between regulatory and adaptor molecules downstream of TLR2 and may reflect specific expression patterns of signaling proteins in micro- and macro-vascular ECs, or can be controlled by additional crosstalk mechanisms between differentially expressed TLR receptors. Further application of the methods of systems biology may be a powerful tool for comprehensive understanding of such signaling differences between micro- and macro-vascular EC.
In conclusion, our results demonstrate the differences in signaling responses induced by Staphylococcus aureus-derived pathogens in pulmonary macrovascular and microvascular human lung endothelium. Although both cell types developed barrier-disruptive response to Gram-positive bacterial compounds, such hyper-permeability was mediated by differential interplay between Rho, p38 MAPK, and NFκB signaling. These observations may have important implications in defining microvasculature-specific therapeutic strategies aimed at the treatment of sepsis and ALI induced by Gram-positive bacterial pathogens.
Acknowledgments
Supported by NHLBI grants HL89257 and HL107920
Sources of Funding: the National Heart, Lung, and Blood Institutes grants HL89257 and HL107920
Footnotes
The authors have no conflicts of interest to declare. All authors have read the journal’s policy on disclosure of potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Melamed A, Sorvillo FJ. The burden of sepsis-associated mortality in the united states from 1999 to 2005: An analysis of multiple-cause-of-death data. Crit Care. 2009;13(1):R28. doi: 10.1186/cc7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matthay MA, Zimmerman GA, Esmon C, et al. Future research directions in acute lung injury: Summary of a national heart, lung, and blood institute working group. Am J Respir Crit Care Med. 2003;167(7):1027–1035. doi: 10.1164/rccm.200208-966WS. [DOI] [PubMed] [Google Scholar]
- 3.Pinner RW, Teutsch SM, Simonsen L, et al. Trends in infectious diseases mortality in the united states. Jama. 1996;275(3):189–193. [PubMed] [Google Scholar]
- 4.Gacouin A, Barbarot N, Camus C, et al. Late-onset ventilator-associated pneumonia in nontrauma intensive care unit patients. Anesth Analg. 2009;109(5):1584–1590. doi: 10.1213/ANE.0b013e3181b6e9b6. [DOI] [PubMed] [Google Scholar]
- 5.Hortal J, Giannella M, Perez MJ, et al. Incidence and risk factors for ventilator-associated pneumonia after major heart surgery. Intensive Care Med. 2009;35(9):1518–1525. doi: 10.1007/s00134-009-1523-3. [DOI] [PubMed] [Google Scholar]
- 6.Lechner AJ, Ryerse JS, Matuschak GM. Acute lung injury during bacterial or fungal sepsis. Microsc Res Tech. 1993;26(5):444–456. doi: 10.1002/jemt.1070260512. [DOI] [PubMed] [Google Scholar]
- 7.Hayashida A, Bartlett AH, Foster TJ, et al. Staphylococcus aureus beta-toxin induces lung injury through syndecan-1. Am J Pathol. 2009;174(2):509–518. doi: 10.2353/ajpath.2009.080394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang JE, Dahle MK, McDonald M, et al. Peptidoglycan and lipoteichoic acid in gram-positive bacterial sepsis: Receptors, signal transduction, biological effects, and synergism. Shock. 2003;20(5):402–414. doi: 10.1097/01.shk.0000092268.01859.0d. [DOI] [PubMed] [Google Scholar]
- 9.Kielian T, Haney A, Mayes PM, et al. Toll-like receptor 2 modulates the proinflammatory milieu in staphylococcus aureus-induced brain abscess. Infect Immun. 2005;73(11):7428–7435. doi: 10.1128/IAI.73.11.7428-7435.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leemans JC, Vervoordeldonk MJ, Florquin S, et al. Differential role of interleukin-6 in lung inflammation induced by lipoteichoic acid and peptidoglycan from staphylococcus aureus. Am J Respir Crit Care Med. 2002;165(10):1445–1450. doi: 10.1164/rccm.2106045. [DOI] [PubMed] [Google Scholar]
- 11.De Kimpe SJ, Kengatharan M, Thiemermann C, et al. The cell wall components peptidoglycan and lipoteichoic acid from staphylococcus aureus act in synergy to cause shock and multiple organ failure. Proc Natl Acad Sci U S A. 1995;92(22):10359–10363. doi: 10.1073/pnas.92.22.10359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Netea MG, van der Graaf C, Van der Meer JW, et al. Toll-like receptors and the host defense against microbial pathogens: Bringing specificity to the innate-immune system. J Leukoc Biol. 2004;75(5):749–755. doi: 10.1189/jlb.1103543. [DOI] [PubMed] [Google Scholar]
- 13.Takeda K, Akira S. Tlr signaling pathways. Semin Immunol. 2004;16(1):3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 14.Mayer AK, Muehmer M, Mages J, et al. Differential recognition of tlr-dependent microbial ligands in human bronchial epithelial cells. J Immunol. 2007;178(5):3134–3142. doi: 10.4049/jimmunol.178.5.3134. [DOI] [PubMed] [Google Scholar]
- 15.Xing J, Moldobaeva N, Birukova AA. Atrial natriuretic peptide protects against staphylococcus aureus-induced lung injury and endothelial barrier dysfunction. J Appl Physiol. 2011;110(1):213–224. doi: 10.1152/japplphysiol.00284.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arbibe L, Mira JP, Teusch N, et al. Toll-like receptor 2-mediated nf-kappa b activation requires a rac1-dependent pathway. Nat Immunol. 2000;1(6):533–540. doi: 10.1038/82797. [DOI] [PubMed] [Google Scholar]
- 17.Dauphinee SM, Karsan A. Lipopolysaccharide signaling in endothelial cells. Lab Invest. 2006;86(1):9–22. doi: 10.1038/labinvest.3700366. [DOI] [PubMed] [Google Scholar]
- 18.Birukova AA, Birukov KG, Gorshkov B, et al. Map kinases in lung endothelial permeability induced by microtubule disassembly. Am J Physiol Lung Cell Mol Physiol. 2005;289(1):L75–84. doi: 10.1152/ajplung.00447.2004. [DOI] [PubMed] [Google Scholar]
- 19.Kiemer AK, Weber NC, Furst R, et al. Inhibition of p38 mapk activation via induction of mkp-1: Atrial natriuretic peptide reduces tnf-alpha-induced actin polymerization and endothelial permeability. Circ Res. 2002;90(8):874–881. doi: 10.1161/01.res.0000017068.58856.f3. [DOI] [PubMed] [Google Scholar]
- 20.Nwariaku FE, Chang J, Zhu X, et al. The role of p38 map kinase in tumor necrosis factor-induced redistribution of vascular endothelial cadherin and increased endothelial permeability. Shock. 2002;18(1):82–85. doi: 10.1097/00024382-200207000-00015. [DOI] [PubMed] [Google Scholar]
- 21.Ruse M, Knaus UG. New players in tlr-mediated innate immunity: Pi3k and small rho gtpases. Immunol Res. 2006;34(1):33–48. doi: 10.1385/IR:34:1:33. [DOI] [PubMed] [Google Scholar]
- 22.Birukova AA, Xing J, Fu P, et al. Atrial natriuretic peptide attenuates lps-induced lung vascular leak: Role of pak1. Am J Physiol Lung Cell Mol Physiol. 2010;299(5):L652–663. doi: 10.1152/ajplung.00202.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xing J, Birukova AA. Anp attenuates inflammatory signaling and rho pathway of lung endothelial permeability induced by lps and tnfalpha. Microvasc Res. 2010;79(1):26–62. doi: 10.1016/j.mvr.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Teusch N, Lombardo E, Eddleston J, et al. The low molecular weight gtpase rhoa and atypical protein kinase czeta are required for tlr2-mediated gene transcription. J Immunol. 2004;173(1):507–514. doi: 10.4049/jimmunol.173.1.507. [DOI] [PubMed] [Google Scholar]
- 25.He F, Peng J, Deng XL, et al. Rhoa and nf-kappab are involved in lipopolysaccharide-induced brain microvascular cell line hyperpermeability. Neuroscience. 2011;188:35–47. doi: 10.1016/j.neuroscience.2011.04.025. [DOI] [PubMed] [Google Scholar]
- 26.Manukyan M, Nalbant P, Luxen S, et al. Rhoa gtpase activation by tlr2 and tlr3 ligands: Connecting via src to nf-kappa b. J Immunol. 2009;182(6):3522–3529. doi: 10.4049/jimmunol.0802280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmeck B, Brunsch M, Seybold J, et al. Rho protein inhibition blocks cyclooxygenase-2 expression by proinflammatory mediators in endothelial cells. Inflammation. 2003;27(2):89–95. doi: 10.1023/a:1023278600596. [DOI] [PubMed] [Google Scholar]
- 28.Vouret-Craviari V, Boquet P, Pouyssegur J, et al. Regulation of the actin cytoskeleton by thrombin in human endothelial cells: Role of rho proteins in endothelial barrier function. Mol Biol Cell. 1998;9(9):2639–2653. doi: 10.1091/mbc.9.9.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Nieuw Amerongen GP, van Delft S, Vermeer MA, et al. Activation of rhoa by thrombin in endothelial hyperpermeability: Role of rho kinase and protein tyrosine kinases. Circ Res. 2000;87(4):335–340. doi: 10.1161/01.res.87.4.335. [DOI] [PubMed] [Google Scholar]
- 30.Fukata Y, Amano M, Kaibuchi K. Rho-rho-kinase pathway in smooth muscle contraction and cytoskeletal reorganization of non-muscle cells. Trends Pharmacol Sci. 2001;22(1):32–39. doi: 10.1016/s0165-6147(00)01596-0. [DOI] [PubMed] [Google Scholar]
- 31.Hocke AC, Temmesfeld-Wollbrueck B, Schmeck B, et al. Perturbation of endothelial junction proteins by staphylococcus aureus alpha-toxin: Inhibition of endothelial gap formation by adrenomedullin. Histochem Cell Biol. 2006;126(3):305–316. doi: 10.1007/s00418-006-0174-5. [DOI] [PubMed] [Google Scholar]
- 32.Tasaka S, Koh H, Yamada W, et al. Attenuation of endotoxin-induced acute lung injury by the rho-associated kinase inhibitor, y-27632. Am J Respir Cell Mol Biol. 2005;32(6):504–510. doi: 10.1165/rcmb.2004-0009OC. [DOI] [PubMed] [Google Scholar]
- 33.Slotta JE, Braun OO, Menger MD, et al. Fasudil, a rho-kinase inhibitor, inhibits leukocyte adhesion in inflamed large blood vessels in vivo. Inflamm Res. 2006;55(9):364–367. doi: 10.1007/s00011-006-6013-2. [DOI] [PubMed] [Google Scholar]
- 34.Birukov KG, Bochkov VN, Birukova AA, et al. Epoxycyclopentenone-containing oxidized phospholipids restore endothelial barrier function via cdc42 and rac. Circ Res. 2004;95(9):892–901. doi: 10.1161/01.RES.0000147310.18962.06. [DOI] [PubMed] [Google Scholar]
- 35.Birukova AA, Cokic I, Moldobaeva N, et al. Paxillin is involved in the differential regulation of endothelial barrier by hgf and vegf. Am J Respir Cell Mol Biol. 2008 doi: 10.1165/rcmb.2008-0099OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Birukova AA, Chatchavalvanich S, Rios A, et al. Differential regulation of pulmonary endothelial monolayer integrity by varying degrees of cyclic stretch. Am J Pathol. 2006;168(5):1749–1761. doi: 10.2353/ajpath.2006.050431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Birukova AA, Moldobaeva N, Xing J, et al. Magnitude-dependent effects of cyclic stretch on hgf- and vegf-induced pulmonary endothelial remodeling and barrier regulation. Am J Physiol Lung Cell Mol Physiol. 2008;295(4):L612–623. doi: 10.1152/ajplung.90236.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Birukova AA, Smurova K, Birukov KG, et al. Role of rho gtpases in thrombin-induced lung vascular endothelial cells barrier dysfunction. Microvasc Res. 2004;67(1):64–77. doi: 10.1016/j.mvr.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 39.Birukova AA, Tian Y, Meliton AY, et al. Stimulation of rho signaling by pathologic mechanical stretch is a “Second hit” To rho-independent lung injury induced by il-6. Am J Physiol Lung Cell Mol Physiol. 2012;302(9):L965–975. doi: 10.1152/ajplung.00292.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu S, Feng G, Wang GL, et al. P38mapk inhibition attenuates lps-induced acute lung injury involvement of nf-kappab pathway. Eur J Pharmacol. 2008;584(1):159–165. doi: 10.1016/j.ejphar.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 41.Birukova AA, Fu P, Chatchavalvanich S, et al. Polar head groups are important for barrier protective effects of oxidized phospholipids on pulmonary endothelium. Am J Physiol Lung Cell Mol Physiol. 2007;292(4):L924–935. doi: 10.1152/ajplung.00395.2006. [DOI] [PubMed] [Google Scholar]
- 42.Fu P, Birukova AA, Xing J, et al. Amifostine reduces lung vascular permeability via suppression of inflammatory signalling. Eur Respir J. 2009;33(3):612–624. doi: 10.1183/09031936.00014808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lowe K, Alvarez D, King J, et al. Phenotypic heterogeneity in lung capillary and extra-alveolar endothelial cells. Increased extra-alveolar endothelial permeability is sufficient to decrease compliance. J Surg Res. 2007;143(1):70–77. doi: 10.1016/j.jss.2007.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cioffi DL, Lowe K, Alvarez DF, et al. Trping on the lung endothelium: Calcium channels that regulate barrier function. Antioxid Redox Signal. 2009;11(4):765–776. doi: 10.1089/ars.2008.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang Q, Yang J, Lin Y, et al. Differential regulation of interleukin 1 receptor and toll-like receptor signaling by mekk3. Nat Immunol. 2004;5(1):98–103. doi: 10.1038/ni1014. [DOI] [PubMed] [Google Scholar]
- 46.Shibolet O, Giallourakis C, Rosenberg I, et al. Akap13, a rhoa gtpase-specific guanine exchange factor, is a novel regulator of tlr2 signaling. J Biol Chem. 2007;282(48):35308–35317. doi: 10.1074/jbc.M704426200. [DOI] [PubMed] [Google Scholar]