

Abstract
For each round of pre-mRNA splicing, a spliceosome is assembled anew on its substrate. RNA–protein remodeling events required for spliceosome assembly, splicing catalysis, and spliceosome disassembly are driven and controlled by a conserved group of ATPases/RNA helicases. The activities of most of these enzymes are timed by their recruitment to the spliceosome. The Brr2 enzyme, however, which mediates spliceosome catalytic activation, is a stable subunit of the spliceosome, and thus, requires special regulation. Recent structural and functional studies have revealed diverse mechanisms whereby an RNaseH-like and a Jab1/MPN-like domain of the Prp8 protein regulate Brr2 activity during splicing both positively and negatively. Reversible Brr2 inhibition might in part be achieved via an intrinsically unstructured element of the Prp8 Jab1/MPN domain, a concept widespread in biological systems. Mutations leading to changes in the Prp8 Jab1/MPN domain, which are linked to a severe form of retinitis pigmentosa, disrupt Jab1/MPN-mediated regulation of Brr2.
Keywords: Pre-mRNA splicing, RNA helicase, RNA-protein complex, retinitis pigmentosa, spliceosome catalytic activation
Pre-mRNA Splicing by the Spliceosome
Most eukaryotic mRNAs are derived from precursors that contain non-coding introns interspersed among coding exons. During mRNA maturation, introns are removed and exons are ligated in a process called pre-mRNA splicing. Each splicing event comprises two consecutive transesterification reactions. In the first step, a conserved branch point adenosine in the intron attacks the phosphodiester bond at the 5′-splice site (SS), liberating the 5′-exon with a free 3′-hydroxyl group. In the second step, the 3′-hydroxyl group of the 5′-exon attacks the 3′-SS, leading to exon ligation and excision of the intron in the form of a lariat.1
Pre-mRNA splicing is catalyzed by the spliceosome, a large and highly dynamic RNA–protein (RNP) enzyme. For each round of splicing, a spliceosome is assembled de novo and in a stepwise manner from five small nuclear ribonucleoprotein particles (snRNPs U1, U2, U4, U5, and U6 in the case of the major spliceosome) and many non-snRNP proteins (Fig. 1A).2-4 Several intermediates during spliceosome assembly and catalysis have been characterized biochemically and in part structurally, showing that transitions between assembly and catalysis stages of the spliceosome are accompanied by profound compositional and conformational remodeling of the spliceosome’s RNA and protein interaction networks.3,4 Spliceosome assembly is initiated by U1 snRNP recognizing the 5′-SS (forming the E complex) and U2 snRNP binding the branch point region (giving rise to the A complex) via base paring interactions between the pre-mRNA and U1 or U2 snRNA, respectively. Subsequently, the spliceosomal B complex is formed by incorporation of the remaining three snRNPs as a preformed U4/U6•U5 tri-snRNP. In the tri-snRNP, the U4 and U6 snRNAs are extensively base paired5-7 and linked to U5 snRNP primarily through protein–protein interactions.8-10 None of the spliceosome’s subunits contains a preformed active center for the splicing transesterification reactions and also the B complex, which contains all snRNPs, is still catalytically inactive. During the subsequent process of catalytic activation, the B complex is first converted into a Bact and then to a B* complex, which carries out the first step of splicing. Further rearrangements after step 1 lead to the C complex, which can catalyze the second transesterification step, after which the spliceosome is disassembled in an ordered fashion.

Figure 1. Pre-mRNA splicing by the spliceosome. (A) Model of pre-mRNA splicing by the spliceosome depicting known assembly and catalysis intermediates. ATPases/RNA helicases and a G-protein (Snu114) are shown where their activities are required (red and pink, respectively). Red traffic lights indicate stages at which Brr2 is expected to be shut off, whereas a green traffic light indicates Brr2 being switched on during catalytic activation. Presently, it is not clear if Brr2 is actively shut off again after spliceosome catalytic activation. (B) Changes in the snRNA–pre-mRNA interaction network during spliceosome catalytic activation, highlighting the presumed substrates of the Prp28 and Brr2 RNA helicases.
Remodeling events during spliceosome assembly and catalysis are mediated by at least eight universally conserved superfamily 2 (SF2) ATPases/RNA helicases (Fig. 1A).11 The most dramatic reorganizations of the spliceosome take place during catalytic activation (Fig. 1B). At this stage, U1 snRNA is displaced from the pre-mRNA 5′-SS, a process that requires the activity of the Prp28 protein.12,13 Concomitantly, the U4/U6 base pairing interaction is disrupted by the Brr2 enzyme,14-16 and U4 snRNA, as well as all U4/U6-associated proteins, are displaced from the spliceosome. U4/U6 disruption allows U6 snRNA to engage in new base pairing interactions with the 5′-SS and with U2 snRNA and to adopt a catalytically important internal stem-loop structure (Fig. 1B).3,4,11 The newly assembled U2/U6/pre-mRNA interaction network forms the heart of the spliceosome's catalytic center.17,18
The Spliceosomal Brr2 RNA Helicase Requires Tight Regulation
Most spliceosomal ATPases/RNA helicases associate only transiently with the spliceosome and their regulation is achieved by the timing of their recruitment to the splicing machinery. Brr2 is a notable exception. As a stable component of the U5 snRNP, it already encounters its U4/U6 substrate outside of the spliceosome in the U4/U6•U5 tri-snRNP, posing the danger of premature U4/U6 dissociation. Indeed, tri-snRNPs enriched in a cellular fraction from yeast can undergo U4 snRNA release in a Brr2- and ATP-dependent manner,15 a reaction that is thought to resemble U4/U6 unwinding during spliceosome catalytic activation. Thus, special mechanisms are required to prevent Brr2 to unwind U4/U6 prematurely in the tri-snRNP or directly upon tri-snRNP association with the spliceosome (Fig. 1A). After catalytic activation, Brr2 remains associated with the spliceosome throughout splicing catalysis until spliceosome disassembly. The enzyme has been suggested to be required again during splicing catalysis19 and spliceosome disassembly,20 but presumably does not act as an ATPase/RNA helicase during these stages.19,21 At least in vitro, the entire catalysis and disassembly steps proceed in the presence of UTP as the only nucleotide tri-phosphate, which cannot be hydrolyzed by Brr2.21 Thus, after spliceosome catalytic activation, Brr2 may be shut off again or may be prevented by other means from unwinding other RNA duplexes it may then encounter (Fig. 1A). On the other hand, Brr2 alone exhibits comparatively weak RNA duplex unwinding activity in vitro,14,22,23 conflicting with its requirement for unwinding U4/U6, the most extensively base-paired and stable RNA duplex of the spliceosome.7 Thus, for efficient spliceosome catalytic activation, Brr2 may also have to be upregulated by specific signals at the correct time (Fig. 1A).
The Special Structure of the Brr2 Helicase
Matching its special regulatory requirements, Brr2 is structurally unique among the spliceosomal ATPases/RNA helicases. It is the only member of this group of enzymes that belongs to the Ski2-like subfamily of SF2.24 Furthermore, Brr2 is part of a unique subclass of nucleic acid helicases that contain tandem helicase cassettes, each with dual RecA-like domains followed by Sec63 homology units.23,25,26 Only the Brr2 N-terminal cassette exhibits ATPase and RNA unwinding activity in vitro,27 and only the enzymatic activity of this cassette is required for splicing in vivo.16 We recently determined the crystal structure of a large fragment of human (h) Brr2 comprising both helicase cassettes (Brr2 helicase region, Brr2HR).27 Both cassettes are similarly structured, with dual RecA-like domains, a winged helix (WH) domain, and a helical bundle (HB) domain surrounding a central tunnel (Fig. 2A–C). The HB domain together with lateral helix-loop-helix (HLH) and immunoglobulin-like (IG) domains form the Sec63 units. The RecA domains of the N-terminal cassette contain conserved sequence motifs for binding and hydrolyzing ATP, and for binding and unwinding an RNA duplex, which are clustered at the RecA interface and around the central tunnel, respectively.27 The structure of a related, prokaryotic SF2 DNA helicase, Hel308, in complex with a DNA substrate28 suggests that the central tunnel in the N-terminal cassette of Brr2 serves for binding and translocating on one RNA strand of a substrate duplex in a 3′-to-5′ direction (Fig. 2C). An extended loop of the RecA-2 domain (the separator loop) extends across the tunnel entrance and may act like a plowshare, separating the two RNA strands of an RNA duplex when one strand is transported through the tunnel (Fig. 2C).27,28 The structure also revealed multiple contacts between the two helicase units that allow these elements to functionally cooperate. Although the C-terminal unit is catalytically inactive on its own, it strongly stimulates the helicase activity of the N-terminal cassette, and thus, serves as an intramolecular helicase cofactor.27

Figure 2. Structures of Brr2HR and Brr2HR–Prp8Jab1 complexes. (A) Domain organization of human Brr2. Domain coloring is conserved in all subsequent panels. Numbers indicate first and last residues of each domain. NE, N-terminal extension; L, linker. (B) Overall structure of human Brr2HR (PDB ID 4F91). SL, separator loop. Domains and functional elements are labeled. (C) Model of a portion of U4/U6 snRNA bound to Brr2HR. Arrow, putative breakpoint between the N-terminal RecA-2 and HB domains to allow RNA loading. Rotated 150° about the vertical axis compared with (B) as indicated. (D) Structure of the human Brr2HR–Prp8Jab1 complex (PDB ID 4KIT). C-terminal cassette orientated as in (B). The Brr2 N-terminal cassette is colored by domain as above. Conserved RNA-binding elements in the tunnel of the N-terminal cassette, brown; C-terminal cassette, light brown; hPrp8Jab1, gold with semi-transparent surface. (E) Structure of the yeast Brr2HR–Prp8Jab1 complex (PDB ID 4BGD). C-terminal cassette orientated as in (B and D). Panels A–C adapted from reference 27 with changes; panel C adapted from reference 42.
Evidence for Positive and Negative Regulation of Brr2 by the Prp8 Protein
Brr2 tightly interacts with the U5 snRNP proteins Prp8, a large regulatory scaffold, and Snu114, an EF-G/eEF2-like G-protein,10,29-31 and both proteins modulate Brr2 activity.20,32-34 Previously, there were conflicting findings as to the effect of Prp8 on Brr2 activity. A C-terminal fragment (CTF) of Prp8, which contains an RNase H-like (RH) domain35-37 followed by a Jab1/MPN-like (Jab1) domain,31,38 was found to stimulate Brr2-mediated U4/U6 unwinding activity in vitro.22,23 On the other hand, genetic studies indicated that Brr2 helicase activity is negatively regulated by Prp8 during splicing.32,33 The underlying mechanism for either effect remained elusive.
The Prp8 RH Domain Can Inhibit Brr2 by Substrate Competition
To further elucidate how Brr2 is regulated by Prp8, we investigated the effect of the RH and Jab1 domains of Prp8 on Brr2-mediated U4/U6 unwinding separately. We found that Brr2 requires a single-stranded region in U4 snRNA 3′ of the first base-paired stem with U6 snRNA (the so-called U4 central domain) to engage and unwind U4/U6 in vitro.39 Consistent with this observation, in vivo crosslinking studies suggested that Brr2 engages the 3′-region of U4 snRNA exclusively via its active N-terminal cassette and targets the first base-paired stem with U6 snRNA.19 In the structure of hBrr2HR, the central RNA-binding tunnel of the N-terminal cassette is sealed on one side by non-covalent contacts between the RecA-2 and HB domains.27 In addition, the 3′-end of U4 snRNA is occluded by secondary structures and the bound Sm proteins.40 These observations suggest that the contacts between the RecA-2 and HB domains in the N-terminal cassette of Brr2 may have to intermittently break open (arrow in Fig. 2C) to position the U4 central domain inside of Brr2’s RNA-binding tunnel next to the separator loop. Structural evidence is in agreement with this scenario, indicating that the RecA-2 domain is a flexible element in the N-terminal cassette of Brr2.27
Interestingly, the Prp8 RH domain also binds U4/U6 at regions overlapping the Brr2-loading sequence,39 suggesting that it could hinder Brr2 from engaging its substrate. Indeed, yPrp8RH directly competes with yBrr2 for binding U4/U6, thereby inhibiting yBrr2-mediated U4/U6 unwinding.39 The Prp8RH-mediated block of Brr2-substrate interaction may thus constitute one principle by which U4/U6 dissociation is impeded in the U4/U6•U5 tri-snRNP and in the spliceosome before catalytic activation.
The Prp8 Jab1 Domain Can Inhibit Brr2 via a Novel Mechanism
Unlike the RH domain, the Prp8 Jab1 domain directly binds to Brr2.22,26,39,41 To investigate how the Prp8 Jab1 domain may influence Brr2 activity, we determined the co-crystal structure of hBrr2HR in complex with hPrp8Jab1.42 In this structure, the globular portion of hPrp8Jab1 rests on the Sec63 unit of the N-terminal hBrr2HR cassette, while contacts to the hBrr2HR C-terminal cassette are lacking (Fig. 2D). The proximal part of an extended C-terminal tail of the Jab1 domain, which was flexible and only partially resolved in structures of the isolated domain,31,38 binds along a cleft between the HLH and HB domains of the N-terminal Sec63 unit. The distal part of this tail runs between the N-terminal RecA-2 and HB domains and inserts into the N-terminal cassette’s central RNA-binding tunnel (Fig. 2D). Within the tunnel, the distal part of the Jab1 tail covers the RNA-binding surfaces of the RecA domains and its very C terminus interacts with the RecA-1, WH, and HB domains (Fig. 2D).
Consistent with this structure, the binding of U4/U6 and short single-stranded RNAs to yBrr2 was inhibited in the presence of yPrp8Jab1, independent of the nucleotide-bound state of yBrr2. In contrast, a variant of yPrp8Jab1 lacking the last 16 amino acids (yPrp8Jab1-ΔC16) stimulated RNA binding to yBrr2.42 Furthermore, the RNA-stimulated ATPase activity of yBrr2 was strongly reduced by Prp8Jab1 but enhanced by yPrp8Jab1-ΔC16.42 Finally, the U4/U6 unwinding activity of both yeast and human Brr2 was reduced in the presence of Prp8Jab1 when Brr2 was in large excess over the U4/U6 snRNA, while yPrp8Jab1-ΔC16 or hPrp8Jab1-Q2321stop (lacking the C-terminal 15 residues) stimulated this activity.42
Together, these structural and functional analyses show that the Prp8 Jab1 domain can inhibit Brr2 by a mechanism that is unprecedented in previously investigated nucleic acid helicases. Prp8Jab1 engages Brr2 primarily via its global domain, allowing its C-terminal tail to intermittently occlude Brr2’s RNA binding tunnel. It thereby efficiently counteracts loading of Brr2 onto the U4 snRNA, leading to the inhibition of Brr2’s RNA-stimulated ATPase and U4/U6 unwinding activities in vitro. Presently, we do not know if this mode of regulation is unique to Brr2 or shared by other Ski2-like helicases or even other helicase (sub-)families. As we found that the inactive C-terminal cassette is required for Prp8Jab1-mediated regulation of Brr2,42 other dual-cassette nucleic acid helicases, such as yeast Slh1p43 or the ASCC3 subunit of the activating signal cointegrator complex,44 may potentially be subject to similar regulatory mechanisms.
Notably, while Prp8RH-mediated inhibition of Brr2 is only expected to function before spliceosome catalytic activation, the very high affinity of Prp8Jab1 with or without its distal tail to Brr2 (Table 1) suggests that it remains bound to Brr2 during and after U4/U6 unwinding, possibly allowing the C-terminal tail of Prp8 to rebind Brr2's RNA binding tunnel after the displacement of U4 snRNA. It could thus also help to switch off Brr2 activity again in later stages of the splicing cycle (Fig. 3).
Table 1. Affinities of Prp8 fragments to full-length Brr2 proteins.
| hPrp8 fragment | Kd (nM) | yPrp8 fragment | Kd (nM) | Type of RP13-linked mutation |
|---|---|---|---|---|
| hPrp8Jab1 | 7.7 | yPrp8Jab1 | 7.6 | - |
| hPrp8Jab1-Q2321stop | 3.2 | - | - | III |
| - | - | yPrp8Jab1-ΔC16 | 11.1 | - |
| hPrp82300–2335 | 3150.0 | yPrp82378–2413 | 930.0 | - |
| hPrp8Jab1-R2310G | 81.3 | - | - | II |
| hPrp8Jab1-R2310K | 46.0 | - | - | II |
| hPrp8Jab1-F2314L | 63.5 | - | - | II |
| hPrp8Jab1-Y2334N | 20.0 | - | - | III |
For experimental details, see legend to Figure 4.
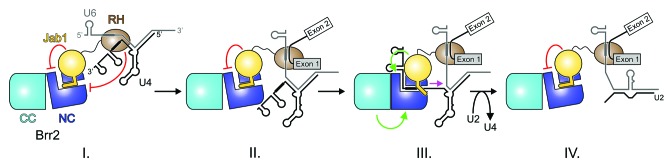
Figure 3. Model for the regulation of Brr2 by Prp8. Model for the regulation of Brr2 by the Prp8 RH and Jab1 domains during pre-mRNA splicing. Red lines, inhibitory effects; green arrows, stimulatory effects. (I) Inhibited state in the U4/U6•U5 tri-snRNP and the B complex achieved by competitive binding of a single-stranded U4 snRNA region upstream of stem I (thick black line) via the RH domain (Mozaffari-Jovin et al., 2012) and by blocking the Brr2 RNA-binding tunnel and disrupting N-terminal cassette (NC)—C-terminal cassette (CC) interactions via the Jab1 domain. (II) State after release of U4 snRNA from RH. Brr2 is still blocked via the Jab1 domain. The conserved U6 ACAGAG box (thick gray line) has taken over base pairing of the 5′ ss from U1 snRNA. (III) Brr2 stimulation during spliceosome catalytic activation via the Jab1 domain and by direct interaction of the NC with CC.27 Magenta arrow—movement of Brr2 on U4 snRNA. (IV) The Jab1 domain may again block Brr2 activity at some point after spliceosome catalytic activation, although there is presently no direct experimental evidence for the direct inhibition of Brr2 during the later stages of splicing. Adapted from reference 42.
A Possible Stepwise Release of the Brr2 Blocks
Apparently, two neighboring domains of Prp8 act in a coordinated manner, but by distinct molecular mechanisms, to keep Brr2 inactive in the U4/U6•U5 tri-snRNP and initially in the spliceosomal B complex. Thus, there appear to be multiple regulatory checkpoints to prevent premature U4/U6 unwinding during the splicing process. For catalytic activation, both blocks to Brr2 loading on U4 snRNA must be relieved. In principle, two separate, inhibitory macromolecular interactions may be easier to disrupt than a continuous interaction surface of the combined size. A similar strategy has been suggested for the sigma factor that guides bacterial RNA polymerase to its promoters but is released upon entry into the transcription elongation phase.45 Furthermore, two separable blocks may render spliceosome catalytic activation dependent on multiple signals, thereby generating additional possibilities for control and possibly supporting a precise order of events.
Affinity purification of spliceosomes with an anti-hPrp31 antibody led to the isolation of a spliceosomal B complex, from which U1 snRNP is released but which has not yet undergone U4/U6 unwinding,46 suggesting that Prp28-mediated U1-5′-SS dissociation may precede U4/U6 unwinding during spliceosome catalytic activation. The Prp8 RH domain is thought to support interaction of U6 with the 5′-SS after Prp28-mediated release of U1.35,47,48 It is conceivable that accommodation of the U6-5′-SS duplex triggers release of U4 from the Prp8 RH domain, thereby facilitating Brr2 engagement with its substrate. A functional link of U1-5′-SS dissociation, which apparently precedes U4/U6 unwinding, and Prp8RH release from U4/U6 suggests that the block by Prp8RH is released before the Jab1-mediated Brr2 inhibition.
Conformational Changes in Brr2 May Lead to Release of Jab1-Mediated Inhibition
Our finding that the Prp8Jab1 tail can inhibit Brr2 suggests that during splicing a specific molecular signal is required at the correct time to remove the tail from Brr2’s RNA-binding tunnel and allow substrate RNA binding and subsequent catalytic activation of the spliceosome. However, the structure of the hBrr2HR–hPrp8Jab1 complex shows that the hPrp8Jab1 C-terminal tail interacts with hBrr2HR via a large surface area (Fig. 2D), which should stabilize this interaction and thus, hinder displacement of the tail. This raises the question as to how an activating signal/trigger can efficiently release the block of Brr2 imposed by Prp8Jab1. The Prp8Jab1 tail contains a large fraction of charged residues (eight of the terminal 26 residues), is amenable to proteolytic degradation in the isolated protein,31 and essentially lacks regular secondary structure in the complex with hBrr2HR, supporting the idea that it constitutes an intrinsically unstructured element in isolation. As is generally the case for intrinsically unstructured elements and proteins,49 immobilization of the Jab1 C-terminal tail on Brr2 must therefore be accompanied by a large loss in conformational entropy. The unfavorable entropy contribution will in part compensate the expected large positive interaction enthalpy. Such entropy–enthalpy compensation should render binding of the Prp8 C-terminal tail on Brr2 less thermodynamically stable than expected for a globular domain interacting via an interface of the same size, but at the same time highly specific.
To test this model, we estimated the relative contributions of the globular Jab1 domain and its C-terminal tail to the overall Brr2–Prp8Jab1 binding free energy by surface plasmon resonance analyses. hPrp8Jab1-Q2321stop-bound Brr2HR with comparable affinity as the hPrp8 Jab1 domain bearing an intact tail (a Kd of 3.2 and 7.7 nM, respectively; Table 1; Fig. 4). In contrast, a peptide comprising only the C-terminal 36 residues of hPrp8 (encompassing the last 11 residues of the globular part and the entire C-terminal tail of the Jab1 domain) bound about three orders of magnitude less tightly (Kd of ca. 3.2 µM; Table 1; Fig. 4). No binding was seen in this assay with a peptide comprising only the last 16 residues of hPrp8 (i.e. the entire distal tail). A very similar result was seen with the corresponding yeast proteins and peptides (Table 1). These results confirm that efficient inhibition of Brr2 activity via the Jab1 C-terminal tail requires anchoring of the tail on the surface of the N-terminal helicase cassette of Brr2 via the globular part of Jab1, which thereby gives rise to a high local concentration of the tail in the vicinity of the Brr2 RNA-binding tunnel. In this situation, the C-terminal Jab1 tail can apparently counteract Brr2 RNA binding but the strength of the inhibition is probably finely tuned to allow efficient release in response to the appropriate signal.

Figure 4. Analysis of Brr2-Prp8 interactions by surface plasmon resonance. (A) Sensorgrams of the interaction of human Brr2 with various human Prp8Jab1 variants (indicated) or a hPrp8 C-terminal peptide (residues 2300–2335). (B) Sensorgrams of the interaction of yeast Brr2 with yeast Prp8Jab1, its tail-deleted variant (yPrp8Jab1-ΔC16) and a yeast Prp8 C-terminal peptide (residues 2378–2413). Concentration ranges of the analytes used are indicated. Proteins and RNAs were produced as described previously.42 N-terminally acetylated peptides comprising the C-terminal 36 residues of hPrp8 (NPKEFYHEVHRPSHFLNFALLQEGEVYSADREDLYA) or yPrp8 (IPLEFYNEMHRPVHFLQFSELAGDEELEAEQIDVFS) and bearing free C-termini were obtained from the Research Group Mass Spectrometry, Leibniz-Institute for Molecular Pharmacology. Surface plasmon resonance analyses were performed using a Biacore 2000 instrument (GE Healthcare) at 20 °C with running buffer containing 10 mM HEPES–NaOH, pH 7.4, 150 mM NaCl, 50 µM EDTA, and 0.005% NP40. Flow cells of the Sensor Chip NTA (GE Healthcare) were washed with 20 µl of regeneration solution (350 mM EDTA) and equilibrated using running buffer at 20 µl/min. The second flow cell was activated with 40 µl of 500 µM NiSO4 at 5 µl/min. Human or yeast full-length Brr2, bearing N-terminal His10-tags, were then immobilized on the second flow cell by injecting the proteins at 100 nM in running buffer and at 10 µl/min for 90 s to achieve capture levels of 10 000–12 000 RU. Afterwards, loosely bound proteins were washed away with running buffer. The Jab1 domain variants or peptides were used as analytes in running buffer. Jab1 domain variants and peptides were injected into both flow cells at increasing concentrations (between 5–800 nM for Jab1 variants and between 10–100 µM for peptides) at a flow rate of 30 µl/min. Binding was monitored for 200 s followed by a 300–900 s delay (buffer alone) to monitor dissociation. The sensor surface was regenerated at the end of each binding cycle with 60 µl of regeneration solution at 20 µl/min using the Extraclean feature to remove the immobilized proteins from the surface. Sensorgrams (resonance units vs. time) from the sample cells were corrected by comparison with the sensorgrams from the corresponding control cells and kinetic parameters were extracted using the Biacore 2000 Evaluation Software (GE Healthcare) using a model of 1:1 (Brr2:Jab1 or peptide) binding.
Presently, the signal upon which the Prp8Jab1-mediated block of Brr2 is released is not known. Interestingly, however, the structure of a yBrr2HR-yPrp8Jab1 complex, which is globally quite similar to our human complex (Fig. 2E), was determined independently in the group of Kiyoshi Nagai.50 Although a Jab1 domain bearing an intact C-terminal tail was also used in that study, the Prp8Jab1 tail in the yeast structure was not bound at the yBrr2HR RNA-binding tunnel and was not visible in the electron density, suggesting that it may have been proteolytically degraded during crystallization or that it adopted a disordered, unbound conformation. Superposition of the two complexes via the Brr2 C-terminal cassettes reveals that the N-terminal cassette-Jab1 sub-complexes are rearranged with respect to the C-terminal cassettes (possibly due to different crystal lattice contacts; Mov. S1). This analysis supports the idea that different conformational states of Brr2 exhibit different affinities for the Prp8Jab1 tail. Therefore, the molecular trigger that leads to derepression of Brr2 during splicing may involve a process that induces a Brr2 conformation in which the RNA-binding tunnel exhibits lower affinity for the Prp8Jab1 tail. In principle, such conformational changes could be elicited by changes in the posttranslational modification status of one of the participating proteins, such as Prp8 ubiquitination/deubiquitination,51,52 or by the binding of other spliceosomal proteins, such as the G-protein Snu114 that can interact with Brr2 and Prp810,31,38 and has been demonstrated to influence Brr2 activity.20
Parallels to the Mechanism of Jab1-Mediated Brr2 Inhibition in Other Systems
In addition to its activity in the spliceosome, recruitment of Brr2 to the U5 snRNP is also regulated, at least in yeast. U5 snRNP assembly in yeast proceeds via a cytoplasmic precursor that lacks Brr2 and instead contains the Aar2 protein.53,54 Structural analyses have shown that Aar2 contains a globular portion and a flexible, C-terminal extension.41,55,56 The globular part of Aar2 binds the linker, endonuclease, and RH domains of Prp8, while its C-terminal extension spirals along the Prp8 reverse transcriptase domain and across the RH domain, where it connects an extended hairpin of the RH domain with the β-barrel core of the Jab1 domain.55,56 The Prp8 Jab1 domain is thereby bound to the RH domain in a manner that sterically precludes binding of Brr2.55,56 Phosphorylation of Aar2 at serine 253, possibly aided by additional phosphorylation at other residues, leads to a conformational change that reduces its affinity for Prp8, and thereby allows Brr2 to bind.41,55 Isothermal titration calorimetry revealed that the Aar2 C-terminal extension contributes little to the overall binding free energy with Prp8.55 Thus, Aar2, a reversible inhibitor of the Brr2–Prp8 interaction, and the Prp8 Jab1 domain, a reversible inhibitor of Brr2-mediated U4/U6 unwinding, are structurally organized in a similar manner. Both proteins encompass globular domains, which primarily mediate binding to the target proteins, and intrinsically unstructured extensions, which represent the inhibitory elements of the proteins. In both cases, the ability of these extensions to carry out their inhibitory functions hinges on the tethering via their globular domains to the target protein. Furthermore, in both cases, the intrinsically unstructured elements elicit reversible inhibition by binding functionally important regions, i.e., at the Brr2 RNA-binding tunnel in the case of Jab1, and between the RH and Jab1 domains in the case of Aar2, with high specificity (due to their large interaction surfaces) but with controlled thermodynamic stability (presumably due to the loss of conformation entropy upon binding). Finally, both inhibitory scenarios may thus be reversed by conformational changes in the regulatory factors or their targets, elicited by phosphorylation in the case of Aar255 and possibly by changes in post-translational modification or interacting proteins in the case of Prp8Jab1.
The principle of reversible inhibition based on intrinsically unstructured elements may be widespread in complex enzymes and assembly chaperones. For example, the proteasome core particle is formed by stacked heptameric rings of peripheral α-subunits and central β-subunits.57 The α-subunits can be regarded as regulators of the proteolytically active β-subunits, which shield access to the inner cavity of the proteasome by forming a peripheral plug via their intertwined, intrinsically unstructured N-terminal tails. Minor perturbation, as could be induced by the 19S regulatory caps of the proteasome, can dissolve this meshwork, allowing proteins destined for degradation to be threaded into the proteolytic cavity.58
As another example, Gemin2 regulates the last stage of Sm core RNP assembly by binding and stabilizing a subcore formed by the Sm proteins D1, D2, F, E, and G. Gemin2 binds to the Sm subcore via a globular domain and uses an intrinsically unstructured N-terminal extension to reach around the subcore’s periphery and into the concave RNA binding pocket, where it sterically interferes with snRNA binding.59,60 Presently, it is not known whether a specific signal is required to release this inhibitory conformation and allow the formation of the complete Sm core RNP.59 It is also conceivable that the intrinsically unstructured Gemin2 extension binds with just enough affinity to counteract binding of non-cognate RNAs but would give way to snRNAs whose Sm sites would have higher affinity to the Sm subcore.
A variation of the above theme is afforded by the pICln protein that also functions during Sm core assembly upstream of Gemin2. pICln interacts with the Sm subcore as an intermediate placeholder of Sm proteins D3 and B. In complex with the subcore, pICln adopts an extended pleckstrin homology (PH) fold, with which it contacts Sm protein D1 on one side and Sm protein G on the other.60 The N-terminal extension segment of the pICln PH domain is intrinsically unstructured in the isolated protein61 but folds into a β-structure upon contacting Sm protein G, allowing formation of a continuous β-sheet around the Sm subcore-pICln complex.60 The entropic loss associated with folding upon binding of this intrinsically unstructured pICln element may lead to a strategic weakening of the pICln–SmG interface, thereby providing a predetermined breakpoint for later removal of pICln to allow completion of Sm core assembly. This is fully in line with other evidence that also supports this scenario.60
The principle of entropy–enthalpy compensation may also be exploited to upregulate the activity of regulatory proteins in a reversible manner. For instance, the binding of splicing regulatory SR proteins to target proteins or RNAs can be increased upon phosphorylation of the proteins’ RS-domains. A recent study has shown that phosphorylation reduces the intrinsic flexibility of RS-domains in isolated SR proteins, thus reducing the entropic loss they undergo when immobilized on interaction partners.62
The Prp8 Jab1 Domain Can Stimulate Brr2 Activity After Substrate Loading
Inhibition of Brr2 by both the Prp8 RH and Jab1 domains conflicts with the previously observed Brr2 activation by the Prp8CTF,22,23 which encompasses both of these motifs. However, when U4/U6 unwinding was assayed at a higher RNA to Brr2 ratio, as used in these previous studies, yPrp8Jab1 no longer inhibited yBrr2, but rather enhanced its activity, while yPrp8Jab1-ΔC16 stimulated Brr2 unwinding activity to an even greater extent.42 Thus, in a purified, ternary in vitro system, increased concentrations of U4 snRNA can apparently out-compete the Prp8Jab1 tail for Brr2’s RNA binding pocket, mitigating the inhibitory effects of the tail on RNA loading. As, under conditions favoring RNA binding in vitro, Prp8Jab1 can also act as a positive regulator of Brr2 helicase activity, it likely plays a similar role after triggered release of the Prp8Jab1 tail during splicing; however, in the complex environment of the spliceosome specific signals likely play important, coordinated roles in switching Brr2 activity on and off (see above). Thus, the C-terminal domain of Prp8 could play dual essential roles, ensuring that Brr2 activity is turned off early in the splicing process, but also turned up to a sufficient level to allow efficient catalytic activation of the spliceosome.
The ability of Prp8Jab1 to stimulate Brr2-mediated U4/U6 unwinding could be due to a number of factors, including the more efficient coupling of ATP hydrolysis to RNA duplex unwinding as suggested previously.22 To further test this idea, we assayed Brr2-mediated U4/U6 unwinding and RNA-stimulated ATP hydrolysis under equivalent conditions that favor RNA binding. Initial rates of Brr2 helicase and ATPase activities were then used to calculate the amount of ATP hydrolyzed per U4/U6 duplex unwound, which reflects the apparent efficiency of coupling ATP hydrolysis to RNA strand separation (Table 2). yBrr2 alone hydrolyzes ~13 000 molecules of ATP per U4/U6 duplex, indicating that a large fraction of the enzyme-substrate complexes formed are unproductive in RNA unwinding— i.e. it hydrolyzes a large amount of ATP upon interaction with RNA but cannot couple this efficiently to strand separation. Upon binding yPrp8Jab1-ΔC16, Brr2 unwinds U4/U6 very fast and a ~10-fold lower amount of ATP is hydrolyzed per U4/U6 di-snRNA unwound (Table 2), indicating more efficient coupling of ATP hydrolysis to RNA strand separation by Brr2. Although yPrp8Jab1 reduced the rate of RNA duplex unwinding compared with Prp8Jab1-ΔC16, it led to an even lower amount of energy (~300 ATP molecules) that was required to unwind a U4/U6 duplex (Table 2). Thus, once RNA loading is accomplished, both the globular part and the C-terminal tail of Prp8Jab1 act in concert to increase the apparent efficiency with which Brr2 unwinds U4/U6.
Table 2. Effects of yPrp8Jab1 variants on the efficiency of yBrr2.
| Proteins | ATP hydrolysis [nM min−1] |
U4/U6 unwinding [nM min−1] |
ATP hydrolyzed per U4/U6 unwound |
Type of RP13-like mutation |
|---|---|---|---|---|
| yBrr2 | 15860 ± 2315 | 1.18 | 13441 ± 1962 | - |
| + yPrp8Jab1 | 864 ± 103 | 2.80 | 313 ± 37 | - |
| + yPrp8Jab1-ΔC16 | 27520 ± 358 | 18.0 | 1528 ± 20 | - |
| + yPrp8Jab1-R2388K | 8926 ± 1083 | 0.50 | 17853 ± 2165 | II |
| + yPrp8Jab1-F2392L | 3202 ± 301 | 0.95 | 3370 ± 317 | II |
| + yPrp8Jab1-A2399stop | 27317 ± 455 | 16.30 | 1676 ± 28 | III |
| + yPrp8Jab1-F2412N | 6868 ± 1994 | 12.95 | 530 ± 153 | III |
ATP hydrolysis and U4/U6 unwinding were monitored as described.42 For comparison of apparent unwinding efficiencies, ATP hydrolysis and U4/U6 unwinding were measured under identical conditions (200 nM yBrr2, 50 nM U4/U6 duplex, without or with 0.75 µM of the indicated yPrp8Jab1 variants, 30 °C).
The exact molecular mechanism underlying the stimulatory effect by Prp8Jab1 on Brr2 is presently not clear. In both the human and yeast Brr2HR-Prp8Jab1 structures,42,50 the proximal part of the Prp8Jab1 C-terminal tail bridges the Brr2 HB and HLH domains. While the HB domain is thought to provide a ratcheting function during RNA translocation, the HLH domain may serve as a surface via which the unwound RNA exits Brr2.27,28 Direct interactions between these domains are missing in the Brr2 apo structure,27 but have been shown to be important in related helicases.63,64 Thus, an HB–HLH interaction mediated by Prp8Jab1 could help couple RNA-driven ATP hydrolysis to the ratcheting movement of Brr2 on the newly emerged single-stranded RNA.
Presently, we do not know the structure of a Brr2–Prp8Jab1 complex in the process of unwinding. As after RNA loading, the Prp8 C-terminal tail contributes to the apparent efficiency of RNA unwinding by Brr2 (see above), it is possible that during unwinding the tail acts like a latch, reinforcing the circular domain arrangement of the Brr2 N-terminal cassette, thereby helping to trap the RNA. As the Jab1 core domain increases Brr2’s affinity for RNA,42 enhanced ATPase–helicase coupling in the presence of Prp8Jab1 and Prp8Jab1-ΔC16 could also rely in part on additional direct RNA contacts of the Jab1 globular part to the unwound RNA strand, consistent with its position neighboring the N-terminal Brr2 HLH domain (Fig. 2D and E), across which the unwound RNA strand may exit Brr2.27
Many RNA helicases require protein cofactors to be turned on or off at the right place and time. Previously, both inhibitory, as well as stimulatory cofactors, acting in cis or trans and by diverse mechanisms, have been characterized in different systems and in different helicase families. For example, a serine protease domain of the NS3 helicase from hepatitis C virus enhances its helicase activity by supporting RNA binding.65 An example of helicase inhibition is afforded by eIF4AIII in the exon junction complex, where a pre-hydrolysis state of the helicase is locked by a multi-protein interaction network.66,67 However, Brr2 represents a unique case, in which the same cofactor, Prp8, is employed as an inhibitor that counteracts RNA loading, as well as an activator once RNA has been accommodated. This dual-mode regulation is ideally suited to meet the requirement for Brr2 to be switched on and off repeatedly during splicing (Fig. 3).
A number of studies have indicated that several other spliceosomal proteins bind Brr2 primarily through the C-terminal helicase cassette.10,30,68 As Brr2 helicase activity can in principle be regulated by ligands binding to the inactive C-terminal cassette,27 it will be interesting to see whether these additional Brr2-binding proteins also regulate Brr2 activity and what the functional consequence of such regulation would be. Recently, the yeast Sad1 protein, an ortholog of the human U4/U6•U5-specific 65K protein that is required for recruitment of the U4/U6•U5 tri-snRNP to the spliceosome,69 was shown to counteract Brr2-mediated ATP-dependent dissociation of the U4/U6•U5 tri-snRNP into its U5 and U4/U6 components.70 However, the functional importance of this type of tri-snRNP disruption is presently unclear and it is also not known whether Sad1 acts by directly inhibiting Brr2 activity or whether it merely stabilizes the tri-snRNP via interactions with other components.
Links to Retinal Disease in Humans
Mutations in several genes coding for essential U4/U6•U5 tri-snRNP-associated splicing factors have been linked to autosomal dominant forms of retinitis pigmentosa,71,72 a progressive degeneration of the retina. Presently, it is unclear how mutations in genes that encode ubiquitously required splicing factors lead to a tissue-specific disorder.
Certain mutations in the gene encoding hPrp8 lead to the severe RP13 form of retinitis pigmentosa.73 Strikingly, all RP13-linked mutations are found in the hPrp8 Jab1 domain and the majority of these mutations lead to residue exchanges or stop codons in the C-terminal 35 amino acids of the protein or to extensions of the tail. We and others have therefore investigated the consequences of RP13-linked changes on (1) the folding of the Jab1 domain, (2) the interaction of Jab1 or Prp8 with Brr2, (3) the Jab1-mediated modulation of RNA binding, ATP hydrolysis and U4/U6 unwinding by Brr2, (4) the assembly of U5 snRNP and U4/U6-U5 tri-snRNP in vivo, and (5) on cell viability and in vivo splicing.22,42,54 Most analyses so far were carried out in the yeast system, as sequence analyses show that the affected hPrp8 residues are highly evolutionarily conserved. In addition, the Jab1 fold and its interaction with Brr2 are very similar in yeast and human (Fig. 2D and E).42,50 Furthermore, there is a correlation between the severity of growth phenotypes in haploid yeast and the severity of retinal degeneration in humans due to the corresponding Prp8 mutations.74 Thus, the molecular mechanisms that underlie the Prp8-associated growth and splicing defects in yeast are presumably also largely responsible for the development of RP13 in humans.
Based on their position in Prp8Jab1 in the Brr2HR–Prp8Jab1 complex structures, the RP13-linked residue exchanges and truncations can be divided into three types (Fig. 5A and B), each likely associated with different functions in Prp8. Type I residues/positions lie within the globular portion of the hPrp8 Jab1 domain (Fig. 5A and B), suggesting that they are important for the fold and stability of that domain, but do not directly contact hBrr2HR. Type II residues/positions map to the proximal region of the Jab1 C-terminal tail, i.e. the transition region between the globular part of the Jab1 domain and the distal part of the C-terminal tail. Residues in this part of the protein may serve dual functions. They could potentially mediate structurally important intra-Jab1 contacts and additionally engage in direct interactions with the N-terminal Sec63 unit of Brr2HR. Finally, type III residues/positions map to the most C-terminal part of Prp8, i.e. the distal region of the C-terminal tail, which interacts with the Brr2 RNA-binding tunnel in the hBrr2HR–hPrp8Jab1 structure. All RP13-linked Prp8 modifications tested so far lead, in varying degrees, to yeast growth defects and accumulation of pre-U3 RNA in vivo, suggesting that the observed growth defects likely arise from defects in pre-mRNA splicing. Strikingly, however, a more detailed investigation of the effects of these mutations in vivo or in vitro, revealed different phenotypes for the various types of affected residues/positions that were consistent with their different suggested functions within Prp8 and/or in Brr2 regulation (Fig. 5C).

Figure 5. Structural basis for RP13 disease mechanisms. (AandB) Location of human RP13-linked residues (A) and yeast RP13-like residues (B) in the structures of human and yeast Brr2HR–Prp8Jab1 complexes. Brr2 is shown in surface representation. RP13-linked (human) or RP13-like (yeast) residues are shown with small spheres and are labeled. Type I residues/positions, black; type II residues/positions and proximal tail, violet; type III residues/positions and distal tail, green. Other colors as before. (C) Scheme illustrating the phenotypes associated with the various types of RP13-linked/RP13-like mutations. Symbols as in Figure 3. Green arrows or “+” indicate functional processes, red lines or “-“ indicate dysfunctional processes; dashed red lines or “-/+” indicate partial functionality. Brr2 inhibition or activation by Prp8Jab1 variants bearing type I exchanges could not be tested as the proteins were insoluble. However, type I exchanges would be expected to also severely interfere with Brr2 regulation in cases where soluble protein is produced in vivo.
Prp8Jab1 domain constructs bearing type I changes, i.e. S2118P/S2197P (human/yeast numbering), P2301T/P2379T, F2304L/F2382L, H2309P/H2387P, H2309R/H2387R could not be produced in soluble form in various expression hosts.42 Furthermore, corresponding changes in Prp8 destabilized or abolished the interaction of Prp8 with Brr2, leading to defects in the maturation of U5 snRNP, and thus, to reduced tri-snRNP levels in yeast.42,54
Based on their positions in the Brr2HR–Jab1 complex structures, we would expect that type II residues contribute to stable binding of Jab1 to Brr2, thus helping to anchor the distal C-terminal Jab1 tail such that it can intermittently occupy the Brr2 RNA-binding tunnel and inhibit the enzyme (Fig. 5A and B). Furthermore, based on their location in the proximal tail, which may bridge the N-terminal HB and HLH domains in Brr2 during U4/U6 unwinding (see above), type II changes would be expected to interfere with Jab1-mediated Brr2 activation after substrate loading (Fig. 5A and B). Functional analyses fully support these hypotheses. Jab1 domain or Prp8CTF variants bearing changes in type II residues/positions (R2310K/R2388K, R2310G/R2388G, F2314L/F2392L) showed reduced interaction with Brr2 in pull-down,54 analytical gel filtration,42 or Biacore assays (Table 1). These lower Brr2 affinities are in line with type II Prp8 variants, leading to reduced levels of U4/U6•U5 tri-snRNP in vivo,42,54 and are accompanied by incomplete inhibition of the RNA-stimulated ATPase activity of yBrr2 by the R2388K and F2392L variants of yPrp8Jab1 compared with wild-type (wt) Prp8Jab1 (Fig. 6A), as well as by defects of type II Prp8Jab1 and Prp8CTF variants in stimulating Brr2 helicase activity (Fig. 6B).22 Even quantitative differences in the Brr2 affinities of the type II Jab1 domain variants correlate with quantitative differences in their effects on Brr2 activity. For example, the F2392L variant of yPrp8Jab1 reduced the steady-state rate of RNA-stimulated ATP hydrolysis by yBrr2 to a greater extent than R2388K (Fig. 6A), reflecting its more stable interaction with yBrr2.42 Consistent with the diminished activation of yBrr2 by type II variants of yPrp8Jab1 (Fig. 6B), the apparent efficiency with which yBrr2 can couple ATP hydrolysis with U4/U6 unwinding in the presence of yPrp8Jab1-R2388K or yPrp8Jab1-F2392L was reduced more than 50-fold and ca. 10-fold, respectively, compared with wt yPrp8Jab1 (Table 2).
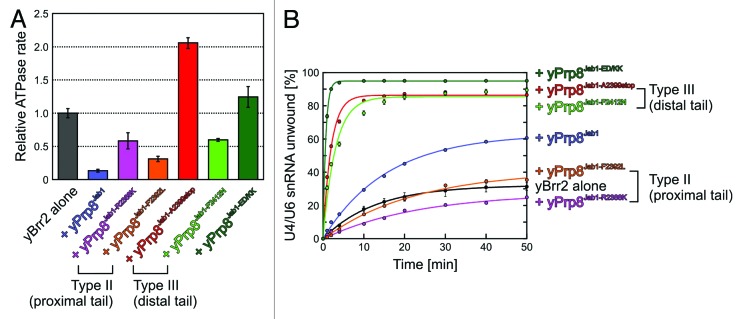
Figure 6. Functional effects of RP13-like mutations. (A) Effect of yPrp8Jab1 mutations on the RNA-stimulated ATPase activity of yBrr2. Error bars represent standard errors of the mean of three independent experiments. yPrp8ED/KK–yPrp8E2407K/D2410K variant, designed based on the structure of hBrr2HR–hPrp8Jab1 to destabilize binding of the distal Prp8Jab1 tail to the RNA-binding tunnel of Brr2, mimicking type III mutations. (B) Effect of yPrp8Jab1 mutations on yBrr2's U4/U6 unwinding activity under conditions favoring RNA binding in vitro (200 nM yBrr2, 50 nM U4/U6, 30 °C). Error bars represent standard errors of the mean of at least two independent experiments. Except for the effects of type II variants of yPrp8Jab1 (yPrp8Jab1-R2388K and yPrp8Jab1-F2392L), the data were reported in reference 42. ATP hydrolysis and U4/U6 unwinding were monitored as described.42
Type III residues/positions represent perhaps the most conspicuous RP13-linked defects, as the associated phenotypes directly support our model of reversible Brr2 inhibition via the Jab1 C-terminal tail. They include a stop mutation at residue Q2321/A2399, which gives rise to a Prp8 variant that is C-terminally truncated by 15 amino acids, and the Y2334N/F2412N point mutation that affects the penultimate residue of Prp8 (Fig. 5A). Decisively, unlike type I and type II RP13-linked mutations, type III Prp8 variants (or designed mutants that reduced binding of the distal Prp8 C-terminal tail to Brr2’s RNA-binding tunnel) did not lead to diminished Brr2 association with U5 snRNP or U4/U6•U5 tri-snRNP and the amount of tri-snRNP relative to U4/U6 di-snRNP was not altered in cells expressing such Prp8 variants.42 Consistent with this observation, the introduction of stop codons at positions Q2321/A2399 in hPrp8Jab1/yPrp8Jab1 had essentially no effect on the interaction of the variant Jab1 domains with the corresponding Brr2 proteins (Table 1).42 At the same time, yPrp8Jab1-A2399stop strongly enhanced the RNA binding, RNA-stimulated ATPase and U4/U6 unwinding activities of yBrr2 (compared with wt yPrp8Jab1), in a manner similar to yPrp8Jab1-ΔC16.42 It also enhanced the efficiency with which Brr2 couples ATP hydrolysis to U4/U6 unwinding, but to a lesser extent than wt Prp8Jab1 (Table 2).
The structure of hBrr2HR–hPrp8Jab1 revealed that the penultimate Y2334 residue of hPrp8Jab1 intimately interacts with residues of the N-terminal RecA-1, WH, and HB domains,42 suggesting that it is important for anchoring the hPrp8Jab1 C-terminal tail in the hBrr2HR RNA-binding tunnel. Other residues at this position may interfere with this anchoring function and fail to fully compensate for the entropic loss associated with immobilization of the variant tail. Binding of a Y2334N variant of hPrp8Jab1 to hBrr2 is somewhat impaired compared with wt or tail-deleted hPrp8Jab1 but still binds hBrr2 with high affinity (Table 1), in agreement with no effect of a corresponding Prp8 mutant on snRNP stabilities.42 Thus, distal tail mutations like Y2334N lead to destabilization of the Prp8 tail in Brr2’s RNA-binding tunnel without severely reducing the overall affinity of Prp8 to Brr2. Consistent with this scenario, yPrp8Jab1-F2412N led to increased RNA binding and RNA-stimulated ATP hydrolysis by yBrr2 compared with wt yPrp8Jab1.42 In addition, the apparent efficiency of coupling ATP hydrolysis to U4/U6 strand separation was greatly enhanced compared with yBrr2 alone, to a similar extent as with wt yPrp8Jab1 (Table 2). The effects of additional yPrp8Jab1 mutations that were designed, based on the structure of the hBrr2HR–hPrp8Jab1 complex, to interfere with stable binding of the Jab1 C-terminal tail in Brr2’s RNA-binding tunnel, further corroborated the conclusion that small alterations in the interaction between Brr2 and the C-terminal tail of Prp8 can have large effects on Prp8Jab1-mediated inhibition of Brr2 helicase activity.42
Possible RP13 Disease Mechanisms
The above results provide clear evidence that different RP13-linked mutations elicit their effects by different molecular mechanisms or combinations thereof (Fig. 5C), either affecting U5 snRNP and U4/U6•U5 tri-snRNP levels (type I and type II), interfering with Brr2 activation (type II and possibly type I), and/or or interfering with Brr2 inhibition (type II, type III, and possibly type I). Type I mutations (in the globular part of the Jab1 domain) are expected to generate folding-defective Prp8 variants in vivo, possibly leading to degradation or aggregation of the mutant protein, and thus, perhaps insufficient levels of active Prp8 in affected cells (Fig. 5C). Even if still produced in soluble form in vivo, type I variants of Prp8 most likely will show decreased interaction with Brr2. All of these scenarios would explain the observed reduced formation of intact U4/U6•U5 tri-snRNPs in the presence of Prp8 bearing type I RP13-related changes, which may have deleterious consequences on cell viability and splicing. Misregulation of Brr2 will likely contribute to the splicing and yeast growth defects in those cases where lower amounts of tri-snRNPs, bearing type I Prp8 variants, are still formed.
Type II variants of Prp8Jab1 can still be produced as soluble, recombinant proteins. Thus, corresponding Prp8 variants are also likely produced in a soluble form in cells bearing the corresponding mutations. Type II mutations (affecting the proximal part of the Prp8 C-terminal tail) still decrease Prp8Jab1 binding to Brr2 by abrogating direct contacts to Brr2 (Fig. 5C), likely explaining the observed snRNP assembly defects in vivo. As in the case of type I mutations, the RP13 phenotype associated with type II Prp8 variants could thus be partially due to less efficient tri-snRNP assembly. However, type II mutations specifically affect the interaction of Prp8Jab1 along the cleft between the HLH and HB domains (Fig. 5A and B). The possible function of the affected amino acids in mediating interactions between the HLH and HB domains in Brr2 suggest that even if assembled into tri-snRNPs, type II variants of Prp8 will interfere with Brr2 activation. At the same time, these variants may also interfere with Brr2 inhibition by failing to stably position the Prp8 C-terminal tail (Fig. 5C).
Finally, type III mutations (in the distal part of the C-terminal tail) have no or mild effects on the binding of Prp8Jab1 to Brr2 and still support Brr2 activation. As a consequence, the corresponding Prp8 variants do not lead to recognizable snRNP assembly defects in vivo (Fig. 5C). Therefore, haploinsufficiency due to reduced tri-snRNP levels, as well as impaired Brr2 activation, are unlikely mechanisms for the disease phenotype in the case of type III mutations. However, type III mutations strongly interfere with the ability of Prp8Jab1 to inhibit Brr2 activity. As lack of Brr2 inhibition is the only clear phenotype associated with all Prp8 type III variants (Fig. 5C), a complete, intermittent block in Brr2 activity is likely crucial for efficient and correct splicing. The lack of such a block is detrimental, presumably due to premature U4/U6 unwinding.
The above results show that, in addition to reduced levels of the active splicing machinery, misregulation of Brr2 activity that disrupts the catalytic activation step of splicing is a bona fide RP13 disease principle. Misregulation of Brr2 could have a general negative effect on splicing in photoreceptor cells, or specifically inhibit the splicing of one or more pre-mRNAs, leading to cell death and an RP disease phenotype.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Christian Becke (Laboratory of Structural Biochemistry, Free University Berlin) for generating Movie S1. This work was supported by the Deutsche Forschungsgemeinschaft (grants SFB 860 to Lührmann R and SFB 740 to Wahl MC). Mozaffari-Jovin S was supported by a scholarship from the International Max Planck Research School (IMPRS) program for Molecular Biology.
References
- 1.Padgett RA, Grabowski PJ, Konarska MM, Seiler S, Sharp PA. Splicing of messenger RNA precursors. Annu Rev Biochem. 1986;55:1119–50. doi: 10.1146/annurev.bi.55.070186.005351. [DOI] [PubMed] [Google Scholar]
- 2.Brow DA. Allosteric cascade of spliceosome activation. Annu Rev Genet. 2002;36:333–60. doi: 10.1146/annurev.genet.36.043002.091635. [DOI] [PubMed] [Google Scholar]
- 3.Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–18. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 4.Will CL, Lührmann R. Spliceosome structure and function. Cold Spring Harb Perspect Biol. 2011;3:1–24. doi: 10.1101/cshperspect.a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bringmann P, Appel B, Rinke J, Reuter R, Theissen H, Lührmann R. Evidence for the existence of snRNAs U4 and U6 in a single ribonucleoprotein complex and for their association by intermolecular base pairing. EMBO J. 1984;3:1357–63. doi: 10.1002/j.1460-2075.1984.tb01977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hashimoto C, Steitz JA. U4 and U6 RNAs coexist in a single small nuclear ribonucleoprotein particle. Nucleic Acids Res. 1984;12:3283–93. doi: 10.1093/nar/12.7.3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brow DA, Guthrie C. Spliceosomal RNA U6 is remarkably conserved from yeast to mammals. Nature. 1988;334:213–8. doi: 10.1038/334213a0. [DOI] [PubMed] [Google Scholar]
- 8.Behrens SE, Lührmann R. Immunoaffinity purification of a [U4/U6.U5] tri-snRNP from human cells. Genes Dev. 1991;5:1439–52. doi: 10.1101/gad.5.8.1439. [DOI] [PubMed] [Google Scholar]
- 9.Makarova OV, Makarov EM, Liu S, Vornlocher HP, Lührmann R. Protein 61K, encoded by a gene (PRPF31) linked to autosomal dominant retinitis pigmentosa, is required for U4/U6*U5 tri-snRNP formation and pre-mRNA splicing. EMBO J. 2002;21:1148–57. doi: 10.1093/emboj/21.5.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu S, Rauhut R, Vornlocher HP, Lührmann R. The network of protein-protein interactions within the human U4/U6.U5 tri-snRNP. RNA. 2006;12:1418–30. doi: 10.1261/rna.55406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Staley JP, Guthrie C. Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell. 1998;92:315–26. doi: 10.1016/S0092-8674(00)80925-3. [DOI] [PubMed] [Google Scholar]
- 12.Staley JP, Guthrie C. An RNA switch at the 5′ splice site requires ATP and the DEAD box protein Prp28p. Mol Cell. 1999;3:55–64. doi: 10.1016/S1097-2765(00)80174-4. [DOI] [PubMed] [Google Scholar]
- 13.Price AM, Görnemann J, Guthrie C, Brow DA. An unanticipated early function of DEAD-box ATPase Prp28 during commitment to splicing is modulated by U5 snRNP protein Prp8. RNA. 2014;20:46–60. doi: 10.1261/rna.041970.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laggerbauer B, Achsel T, Lührmann R. The human U5-200kD DEXH-box protein unwinds U4/U6 RNA duplices in vitro. Proc Natl Acad Sci U S A. 1998;95:4188–92. doi: 10.1073/pnas.95.8.4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raghunathan PL, Guthrie C. RNA unwinding in U4/U6 snRNPs requires ATP hydrolysis and the DEIH-box splicing factor Brr2. Curr Biol. 1998;8:847–55. doi: 10.1016/S0960-9822(07)00345-4. [DOI] [PubMed] [Google Scholar]
- 16.Kim DH, Rossi JJ. The first ATPase domain of the yeast 246-kDa protein is required for in vivo unwinding of the U4/U6 duplex. RNA. 1999;5:959–71. doi: 10.1017/S135583829999012X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fica SM, Tuttle N, Novak T, Li NS, Lu J, Koodathingal P, Dai Q, Staley JP, Piccirilli JA. RNA catalyses nuclear pre-mRNA splicing. Nature. 2013;503:229–34. doi: 10.1038/nature12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anokhina M, Bessonov S, Miao Z, Westhof E, Hartmuth K, Lührmann R. RNA structure analysis of human spliceosomes reveals a compact 3D arrangement of snRNAs at the catalytic core. EMBO J. 2013;32:2804–18. doi: 10.1038/emboj.2013.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hahn D, Kudla G, Tollervey D, Beggs JD. Brr2p-mediated conformational rearrangements in the spliceosome during activation and substrate repositioning. Genes Dev. 2012;26:2408–21. doi: 10.1101/gad.199307.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Small EC, Leggett SR, Winans AA, Staley JP. The EF-G-like GTPase Snu114p regulates spliceosome dynamics mediated by Brr2p, a DExD/H box ATPase. Mol Cell. 2006;23:389–99. doi: 10.1016/j.molcel.2006.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fourmann JB, Schmitzová J, Christian H, Urlaub H, Ficner R, Boon KL, Fabrizio P, Lührmann R. Dissection of the factor requirements for spliceosome disassembly and the elucidation of its dissociation products using a purified splicing system. Genes Dev. 2013;27:413–28. doi: 10.1101/gad.207779.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maeder C, Kutach AK, Guthrie C. ATP-dependent unwinding of U4/U6 snRNAs by the Brr2 helicase requires the C terminus of Prp8. Nat Struct Mol Biol. 2009;16:42–8. doi: 10.1038/nsmb.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pena V, Jovin SM, Fabrizio P, Orlowski J, Bujnicki JM, Lührmann R, Wahl MC. Common design principles in the spliceosomal RNA helicase Brr2 and in the Hel308 DNA helicase. Mol Cell. 2009;35:454–66. doi: 10.1016/j.molcel.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 24.Fairman-Williams ME, Guenther UP, Jankowsky E. SF1 and SF2 helicases: family matters. Curr Opin Struct Biol. 2010;20:313–24. doi: 10.1016/j.sbi.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ponting CP. Proteins of the endoplasmic-reticulum-associated degradation pathway: domain detection and function prediction. Biochem J. 2000;351:527–35. doi: 10.1042/0264-6021:3510527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang L, Xu T, Maeder C, Bud LO, Shanks J, Nix J, Guthrie C, Pleiss JA, Zhao R. Structural evidence for consecutive Hel308-like modules in the spliceosomal ATPase Brr2. Nat Struct Mol Biol. 2009;16:731–9. doi: 10.1038/nsmb.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Santos KF, Jovin SM, Weber G, Pena V, Lührmann R, Wahl MC. Structural basis for functional cooperation between tandem helicase cassettes in Brr2-mediated remodeling of the spliceosome. Proc Natl Acad Sci U S A. 2012;109:17418–23. doi: 10.1073/pnas.1208098109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Büttner K, Nehring S, Hopfner KP. Structural basis for DNA duplex separation by a superfamily-2 helicase. Nat Struct Mol Biol. 2007;14:647–52. doi: 10.1038/nsmb1246. [DOI] [PubMed] [Google Scholar]
- 29.Achsel T, Ahrens K, Brahms H, Teigelkamp S, Lührmann R. The human U5-220kD protein (hPrp8) forms a stable RNA-free complex with several U5-specific proteins, including an RNA unwindase, a homologue of ribosomal elongation factor EF-2, and a novel WD-40 protein. Mol Cell Biol. 1998;18:6756–66. doi: 10.1128/mcb.18.11.6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Nues RW, Beggs JD. Functional contacts with a range of splicing proteins suggest a central role for Brr2p in the dynamic control of the order of events in spliceosomes of Saccharomyces cerevisiae. Genetics. 2001;157:1451–67. doi: 10.1093/genetics/157.4.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pena V, Liu S, Bujnicki JM, Lührmann R, Wahl MC. Structure of a multipartite protein-protein interaction domain in splicing factor prp8 and its link to retinitis pigmentosa. Mol Cell. 2007;25:615–24. doi: 10.1016/j.molcel.2007.01.023. [DOI] [PubMed] [Google Scholar]
- 32.Kuhn AN, Brow DA. Suppressors of a cold-sensitive mutation in yeast U4 RNA define five domains in the splicing factor Prp8 that influence spliceosome activation. Genetics. 2000;155:1667–82. doi: 10.1093/genetics/155.4.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuhn AN, Reichl EM, Brow DA. Distinct domains of splicing factor Prp8 mediate different aspects of spliceosome activation. Proc Natl Acad Sci U S A. 2002;99:9145–9. doi: 10.1073/pnas.102304299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bartels C, Klatt C, Lührmann R, Fabrizio P. The ribosomal translocase homologue Snu114p is involved in unwinding U4/U6 RNA during activation of the spliceosome. EMBO Rep. 2002;3:875–80. doi: 10.1093/embo-reports/kvf172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pena V, Rozov A, Fabrizio P, Lührmann R, Wahl MC. Structure and function of an RNase H domain at the heart of the spliceosome. EMBO J. 2008;27:2929–40. doi: 10.1038/emboj.2008.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ritchie DB, Schellenberg MJ, Gesner EM, Raithatha SA, Stuart DT, Macmillan AM. Structural elucidation of a PRP8 core domain from the heart of the spliceosome. Nat Struct Mol Biol. 2008;15:1199–205. doi: 10.1038/nsmb.1505. [DOI] [PubMed] [Google Scholar]
- 37.Yang K, Zhang L, Xu T, Heroux A, Zhao R. Crystal structure of the beta-finger domain of Prp8 reveals analogy to ribosomal proteins. Proc Natl Acad Sci U S A. 2008;105:13817–22. doi: 10.1073/pnas.0805960105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang L, Shen J, Guarnieri MT, Heroux A, Yang K, Zhao R. Crystal structure of the C-terminal domain of splicing factor Prp8 carrying retinitis pigmentosa mutants. Protein Sci. 2007;16:1024–31. doi: 10.1110/ps.072872007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mozaffari-Jovin S, Santos KF, Hsiao HH, Will CL, Urlaub H, Wahl MC, Lührmann R. The Prp8 RNase H-like domain inhibits Brr2-mediated U4/U6 snRNA unwinding by blocking Brr2 loading onto the U4 snRNA. Genes Dev. 2012;26:2422–34. doi: 10.1101/gad.200949.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leung AK, Nagai K, Li J. Structure of the spliceosomal U4 snRNP core domain and its implication for snRNP biogenesis. Nature. 2011;473:536–9. doi: 10.1038/nature09956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weber G, Cristão VF, de L Alves F, Santos KF, Holton N, Rappsilber J, Beggs JD, Wahl MC. Mechanism for Aar2p function as a U5 snRNP assembly factor. Genes Dev. 2011;25:1601–12. doi: 10.1101/gad.635911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mozaffari-Jovin S, Wandersleben T, Santos KF, Will CL, Lührmann R, Wahl MC. Inhibition of RNA helicase Brr2 by the C-terminal tail of the spliceosomal protein Prp8. Science. 2013;341:80–4. doi: 10.1126/science.1237515. [DOI] [PubMed] [Google Scholar]
- 43.Martegani E, Vanoni M, Mauri I, Rudoni S, Saliola M, Alberghina L. Identification of gene encoding a putative RNA-helicase, homologous to SKI2, in chromosome VII of Saccharomyces cerevisiae. Yeast. 1997;13:391–7. doi: 10.1002/(SICI)1097-0061(19970330)13:4<391::AID-YEA92>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 44.Dango S, Mosammaparast N, Sowa ME, Xiong LJ, Wu F, Park K, Rubin M, Gygi S, Harper JW, Shi Y. DNA unwinding by ASCC3 helicase is coupled to ALKBH3-dependent DNA alkylation repair and cancer cell proliferation. Mol Cell. 2011;44:373–84. doi: 10.1016/j.molcel.2011.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murakami KS, Darst SA. Bacterial RNA polymerases: the wholo story. Curr Opin Struct Biol. 2003;13:31–9. doi: 10.1016/S0959-440X(02)00005-2. [DOI] [PubMed] [Google Scholar]
- 46.Boehringer D, Makarov EM, Sander B, Makarova OV, Kastner B, Lührmann R, Stark H. Three-dimensional structure of a pre-catalytic human spliceosomal complex B. Nat Struct Mol Biol. 2004;11:463–8. doi: 10.1038/nsmb761. [DOI] [PubMed] [Google Scholar]
- 47.Reyes JL, Gustafson EH, Luo HR, Moore MJ, Konarska MM. The C-terminal region of hPrp8 interacts with the conserved GU dinucleotide at the 5′ splice site. RNA. 1999;5:167–79. doi: 10.1017/S1355838299981785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reyes JL, Kois P, Konforti BB, Konarska MM. The canonical GU dinucleotide at the 5′ splice site is recognized by p220 of the U5 snRNP within the spliceosome. RNA. 1996;2:213–25. [PMC free article] [PubMed] [Google Scholar]
- 49.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 50.Nguyen TH, Li J, Galej WP, Oshikane H, Newman AJ, Nagai K. Structural basis of Brr2-Prp8 interactions and implications for U5 snRNP biogenesis and the spliceosome active site. Structure. 2013;21:910–9. doi: 10.1016/j.str.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bellare P, Kutach AK, Rines AK, Guthrie C, Sontheimer EJ. Ubiquitin binding by a variant Jab1/MPN domain in the essential pre-mRNA splicing factor Prp8p. RNA. 2006;12:292–302. doi: 10.1261/rna.2152306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bellare P, Small EC, Huang X, Wohlschlegel JA, Staley JP, Sontheimer EJ. A role for ubiquitin in the spliceosome assembly pathway. Nat Struct Mol Biol. 2008;15:444–51. doi: 10.1038/nsmb.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gottschalk A, Kastner B, Lührmann R, Fabrizio P. The yeast U5 snRNP coisolated with the U1 snRNP has an unexpected protein composition and includes the splicing factor Aar2p. RNA. 2001;7:1554–65. [PMC free article] [PubMed] [Google Scholar]
- 54.Boon KL, Grainger RJ, Ehsani P, Barrass JD, Auchynnikava T, Inglehearn CF, Beggs JD. prp8 mutations that cause human retinitis pigmentosa lead to a U5 snRNP maturation defect in yeast. Nat Struct Mol Biol. 2007;14:1077–83. doi: 10.1038/nsmb1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weber G, Cristão VF, Santos KF, Jovin SM, Heroven AC, Holton N, Lührmann R, Beggs JD, Wahl MC. Structural basis for dual roles of Aar2p in U5 snRNP assembly. Genes Dev. 2013;27:525–40. doi: 10.1101/gad.213207.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Galej WP, Oubridge C, Newman AJ, Nagai K. Crystal structure of Prp8 reveals active site cavity of the spliceosome. Nature. 2013;493:638–43. doi: 10.1038/nature11843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Groll M, Ditzel L, Löwe J, Stock D, Bochtler M, Bartunik HD, Huber R. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature. 1997;386:463–71. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 58.Groll M, Bajorek M, Köhler A, Moroder L, Rubin DM, Huber R, Glickman MH, Finley D. A gated channel into the proteasome core particle. Nat Struct Biol. 2000;7:1062–7. doi: 10.1038/80992. [DOI] [PubMed] [Google Scholar]
- 59.Zhang R, So BR, Li P, Yong J, Glisovic T, Wan L, Dreyfuss G. Structure of a key intermediate of the SMN complex reveals Gemin2’s crucial function in snRNP assembly. Cell. 2011;146:384–95. doi: 10.1016/j.cell.2011.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grimm C, Chari A, Pelz JP, Kuper J, Kisker C, Diederichs K, Stark H, Schindelin H, Fischer U. Structural basis of assembly chaperone- mediated snRNP formation. Mol Cell. 2013;49:692–703. doi: 10.1016/j.molcel.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 61.Fürst J, Schedlbauer A, Gandini R, Garavaglia ML, Saino S, Gschwentner M, Sarg B, Lindner H, Jakab M, Ritter M, et al. ICln159 folds into a pleckstrin homology domain-like structure. Interaction with kinases and the splicing factor LSm4. J Biol Chem. 2005;280:31276–82. doi: 10.1074/jbc.M500541200. [DOI] [PubMed] [Google Scholar]
- 62.Xiang S, Gapsys V, Kim HY, Bessonov S, Hsiao HH, Möhlmann S, Klaukien V, Ficner R, Becker S, Urlaub H, et al. Phosphorylation drives a dynamic switch in serine/arginine-rich proteins. Structure. 2013;21:2162–74. doi: 10.1016/j.str.2013.09.014. [DOI] [PubMed] [Google Scholar]
- 63.Richards JD, Johnson KA, Liu H, McRobbie AM, McMahon S, Oke M, Carter L, Naismith JH, White MF. Structure of the DNA repair helicase hel308 reveals DNA binding and autoinhibitory domains. J Biol Chem. 2008;283:5118–26. doi: 10.1074/jbc.M707548200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Woodman IL, Briggs GS, Bolt EL. Archaeal Hel308 domain V couples DNA binding to ATP hydrolysis and positions DNA for unwinding over the helicase ratchet. J Mol Biol. 2007;374:1139–44. doi: 10.1016/j.jmb.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 65.Beran RK, Serebrov V, Pyle AM. The serine protease domain of hepatitis C viral NS3 activates RNA helicase activity by promoting the binding of RNA substrate. J Biol Chem. 2007;282:34913–20. doi: 10.1074/jbc.M707165200. [DOI] [PubMed] [Google Scholar]
- 66.Bono F, Ebert J, Lorentzen E, Conti E. The crystal structure of the exon junction complex reveals how it maintains a stable grip on mRNA. Cell. 2006;126:713–25. doi: 10.1016/j.cell.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 67.Andersen CB, Ballut L, Johansen JS, Chamieh H, Nielsen KH, Oliveira CL, Pedersen JS, Séraphin B, Le Hir H, Andersen GR. Structure of the exon junction core complex with a trapped DEAD-box ATPase bound to RNA. Science. 2006;313:1968–72. doi: 10.1126/science.1131981. [DOI] [PubMed] [Google Scholar]
- 68.Hegele A, Kamburov A, Grossmann A, Sourlis C, Wowro S, Weimann M, Will CL, Pena V, Lührmann R, Stelzl U. Dynamic protein-protein interaction wiring of the human spliceosome. Mol Cell. 2012;45:567–80. doi: 10.1016/j.molcel.2011.12.034. [DOI] [PubMed] [Google Scholar]
- 69.Makarova OV, Makarov EM, Lührmann R. The 65 and 110 kDa SR-related proteins of the U4/U6.U5 tri-snRNP are essential for the assembly of mature spliceosomes. EMBO J. 2001;20:2553–63. doi: 10.1093/emboj/20.10.2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang YH, Chung CS, Kao DI, Kao TC, Cheng SC. Sad1 counteracts Brr2-mediated dissociation of U4/U6.U5 in tri-snRNP homeostasis. Mol Cell Biol. 2014;34:210–20. doi: 10.1128/MCB.00837-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mordes D, Luo X, Kar A, Kuo D, Xu L, Fushimi K, Yu G, Sternberg P, Jr., Wu JY. Pre-mRNA splicing and retinitis pigmentosa. Mol Vis. 2006;12:1259–71. [PMC free article] [PubMed] [Google Scholar]
- 72.Singh RK, Cooper TA. Pre-mRNA splicing in disease and therapeutics. Trends Mol Med. 2012;18:472–82. doi: 10.1016/j.molmed.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McKie AB, McHale JC, Keen TJ, Tarttelin EE, Goliath R, van Lith-Verhoeven JJ, Greenberg J, Ramesar RS, Hoyng CB, Cremers FP, et al. Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13) Hum Mol Genet. 2001;10:1555–62. doi: 10.1093/hmg/10.15.1555. [DOI] [PubMed] [Google Scholar]
- 74.Towns KV, Kipioti A, Long V, McKibbin M, Maubaret C, Vaclavik V, Ehsani P, Springell K, Kamal M, Ramesar RS, et al. Prognosis for splicing factor PRPF8 retinitis pigmentosa, novel mutations and correlation between human and yeast phenotypes. Hum Mutat. 2010;31:E1361–76. doi: 10.1002/humu.21236. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.