

Abstract
Several cellular microRNAs show substantial changes in expression during HIV-1 infection and their active role in the viral life cycle is progressively emerging.
In the present study, we found that HIV-1 infection of Jurkat T cells significantly induces the expression of miR-222. We show that this induction depends on HIV-1 Tat protein, which is able to increase the transcriptional activity of NFkB on miR-222 promoter. Moreover, we demonstrate that miR-222 directly targets CD4, a key receptor for HIV-1, thus reducing its expression. We propose that Tat, by inducing miR-222 expression, complements the CD4 downregulation activity exerted by other viral proteins (i.e., Nef, Vpu, and Env), and we suggest that this represents a novel mechanism through which HIV-1 efficiently represses CD4 expression in infected cells.
Keywords: microRNAs, HIV-1, post-transcriptional regulation, CD4, NFkB
Introduction
MicroRNAs (miRNAs) are important post-transcriptional negative regulators, which are able to fine-tune the expression of more than half of all gene transcripts.1 It is now apparent that miRNAs play a key role in many aspects of normal cellular development and function, and moreover, that dysregulation of their expression or function is associated with numerous disease states. MiRNAs work in RNA silencing by driving the RNA-induced silencing complex (RISC) to inhibit translation or destroy target mRNAs. Especially important for miRNA targeting is the “seed” sequence (nucleotide 2–8 of the mature miRNA), which generally binds with perfect Watson-Crick base pairing to target mRNAs. The role of the “seed” sequence suggests that any given miRNA can bind to a broad spectrum of different mRNAs, and so holds a vast regulatory potential.1
Several cellular miRNAs have now emerged that either repress or enhance HIV-1 replication and that show substantial changes in expression upon viral infection.2 Recent studies have shown that miRNAs can regulate the outcome of HIV infection, either via targeting viral RNA directly,3,4 or via the repression of cellular key factors that regulate viral replication or host defense.5-7
Here, we investigated the expression of miR-222 in the context of HIV-1 infection, focusing on the role of Tat and on the impact of a newly identified miR-222 target that encodes the primary HIV-1 receptor, CD4 mRNA. MiR-222 is widely overexpressed in a large variety of human cancers, where it was shown to play its oncogenic roles via the downregulation of several tumor suppressors such as p27, p57, PTEN, and apoptotic factors.8-10 However, miR-222 involvement in viral infection is much less investigated. In the present study, we provide a new role for miR-222 and take a further step in the comprehension of how HIV-1 affects the host miRNA pathway.
Results and Discussion
To determine the effect of HIV-1 infection on cellular miR-222 expression, we infected the Jurkat lymphoblastoma T cell line with HIV-1 (NL4-3 strain) and then measured miR-222 levels by quantitative real-time PCR (qRT-PCR) (Fig. 1). Productively infected cells appeared at day 1 post-infection (p.i.), and reached a maximum at day 2 p.i, as determined by flow cytometric analysis of intracellular p24 Gag capsid antigen accumulation (Fig. 1A, data not shown). Concomitantly, we observed a progressive upregulation of cellular miR-222 that was induced up to 20-folds at day 2 p.i. (Fig. 1B).
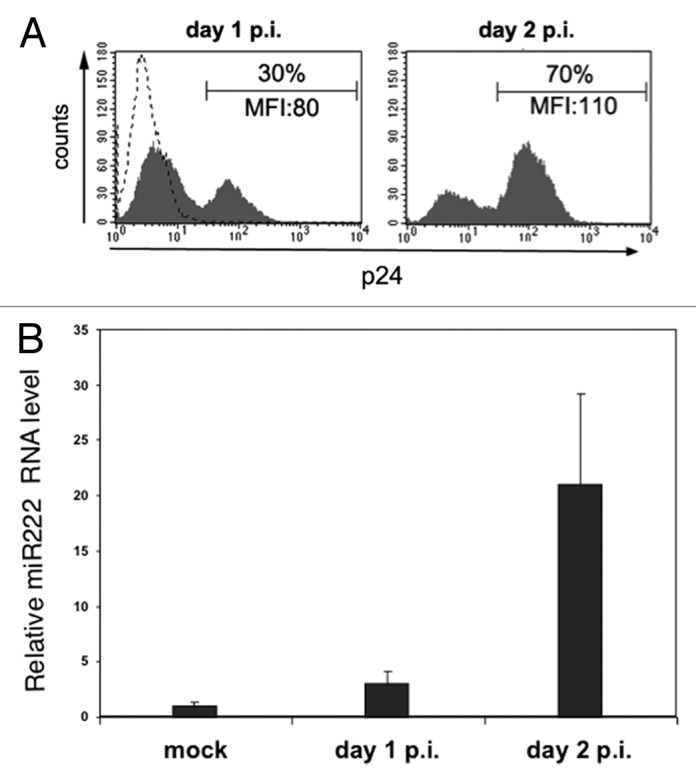
Figure 1. HIV-1 infection induces miR-222 expression. Jurkat cells were infected with HIV-1 by standard spin-infection method or not infected (mock) and analyzed at day 1 and 2 post-infection (p.i.) by flow cytometry (A) and qRT-PCR (B). (A) Histograms show the fluorescence of cells labeled with PE-conjugated anti-p24 mAb. The mean fluorescence intensity (MFI) values and the percentage of p24+ cells are indicated. Dashed line represents labeled mock-infected cells. (B) qRT-PCR was used to measure endogenous miR-222 expression level. Relative miR-222 level in mock Jurkat cells was set to 1. Error bars are SD of one experiment performed in triplicate. One representative experiment out of three is shown.
In order to decipher the molecular mechanisms of miR-222 upregulation induced by HIV-1, we investigated the role of the Tat viral protein, based on two previously reported lines of evidence. First, in glioblastoma and prostate cancer, miR-222 is activated by NFkB and AP-1,11 two very well-characterized factors that regulate HIV-1 transcription. Second, a recent paper reported that Tat associates with the p65 subunit of NFkB and increases both the DNA-binding affinity and the transcriptional activity of p65.12 Thus, we transduced HeLa cells with a Flag-tagged Tat-expressing construct and measured the levels of miR-222 by qRT-PCR. The levels of Mip-1a, a NFkB responsive gene activated by Tat,12 were analyzed as a positive control. Figure 2A indicates that the ectopic expression of Tat resulted in a ~5-fold upregulation of miR-222. Of note, the expression of a mutated Tat protein lacking the binding site for p65 (Flag-TatCA)12 did not significantly modify miR-222 levels, indicating that Tat induces miR-222 in a NFkB-dependent manner.

Figure 2. Tat increases miR-222 levels in a NFkB-dependent manner. (A) HeLa cells were transfected with the empty vector (C), Flag-Tat or Flag-Tat CA expression constructs. Forty-eight hours post-transfection, total RNA was collected, and expression of miR-222 was analyzed by qRT-PCR. As a control, we checked the expression of MIP-1a, a NFkB-dependent gene that was shown to be sensitive to Tat.12 Results shown represent the average of three independent experiments, all performed in triplicate. (B) ChIP assay of chromatin isolated from HeLa cells transfected with empty plasmid (C), Flag Tat, Flag-TatCA, or Flag-Tat plus pCMV-IkBam plasmids (Flag-Tat+IkBa), and immunoprecipitated by anti-Flag or control IgGs, followed by qPCR analysis with primers targeted either to miR-222 enhancer region, or to a sequence on chromosome 1 used as the negative control,11 or to Mip-1a promoter used as positive control.12 The data show occupancy relative to control IgGs and represent mean ± SD of three independent experiments. (C) Nuclear protein extracts from HeLa cells transfected with empty plasmid (C), Flag Tat, Flag-Tat plus pCMV-IkBam plasmids (Flag-Tat+IkBa), were harvested and p65 NFκB activity was measured by transAM p65 protein kit. *, P < 0.05; **, P < 0.01 by Student's t test
We then took advantage of recent results obtained by combining chromatin immunoprecipitation (ChIP) with next-generation sequencing to analyze HIV-1-infected Jurkat cells,13 showing that Tat binds a region located upstream of the miR-222 transcriptional unit (Kurdistani SK, personal communication). Notably, this region matches a genomic sequence we previously described to bind the p65 subunit of NFkB complex and behave as a transcriptional enhancer.11 To further dissect the interplay between Tat, NFkB, and the miR-222 enhancer region, we transduced HeLa cells with a Flag-Tat construct and performed a ChIP assay. Isolated chromatin was immunoprecipitated with anti-Flag antibody or control IgG, followed by qPCR analysis using primers targeted to the enhancer region, to a region on chromosome 1 that served as negative control,14 and to Mip1α promoter used as a positive control. As shown in Figure 2B, Tat binding was remarkably enriched in the miR-222 enhancer region. The ChIP assay also showed that the Flag-TatCA mutant does not bind the same region, indicating that the Tat binding affinity is mediated by p65 (Fig. 2B). This latter observation was confirmed by experiments of functional knockdown, in which the Flag-Tat construct was co-trasfected with pCMV-IkBαM, a plasmid-encoding a dominant-negative mutant of IκBα that blocks the NFkB pathway. pCMV–IkBaM efficiency was measured in nuclear protein extracts of transduced cell by the TransAM NFkB p65 kit, an ELISA-based method designed to specifically detect and quantify NFkB p65 subunit activation (Fig. 2C). As expected, NFkB blocking decreased Tat enrichment on miR-222 enhancer region (Fig. 2B, dark gray columns).
Altogether, these results indicate that Tat has the capacity to induce the p65-dependent activation of miR-222 expression through the occupancy of the miR-222 enhancer region.
To study the possible biological effects of miR-222 on host cellular gene expression during HIV-1 infection, we searched for potential miR-222 targets by bioinformatics prediction algorithm analysis, with a special attention toward mRNAs coding for cellular proteins involved in the HIV-1 cell cycle. We found potential binding site for miR-222 within the 3′ UTR of the CD4 glycoprotein, as predicted by microRNA.org (www.microRNA.org). CD4 acts as primary receptor for HIV-1 on target cells but, soon after viral particles entry, CD4 levels are progressively reduced on the surface of infected cells due to the activity of three viral proteins: Nef, Vpu, and Env.15-17 To experimentally validate that the 3′UTR of CD4 mRNA indeed interacts with miR-222, we employed a dual luciferase reporter assay. We cloned the whole 3′UTR region downstream of the luciferase open reading frame (p3′UTR-CD4), and we used this reporter construct to transfect HeLa cells that exhibit constitutively low levels of miR-222, with or without a plasmid-expressing miR-222. Figure 3A shows that the presence of miR-222 strongly affected luciferase expression based on relative luciferase activity (~50% inhibition). On the contrary, when we used a reporter plasmid harboring the 3′UTR of CD4 mRNA where the binding site for miR-222 has been inactivated by site-directed mutagenesis (p3′UTRmut-CD4), we observed no changes in luciferase activity with or without miR-222 overexpression (Fig. 3A). These results confirmed the bioinformatic prediction indicating the 3′UTR of CD4 mRNA as a bona fide target for miR-222. Moreover, results denote that the here-identified matching site in the 3′UTR of CD4 mRNA strongly contributes to the association with miRNA-222, an interaction that should predictably result in the post-transcriptional inhibition of CD4 expression.

Figure 3. CD4 mRNA is a target of miR-222. (A) p3’UTR-CD4 or p3’UTRmut-CD4 luciferase constructs containing a wild-type (black columns) or a mutated (gray columns) CD4 3’UTR were co-transfected into HeLa cells with empty vector (C), or plasmid expressing miR-222 (p-222).8 Luciferase activity was determined 48 h after transfection. The ratio of normalized sensor to control luciferase activity is shown. Error bars represent SD and were obtained from three independent experiments. (B) Jurkat miR-222 and Jurkat C cells treated or not with DOX for 48 h, were analyzed by qRT-PCR to measure miR-222 expression level. Relative miR-222 level in Jurkat miR-222 cells not induced by DOX was set to 1. Error bars are SD of one experiment performed in triplicate. (C–F) Jurkat miR-222 cells treated as described in (B) were analyzed by flow cytometry (C and D), western blotting (E), and qRT-PCR (F). (C) Histograms show the fluorescence distribution of cells labeled with PE-conjugated anti-CD4 (left and right panels) or anti-HLA-I (middle panel) mAbs. Also, parental Jurkat cells with and without DOX were analyzed for cell-surface CD4 expression (right panel). Solid lines and filled gray histograms represent cells with and without DOX, respectively. Staining with control IgG is also shown (thin line). (D) The cell-surface CD4 MFI ± SEM obtained in three independent experiments like the one in panel (A) is shown. (E) Equal amounts of total cell lysates (20 μg) were separated by 10% SDS-PAGE followed by immunoblotting with anti-CD4 and anti-GAPDH antibodies. Molecular weight standards are indicated (kDa). The CD4 levels were measured by densitometry, normalized by GAPDH, and expressed by setting at 1 the value found in untreated Jurkat miR-222 cells. (F) qRT-PCR was used to assess CD4 mRNA expression. Relative mRNA level in untreated Jurkat miR-222 cells was set at 1. Error bars are SD of an experiment performed in triplicate. Results shown in panels (C–F) are representative of three independent experiments. *P < 0.05 by Student’s t test.
To test whether miR-222 actually represses CD4 expression, we generated Jurkat cells stably expressing miR-222 under a Doxycycline (DOX)-inducible promoter (Jurkat miR-222). DOX treatment of Jurkat miR-222 cells increased the levels of the miRNA as compared with the untreated cells or with Jurkat cells transformed with the empty vector (Jurkat C) treated or not with DOX (Fig. 3B). Concomitantly, DOX treatment resulted in significantly reduced CD4 expression on the membrane of Jurkat miR-222 cells (Fig. 3C and D). A non-specific effect of DOX on the expression of cell-surface molecules not related to miR-222 (e.g., HLA-I) or on cell viability could be excluded (Fig. 3C, data not shown). In addition, CD4 downregulation by DOX was dependent on miR-222 overexpression because it did not occur in DOX-treated parental Jurkat cells (Fig. 3C). Finally, DOX-induced CD4 downregulation on the membrane of Jurkat miR-222 cells was associated with a dramatic decrease of overall CD4 protein and mRNA levels, as determined by western blotting and qRT-PCR, respectively (Fig. 3E and F). We also observed a decrease in CD4 mRNA level in HIV-1-infected Jurkat cells at day 2 p.i. (data not shown), when miR-222 is strongly upregulated (Fig. 1B). This is in agreement with other transcriptome-based studies reporting reduced CD4 mRNA levels following HIV-1 infection.18,19
Our results indicate that CD4 is a direct target of miR-222, and suggest that miR-222, by promoting CD4 mRNA degradation, and thus, preventing CD4 translation, can contribute to the well-known dramatic reduction of CD4 expression on the surface of HIV-1-infected cells. In fact, once infection has occurred, the presence of CD4 poses problems for the virus life cycle, such as the possibility of super-infection that would be toxic to cells, or inhibition of assembly, release, and infectivity of progeny viral particles.15-17 Soon after infection, the early viral Nef protein efficiently downregulates cell-surface CD4 by increasing the rate of CD4 internalization and targeting internalized molecules toward lysosomal compartments for degradation.20 Then, the late proteins Vpu and Env interfere with the anterograde transport of newly synthesized molecules to the cell surface.15,21 We propose that Tat, by inducing miR-222 expression, complements the function of Nef, Vpu, and Env on CD4 downregulation by acting at a different level, that is degradation of CD4 transcripts. The identification of an additional mechanism developed by HIV-1 to eliminate CD4 at every possible step of its biosynthetic pathway supports the general consensus on CD4 being a serious threat that the virus must circumvent.
Materials and Methods
Plasmid construction
p3′UTR-CD4: the full-length 3′UTR of CD4 was PCR-amplified from human genomic DNA and cloned into psiCHECK-2 dual-luciferase reporter plasmid immediately downstream of the stop codon of the Renilla luciferase gene.
p3′UTRmut-CD4: four nucleotides of 3′UTR of CD4, which are perfect binding sites for miR-222 seed sequence, were mutated from AUGUAGC to AUcacaC by site-directed PCR mutagenesis.
p3xFLAG-CMV-Tat and p3xFLAG-CMV-Tat CA were previously described.12
epB-Puro-miR-222: the sequence corresponding to pri-miR-222 was PCR-amplified from human genomic DNA and cloned into epB-Puro-TT derived plasmids.22
Cell lines, treatments, and transduction
HeLa and Jurkat cells were maintained in Dulbecco’s modified Eagle’s and RPMI 1640 medium, respectively, supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin-streptomycin (Euroclone and Gibco-BRL). Transfections were performed by Lipofectamine 2000 (Invitrogen) as recommended by the manufacturer. For the generation of stable Jurkat cells expressing miR-222, upon co-transfection of plasmid epB-Puro-miR-222 and epiggyBac transposase vector, cells were selected by Puromycin (1 μg/ml) treatment and the expression of the microRNA was induced by adding Dox (0.2 μg/ml) to the culture medium. NFkB activity was measured in nuclear protein extracts (15 μg) by the TransAMTM NFkB p65 protein assay (Active Motif),
HIV-1 infection
Stocks of VSV-G-pseudotyped HIV-1 (NL4-3 strain) were prepared and used to infect Jurkat cells with 50 ng of p24/106 cells as described previously.23
Flow cytometry
All procedures were performed at 4 °C in PBS containing 0.5% BSA and 0.1% sodium azide. HIV-1-infected or uninfected cells (2 × 105) were incubated with specific mAbs: phycoerythrin (PE)-conjugated anti-CD4, anti-HLA-I, or isotype control IgG (BD Biosciences). For detection of intracellular p24, cells were fixed and permeabilized with BD Biosciences reagents and incubated at room temperature with fluorescein isothiocyanate (FITC) anti-p24 mAb (KC57; Beckman Coulter). Finally, cells were washed, fixed in 1% paraformaldehyde (PFA), and analyzed (FACSCanto, BD Biosciences).
Western blot analysis
Total lysates of Jurkat cells were separated by 10% SDS-PAGE and analyzed by standard immunoblotting analysis with appropriate primary (anti-CD4, H370, Santa Cruz Biotechnology; anti-GAPDH, MAB374, Millipore) and secondary antibody and Pierce ECL substrate (Thermo Scientific).
Quantitative real-time polymerase chain reaction (qPCR)
Comparative real-time PCR was performed in triplicate with the use of the Taqman Universal PCR Master Mix (Life Technologies) Normalization was performed by simultaneous quantification of snRNA U6B, and relative expression was calculated employing the comparative CT method.
Chromatin immunoprecipitation assay (ChIP)
Chromatin was prepared, immunoprecipitated, and analyzed as previously described.12 Enhancer region amplification was performed with oligos fw 5′-ATCTTGTGCC AACCAGTCCC TTCT-3′, rv 5′-AACATTCTCT GGGACGTTCC TGCT-3′, Control amplifications with oligos 5′-AAACCACCCA TCCAGAAGGG-3′, and 5′-CGTGGCAGCA CTCGTAAGAC T-3′ (chr1:204,366,822-204,366,872.) The relative occupancy of the immunoprecipitated factor at a locus was estimated by using the comparative threshold method.
Luciferase reporter assay
For luciferase reporter assay, HeLa cells were co-transfected with p-222 plasmid8 and psiCHECK-2-vector (Promega), pLuc-CD4-WT, pLucCD4-Mut. After 48 h infection, cells were lysed, and luciferase activity was measured using a dual-luciferase reporter assay system (catalog no.E1960; Promega) following the manufacturer's instructions. Transfection efficiency was normalized to thymidine kinase-driven Renilla luciferase activity. All experiments were performed in triplicate at least.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grants of the Ministry of Health, Ricerca Finalizzata, and Ricerca Corrente co-funded by the Italian 5 × 1000 contribution to Doria M and by grants of the Ministry of Health, “Programma Nazionale di Ricerca su AIDS” in collaboration with ISS to Michienzi A.
Acknowledgments
We are grateful to Ileana Quinto for providing Flag-Tat plasmids.
References
- 1.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Swaminathan S, Murray DD, Kelleher AD. miRNAs and HIV: unforeseen determinants of host-pathogen interaction. Immunol Rev. 2013;254:265–80. doi: 10.1111/imr.12077. [DOI] [PubMed] [Google Scholar]
- 3.Huang J, Wang F, Argyris E, Chen K, Liang Z, Tian H, Huang W, Squires K, Verlinghieri G, Zhang H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat Med. 2007;13:1241–7. doi: 10.1038/nm1639. [DOI] [PubMed] [Google Scholar]
- 4.Nathans R, Chu CY, Serquina AK, Lu CC, Cao H, Rana TM. Cellular microRNA and P bodies modulate host-HIV-1 interactions. Mol Cell. 2009;34:696–709. doi: 10.1016/j.molcel.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Triboulet R, Mari B, Lin YL, Chable-Bessia C, Bennasser Y, Lebrigand K, Cardinaud B, Maurin T, Barbry P, Baillat V, et al. Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science. 2007;315:1579–82. doi: 10.1126/science.1136319. [DOI] [PubMed] [Google Scholar]
- 6.Sung TL, Rice AP. miR-198 inhibits HIV-1 gene expression and replication in monocytes and its mechanism of action appears to involve repression of cyclin T1. PLoS Pathog. 2009;5:e1000263. doi: 10.1371/journal.ppat.1000263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiang K, Liu H, Rice AP. miR-132 enhances HIV-1 replication. Virology. 2013;438:1–4. doi: 10.1016/j.virol.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galardi S, Mercatelli N, Giorda E, Massalini S, Frajese GV, Ciafrè SA, Farace MG. miR-221 and miR-222 expression affects the proliferation potential of human prostate carcinoma cell lines by targeting p27Kip1. J Biol Chem. 2007;282:23716–24. doi: 10.1074/jbc.M701805200. [DOI] [PubMed] [Google Scholar]
- 9.Ciafrè SA, Galardi S. microRNAs and RNA-binding proteins: a complex network of interactions and reciprocal regulations in cancer. RNA Biol. 2013;10:935–42. doi: 10.4161/rna.24641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhong S, Li W, Chen Z, Xu J, Zhao J. MiR-222 and miR-29a contribute to the drug-resistance of breast cancer cells. Gene. 2013;531:8–14. doi: 10.1016/j.gene.2013.08.062. [DOI] [PubMed] [Google Scholar]
- 11.Galardi S, Mercatelli N, Farace MG, Ciafrè SA. NF-kB and c-Jun induce the expression of the oncogenic miR-221 and miR-222 in prostate carcinoma and glioblastoma cells. Nucleic Acids Res. 2011;39:3892–902. doi: 10.1093/nar/gkr006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fiume G, Vecchio E, De Laurentiis A, Trimboli F, Palmieri C, Pisano A, Falcone C, Pontoriero M, Rossi A, Scialdone A, et al. Human immunodeficiency virus-1 Tat activates NF-κB via physical interaction with IκB-α and p65. Nucleic Acids Res. 2012;40:3548–62. doi: 10.1093/nar/gkr1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marban C, Su T, Ferrari R, Li B, Vatakis D, Pellegrini M, Zack JA, Rohr O, Kurdistani SK. Genome-wide binding map of the HIV-1 Tat protein to the human genome. PLoS One. 2011;6:e26894. doi: 10.1371/journal.pone.0026894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, Dang CV, Thomas-Tikhonenko A, Mendell JT. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008;40:43–50. doi: 10.1038/ng.2007.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lama J. The physiological relevance of CD4 receptor down-modulation during HIV infection. Curr HIV Res. 2003;1:167–84. doi: 10.2174/1570162033485276. [DOI] [PubMed] [Google Scholar]
- 16.Doria M. Role of the CD4 down-modulation activity of Nef in HIV-1 infectivity. Curr HIV Res. 2011;9:490–5. doi: 10.2174/157016211798842125. [DOI] [PubMed] [Google Scholar]
- 17.Levesque K, Finzi A, Binette J, Cohen EA. Role of CD4 receptor down-regulation during HIV-1 infection. Curr HIV Res. 2004;2:51–9. doi: 10.2174/1570162043485086. [DOI] [PubMed] [Google Scholar]
- 18.Chang ST, Sova P, Peng X, Weiss J, Law GL, Palermo RE, Katze MG. Next-generation sequencing reveals HIV-1-mediated suppression of T cell activation and RNA processing and regulation of noncoding RNA expression in a CD4+ T cell line. MBio. 2011;2:e00134–11. doi: 10.1128/mBio.00134-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mohammadi P, Desfarges S, Bartha I, Joos B, Zangger N, Muñoz M, Günthard HF, Beerenwinkel N, Telenti A, Ciuffi A. 24 hours in the life of HIV-1 in a T cell line. PLoS Pathog. 2013;9:e1003161. doi: 10.1371/journal.ppat.1003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindwasser OW, Chaudhuri R, Bonifacino JS. Mechanisms of CD4 downregulation by the Nef and Vpu proteins of primate immunodeficiency viruses. Curr Mol Med. 2007;7:171–84. doi: 10.2174/156652407780059177. [DOI] [PubMed] [Google Scholar]
- 21.Dubé M, Bego MG, Paquay C, Cohen ÉA. Modulation of HIV-1-host interaction: role of the Vpu accessory protein. Retrovirology. 2010;7:114. doi: 10.1186/1742-4690-7-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lacoste A, Berenshteyn F, Brivanlou AH. An efficient and reversible transposable system for gene delivery and lineage-specific differentiation in human embryonic stem cells. Cell Stem Cell. 2009;5:332–42. doi: 10.1016/j.stem.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 23.Matusali G, Potestà M, Santoni A, Cerboni C, Doria M. The human immunodeficiency virus type 1 Nef and Vpu proteins downregulate the natural killer cell-activating ligand PVR. J Virol. 2012;86:4496–504. doi: 10.1128/JVI.05788-11. [DOI] [PMC free article] [PubMed] [Google Scholar]