

Abstract
The balance between controlling infection and limiting inflammation is particularly precarious in the brain because of its unique vulnerability to the toxic effects of inflammation. Astrocytes have been implicated as key regulators of neuroinflammation in CNS infections, including infection with Toxoplasma gondii, a protozoan parasite that naturally establishes a chronic CNS infection in mice and humans. In CNS toxoplasmosis, astrocytes are critical to controlling parasite growth. They secrete pro-inflammatory cytokines and physically encircle parasites. However, the molecular mechanisms used by astrocytes to limit neuroinflammation during toxoplasmic encephalitis have not yet been identified. Transforming growth factor beta (TGFβ) signaling in astrocytes is of particular interest because TGFβ is universally upregulated during CNS infection and serves master regulatory and primarily anti-inflammatory functions. We report here that TGFβ signaling is activated in astrocytes during toxoplasmic encephalitis, and that inhibition of astrocytic TGFβ signaling increases immune cell infiltration, uncouples pro-inflammatory and chemokine production from CNS parasite burden, and increases neuronal injury. Remarkably, we show that the effects of inhibiting astrocytic TGFβ signaling are independent of parasite burden and the ability of GFAP+ astrocytes to physically encircle parasites.
INTRODUCTION
Astrocytes have recently gained prominence as essential mediators of the brain’s innate immune response to a variety of brain insults including infection. During brain infections, astrocytes secrete pro-inflammatory cytokines and express key immune receptors, such as toll-like receptors and cytokine receptors, enabling them to mount a pro-inflammatory response to a number of signals (1–3). In addition, astrocytes upregulate the intermediate filament glial fibrillary acidic protein (GFAP) and form a physical barrier around the microbes and the infiltrating immune cells. This barrier function is a key characteristic of astrocytes during infection and serves to protect the brain from immune cell infiltration and neuronal injury (4, 5). Protecting the brain from neuroinflammation is critical as the brain is uniquely vulnerable to the toxic effects of inflammation due to its limited regenerative capacity and its confined space (6, 7).
Given the importance of protecting the brain from inflammatory responses and the recognition that astrocytes play a critical role in this process, it is surprising how little is known in vivo about what role astrocytes might play in dampening neuroinflammation through their response to anti-inflammatory cytokines. Transforming growth factor beta (TGFβ) signaling in astrocytes is of particular interest because TGFβ s are master regulatory and primarily anti-inflammatory cytokines that are universally increased during CNS infection and injury (8–10). While TGFβ is directly neuroprotective (11), it can also signal to all major brain cell types, including astrocytes (9, 11–13). In addition, astrocytic TGFβ signaling after stroke decreases neuroinflammation and preserves neuronal function (14). Thus, we hypothesized that astrocytic TGFβ signaling might be a key pathway for limiting brain inflammation during CNS infection. To test this hypothesis, we used the naturally neurotropic parasite Toxoplasma gondii to infect transgenic mice in which astrocytic TGFβ signaling was selectively inhibited, and then compared the inflammatory outcomes to infected wildtype littermates.
Toxoplasma is an obligate intracellular parasite that naturally establishes a chronic CNS infection in mice and humans and is known to increase CNS TGFβ expression (15). Astrocytes are known to play a critical pro-inflammatory role in controlling murine CNS toxoplasmosis. In vitro, astrocytes infected with Toxoplasma limit the intracellular growth of the parasite after stimulation with pro-inflammatory cytokines such as IFNγ (16). In vivo, they express pro-inflammatory cytokines and chemokines that likely both limit Toxoplasma growth and also attract immune cells (16–18). Astrocytes also clearly form a physical barrier by upregulating GFAP early in toxoplasmic encephalitis andphysically surrounding Toxoplasma and leukocyte infiltrates (17, 19).
Numerous studies have shown that GFAP+ astrocytes surround sites of CNS infection and inflammation and that when there are fewer GFAP+ astrocytes, infection and inflammation becomes more diffuse (5, 17, 19, 20). GFAP knockout mice infected with Toxoplasma exhibit an exacerbated brain parasite burden, an increased immune response and an increased mortality (19). Toxoplasma infection produces a similar phenotype in transgenic mice that lack astrocytic gp130, a cytokine receptor that mediates the signaling of the IL-6 cytokine family (17). However, potential anti-inflammatory functions of astrocytes during Toxoplasma infection are poorly understood.
We report here that TGFβ signaling is activated in astrocytes during toxoplasmic encephalitis and that inhibition of astrocytic TGFβ signaling increases immune cell infiltration, uncouples pro-inflammatory cytokine and chemokine production from CNS parasite burden, and increases neuronal injury. Remarkably, we show that the effects of inhibiting astrocytic TGFβ signaling are independent of parasite burden and the ability of GFAP+ astrocytes to physically encircle parasites and support the notion that astrocytes play a critical role in targeting the adaptive immune response to sites of infection.
MATERIALS AND METHODS
Mice
Animal experiments were performed in compliance with the NIH Guide for Care and Use of Animals and were approved by the Stanford University and University of Arizona Institutional Animal Care and Use Committees and the NIH Guide for Care and Use of Animals. Ast-Tbr2DN transgenic mice were double transgenic mice bred from B6.FVB-Tg(tetO-EGFP,-Tgfbr2)8Mcle/J (JAX #005738) and B6.Cg-Tg(GFAP-tTA)110Pop/J (JAX #005964). All experiments were done using three month-old males. Singly transgenic B6.FVB-Tg(tetO-EGFP,-Tgfbr2)8Mcle/J littermates were used as wildtype controls.
Toxoplasma gondii infection
Type II (Prugniaud) parasites expressing mCherry were maintained and purified as previously described (21). Experimental mice were orally infected with Toxoplasma cysts as previously described (22). In brief, Ast-Tbr2DN or wildtype mice were starved overnight and then placed in individual cages and offered mouse chow soaked in brain homogenates that contained 150–180 Toxoplasma cysts. The mice remained in the individual cage until all of the homogenate was eaten (< 3 hours.) In each experiment, the same number of cysts was fed to wildtype and Ast-Tbr2DN mice. The brain homogenates containing cysts were derived from female CBA/J mice that had been infected with 2.5×104 parasites intraperitoneally 3 weeks prior to harvest. Orally infected mice were sacrificed at 2 or 4 weeks post infection (wpi).
Perfusion and brain processing for immunohistochemistry
Mice were anesthetized with a ketamine/xylazine cocktail (24 mg/ml and 4.8 mg/ml, respectively) and terminally perfused with 0.9% NaCl containing 10 U/mL heparin. Brains were fixed in 4% paraformaldehyde in phosphate buffer for 24 hours, rinsed with phosphate-buffered saline (PBS), then sunk in 30% sucrose in PBS. Using a freezing sliding microtome (Microm HM430), 40 μm thick, free-floating sagittal brain sections were cut into 24 sequential tubes, so that each tube contained every 24th section spaced 960 μm apart. Sections were stored in cryoprotective medium at −20°C.
Immunohistochemisty
Immunohistochemistry was performed using standard techniques (9), including no-primary antibody controls for each antibody. We used the following primary antibodies for immunohistochemistry: anti-phosphorylated Smad2 (rabbit, 1:1000, Millipore, #AB3849), anti-eGFP (chicken, 1:200, Millipore, #AB16901), anti-GFAP (rat, 1:10,000, Invitrogen, #12-0300), anti-GFAP (rabbit, 1:1000, Dako, #Z0334) and biotinylated anti-GFAP (1:200, Abcam, #ab79203), biotinylated anti-NeuN (mouse, 1:200, Millipore, #MAB377B), anti-MAP2 (mouse, 1:200, Sigma, #M2320, biotinylated using Thermo Scientific Pierce EZ-Link Micro Biotinylation kit, #PI-21935), anti-CD68 (rat, 1:1000, Serotec, #MCA1957S), anti-CD3 (hamster, 1:500, BD Biosciences, #550277), anti-CCL5 (goat, 1:200, R&D Systems, #AF478) and anti-NF-κB p65 (rabbit, 1:200, Santa Cruz, sc-372).
We used the following secondary antibodies for immunohistochemistry: AlexaFluor-405 goat anti-rabbit (Invitrogen, #A-31556), AlexaFluor-488 donkey anti-chicken (Jackson Immunoresearch, #703-096-155), AlexaFluor-555 streptavidin (Invitrogen/Molecular Probes, #S-32355), AlexaFluor-555 donkey anti-rabbit (Invitrogen, #A31572), AlexaFluor-647 donkey anti-rabbit (Invitrogen, #A-31573), AlexaFluor-647 streptavidin (Invitrogen/Molecular Probes, #S-21374), AlexaFluor-647 donkey anti-rat (Jackson Immunoresearch, #712-606-153), biotinylated rabbit anti-rat (Vector Laboratories, #BA-4001), biotinylated rabbit anti-goat (Vector Laboratories, #BA-5000) and biotinylated goat anti-hamster (Vector Laboratories, #BA-9100). All fluorescent antibodies were used at 1:200 and all biotinylated secondary antibodies at 1:500. 3,3′-Diaminobenzidine(DAB) was used to detect biotinylated antibodies. Primary anti-CCL5 antibody was used with secondary biotinylated rabbit anti-goat antibody and streptavidin-555 in sequence.
Image analysis
Images were obtained using 10x lens and 40x oil lens on a Leica TCS SPE confocal microscope, or 10x lens on an upright fluorescence microscope (Zeiss Axio Imager M1 with CCD camera). Images were analyzed using MetaMorph or ImageJ software. All co-localization studies were performed in 3 images per section in 2 sagittal sections per mouse, spaced 480 μm apart. Image acquisition, processing, and analysis were uniformly done blinded to genotype. For co-localization of pSmad2, CCL5 or NF-κB p65 with GFAP and eGFP, markers were scored according to their co-localization with GFAP prior to assessing eGFP expression. CD68, CD3, NeuN and MAP2 immunostaining was quantified in 5 sagittal sections per mouse 480 μm apart, tracing the entire cortex for NeuN and MAP2, and using representative image per section for CD68. GFAP expression was quantified in 2 sagittal sections per mouse 960 μm apart. Six to ten animals per genotype were used for all image analysis and quantification experiments.
Stereology
Brain sections were stained for CD3+ cells using antibodies listed above and detected with DAB. The number of CD3+ cells was quantified by analyzing digital images collected using SimplePCI software (Hamamatsu Inc., Sewickley, PA) on an Olympus IMT-2 inverted light microscope. For each section, 12 fields of view were randomly sampled throughout the cortex, beginning with the frontal cortex and moving posteriorly. We used a computerized threshold to detect only the CD3+ antibody staining. All quantifications were made in 5 sagittal sections per mouse 480 μm apart, using six to ten animals per genotype.
Flow cytometry
We performed flow cytometry on whole hemisphere tissue samples using six animals per genotype. We mechanically dissociated and digested tissue using 1 ml/hemisphere collagenase D (Roche, #11-088-874-103) with 5 μl/hemisphere DNAseI (Sigma, # AMP-D1) for 1 hour at 37°C. Cells were passed through a 70 μm cell strainer, isolated on a 70%/40% Percoll gradient (Sigma, #P1644), and blocked using Fc block (BioLegend, #101302). Cells were then stimulated with PMA (Sigma, #P8139), ionomycin (Sigma, #I3909) and GolgiStop (BD Biosciences, #554724) for 4h and surface-stained with fluorochrome-labeled antibodies against CD3, CD4 and CD8 (BD Biosciences). For intracellular IFNγ and IL-17 staining, cells were surface stained with anti-CD3, then fixed and permeabilized with Cytofix/Cytoperm (BD Biosciences, #554714) and stained with fluorochrome-labeled antibodies against IFNγ and IL-17 (BD Biosciences). The data were collected using FACScan (BD Biosciences) and analyzed using FlowJo software (TreeStar). T cell subsets were analyzed by gating on lymphocytes defined by forward scatter and side scatter profile, followed by gating on CD3. The CD3+ population was then gated on CD4, CD8 and IFNγ expression. We repeated and confirmed all flow cytometry analysis for infected tissue using a similar protocol for tissue preparation and cell staining (23) except that the protocol does not use collagenase/DNAse digestion or stimulation.
We also performed flow cytometry on whole spleen tissue samples using the same preparation, but without collagenase/DNAse digestion, stimulation or intracellular staining. We surface-stained the samples using fluorochrome-labeled antibodies against CD3, CD4, CD8, CD25 and CD19 (BD Biosciences) as well as CD11b. CD11c, Gr1 and F4/80 (eBioscience). Immune cell subsets were gated on lymphocytes and myeloid cells defined by forward scatter and side scatter profile, followed by gating on Live/Dead AquaAmine stain (Molecular Probes). Then lymphocytes were gated on CD3 and CD19 expression. CD3+ population was further gated on CD4 and CD8 expression, and CD4+ population was further gated on CD25 expression. Myeloid cells were gated on CD11b expression, followed by gating on CD11c, Gr1 and F4/80 expression. All data were collected using LSRII cytometer (BD Biosciences) and analyzed using FlowJo software (TreeStar).
Protein quantification
After perfusion, brain tissue was flash frozen and kept at −80°C. Tissue was homogenized in cell lysis buffer with Complete Mini protease inhibitor (Roche, #11836153001) and 0.1% Na3VO4. Samples were sonicated for 10 seconds, centrifuged at 14000 rpm for 10 minutes, and protein concentration in the supernatant equalized using a BCA Pierce protein assay kit (ThermoScientific, #23227). Multiplex luminex assay was performed by the Human Immune Monitoring Center at Stanford University (himc.stanford.edu). Each sample was measured in duplicate. Plates were read using a Luminex 200 instrument with a lower bound of 100 beads per sample per cytokine. Results were analyzed using Cluster 3.0 and visualized using JavaTreeView software. CCL5 levels were measured using a mouse CCL5 Quantikine ELISA kit (R&D Systems, #MMR00) on six animals per genotype.
Quantitative RT-PCR
We isolated mRNA and performed qPCR as previously described (24). RNA was isolated from flash frozen tissue, which was kept at −80°C until homogenization in TRIzol reagent (Invitrogen, #15596-026). Samples were ground using a vibrating pestle until dissolved, mixed with chloroform at 5:1 ratio and centrifuged at 14000 rpm for 15 minutes. Supernatant was mixed 1:1 with isopropanol and centrifuged at 14,000 rpm for 10 minutes. Pellets were washed with 80% ethanol three times and air-dried. RNA concentration and purity was measured based on light absorbance at 260 nm and 280 nm using a spectrophotometer. We used RNA with a 260nm:280nm ratio of at least 1.8 and concentration of at least 200 ng/μl. Any genomic DNA was digested using a DNAse kit (Invitrogen, #18068-015), and mRNA was converted into cDNA using Omniscript Reverse Transcriptase kit (Qiagen, #205113) with random primers (Promega, #C118A) and RNAse inhibitor (Roche, #N808-119), according to manufacturer’s instructions.
For quantification of parasite burden, whole brain genomic DNA was isolated using a DNeasy blood and tissue kit (Qiagen, #69504) and following the manufacturer’s protocol. Genomic DNA or cDNA samples were mixed with SYBR green reagent (Qiagen, #204143) and primers according to manufacturer’s instructions and run on Peltier Thermal Cycler PTC-200 using Opticon Monitor 3 software. Six to eleven animals per group were used for all QPCR assays. We quantified the expression of B1 gene (forward primer: 5′-TCCCCTCTGCTGGCGAAAAGT-3′, reverse primer: 5′-AGCGTTCGTGGT CAACTATCGATTG-3′) (25), with GAPDH control (forward primer: 5′-AGGTCGGTGTGAACGGATTTG-3′, reverse primer: 5′-TGTAGACCATGTAGTTGA GGTCA-3′). Parasite DNA levels were normalized using GAPDH (to control for the amount of brain tissue). Results were calculated as previously described (26).
Cyst counts
2 and 4 wpi brain sections were stained with fluorescein labeled Dolichos biflorus agglutinin (Vector Labs #1031), which binds sugars of the cyst wall. The number of cysts was quantified by confocal microscopy (Leica TCS SPE confocal microscope). Five sagittal sections were counted per mouse for 2 wpi samples and four sagittal sections were counted per mouse for 4 wpi samples. These counts represented approximately one-twelfth of the brain. There were seven to nine mice per genotype for 2 wpi samples, and eleven to twelve mice per genotype for 4 wpi samples. For images shown in Fig. 3, A and B, brain sections were stained with biotinylated Dolichos biflorus agglutinin (1:500, Vector labs, #B-1035), which was then detected by use of streptavidin-Cy5 (1:200, Invitrogen, #434316).
Figure 3. Inhibiting astrocytic TGFβ signaling does not affect Toxoplasma parasite burden in the brain, weight loss, or mortality.

A–F. Toxoplama burden in the brain during acute (2wpi) and chronic (4wpi) infection. A, B. Representative images of brain cysts at 2 wpi and 4wpi. Panels: mCherry signal from parasites (upper left), Dolichos biflorus agglutinin (upper right) which stains the cyst wall, DAPI (lower left) and merge (lower right). Arrowheads point to cysts. Arrows point to free tachyzoites (2 wpi). No free tachyzoites were seen in 4 wpi brain sections. Scale bars, 20 μm. C, D. mCherry+ Dolichos+ Toxoplasma cysts were counted at 2wpi and 4wpi. Bars, mean ± SEM. N=7–9 mice per genotype at 2 wpi, 11–12 mice per genotype at 4 wpi. E, F. At 2 and 4 wpi brain homogenates were used to isolate Toxoplasma and mouse genomic DNA for quantitative real-time PCR for the Toxoplasma specific gene B1 and loading control GAPDH for quantification of Toxoplasma genomic DNA. Bars, mean ± SEM. N=6 mice per genotype at 2 wpi, 11 mice per genotype 4 wpi. G, H. Weight loss in Ast-Tbr2DN mice and wildtype controls. Bars, mean ± SEM. G. Combined data from three independent experiments, N=19–21 mice per genotype. H. N=11–12 mice per genotype. I, J. Mortality in Ast-Tbr2DN mice and wildtype controls. Mantel-Cox (log-rank) test to compare mortality between genotypes. I. Combined data from five independent experiments, N=34 mice per genotype. (J) N=12 mice per genotype.
Statistical analysis
All experimental data were acquired in a blinded and unbiased fashion. Statistical analyses were performed using Prism 6 software. Means between two groups were compared using two-tailed, unpaired Student’s t-test for parametric data and Mann-Whitney test for non-parametric data. Means between three or more groups were compared using 1-way ANOVA with Bonferroni correction for multiple comparisons for parametric data and Kruskal-Wallis test with Dunn’s correction for multiple comparisons for non-parametric data. Linear correlation between Toxoplasma cDNA and cytokine expression was computed using Prism 6 software and expressed as R2 and p-value compared to zero-slope line. Weight loss between genotypes was compared using 1-way repeated measures ANOVA. Mortality was compared using Mantel-Cox log-rank test.
RESULTS
Activated astrocytes exhibit TGFβ signaling during Toxoplasma infection
To verify that wildtype astrocytes surround Toxoplasma infiltrates and respond to TGFβ during acute CNS Toxoplasma infection, we orally infected control mice with Toxoplasma cysts derived from parasites that express the fluorescent protein mCherry. As expected, we observed GFAP+ reactive astrocytes surrounding active sites of infection at two weeks post infection (wpi) (Fig. 1A). We next immunostained for phosphorylated Smad2 (pSmad2), a readout of TGFβ signaling (27). Unsurprisingly, many cells demonstrated pSmad2 immunostaining, and that population included GFAP+ astrocytes localized around mCherry+ Toxoplasma infiltrates (Fig 1B). Thus, we verified that GFAP+ astrocytes do exhibit TGFβ pathway activation during Toxoplasma infection.
Figure 1. TGFβ signaling is activated in brain cells, including astrocytes during CNS toxoplasmosis.
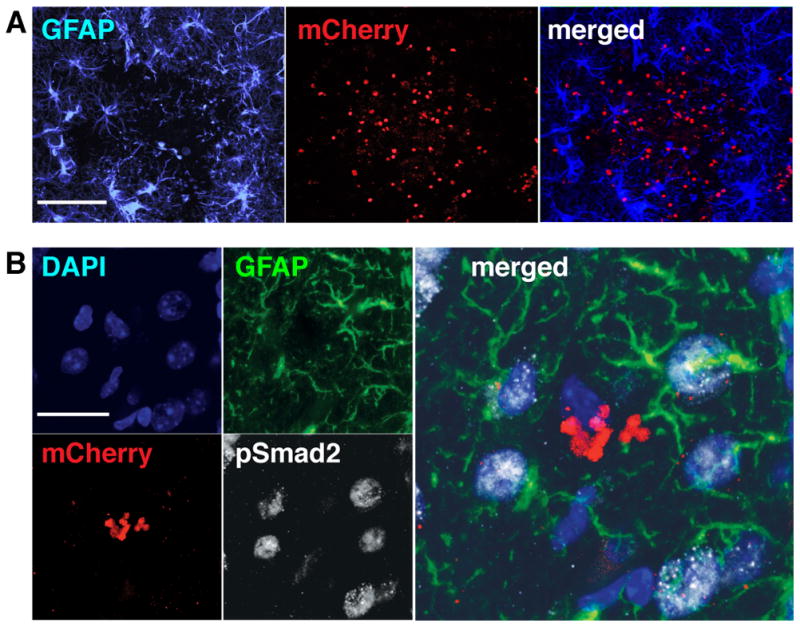
Brain sections from wildtype mice infected with mCherry-expressing Toxoplasma were stained as indicated and then examined by confocal microscopy. A. Representative images showing astrocytes that immunostain for GFAP+ (left panel, blue), mCherry+ parasites (middle panel, red), and the merged image (right panel). Scale bar, 200 μm. B. Representative images of pSmad2 and GFAP co-immunostaining in areas surrounding mCherry+ Toxoplasma infiltrates. Panels: DAPI-stained nuclei (upper left panel, blue), GFAP+ astrocytes (upper right panel, green), mCherry+ parasites (lower left panel, red), pSmad2 staining (lower right panel, white), and merge of all 4 channels (right panel). Arrowheads highlight nuclei associated with the GFAP+ astrocytes seen in these images. Scale bar, 20 μm.
Astrocytic TGFβ signaling is inhibited in the “Ast-Tbr2DN” mouse model
Having verified that astrocytic TGFβ signaling occurs during CNS Toxoplasma infection, we sought to study its functions by infecting transgenic mice in which TGFβ signaling is inhibited specifically in GFAP+ astrocytes (Astrocytic Tbr2 Dominant Negative or Ast-Tbr2DN mice) (Fig. 2A). To engineer Ast-Tbr2DN mice, we crossed two previously described mouse lines, both of which are on a C57BL/6J genetic background and have been extensively characterized and used in other transgenic combinations (28–36). The first line (28) carries a bi-tetO promoter which, when bound by the tetracycline transactivator protein (tTA), drives the expression of two genes: a dominant negative mutant Type II TGFβ receptor, and enhanced green fluorescent protein (eGFP). The type II receptor is required for all intracellular TGFβ signaling (27), and the dominant negative mutant receptor is truncated before the kinase domain, preventing the initiation of downstream signaling when it dimerizes with either another truncated receptor or a full-length native receptor. The second transgenic line expresses GFAP-tTA, which directs bi-tetO promoter-mediated expression of the truncated receptor as well as eGFP to GFAP+ astrocytes (32). Single-transgenic bi-tetO mice do not express either transgene and serve as wildtype littermate controls.
Figure 2. During Toxoplasma infection TGFβ signaling is inhibited specifically in eGFP+ astrocytes in Ast-Tbr2DN mice.
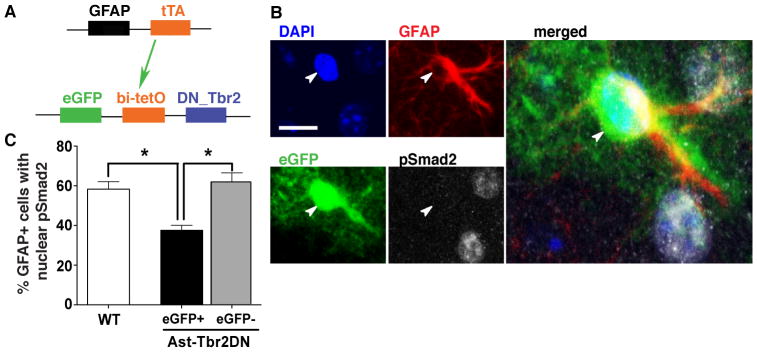
A. Schematic representation of the double-transgenic Astrocytic Tbr2 Dominant Negative mouse (Ast-Tbr2DN) line. The first transgene encodes the tetracycline transactivator protein (tTA), driven by a GFAP promoter. tTA binds the bidirectional bi-tetO promoter on the second transgene to stimulate expression of both eGFP and a dominant negative mutant type II TGFβ receptor which cannot initiate downstream signaling. B. Representative images of 2 wpi Ast-Tbr2DN brain section, stained as indicated. Arrowhead denotes the nucleus of the GFAP+/eGFP+ cell. Scale bar, 20 μm. Large image: Enlargement of merged image (all 4 channels). C. Percentage of GFAP+ cells in wildtype or Ast-Tbr2DN brain sections that showed nuclear pSmad2 expression at 2 wpi, as assessed by confocal microscopy. Note that in Ast-Tbr2DN mice the GFAP+ cells are subdivided into GFAP+/eGFP+ and GFAP+/eGFP− cells. N=4 mice per genotype, 100 GFAP+ cells per mouse. Bars, mean ± SEM. *P<0.05, Kruskal-Wallis test with Dunn’s correction for multiple comparisons.
To assess the cell specificity of transgene expression and verify downregulation of TGFβ signaling in GFAP+/eGFP+ astrocytes in Ast-Tbr2DN mice we co-immunostained for pSmad2, eGFP and GFAP. Although we examined uninfected mice, we did not quantify expression in them because at rest cortical astrocytes do not highly express GFAP, and thus uninfected Ast-Tbr2DN mice have very few GFAP+ astrocytes at baseline. Transgene expression was completely specific to astrocytes in both infected and uninfected Ast-Tbr2DN brain, and the eGFP marker was expressed in approximately 40% of GFAP+ astrocytes 2 weeks post Toxoplasma infection. eGFP was not expressed in any other cell types including monocytic or lymphocytic cells (data not shown).
Consistent with both eGFP and the mutant type II TGFβ receptor being driven by the same promoter, at 2 wpi, GFAP+/eGFP+ astrocytes in Ast-Tbr2DN mice were significantly less likely to immunostain for nuclear pSmad2 than astrocytes in wildtype mice (40% vs. 60% respectively, Fig. 2, B and C), indicating reduced TGFβ signaling. However, some GFAP+/eGFP+ astrocytes did show pSmad2 immunostaining, which might be due to the expression of a functional, wildtype TGFβ receptor complex in addition to the mutant receptor. By comparison, GFAP+/eGFP− astrocytes, which would be expected to express very little if any of the mutant receptor, were as likely to exhibit nuclear pSmad2 as GFAP+ astrocytes in wildtype mice and could therefore be considered an internal control (Fig. 2C). Collectively, our data indicate that after Toxoplasma infection the mutant TGFβ receptor is co-expressed with eGFP and reduces TGFβ signaling in a significant population of Ast-Tbr2DN mouse astrocytes.
Inhibiting astrocytic TGFβ signaling has no effect on parasite burden
To study the effects of astrocytic TGFβ signaling on inflammation after Toxoplasma infection, we first investigated whether it might indirectly affect neuroinflammation by altering the ability of astrocytes to contain parasites or altering total parasite burden. To address these questions, we assessed the amount and distribution of Toxoplasma in Ast-Tbr2DN and wildtype mice at 2 and 4 wpi using tissue fluorescence, immunohistochemistry, and DNA-based quantification.
Gross scoring of mCherry+ parasites in 2 wpi brain sections did not reveal any differences between genotypes (data not shown). In addition, at both 2 and 4 wpi, (Fig. 3, A and B) we observed that parasites formed clusters that stained for Dolichos biflorus agglutinin (a marker of cysts) in the cortex of both Ast-Tbr2DN mice and wildtype controls and that the number of cysts did not differ between genotypes (Fig. 3, C and D). At both time points, we further quantified parasite burden in brain homogenates by quantitative PCR for the Toxoplasma-specific gene B1, a method that ensures we accounted for all parasites, even those not in cysts (25). Again, there was no difference in normalized B1 expression between genotypes at either time point (Fig. 3, E and F). Thus, based both on direct counts and on quantitative PCR, inhibiting astrocytic TGFβ signaling did not affect Toxoplasma burden or distribution in the brain during acute or chronic infection. Consistent with these findings, there was no significant difference in weight loss or mortality between the Ast-Tbr2DN and wildtype mice at any timepoint (Fig. 3, G – J).
Inhibiting astrocytic TGFβ signaling increases astrocyte and myeloid cell activation during acute infection
Having verified that astrocytic TGFβ signaling does not affect parasite burden, we next evaluated whether Ast-Tbr2DN mice showed a difference in the immune response compared to wildtype mice. The main CNS resident cells that participate in neuroinflammation are astrocytes and microglia, and during infection they are joined by infiltrating macrophages (17, 19, 37, 38). As GFAP upregulation is a hallmark of astrocyte activation (39), we quantified the area covered by GFAP immunostaining to measure the effects of inhibiting TGFβ signaling on astrocyte activation. In uninfected mice, we did not observe any differences in the percent of cortical area covered by GFAP+ immunostaining between Ast-Tbr2DN mice and wildtype controls (data not shown). However at 2 wpi, Ast-Tbr2DN mice exhibited an approximately two-fold increase in the cortical area covered by GFAP immunostaining compared to wildtype mice (Fig. 4, A and B). To determine if this increase in GFAP was related to an increase in proliferative capacity of astrocytes that lacked TGFβ signaling, we quantified the co-localization of GFAP with the proliferation marker Ki67 in WT and Ast-Tbr2DN brain sections. These studies showed no genotype-specific difference in Ki67/GFAP colocalization (wildtype, 5.60 ± 0.79%, Ast-Tbr2DN, 8.15 ± 1.47%, p = 0.14, Student’s t-test). In addition, there was no difference in Ki67 immunostaining in eGFP+ vs eGFP− astrocytes within Ast-Tbr2DN mice (GFAP+/eGFP+, 8.04 ± 1.64, GFAP+/eGFP−, 8.17 ± 1.53, p = 0.96, Student’s t-test) and no difference in GFAP+ cell number (wildtype, 200.8 ± 10.8, Ast-Tbr2DN, 202.8 ± 12.8 cells per 0.34 mm2 region, p = 0.90, Student’s t-test). We then confirmed the GFAP increase in Ast-Tbr2DN mice by quantifying GFAP protein and mRNA expression in a separate cohort of mice 2wpi. We observed a 1.5 fold increase in GFAP protein level in Ast-Tbr2DN compared to WT mice but no difference in mRNA levels at 2 wpi (Supplementary Fig. 1).
Figure 4. Lack of astrocytic TGFβ signaling increases the reactive astrogliosis and microglial/macrophage infiltration to acute Toxoplasma infection.

Brain sections from 2 wpi wildtype (WT) or Ast-Tbr2DN mice were stained as indicated and examined by light microscopy. For (B) and (D) quantification was done by stereology. A, B. Representative images (A) and quantification (B) of cortical area covered by GFAP+ reactive astrocytes in Ast-Tbr2DN and wildtype mice. C, D. Representative images (C) and quantification (D) of cortical area covered by CD68+ activated microglia and infiltrating macrophages in Ast-Tbr2DN and wildtype mice. Scale bar, 200 μm. N=7–9 mice per genotype, 2 images per mouse (B) and 5 images per mouse (D) 480 μm apart. Bars, mean ± SEM. *P<0.05, **P<0.01, Student’s t test.
To investigate if inhibiting TGFβ signaling in astrocytes affects myeloid cell activation as well, we stained for CD68, which is a marker for activated microglia and macrophages. Since CD68+ cells showed significant spatial overlap in the infected cortex, we quantified their presence as the percentage of cortical area instead of attempting to count individual cells. We found that similarly to GFAP, CD68 immunostaining was not different between Ast-Tbr2DN mice and wildtype controls in uninfected mice (data not shown). However, at 2 wpi Ast-Tbr2DN mice exhibited a two-fold increase in the cortical area covered by CD68+ cells compared to wildtype controls (Fig. 4, C and D). Taken together, these results demonstrate that CNS Toxoplasma infection induces more astrocyte and myeloid cell activation when astrocytic TGFβ signaling is inhibited.
Inhibiting astrocytic TGFβ signaling exacerbates the adaptive immune response to acute infection
As in other organs, the T lymphocyte response in the brain is the major mediator of the adaptive immune response to Toxoplasma infection (40). Using immunohistochemistry for the T lymphocyte marker CD3, we observed that some CD3+ cells formed clusters, which have previously been shown to correspond to areas of active Toxoplasma infection (41), and other CD3+ cells diffusely infiltrated cortical area between clusters (Fig. 5, A and B).
Figure 5. Lack of astrocytic TGFβ signaling exacerbates the T cell response to acute Toxoplasma infection.

Brain sections from 2 wpi wildtype (WT) or Ast-Tbr2DN mice were stained for CD3, examined by light microscopy and quantified using stereology. A. Representative images of CD3+ cells in the cortex of Ast-Tbr2DN and wildtype mice. Scale bar, 200 μm. B. Enlargement of boxed area of A. Scale bar, 50 μm. C. Numbers of discrete CD3+ cortical infiltrates in Ast-Tbr2DN and wildtype mice as quantified by 2 observers using light microscopy. N = 7–9 mice per genotype D. Stereology was used to quantify the total counts of CD3+ cells in cortex of Ast-Tbr2DN and wildtype mice. N=5 sections per mouse 480 μm apart, 7–9 mice per genotype. E–H. At 2 wpi, T cell lymphocytes were isolated from Ast-Tbr2DN and wildtype (WT) mice and analyzed by flow cytometery for the percentage of CD4+, CD8+ and CD3+IFNγ+ cells E. A representative analysis of the CD3+ cells isolated from the brain homogenate of a single mouse. F–H. Quantification of % CD3+ cell populations that are CD4+ (F) and CD8+ (G) in Ast-Tbr2DN and wildtype mice. H. Quantification of % CD3+ cell population that are IFNγ+ in Ast-Tbr2DN mice and wildtype controls. N=6 mice per genotype. Bars, mean ± SEM. **P<0.01, Student’s t test.
Consistent with the finding of equivalent parasite burden in both groups there was no difference in the number of CD3+ clusters between genotypes (Fig. 5C). However, we observed that Ast-Tbr2DN mice showed significantly more CD3+ cells diffusely infiltrating the area between the distinct clusters (Fig. 5B). To quantify this difference, we used stereology to determine an unbiased estimate of total CD3+ cells in the cortex of Ast-Tbr2DN and wildtype mice. This verified a 2.5-fold increase in the number of CD3+ cells in Ast-Tbr2DN cortex (Fig. 5D). Thus, inhibition of astrocytic TGFβ signaling not only increases the innate immune response to CNS toxoplasmosis, it also strongly increases the number of infiltrating T lymphocytes.
To investigate whether inhibiting astrocytic TGFβ signaling affects the phenotype of the adaptive immune response, we characterized T cells isolated from the brain using flow cytometry. Figure 5E shows our gating strategy. We did not observe any differences between genotypes in the proportion of CD3+ T lymphocytes that were CD4+ or CD8+ (Fig. 5, F and G). Furthermore, we did not observe any differences in the percent of CD3+ T cells that expressed IFNγ (Fig. 5H). The numbers of CD3+ IL-17 expressing lymphocytes were negligible in both genotypes (data not shown).
To determine if inhibition of astrocytic TGFβ signaling increases T cell number by promoting T cell proliferation, we quantified the percentage of proliferating CD3+ T cells between genotypes by co-staining sections for CD3 and Ki67 (Supplementary Fig. 2). This method again revealed an increase in T cell number, but there was no genotypic difference in the percentage of CD3+ cells co-localizing with Ki67 (Supplementary Fig. 2B). Thus, inhibiting astrocytic TGFβ signaling primarily increased the number of infiltrating CD3+ T cells rather than skewing their differentiation to CD4+, CD8+, Th1 or Th17 lymphocytes or affecting their proliferation.
To ensure that the numerical difference in the Ast-Tbr2DN CNS was not secondary to a baseline difference in Ast-Tbr2DN versus wildtype mice, and to determine whether the effects of inhibiting astrocytic TGFβ signaling were confined to the CNS, we used flow cytometry to measure splenic immune cells in uninfected and Toxoplasma infected Ast-Tbr2DN and wildtype mice. In the uninfected Ast-Tbr2DN and wildtype mice, we found no baseline differences in the spleen immune cell populations we characterized: dendritic cells (CD11b+/CD11c+/Gr1−/F4/80−), macrophages (CD11b+/F4/80+/CD11c−/Gr1−), granulocytes (CD11b+/Gr1+/CD11c−/F4/80−), CD4+ T cells (CD3+/CD4+), CD8+ T cells (CD3+/CD8+), regulatory T cells (CD3+/CD4+/CD25+) and B cells (CD19+)(Supplementary Table I). Similarly, we found no difference in splenic T cell populations between wildtype controls and infected Ast-Tbr2DN mice: CD4+ cells (wildtype, 60.68 ± 1.65, Ast-Tbr2DN, 62.49 ± 0.82), CD8+ cells (wildtype, 27.17 ± 1.36, Ast-Tbr2DN, 27.79 ± 0.72), CD3+/CD4+/IFNγ+ Th1 cells (wildtype, 5.13 ± 0.79, Ast-Tbr2DN, 4.85 ± 0.54) and CD3+/CD4+/IL-17+ Th17 cells (wildtype, 0.10 ± 0.02, Ast-Tbr2DN, 0.13 ± 0.03).
In addition, we used a multiplex luminex platform to analyze the expression of 26 mouse cytokines and chemokines in serum from Ast-Tbr2DN and wildtype mice 2 weeks after Toxoplasma infection. Again we did not observe any differences between genotypes (Supplementary Table IIA). Thus, we found no evidence of changes in the immune response at baseline or outside the CNS in infected Ast-Tbr2DN mice. Collectively, these data show that inhibiting astrocytic TGFβ signaling influences the number but not the subtypes of T lymphocytes responding to CNS toxoplasmosis and that this difference only occurs in the CNS.
Inhibiting astrocytic TGFβ signaling increases astrocytic NF-κB activation
Prior studies have indicated that a major mechanism by which TGFβ reduces the immune response is by inhibiting the activation and nuclear translocation of the pro-inflammatory transcription factor NF-κB (42, 43). If the NF-κB pathway were similarly inhibited by TGFβ in astrocytes during a Toxoplasma infection, we would expect an increase in NF-κB signaling only in GFAP+/eGFP+ astrocytes in Ast-Tbr2DN mice. To test this, we quantified the percentage of GFAP+ astrocytes that exhibited nuclear localization of the NF-κB subunit p65, since its nuclear translocation is a marker of NF-κB activation (44). In Ast-Tbr2DN mice, we observed a significantly higher proportion of GFAP+/eGFP+ astrocytes with nuclear NF-κB p65 immunostaining, compared to wildtype GFAP+ astrocytes (80% vs. 40% respectively, Fig. 6).
Figure 6. Lack of astrocytic TGFβ signaling increases NF-κB pathway activation in eGFP+ astrocytes.

A., B. Representative images of 2 wpi wildtype (WT) (A) and Ast-Tbr2DN (B) brain sections stained as indicated. Arrowheads denote the nuclei of the associated GFAP+ cells. C. Quantification of % of GFAP+ astrocytes that immunostain for nuclear NF-κB p65 in wildtype and Ast-Tbr2DN mice. Note that in Ast-Tbr2DN mice the GFAP+ cells are subdivided into GFAP+/eGFP+ and GFAP+/eGFP− cells. Scale bars, 20μm. N=6 mice per genotype. Bars, mean ± SEM. ****P<0.0001, 1-way ANOVA with Bonferroni correction for multiple comparisons.
GFAP+/eGFP− astrocytes in Ast-Tbr2DN mice, which do not express transgene at detectable levels and show wildtype TGFβ signaling (Fig. 2C), showed the same probability of immunostaining for nuclear NF-κB p65 as wildtype astrocytes. Thus, consistent with the prior studies that have linked TGFβ signaling with inhibition of NF-κB signaling, GFAP+/eGFP+ astrocytes in Ast-Tbr2DN mice that exhibit less TGFβ signaling also exhibit more astrocytic NF-κB pathway activation.
Increased NF-κB activation is associated with astrocytic CCL5 expression in Ast-Tbr2DN mice
The NF-κB signaling pathway is associated with pro-inflammatory cytokine production and activation of both innate and adaptive immune responses (45). Therefore, we screened a panel of pro- and anti-inflammatory cytokines and chemokines in the cortex of Ast-Tbr2DN mice and wildtype controls at 2wpi.
Surprisingly, there was no significant genotypic difference in the level of the majority of these cytokines until we took into account the inter-mouse variability in Toxoplasma burden (Fig. 3). We correlated the Toxoplasma burden in each mouse, as measured by Toxoplasma DNA content, with that mouse’s brain cytokine expression. This method allowed us to take into consideration the variability of infection rates between individual mice within the same genotype. We observed that in wildtype mice there was a strong linear correlation between Toxoplasma burden and the expression of the major Th1 cytokines IFNγ, IL-1α, IL-6 and IL-12p40, as well as the T cell chemoattractants CCL5 and CXCL10, and myeloid cell chemoattractants CCL2 and CXCL1 (blue dots, Fig. 7A–H). In marked contrast, the levels of pro-inflammatory cytokines and chemokines in Ast-Tbr2DN mouse brain were not linearly correlated with the parasite burden, and trended towards abnormally high expression for a given parasite burden (red squares, Fig. 7A–H). This lack of correlation between pro-inflammatory cytokine levels and parasite burden in Ast-Tbr2DN mouse brain could be caused by an increase in pro-inflammatory cytokines produced by Ast-Tbr2DN astrocytes that exhibit more NF-κB activation, or could simply be a result of the increased number of activated immune cells found in the Ast-Tbr2DN mice cortex, or both.
Figure 7. Inhibition of astrocytic TGFβ signaling leads to loss of correlation of Th1 cytokine and chemokine levels to acute CNS Toxoplasma burden.

At 2 wpi, brain homogenates from Ast-Tbr2DN and wildtype (WT) mice were used to quantify multiple cytokines and chemokines via multiplex cytokine assay and Toxoplasma burden (measured as B1 gene DNA, normalized to GADPH control gene DNA and expressed as fold over wildtype mean) via quantitative-PCR. A–H. Linear regressions of the Toxoplasma burden plotted against the levels of T cell chemokines CCL5 (A) and CXCL10 (B); Th1 cytokines IFNγ(C), IL-12p40 (D), IL-1a (E) and IL-6 (F); and myeloid cell chemoattractants CCL2 (G) and CXCL1 (H). Blue circles, wildtype mice. Red squares, Ast-Tbr2DN mice. There is a statistically significant linear correlation between Toxoplasma load and Th1 cytokines and chemokines in wildtype controls (blue line), but it is lost in Ast-Tbr2DN mice (red line). I, Mean CCL5 levels were approximately twice normal in Ast-Tbr2DN mouse brains. N=6 mice per genotype. Bars, mean ± SEM. *P<0.05, Student’s t test.
The remaining cytokines and chemokines (IL-1β, IL-2, IL-3, IL-4, IL-6, IL-10, IL-12p70, IL-13, IL-17, IL-23, GM-CSF, G-CSF, CCL3, CCL7, CCL11 and VEGF) were not linearly correlated with Toxoplasma burden in either genotype and were not affected by astrocytic TGFβ signaling. At 4 wpi, when a chronic CNS infection is well-established and the majority of the parasites are encysted bradyzoites (Fig 3D), none of the 26 chemokines and cytokines we measured linearly correlated with Toxoplasma burden nor were any affected by astrocytic TGFβ signaling (Supplementary Table IIB).
Remarkably, CCL5 exhibited the most change and was 2.4 fold higher in Ast-Tbr2DN mice by multiplex luminex assay at 2wpi. A significant increase was confirmed with a CCL5-specific ELISA (Fig. 7I). Given the abnormally high level of CCL5 in the Ast-Tbr2DN mouse brain and that CCL5 is a well-established chemoattractant for lymphocytes and macrophages, we sought to define which cells were producing excessive CCL5. As CCL5 can be secreted by a number of cells, including astrocytes (46), macrophages/microglia (18), and lymphocytes (47), we co-immunostained brain sections from Ast-Tbr2DN and wildtype mice for CCL5 and either the astrocyte marker GFAP or the activated microglia/macrophage marker CD68. In both genotypes, we found that during acute infection, astrocytes were the primary cells that co-localized with CCL5. Activated CD68+ microglia and macrophages did not co-localize with CCL5 and we did not observe any CCL5-expressing cells that exhibited typical lymphocyte morphology (data not shown). These data suggested that in our model, regardless of genotype, astrocytes were the primary producers of CCL5 in acute CNS infection with Toxoplasma.
We hypothesized that there might be a direct link between astrocytic TGFβ signaling and CCL5 expression in Ast-Tbr2DN mice in vivo, because previous in vitro studies have shown that TGFβ can inhibit astrocytic production of CCL5 (48, 49). To evaluate this possibility, we determined the percentage of GFAP+ cells that co-immunostained with CCL5 in wildtype and Ast-Tbr2DN mice. Consistent with our hypothesis, we found that GFAP+/eGFP+ astrocytes were 1.5 times more likely to immunostain for intracellular CCL5 than GFAP+ astrocytes in wildtype mice (Fig. 8). Again GFAP+/eGFP− astrocytes in Ast-Tbr2DN mice served as an internal control and were as likely to immunostain for CCL5 as wildtype astrocytes. This higher probability of immunostaining for CCL5 in astrocytes with inhibited TGFβ signaling is consistent with a direct link between TGFβ signaling and inhibition of CCL5 production in astrocytes in vivo.
Figure 8. Ast-Tbr2DN mice exhibit increased immunostaining for CCL5 expression in eGFP+ astrocytes.

A., B. Representative images 2 wpi from wildtype (WT) (A) and Ast-Tbr2DN (B) brain sections stained as indicated. Arrowheads denote the nuclei of the associated GFAP+ cells. C. Quantification of % of GFAP+ astrocytes that immunostain for CCL5 in wildtype controls and Ast-Tbr2DN mice. Note that in Ast-Tbr2DN mice the GFAP+ cells are subdivided into GFAP+/eGFP+ and GFAP+/eGFP− cells. Scale bars, 20μm. N=6 mice per genotype. Bars, mean ± SEM. **P<0.01, 1-way ANOVA with Bonferroni correction for multiple comparisons.
Inhibiting astrocytic TGFβ signaling increases neuronal injury during Toxoplasma infection
Finally, to evaluate if this excessive inflammation in Ast-Tbr2DN mice correlated with an increase in tissue damage, we quantified neuronal injury in Ast-Tbr2DN mice and wildtype controls at two timepoints: 2 wpi (acute stage) and 4 wpi (chronic stage). To assess neuronal injury and damage, we immunostained for two neuronal markers: MAP2, which labels dendrites, and NeuN, which labels neuronal nuclei. We then used light microscopy to evaluate the neocortex for loss of staining in wildtype or Ast-Tbr2DN mice. Ast-Tbr2DN mice showed significantly reduced MAP2 immunostaining at both 2 and 4 wpi consistent with an increase in dendritic injury (Fig. 9, A and B). In addition, by 4 wpi, Ast-Tbr2DN mice exhibited significantly reduced NeuN immunostaining and disturbed cortical architecture consistent with more neuronal nuclear damage (Fig. 9, C and D). Astrocyte loss, as assessed by GFAP staining, did not occur within these areas (Fig. 9E). Thus, the increased inflammation in Ast-Tbr2DN mouse brain was associated with both excessive dendritic loss and neuronal death in chronic toxoplasmic encephalitis.
Figure 9. Inhibiting astrocytic TGFβ signaling increases neuronal damage during Toxoplasma infection.

2 and 4 wpi Ast-Tbr2DN and wildtype (WT) brain sections were stained as indicated and examined by light microscopy. A, C. Representative images of loss of MAP2 (A) or NeuN (C) staining in Ast-Tbr2DN or WT brain sections at 4 wpi. Yellow line represents area observed to have loss of stain. B., D. Quantification of the percentage of cortical areas denoted to have loss of MAP2 (B) or NeuN (D) staining in Ast-Tbr2DN or WT mice. E. Representative images of eGFP+/GFAP+ astrocytes in areas of neuronal damage in an Ast-Tbr2DN mouse 4wpi. Panels: DAPI-stained nuclei (upper left panel, blue), neuronal markers, which are composed of MAP2+ dendrites together with NeuN+ neuronal nuclei(upper right panel, red), GFAP+ astrocytes (lower left panel, white), eGFP+ transgenic astrocytes (lower right panel, green), and merge of all 4 channels (right panel). Yellow line represents area observed to have loss of neuronal marker immunostaining. Scale bars, 200 μm. N=5 images per mouse, 6 mice per genotype 2 wpi, 11–12 mice per genotype 4 wpi. Bars, mean ± SEM. *P<0.05, Student’s t test to compare with wildtype at same time point. In uninfected mice, there are no differences in gross neuroanatomical structures between Ast-Tbr2DN and wildtype littermates.
DISCUSSION
In this study, we sought to determine if astrocytic TGFβ signaling was important in directly inhibiting the neuroinflammatory response to CNS toxoplasmosis. To do this, we used Toxoplasma to infect a transgenic mouse model in which TGFβ signaling is inhibited specifically in a significant portion of GFAP+ cells. We demonstrate here that inhibiting TGFβ signaling in GFAP+ astrocytes did not affect Toxoplasma’s ability to enter or persist in the brain, but it did increase innate immune cell infiltration and activation, and adaptive immune cell infiltration early in CNS infection. Unlike wildtype mice in which pro-inflammatory cytokines and chemokines levels were tightly correlated to parasite burden, Ast-Tbr2DN mice exhibited higher levels of pro-inflammatory cytokines and chemokines that were not correlated with parasite burden. Additionally, we show that a higher proportion of astrocytes with reduced TGFβ signaling demonstrated pro-inflammatory transcription factor NF-κB activation and pro-inflammatory chemokine CCL5 expression. Finally, inhibition of astrocytic TGFβ signaling exacerbated neuronal damage both early and late in the CNS infection.
One of the principal findings of this study is that astrocytic TGFβ signaling is a novel molecular mechanism whereby astrocytes limit the immune response to prevent excessive tissue damage after Toxoplasma infection. This astrocytic TGFβ-mediated mechanism for controlling immune cell infiltration is distinct from the astrocytic barrier to pathogen infiltration that had been previously described during Toxoplasma infection (17, 19). In our study, Ast-Tbr2DN mice exhibited more diffuse infiltration of T lymphocytes without demonstrating a decrease in the cortical area covered by GFAP+ cells. Instead we observed the opposite – an increase in GFAP immunostaining.
The increase in GFAP by CNS Toxoplasma in Ast-Tbr2DN mice was higher when measured by immunohistochemistry than by Western blot (2.5 fold vs. 1.5 fold). This difference is likely due to immunohistochemistry methods being more sensitive to changes in cellular localization, as activated astrocytes exhibit more cytoplasmic GFAP (39). It may also reflect biological variability in parasite burden (and therefore variability in GFAP upregulation) between cohorts, given that the immunohistochemistry was performed on a different cohort than qPCR and Western blots. Interestingly, we did not observe any genotype-dependent differences in GFAP mRNA levels despite a clear increase in GFAP protein in Ast-Tbr2DN mice. A likely explanation for this finding is that since GFAP protein has a very long half life, (weeks in vivo) and its half-life is regulated by phosphorylation (50, 51), it is possible that astrocytic TGFβ signaling either regulates mRNA expression earlier than 2wpi, or that it regulates GFAP protein stability. Finally, we did not observe any GFAP+ astrocytes in either genotype that were infected with mCherry+ Toxoplasma, indicating that astrocyte activation is unlikely to be caused by direct infection and instead is more likely to represent an increased inflammatory response to the infection.
Overall, these findings show no decrease in the physical barrier formed by astrocytes, suggesting that astrocytic TGFβ signaling does not limit neuroinflammation by strengthening the physical barrier. Instead it could be considered to induce a “secretory barrier” that consists of astrocytic cytokines and chemokines and ensures that immune cells target and home only to infected areas.
A major advantage of our study compared to prior in vivo studies on astrocytes and CNS toxoplasmosis is that inhibition of astrocytic TGFβ signaling did not change the acute or chronic CNS parasite load. Therefore, the differences we found in neuroinflammation are unlikely to be driven by parasite burden and are most likely related to the change in astrocytic TGFβ signaling. By contrast, the two previous in vivo studies that examined toxoplasmic encephalitis in the setting of modified astrocytes (GFAP knockout mice and astrocytic gp130 knockout mice (17, 19)) both showed an increase in neuroinflammation and an increased CNS parasite load, making it impossible to determine if the increase in parasite burden or the astrocytic manipulation or both drove the increase in neuroinflammation.
A second advantage of our study is that in the transgenic Ast-Tbr2DN mice eGFP is co-expressed with the dominant negative type II receptor. As shown in Figure 2, this allows us to use eGFP as a marker to determine which astrocytes have reduced TGFβ signaling and compare those astrocytes to the neighboring eGFP− astrocytes which show the same likelihood of downstream TGFβ signaling as astrocytes in the wildtype mice. This ability to distinguish which astrocytes have reduced TGFβ signaling at the level of a single cell enabled us to determine that it is specifically the astrocytes with reduced TGFβ signaling that are more likely to exhibit activation of the NF-κB pathway and produce CCL5. These data lead us to propose that NF-κB is the molecular pathway that TGFβ utilizes in astrocytes to limit astrocytic CCL5 production. This model (Fig. 10) is consistent with previous studies that have shown that NF-κB stimulates CCL5 production in other cell types (43, 52) and that TGFβ inhibits NF-κB activity in multiple, non-astrocyte cell types (42, 53). However, we cannot rule out the alternate possibility that the increases in NF-κB signaling and CCL5 production in astrocytes with decreased TGFβ signaling are independent of each other.
Figure 10.
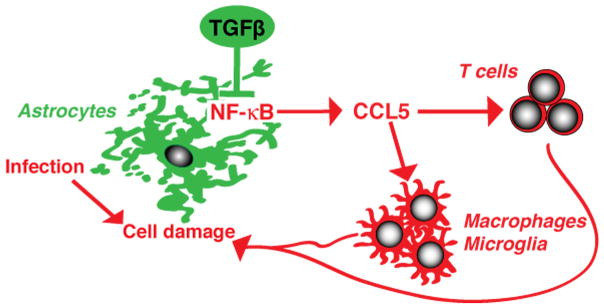
Proposed model of the functions of endogenous astrocytic TGFβ signaling during CNS Toxoplasma infection.
Several lines of evidence suggest that the increased CCL5 production by astrocytes is a potential mechanism that mediates increased CNS inflammation in Ast-Tbr2DN mice. CCL5 was the only cytokine of 26 cytokines assayed that was substantially different in brain homogenates from Ast-Tbr2DN mice compared to wildtype mice. CCL5 is a well-established chemoattractant for T lymphocytes and macrophages (47, 54), so an increase in CCL5 would be expected to attract more lymphocytes and macrophages. Finally, our observation that the CD3+ T lymphocytes in the brains of Ast-Tbr2DN mice proliferate at the same rate as those in WT mice makes it unlikely that changes in T cell proliferation in the CNS account for the increased number of T lymphocytes seen in the Ast-Tbr2DN mice. These data are more consistent with the hypothesis that Ast-Tbr2DN astrocytes that lack TGFβ and have increased expression of CCL5 cause an increase in brain CCL5 levels, which in turn then recruits more T lymphocytes to the brain.
For both genotypes, our co-localization studies clearly showed CCL5 immunostaining primarily in astrocytes and not in immune cells. This finding conflicts with two previous studies from the same group that reported that immune cells produced the majority of the CCL5 in mouse brain chronically infected with Toxoplasma and found little CCL5 production in astrocytes (18, 55). In support of our data, astrocytic CCL5 expression has been reported during other infections in vitro (56) and in vivo (46, 57) and TGFβ has been shown to reduce CCL5 production in astrocyte cultures under pro-inflammatory conditions (48, 49). The discrepancies between the prior studies (18, 55) and our results may be secondary to many factors including different techniques to examine CCL5 (protein vs. mRNA), timing (2 wpi vs. 30 dpi), different Toxoplasma strains, and different mouse strains.
We also report here that inhibiting astrocytic TGFβ signaling exacerbates neuronal damage. The Ast-Tbr2DN mice first show changes in neuronal dendritic arbors/MAP2 staining and then show loss of NeuN staining. We suspect that this increase in neuronal damage in Ast-Tbr2DN mice is directly associated with the exacerbated immune response. Indeed, inhibition of astrocytic NF-κB has been shown to be neuroprotective (58, 59). However, we cannot rule out that astrocytic TGFβ signaling also plays an independent role in neuronal protection, for example, by stimulating the production of neurotrophic molecules (60).
Finally, although we have demonstrated an important role for astrocytic TGFβ signaling, we did not investigate which cell types produced TGFβ during toxoplasmic encephalitis. Cell-specific TGFβ production could be observed by using immunohistochemistry with an anti-TGFβ antibody, but this technique would not achieve definitive results because it might also represent co-localization of TGFβ and its receptors at the cell surface or after internalization (61). However, studies in other infection and injury models have demonstrated that all CNS cell types can produce and respond to TGFβ s (reviewed in (8)). Previous work has demonstrated that TGFβ production is indeed increased in toxoplasmic encephalitis (15) and that murine myeloid cells synthesize it when infected with Toxoplasma in vitro (62).
In summary, our results show that astrocytic TGFβ signaling plays an important role in how astrocytes control the neuroinflammatory response during infection. We propose a model (Fig. 10) in which endogenous astrocytic TGFβ signaling inhibits CCL5 production via the NF-κB signaling pathway and in this way reduces CCL5-mediated recruitment of macrophages and T cells to uninfected areas within the infected brain. In our model, inhibiting TGFβ signaling in a significant proportion of GFAP+ astrocytes uncouples inflammation from parasite burden, perhaps because the excessive inflammation is not appropriately targeted to the invading pathogen. This excessive inflammation then leads to neuronal damage in tissue that would normally be spared. Given that TGFβ is universally upregulated during CNS inflammation, astrocytic TGFβ signaling may reflect a common pathway that induces anti-inflammatory and neuroprotective functions in astrocytes in a broad variety of CNS injuries.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Yael Rosenberg-Hasson of the Stanford Human Immune Monitoring Center for technical assistance with data collection. We thank the entire Koshy, Buckwalter, and Boothroyd labs for helpful discussions. We thank Tonya Bliss, Brenda Porter and Ami Okada for critical review of the manuscript.
Footnotes
Grant support: NIH-NIDKK Stanford Digestive Disease Center Pilot/Feasibility Grant (MSB), NINDS R01 NS067132 (MSB), Stanford ITI Faculty Seed Grant (MSB), NINDS K08 NS065116 (AAK) and Stanford Graduate Fellowship (EC).
Conflict of Interest: the authors declare no competing financial interests.
References
- 1.Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007;28:138–145. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 2.Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol. 2012;12:623–635. doi: 10.1038/nri3265. [DOI] [PubMed] [Google Scholar]
- 3.Carpentier PA, Begolka WS, Olson JK, Elhofy A, Karpus WJ, Miller SD. Differential activation of astrocytes by innate and adaptive immune stimuli. Glia. 2005;49:360–374. doi: 10.1002/glia.20117. [DOI] [PubMed] [Google Scholar]
- 4.Wanner IB, Anderson MA, Song B, Levine J, Fernandez A, Gray-Thompson Z, Ao Y, Sofroniew MV. Glial Scar Borders Are Formed by Newly Proliferated, Elongated Astrocytes That Interact to Corral Inflammatory and Fibrotic Cells via STAT3-Dependent Mechanisms after Spinal Cord Injury. J Neurosci. 2013;33:12870–12886. doi: 10.1523/JNEUROSCI.2121-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron. 1999;23:297–308. doi: 10.1016/s0896-6273(00)80781-3. [DOI] [PubMed] [Google Scholar]
- 6.Donkin JJ, Vink R. Mechanisms of cerebral edema in traumatic brain injury: therapeutic developments. Curr Opin Neurol. 2010;23:293–299. doi: 10.1097/WCO.0b013e328337f451. [DOI] [PubMed] [Google Scholar]
- 7.Zipp F, Aktas O. The brain as a target of inflammation: common pathways link inflammatory and neurodegenerative diseases. Trends Neurosci. 2006;29:518–527. doi: 10.1016/j.tins.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 8.Buckwalter M, Wyss-Coray T. Modelling neuroinflammatory phenotypes in vivo. J Neuroinflammation. 2004;1:10. doi: 10.1186/1742-2094-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doyle KP, Cekanaviciute E, Mamer LE, Buckwalter MS. TGFbeta signaling in the brain increases with aging and signals to astrocytes and innate immune cells in the weeks after stroke. J Neuroinflammation. 2010;7:62. doi: 10.1186/1742-2094-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Finch CE, Laping NJ, Morgan TE, Nichols NR, Pasinetti GM. TGF-b1 is an organizer of responses to neurodegeneration. J Cell Biochem. 1993;53:314–322. doi: 10.1002/jcb.240530408. [DOI] [PubMed] [Google Scholar]
- 11.Zhu Y, Yang GY, Ahlemeyer B, Pang L, Che XM, Culmsee C, Klumpp S, Krieglstein J. Transforming growth factor-b1 increases bad phosphorylation and protects neurons against damage. J Neurosci. 2002;22:3898–3909. doi: 10.1523/JNEUROSCI.22-10-03898.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Motizuki M, Isogaya K, Miyake K, Ikushima H, Kubota T, Miyazono K, Saitoh M, Miyazawa K. Oligodendrocyte Transcription Factor 1 (Olig1) Is a Smad Cofactor Involved in Cell Motility Induced by Transforming Growth Factor-beta. J Biol Chem. 2013;288:18911–18922. doi: 10.1074/jbc.M113.480996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arnold TD, Ferrero GM, Qiu H, Phan IT, Akhurst RJ, Huang EJ, Reichardt LF. Defective retinal vascular endothelial cell development as a consequence of impaired integrin alphaVbeta8-mediated activation of transforming growth factor-beta. J Neurosci. 2012;32:1197–1206. doi: 10.1523/JNEUROSCI.5648-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cekanaviciute E, Fathali N, Doyle KP, Williams AM, Han J, Buckwalter MS. Astrocytic transforming growth factor-beta signaling reduces subacute neuroinflammation after stroke in mice. Glia. 2014 doi: 10.1002/glia.22675. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hunter CA, Abrams JS, Beaman MH, Remington JS. Cytokine mRNA in the central nervous system of SCID mice infected with Toxoplasma gondii: importance of T-cell-independent regulation of resistance to T. gondii. Infect Immun. 1993;61:4038–4044. doi: 10.1128/iai.61.10.4038-4044.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilson EH, Hunter CA. The role of astrocytes in the immunopathogenesis of toxoplasmic encephalitis. Int J Parasitol. 2004;34:543–548. doi: 10.1016/j.ijpara.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 17.Drogemuller K, Helmuth U, Brunn A, Sakowicz-Burkiewicz M, Gutmann DH, Mueller W, Deckert M, Schluter D. Astrocyte gp130 expression is critical for the control of Toxoplasma encephalitis. J Immunol. 2008;181:2683–2693. doi: 10.4049/jimmunol.181.4.2683. [DOI] [PubMed] [Google Scholar]
- 18.Strack A, V, Asensio C, Campbell IL, Schluter D, Deckert M. Chemokines are differentially expressed by astrocytes, microglia and inflammatory leukocytes in Toxoplasma encephalitis and critically regulated by interferon-gamma. Acta Neuropathol. 2002;103:458–468. doi: 10.1007/s00401-001-0491-7. [DOI] [PubMed] [Google Scholar]
- 19.Stenzel W, Soltek S, Schluter D, Deckert M. The intermediate filament GFAP is important for the control of experimental murine Staphylococcus aureus-induced brain abscess and Toxoplasma encephalitis. J Neuropathol Exp Neurol. 2004;63:631–640. doi: 10.1093/jnen/63.6.631. [DOI] [PubMed] [Google Scholar]
- 20.Voskuhl RR, Peterson RS, Song B, Ao Y, Morales LB, Tiwari-Woodruff S, Sofroniew MV. Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J Neurosci. 2009;29:11511–11522. doi: 10.1523/JNEUROSCI.1514-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koshy AA, Dietrich HK, Christian DA, Melehani JH, Shastri AJ, Hunter CA, Boothroyd JC. Toxoplasma co-opts host cells it does not invade. PLoS pathogens. 2012;8:e1002825. doi: 10.1371/journal.ppat.1002825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim SK, Karasov A, Boothroyd JC. Bradyzoite-specific surface antigen SRS9 plays a role in maintaining Toxoplasma gondii persistence in the brain and in host control of parasite replication in the intestine. Infect Immun. 2007;75:1626–1634. doi: 10.1128/IAI.01862-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uhrlaub JL, Brien JD, Widman DG, Mason PW, Nikolich-Zugich J. Repeated in vivo stimulation of T and B cell responses in old mice generates protective immunity against lethal West Nile virus encephalitis. J Immunol. 2011;186:3882–3891. doi: 10.4049/jimmunol.1002799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi J, Johansson J, Woodling NS, Wang Q, Montine TJ, Andreasson K. The prostaglandin E2 E-prostanoid 4 receptor exerts anti-inflammatory effects in brain innate immunity. J Immunol. 2010;184:7207–7218. doi: 10.4049/jimmunol.0903487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Noor S, Habashy AS, Nance JP, Clark RT, Nemati K, Carson MJ, Wilson EH. CCR7-dependent immunity during acute Toxoplasma gondii infection. Infect Immun. 2010;78:2257–2263. doi: 10.1128/IAI.01314-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caffaro CE, Koshy AA, Liu L, Zeiner GM, Hirschberg CB, Boothroyd JC. A nucleotide sugar transporter involved in glycosylation of the Toxoplasma tissue cyst wall is required for efficient persistence of bradyzoites. PLoS pathogens. 2013;9:e1003331. doi: 10.1371/journal.ppat.1003331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 28.Frugier T, Koishi K, Matthaei KI, McLennan IS. Transgenic mice carrying a tetracycline-inducible, truncated transforming growth factor beta receptor (TbetaRII) Genesis. 2005;42:1–5. doi: 10.1002/gene.20115. [DOI] [PubMed] [Google Scholar]
- 29.Li F, Zhou M. Conditional expression of the dominant-negative TGF-beta receptor type II elicits lingual epithelial hyperplasia in transgenic mice. Dev Dyn. 2013;242:444–455. doi: 10.1002/dvdy.23933. [DOI] [PubMed] [Google Scholar]
- 30.Lin HY, Yang LT. Differential response of epithelial stem cell populations in hair follicles to TGF-beta signaling. Dev Biol. 2013;373:394–406. doi: 10.1016/j.ydbio.2012.10.021. [DOI] [PubMed] [Google Scholar]
- 31.Rani R, Smulian AG, Greaves DR, Hogan SP, Herbert DR. TGF-beta limits IL-33 production and promotes the resolution of colitis through regulation of macrophage function. Eur J Immunol. 2011;41:2000–2009. doi: 10.1002/eji.201041135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–116. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- 33.Lin W, Kemper A, McCarthy KD, Pytel P, Wang JP, Campbell IL, Utset MF, Popko B. Interferon-gamma induced medulloblastoma in the developing cerebellum. J Neurosci. 2004;24:10074–10083. doi: 10.1523/JNEUROSCI.2604-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lopez ME, Klein AD, Dimbil UJ, Scott MP. Anatomically defined neuron-based rescue of neurodegenerative Niemann-Pick type C disorder. J Neurosci. 2011;31:4367–4378. doi: 10.1523/JNEUROSCI.5981-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Halassa MM, Florian C, Fellin T, Munoz JR, Lee SY, Abel T, Haydon PG, Frank MG. Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron. 2009;61:213–219. doi: 10.1016/j.neuron.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Florian C, Vecsey CG, Halassa MM, Haydon PG, Abel T. Astrocyte-derived adenosine and A1 receptor activity contribute to sleep loss-induced deficits in hippocampal synaptic plasticity and memory in mice. J Neurosci. 2011;31:6956–6962. doi: 10.1523/JNEUROSCI.5761-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pifer R, Yarovinsky F. Innate responses to Toxoplasma gondii in mice and humans. Trends Parasitol. 2011;27:388–393. doi: 10.1016/j.pt.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller CM, Boulter NR, Ikin RJ, Smith NC. The immunobiology of the innate response to Toxoplasma gondii. Int J Parasitol. 2009;39:23–39. doi: 10.1016/j.ijpara.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 39.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dupont CD, Christian DA, Hunter CA. Immune response and immunopathology during toxoplasmosis. Seminars in immunopathology. 2012;34:793–813. doi: 10.1007/s00281-012-0339-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ferguson DJ, Graham DI, Hutchison WM. Pathological changes in the brains of mice infected with Toxoplasma gondii: a histological, immunocytochemical and ultrastructural study. International journal of experimental pathology. 1991;72:463–474. [PMC free article] [PubMed] [Google Scholar]
- 42.Ghafoori P, Yoshimura T, Turpie B, Masli S. Increased IkappaB alpha expression is essential for the tolerogenic property of TGF-beta-exposed APCs. Faseb J. 2009;23:2226–2234. doi: 10.1096/fj.08-124545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cho ML, Min SY, Chang SH, Kim KW, Heo SB, Lee SH, Park SH, Cho CS, Kim HY. Transforming growth factor beta 1(TGF-beta1) down-regulates TNFalpha-induced RANTES production in rheumatoid synovial fibroblasts through NF-kappaB-mediated transcriptional repression. Immunology letters. 2006;105:159–166. doi: 10.1016/j.imlet.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 44.Crawford DK, Mangiardi M, Song B, Patel R, Du S, Sofroniew MV, Voskuhl RR, Tiwari-Woodruff SK. Oestrogen receptor beta ligand: a novel treatment to enhance endogenous functional remyelination. Brain. 2010;133:2999–3016. doi: 10.1093/brain/awq237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol. 2011;12:715–723. doi: 10.1038/ni.2060. [DOI] [PubMed] [Google Scholar]
- 46.Cota M, Kleinschmidt A, Ceccherini-Silberstein F, Aloisi F, Mengozzi M, Mantovani A, Brack-Werner R, Poli G. Upregulated expression of interleukin-8, RANTES and chemokine receptors in human astrocytic cells infected with HIV-1. J Neurovirol. 2000;6:75–83. doi: 10.3109/13550280009006384. [DOI] [PubMed] [Google Scholar]
- 47.Appay V, Rowland-Jones SL. RANTES: a versatile and controversial chemokine. Trends Immunol. 2001;22:83–87. doi: 10.1016/s1471-4906(00)01812-3. [DOI] [PubMed] [Google Scholar]
- 48.Oh JW, Schwiebert LM, Benveniste EN. Cytokine regulation of CC and CXC chemokine expression by human astrocytes. J Neurovirol. 1999;5:82–94. doi: 10.3109/13550289909029749. [DOI] [PubMed] [Google Scholar]
- 49.Guo H, Jin YX, Ishikawa M, Huang YM, van der Meide PH, Link H, Xiao BG. Regulation of beta-chemokine mRNA expression in adult rat astrocytes by lipopolysaccharide, proinflammatory and immunoregulatory cytokines. Scandinavian journal of immunology. 1998;48:502–508. doi: 10.1046/j.1365-3083.1998.00422.x. [DOI] [PubMed] [Google Scholar]
- 50.DeArmond SJ, Lee YL, Kretzschmar HA, Eng LF. Turnover of glial filaments in mouse spinal cord. J Neurochem. 1986;47:1749–1753. doi: 10.1111/j.1471-4159.1986.tb13084.x. [DOI] [PubMed] [Google Scholar]
- 51.Takemura M, Gomi H, Colucci-Guyon E, Itohara S. Protective role of phosphorylation in turnover of glial fibrillary acidic protein in mice. J Neurosci. 2002;22:6972–6979. doi: 10.1523/JNEUROSCI.22-16-06972.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tai K, Iwasaki H, Ikegaya S, Ueda T. Minocycline modulates cytokine and chemokine production in lipopolysaccharide-stimulated THP-1 monocytic cells by inhibiting IkappaB kinase alpha/beta phosphorylation. Translational research: the journal of laboratory and clinical medicine. 2013;161:99–109. doi: 10.1016/j.trsl.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 53.Haller D, Holt L, Kim SC, Schwabe RF, Sartor RB, Jobin C. Transforming growth factor-beta 1 inhibits non-pathogenic Gram negative bacteria-induced NF-kappa B recruitment to the interleukin-6 gene promoter in intestinal epithelial cells through modulation of histone acetylation. J Biol Chem. 2003;278:23851–23860. doi: 10.1074/jbc.M300075200. [DOI] [PubMed] [Google Scholar]
- 54.Huang WC, Yen FC, Shie FS, Pan CM, Shiao YJ, Yang CN, Huang FL, Sung YJ, Tsay HJ. TGF-beta1 blockade of microglial chemotaxis toward Abeta aggregates involves SMAD signaling and down-regulation of CCL5. J Neuroinflammation. 2010;7:28. doi: 10.1186/1742-2094-7-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Strack A, Schluter D, Asensio VC, Campbell IL, Deckert M. Regulation of the kinetics of intracerebral chemokine gene expression in murine Toxoplasma encephalitis: impact of host genetic factors. Glia. 2002;40:372–377. doi: 10.1002/glia.10104. [DOI] [PubMed] [Google Scholar]
- 56.McKimmie CS, Graham GJ. Astrocytes modulate the chemokine network in a pathogen-specific manner. Biochem Biophys Res Commun. 2010;394:1006–1011. doi: 10.1016/j.bbrc.2010.03.111. [DOI] [PubMed] [Google Scholar]
- 57.Ovanesov MV, Ayhan Y, Wolbert C, Moldovan K, Sauder C, Pletnikov MV. Astrocytes play a key role in activation of microglia by persistent Borna disease virus infection. J Neuroinflammation. 2008;5:50. doi: 10.1186/1742-2094-5-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brambilla R, Hurtado A, Persaud T, Esham K, Pearse DD, Oudega M, Bethea JR. Transgenic inhibition of astroglial NF-kappa B leads to increased axonal sparing and sprouting following spinal cord injury. J Neurochem. 2009;110:765–778. doi: 10.1111/j.1471-4159.2009.06190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brambilla R, Dvoriantchikova G, Barakat D, Ivanov D, Bethea JR, Shestopalov VI. Transgenic inhibition of astroglial NF-kappaB protects from optic nerve damage and retinal ganglion cell loss in experimental optic neuritis. J Neuroinflammation. 2012;9:213. doi: 10.1186/1742-2094-9-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hahn M, Lorez H, Fischer G. Effect of calcitriol in combination with corticosterone, interleukin-1beta, and transforming growth factor-beta1 on nerve growth factor secretion in an astroglial cell line. J Neurochem. 1997;69:102–109. doi: 10.1046/j.1471-4159.1997.69010102.x. [DOI] [PubMed] [Google Scholar]
- 61.Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat Cell Biol. 2003;5:410–421. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]
- 62.Bermudez LE, Covaro G, Remington J. Infection of murine macrophages with Toxoplasma gondii is associated with release of transforming growth factor beta and downregulation of expression of tumor necrosis factor receptors. Infect Immun. 1993;61:4126–4130. doi: 10.1128/iai.61.10.4126-4130.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.