

Abstract
Insights into mechanisms governing resolution of inflammatory pain are of great importance for many chronic pain–associated diseases. Here we investigate the role of macrophages/monocytes and the anti-inflammatory cytokine interleukin-10 (IL-10) in the resolution of transient inflammatory pain. Depletion of mice from peripheral monocytes/macrophages delayed resolution of intraplantar IL-1β- and carrageenan-induced inflammatory hyperalgesia from 1 to 3 days to >1 week. Intrathecal administration of a neutralizing IL-10 antibody also markedly delayed resolution of IL-1β- and carrageenan-induced inflammatory hyperalgesia. Recently, we showed that IL-1β- and carrageenan-induced hyperalgesia is significantly prolonged in LysM-GRK2+/− mice, which have reduced levels of G-protein-coupled receptor kinase 2 (GRK2) in LysM+ myeloid cells. Here we show that adoptive transfer of wild-type, but not of GRK2+/−, bone marrow-derived monocytes normalizes the resolution of IL-1β-induced hyperalgesia in LysM-GRK2+/− mice. Adoptive transfer of IL-10−/− bone marrow-derived monocytes failed to normalize the duration of IL-1β-induced hyperalgesia in LysM-GRK2+/− mice. Mechanistically, we show that GRK2+/− macrophages produce less IL-10 in vitro. In addition, intrathecal IL-10 administration attenuated IL-1β-induced hyperalgesia in LysM-GRK2+/− mice, whereas it had no effect in wild-type mice. Our data uncover a key role for monocytes/macrophages in promoting resolution of inflammatory hyperalgesia via a mechanism dependent on IL-10 signaling in dorsal root ganglia.
Perspective
We show that IL-10-producing monocytes/macrophages promote resolution of transient inflammatory hyperalgesia. Additionally, we show that reduced monocyte/macrophage GRK2 impairs resolution of hyperalgesia and reduces IL-10 production. We propose that low GRK2 expression and/or impaired IL-10 production by monocytes/macrophages represent peripheral biomarkers for the risk of developing chronic pain after inflammation.
Keywords: Monocytes/macrophages, G-protein-coupled receptor kinase 2, interleukin-10, inflammatory pain
According to a recent study by the Institute of Medicine, more than 100 million Americans suffer from chronic pain.14 One of the limitations for development of novel interventions identified in this report is the limited understanding of the neurobiological pathways leading to transition from acute to chronic pain.
Studies in rodents have revealed that spinal cord microglia, the resident macrophages of the central nervous system, play an important role in the development of chronic pain in models of nerve damage–induced neuropathic pain, diabetic neuropathy, and chronic inflammatory pain.3,6,27,32,40,45 In addition, it has been hypothesized that infiltration of peripheral macrophages into the spinal cord enhances the hyperalgesia in models of chronic pain.7 A common finding is that proinflammatory cytokines released by activated spinal cord microglia and/or infiltrating macrophages contribute to chronic hyperalgesia, which is a hallmark of these animal models of chronic pain.5,27,35 Several studies have shown that inhibition of spinal cord proinflammatory cytokine activity or an increase in anti-inflammatory cytokines reduces hyperalgesia in models of chronic pain. In particular, chronic administration of the anti-inflammatory cytokine interleukin-10 (IL-10) has been shown to reduce hyperalgesia in models of neuropathic pain.16,19,25 However, the role of peripheral monocytes/macrophages and IL-10 in spontaneous resolution of transient inflammatory hyperalgesia has yet to be unraveled.
We recently showed that mice with a cell-specific 50% reduction of G-protein-coupled receptor kinase 2 (GRK2) in lysozyme (Lys)M-positive macrophages/microglia develop markedly prolonged hyperalgesia in response to an intraplantar injection of the cytokine IL-1β, the chemokine CC-chemokine ligand 3 (CCL3), or the inflammatory agent carrageenan. For example, thermal hyperalgesia and mechanical allodynia induced by a single intraplantar injection of the proinflammatory cytokine IL-1β resolves within 1 day in wild-type (WT) mice, but lasts at least 8 days in LysM-GRK2+/− mice.42,43 Intrathecal (i.t.) administration of the microglial/macrophage inhibitor minocycline reversed this prolongation of hyperalgesia in LysM-GRK2+/− mice, indicating a contribution of spinal cord and/or dorsal root ganglion (DRG) microglia/macrophages in the transition to persistent hyperalgesia.8,42 The pathophysiological relevance of a reduced level of GRK2 in microglia/macrophages is exemplified by our recent findings that spinal cord microglia/macrophage GRK2 levels are reduced by approximately 40% during chronic inflammatory hyperalgesia and neuropathic pain in WT mice.8,42 In addition, in patients with the painful chronic inflammatory disease rheumatoid arthritis, the level of GRK2 in circulating mononuclear cells is reduced by 40 to 60%.18
Here we investigated the contribution of peripheral monocyte/macrophages in regulating transient inflammatory hyperalgesia using depletion and adoptive transfer strategies on inflammatory hyperalgesia in WT mice. In addition, we investigated the role of peripheral macrophages/monocytes and the anti-inflammatory cytokine IL-10 in the delayed resolution of inflammatory hyperalgesia in LysM-GRK2+/− mice.
Methods
Animals
We used female (aged 10–14 weeks) WT C57BL/6 mice or C57BL/6 mice (Harlan, Horst, The Netherlands) with cell-specific reduction of GRK2 in LysM-positive cells (LysM-GRK2+/−).8,43 For adoptive transfer experiments, WT and GRK2-deficient green fluorescent protein (GFP)-positive bone marrow-derived monocytes (BMDM) were obtained by breeding GRK2+/− mice with CX3CR1gfp/gfp mice (Jackson Laboratories, Bar Harbor, ME). In addition, BMDM from IL-10−/− mice (Jackson Laboratories) were used. Experiments were performed in accordance with international guidelines and approved by the institutional experimental animal committees.
Mice received an intraplantar injection in the hind paw of 5 μL recombinant murine IL-1β (200 ng/mL in saline; PeproTech, Rocky Hill, NJ) or 5 μL λ-carrageenan (1% w/v; Sigma-Aldrich, St. Louis, MO).8,43 Heat withdrawal latency times were determined using the Hargreaves test (IITC Life Science, Woodland Hills, CA).10 Mechanical thresholds were determined using the von Frey test with the up-and-down method, as we described.8,43 All experiments were performed by an experimenter (N.E. or H.W.) blinded to genotype and treatment.
Drug Administration
I.t. injections (5 μL) with goat anti-mouse IL-10 (10 μg in phosphate-buffered saline [PBS]; Sigma-Aldrich), normal goat immunoglobulin G (IgG) (10 μg in PBS; R&D systems, Minneapolis, MN), or human recombinant IL-10 (.5 μg in PBS; Sigma-Aldrich) were performed under light isoflurane anaesthesia as described previously.8
Cell Depletion
Mice received intraperitoneal injections with 100 μL anti-CCR2 (MC21; .2 μg/μL20) or IgG2b control (BD Biosciences, Franklin Lakes, NJ) at 24 hours and .5 hours before and 10 hours after intraplantar IL-1β or 24 hours and .5 hours before and 24 hours after intraplantar carrageenan. Alternatively, mice received intravenous (i.v.) injections with 200 μL (7 mg/mL) clodronate-liposomes37 or PBS-liposomes at 24 hours and .5 hours before intraplantar IL-1β.
Adoptive Transfer
BMDM were isolated as described recently.36 Following Ficoll (GE Healthcare, Pittsburgh, PA) density gradient centrifugation of bone marrow from femora and tibiae, CD115+ monocytes were isolated with biotin labeled anti-CD115 antibodies and streptavidin-coupled magnetic beads following the manufacturer’s instructions (Miltenyi Biotec, San Diego, CA). IL10−/−, WT-CX3CR1gfp/+, or GRK2+/−-CX3CR1gfp/+ BMDM were i.v. injected (3.5 − 106 cells per mouse) or i.t. injected (15,000 cells per mouse). For some experiments, BMDM were labeled with 5 μM carboxyfluorescein succinimidyl ester (Sigma-Aldrich) according to the manufacturer’s instructions.
Flow Cytometry
BMDM, blood leukocytes, and cells isolated from the peritoneum were stained with anti-CD115 (eBioscience, San Diego, CA), anti-CCR2 (R&D systems), and anti-CD45. Cells were analyzed on a FACSCanto II flow cytometer using FacsDiva software (BD Biosciences).
In Vitro Culture
Peritoneal macrophages were collected from the peritoneal cavity of naive mice by washing with 3 mL RPMI- 1640. Cells were cultured (.2 − 106 cells per well) in RPMI-1640 with 10% fetal calf serum, 2 mM glutamine, and 50 μM β-mercaptoethanol (all Gibco; Life Technologies, Carlsbad, CA) and stimulated with 10 ng/mL lipopolysaccharide (LPS) (Sigma-Aldrich) for 18 hours (RNA) or 24 hours (protein). IL-10 and TNF-α concentration in the supernatant was determined with enzyme-linked immunosorbent assay (U-CyTech, Utrecht, The Netherlands). To determine IL-1β with enzyme-linked immunosorbent assay (BD OpTEIA; BD Biosciences), cultured cells were exposed for 60 minutes to 3 mM adenosine triphosphate to induce IL-1β secretion.
Immunohistochemistry
Spinal cord and DRG were post-fixed in 4% paraformaldehyde and cryoprotected in sucrose. Cryosections (10-μm) of lumbar DRG and of lumbar segments L2–L5 were stained. We used 1:500 rat antimouse CD16/32 (BD Bioscience) or 1:500 rabbit anti-Iba- 1 (Wako Pure Chemical Industries, Richmond, VA), 1:100 rabbit anti-GFP (GeneTex, San Antonio, TX), and 1:200 rat anti-CD45 (BD Bioscience), followed by alexafluor 488-conjugated streptavidin or alexafluor 488- or 594-conjugated secondary antibodies. Photographs were taken with a Zeiss Axio Observer microscope (Zeiss, Oberkochen, Germany).
Microglia/Macrophage Isolation
Microglia were isolated from brains and spinal cord by Percoll (GE Healthcare) density gradient centrifugation as described previously.43 Macrophages were collected from the peritoneal cavity of naive mice by washing with 3 mL RPMI-1640 (Gibco). CD11b+ macrophages were isolated from peritoneal lavage of naive mice followed by magnetic beads separation according to the manufacturer’s instructions (BD IMag). GRK2 protein levels in microglia and peritoneal macrophages were determined by Western blot as described previously.42 GRK2 mRNA levels in spinal cord microglia were analyzed by real-time reverse transcriptase polymerase chain reaction.
Real-Time Reverse Transcriptase Polymerase Chain Reaction
Total RNA from freshly isolated microglia was isolated using RNeasy mini kit (Qiagen, Valencia, CA). Total RNA from paw biopsies was isolated with Trizol, and cDNA was synthesized using Superscript Reverse Transcriptase (Invitrogen, Carlsbad, CA). Quantitative real-time polymerase chain reaction was performed with an I-cycler iQ5 (Bio-Rad, Hercules, CA) using the following primers: GRK2 forward: CgggACTTCTgCCTgAACCATCTg, reverse: CTCggCTgCggACCACACg; CX3CR1 forward: TgTCCA CCTCCTTCCCTgAA, reverse: TCgCCCAAATAACAggCC; IL- 1β forward: CAACCAACAAgTgATATTCTCCATg, reverse: gATCCACACTCTCCAgCTgCA; TNF-α forward: gCggTg CCTATgTCTCAg, reverse: gCCATTTgggAACTTCTCATC; and IL-10 forward: gCACCCACTTCCCAgTCg, reverse: gCATTAAggAgTCggTTAgCAg. Data were normalized for GAPDH and β-actin expression. GAPDH forward: TgAAg- CAggCATCTgAggg, reverse: CgAAggTggAAgAgTgggAg; Actin forward: AgAgggAAATCgTgCgTgAC, reverse: CAATAgTgATgACCTggCCgT.
Statistical Analysis
All data are presented as mean ± standard error of the mean (SEM) and were analyzed using Student’s t-test or 2-way analysis of variance with Bonferroni post hoc tests.
Results
Effect of Peripheral Monocyte/Macrophage Depletion
To investigate the contribution of peripheral monocytes/macrophages to the course of inflammatory hyperalgesia, we utilized 2 different monocyte/macrophage depletion strategies. First, we administered anti-CCR2 (MC21) monoclonal antibody intraperitoneally at 24 hours and 30 minutes before and 10 hours after intraplantar IL-1β. The anti-CCR2 antibody MC21 is well known to specifically deplete Ly6C+(GR1+)CCR2+ monocytes by an antibody-dependent cellular cytotoxicity– mediated mechanism, whereas granulocyte numbers remain unchanged after MC21 treatment.13,23,36 As depicted in Fig 1A, anti-CCR2 treatment prolonged IL-1β-induced thermal hyperalgesia to at least 13 days (Fig 1A). In mice treated with control IgG, IL-1β-induced hyperalgesia resolved within 24 hours, which is consistent with our earlier findings in mice receiving intraplantar IL-1β only.8,42 Baseline thermal sensitivity was not affected by antibody injections (decrease in heat withdrawal latency; control IgG: 7.66 ± .15; MC21: 7.88 ± .17, n = 4 per group). Flow cytometric analysis of peritoneal lavage and peripheral blood confirmed monocyte/macrophage depletion in anti-CCR2-treated mice at 24 hours after intraplantar IL-1β (Fig 1B).
Figure 1.

Effect of monocyte/macrophage depletion on the duration of IL-1β-induced hyperalgesia. (A) Thermal sensitivity was assessed over time after intraplantar injection of 1 ng IL-1β. To deplete monocytes/macrophages, mice received intraperitoneal injections of 20 μg anti-CCR2 antibody (MC21) or control IgG at 24 hours and .5 hours before as well as 10 hours after intraplantar IL-1β (n = 4 per group). (B) Flow cytometric analysis shows successful depletion of CD115+/CCR2+ monocytes in peritoneal cavity washes and peripheral blood at 24 hours after intraplantar IL-1β. (C) Mice received i.v. injections of 200 μL (7 mg/mL) clodronate-liposomes or PBS-liposomes at 24 hours and .5 hours before intraplantar IL-1β (n = 8 per group). Thermal sensitivity was followed over time after intraplantar injection of IL-1β (1 ng). Data are expressed as mean ± SEM. *P < .05, **P < .01, ***P < .001.
In a second set of experiments mice received i.v. injections of clodronate-liposomes at 24 hours and 30 minutes before intraplantar IL-1β. Clodronate-liposomes are known to deplete peritoneal macrophages/monocyte without affecting microglial numbers.30,34,37 Clodronate-mediated depletion of peripheral monocytes/macrophages prolonged the hyperalgesic response to intraplantar IL-1β injection with 2 days (Fig 1C). Baseline sensitivity was not affected by clodronate treatment (decrease in heat withdrawal latency; PBS liposomes: 7.99 ± .44; clodronate liposomes: 8.32 ± .15, n = 8 per group). Flow cytometric analysis of peripheral blood obtained 24 hours after the first clodronate-liposome injection (at the time of IL-1β injection) confirmed monocyte/macrophage depletion (Supplementary Figure 1).
To test if peripheral monocytes/macrophages also control resolution of hyperalgesia in the carrageenan model of transient inflammatory hyperalgesia, peripheral monocyte/macrophages were depleted by administering anti-CCR2 antibody at 24 hours and .5 hours before and 24 hours after a single intraplantar injection of 1% carrageenan. The results in Figs 2A and 2B show that monocyte/macrophage depletion delayed resolution of carrageenan-induced thermal and mechanical hyperalgesia from 2 days in IgG-treated control mice to at least 4 days in anti-CCR2-treated mice (Figs 2A and 2B). At day 4, spinal cords were analyzed to determine whether anti-CCR2 treatment alters carrageenan-induced microglia activation. The results in Fig 2C show that MC21 treatment significantly increased spinal cord dorsal horn Iba-1 expression after intraplantar carrageenan injection in WT mice. At day 4, paw mRNA expression of IL-1β and CX3CR1 (as a measure of monocytes/macrophages) in anti-CCR2-treated mice was indistinguishable from mice that had received control IgG (Fig 2D). Tumor necrosis factor (TNF)-α and IL-10 mRNA production was undetectable in both groups. Collectively, these findings indicate that monocyte depletion did not prolong local inflammation in the paw.
Figure 2.

Effect of peripheral monocyte/macrophage depletion on the duration of carrageenan-induced hyperalgesia. Mice received intraperitoneal injections of 20 μg anti-CCR2 antibody (MC21) or control IgG at 24 hours and .5 hours before as well as 24 hours after intraplantar injection of 5 μL carrageenan (1% w/v), and (A) thermal sensitivity and (B) mechanical sensitivity were determined (n = 4 per group). Four days after intraplantar carrageenan (C), iba-1 expression in the dorsal horn of the lumbar spinal cord was analyzed (D) IL-1β and CX3CR1 mRNA was determined in paw biopsies after anti-CCR2 of IgG treatment (n = 8 per group). Data are expressed as mean ± SEM. *P < .05, **P < .01, ***P < .001.
Role of IL-10 in the Regulation of IL-1βand Carrageenan-Induced Hyperalgesia
Next we tested the hypothesis that IL-10 at the level of the DRG and/or spinal cord plays an important role in the resolution of inflammatory hyperalgesia. I.t. injection of a neutralizing anti-IL-10 antibody significantly delayed resolution of IL1β-induced hyperalgesia to at least 7 days. In mice treated with control IgG, IL-1β-induced hyperalgesia resolved within 24 hours (Fig 3A). Similarly, carrageenan-induced thermal hyperalgesia resolved within 2 to 3 days in control IgG-treated mice, but lasted for 7 days in mice treated with i.t. anti-IL10 (Fig 3B). To control for the possibility that part of the effect of i.t. anti-IL-10 treatment is exerted in the paw by anti-IL-10 reaching the periphery, we applied the same dose of anti-IL-10 or control IgG intraplantarly. The data in Supplementary Figure 2 demonstrate that the severity or time course of IL-1β-induced hyperalgesia was indistinguishable between mice treated with intraplantar IgG- and those treated with anti-IL10.
Figure 3.

Role of IL-10 in the regulation of IL-1β- and carrageenan-induced hyperalgesia. Mice received i.t. injection( s) of 10 μg anti-IL-10 or control IgG (A) 15 minutes prior intraplantar IL-1β or (B) 15 minutes prior and 24 hours and 48 hours after intraplantar carrageenan (n = 4 per group), and thermal sensitivity was followed over time. Data are expressed as mean ± SEM. *P < .05, **P < .01, ***P < .001.
Adoptive Transfer of WT BMDM to LysM-GRK2+/− Mice
The findings in Figs 1 and 2 indicate that peripheral monocytes/macrophages are key to preventing the transition from acute to persistent inflammatory hyperalgesia. We previously described that mice with reduced GRK2 levels in LysM+ myeloid cells (LysMGRK2 +/−) develop persistent hyperalgesia after intraplantar IL-1β or carrageenan.8,42,43 Supporting our hypothesis that GRK2 deficiency in peripheral monocytes/macrophages is key to the prolongation of hyperalgesia in LysM-GRK2+/− mice, the data in Supplementary Figure 3 show that GRK2 protein levels are reduced by −50% in macrophages of these mice, whereas GRK2 protein or mRNA levels did not differ between naive freshly isolated microglia from WT and LysM-GRK2+/− mice. In addition, spinal cord microglial GRK2 mRNA levels were not decreased 24 hours after intraplantar IL-1β (Supplementary Figure 4A), and GRK2 mRNA expression levels did not differ between microglia from WT and LysMGRK2 +/− mice 24 hours after intraplantar IL-1β, indicating that the LysM promotor is not activated at this time point (Supplementary Figure 4B). To determine the contribution of the GRK2-deficient peripheral monocytes/macrophages to the marked prolongation of hyperalgesia in the LysM-GRK2+/− mouse model, we adoptively transferred WT BMDM to LysM GRK2+/− mice. LysM-GRK2+/− mice were injected i.v. with 3.5 − 106 WT BMDM (based on36) 10 minutes prior to intraplantar IL-1β injection. Transfer of WT BMDM to LysM-GRK2+/− mice completely prevented the development of persistent IL-1β hyperalgesia and led to resolution of IL-1β hyperalgesia in LysMGRK2 +/− mice within a time frame similar to that observed in WT mice (Figs 1A vs 4A). Transfer of GRK2-deficient BMDM to LysM-GRK2+/− did not have any effect on IL-1β-induced hyperalgesia (Fig 4B). Transfer of WT BMDM to WT mice prior to intraplantar IL-1β did not affect the spontaneous resolution of hyperalgesia in these mice (Fig 4C). Flow cytometric analysis showed that the percentage of CD115+/CD45+ and CCR2+ cells in BMDM was similar in WT and GRK2+/− mice (Supplementary Figure 5).
Figure 4.

Adoptive transfer of WT BMDM prevents and treats persistent IL-1β-induced hyperalgesia in LysM-GRK2+/− mice. (A–D) Thermal sensitivity was assessed over time after intraplantar IL-1β. (A and B) LysM-GRK2+/− mice were injected i.v. with (A) WT BMDM (3.5 − 106 cells per mouse; n = 10), or (B) GRK2+/− BMDM (3.5 − 106 cells per mouse; n = 8 or vehicle (n = 8) 10 minutes before intraplantar IL-1β. (C) WT mice received i.v. WTBMDM(3.5−106 cells per mouse) or vehicle (n = 10 per group) 10 minutes before intraplantar IL-1β. (D) LysM-GRK2+/− received WT or GRK2+/− BMDM (15,000 per mouse) or vehicle i.t. (n = 6 per group) 10 minutes before intraplantar IL-1β. (E) Representative pictures of DRG of LysM-GRK2+/− mice treated with vehicle or WT GFP-BMDM (3.5 − 106 cells per mouse) 10 minutes before intraplantar IL-1β. DRG were isolated 8 hours after intraplantar IL-1β administration and stained for GFP and CD45 to identify transplanted cells. Scale bar indicates 40 μm. (F) WT or GRK2+/− BMDM (3.5 − 106 cells per mouse) or vehicle were administered i.v. 24 hours after intraplantar IL-1β, that is, during already established persistent hyperalgesia in LysM-GRK2+/− mice. Thermal sensitivity was assessed over time (n = 4–8). Data are expressed as mean ± SEM. **P < .01, ***P < .001 WT BMDM vs vehicle. #P < .05, ##P < .01 GRK2+/− BMDM vs vehicle.
I.t. administration of 15,000 WT BMDM to LysMGRK2 +/− mice promoted a more rapid resolution of IL-1β hyperalgesia as compared to i.v. administration (Figs 4A vs 4D), indicating that the transplanted WT macrophages are likely to act locally in the DRG or spinal cord. We detected GFP+-BMDM in lumbar DRG after i.v. administration (Fig 4E). However, we did not detect donor GFP+-BMDM in lumbar spinal cord.
To determine whether the lack of effect of transplanted GRK2-deficient BMDM was due to a migration defect rather than a functional deficit, we administered GRK2+/− BMDM i.t. This administration of GRK2+/− BMDM did not promote resolution of IL-1β hyperalgesia in LysM-GRK2+/− mice (Fig 4D). However, both WT and GRK2+/− BMDM were present in the DRG at 24 hours after i.t. administration (Supplementary Figure 6), indicating that GRK2-deficient BMDM do migrate to the DRG normally. We only detected infiltration of i.t. injected BMDM into DRG after intraplantar IL-1β and not in naive mice (data not shown). The data in Fig 4F show that i.v. administration of WT BMDM at 24 hours after intraplantar IL-1β also reduces already persistent hyperalgesia in LysM-GRK2+/− mice. Transfer of GRK2+/− BMDM at 24 hours after IL-1β increased hyperalgesia at 3 and 18 hours after transfer (Fig 4F).
Role of IL-10 in the Regulation of IL-1β-Induced Hyperalgesia in LysM-GRK2+/− Mice
In WT mice, IL-10 is crucial for resolution of transient inflammatory hyperalgesia (Fig 3). Therefore, we tested the capacity of GRK2+/− macrophages to produce IL-10. Both the level of IL-10 mRNA and the amount of IL-10 secreted were significantly lower in LPS-stimulated cultures of GRK2+/− compared to WT macrophages (Figs 5A and 5B). Conversely, GRK2+/− macrophages released higher levels of the proinflammatory cytokines TNF-α and IL-1β (Fig 5C) after stimulation with LPS.31
Figure 5.
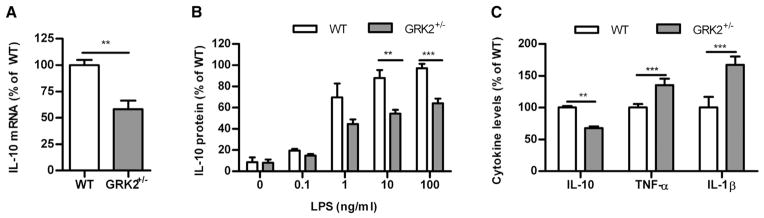
IL-10 production by WT and GRK2+/− macrophages. (A) Freshly isolated WT and GRK2+/− peritoneal macrophages were stimulated with 10 ng/mL LPS and after 18 hours IL-10mRNAexpression was determined. (B)WT and GRK2+/− macrophages were stimulated for 24 hours with different doses of LPS, and IL-10 protein content in supernatant was determined. Data were normalized against the maximal response of WT macrophages induced by 100 ng/mL LPS. (C) WT and GRK2+/− monocytes were stimulated with 10 ng/mL LPS for 24 hours, and IL-10 (100% represents 147 ± 10 pg/mL), TNF-α (100% represents 732 ± 49 pg/mL), and IL-1β (100% represents 944 ± 138 pg/mL) protein content in supernatant was determined (n = 4–8 per group). Data are expressed as mean ± SEM. **P < .01, ***P < .001.
In line with these in vitro observations, our in vivo study shows that i.t. IL-10 injection at 6 hours after intraplantar IL-1β transiently reduced IL-1β hyperalgesia in LysM-GRK2+/− mice without having any effect in WT mice (Fig 6A). This transient effect of IL-10 is likely due to the reported short half-life of IL-10.23 Conversely, i.t. anti-IL-10 did not have any effect on IL-1β-induced hyperalgesia in LysM-GRK2+/− mice (Supplementary Figure 7), whereas anti-IL-10 treatment delayed resolution of inflammatory hyperalgesia in WT mice (Figs 3A and 3B).
Figure 6.

Role of IL-10 in the regulation of IL-1β-induced hyperalgesia. Thermal sensitivity was determined over time after intraplantar IL-1β in LysM-GRK2+/− mice. (A) WT and LysM-GRK2+/− mice received an i.t. injection of .5 μg recombinant IL-10 (n = 8) or vehicle (n = 4) 6 hours after intraplantar IL-1β. (B) IL10−/− BMDM or WT BMDM (3.5 − 106 cells per mouse) were injected i.v. 10 minutes prior to intraplantar IL-1β in LysM-GRK2+/− mice (n = 6–8 per group). (C and D) Twenty-four hours after adoptive transfer of WT or IL-10−/− BMDM, lumbar dorsal horn spinal cord sections were stained for the M1-type microglia/macrophage activation marker CD16/32. (C) Quantification of the CD16/32 immunofluorescence (n = 4) and (D) representative sections of lumbar dorsal horn spinal cord stained for CD16/CD32. Scale bar indicates 40 μm. All data are expressed as mean ± SEM. *P < .05, ***P < .001.
Finally, we determined whether IL-10 production by transferred BMDM is required to normalize the prolonged hyperalgesic response in LysM-GRK2+/− mice. The data in Fig 6B show that transfer of IL-10−/− BMDM into LysM-GRK2+/− mice did not prevent the transition to persistent hyperalgesia induced by IL-1β, whereas transfer of the same number of WT BMDM completely prevented the transition to persistent hyperalgesia in LysM-GRK2+/− mice (Fig 4A). These results indicate that IL-10-producing BMDM promote resolution of transient inflammatory hyperalgesia.
We previously reported that the persistent IL-1βinduced hyperalgesia in LysM-GRK2+/− mice is associated with a proinflammatory M1 microglia/macrophage phenotype in the spinal cord.43 Here we show that transfer of WT, but not of IL-10−/−, monocytes reduces expression of the M1 marker CD16/32 in the spinal cord of intraplantar IL1β-treated LysM-GRK2+/− mice (Figs 6C and 6D).
Discussion
Here we present a so far unrecognized and crucial role for IL-10-producing peripheral monocytes/macrophages in the spontaneous resolution of transient inflammatory hyperalgesia. Specifically, depletion of peripheral monocytes/macrophages in WT mice causes transition from transient IL-1β- or carrageenan-induced hyperalgesia into persistent hyperalgesia. Moreover, we show here that i.t. administration of anti-IL-10 also caused a transition to persistent inflammatory hyperalgesia. The role of peripheral monocytes/macrophages and IL-10 was further substantiated in our recently described mouse model in which reduced GRK2 levels in LysM+ monocytes/macrophages (LysM-GRK2+/−) causes transition to persistent hyperalgesia after a mild inflammatory stimulus. Adoptive transfer of WT BMDM, but not of IL-10−/− BMDM, to LysM-GRK2+/− mice prevents and reverses the persistent hyperalgesia that develops in these mice. Collectively, our findings identify a crucial role for peripheral monocytes/macrophages with normal GRK2 levels, which produce IL-10 in the DRG to promote resolution of IL-1β-induced hyperalgesia.
We are the first to show that peripheral monocytes/macrophages are required for the spontaneous resolution of hyperalgesia in response to a transient inflammatory stimulus. We demonstrate that anti- CCR2- or clodronate-mediated peripheral monocyte/macrophage depletion prolongs hyperalgesia in response to 2 different transient peripheral inflammatory stimuli (intraplantar IL-1β and carrageenan). Although both depletion strategies significantly prolonged IL-1β induced hyperalgesia, the effect of anti-CCR2 was more pronounced than that of clodronate liposomes. Interestingly, the Ly6C+(GR1+)CCR2+ monocytes/macrophage subset that is depleted by the anti-CCR2 antibody is the subset known to preferably infiltrate into tissues and to have increased expression levels of anti-inflammatory cytokines like IL-10 and transforming growth factor beta.9,13,39 Therefore, we suggest that the more pronounced effect of anti-CCR2 is due to the depletion of the anti-inflammatory effect–producing Ly6C+CCR2+ subset of monocytes/macrophages as opposed to the more general depletion of all monocytes/macrophages when using clodronate liposomes. An alternative possibility would be that the depletion with clodronate liposomes is less efficient or that repopulation is faster after depletion with clodronate liposomes than after depletion with anti-CCR2. Overall, both data sets indicate that peripheral monocytes/macrophages are key to inflammatory hyperalgesia and thereby may contribute to preventing the transition to persistent pain.
The few earlier studies that used depletion strategies to determine the contribution of peripheral monocytes/macrophages reported controversial results. Rutkowski et al described that persistent nerve injury–induced mechanical allodynia was not altered by a single i.v. injection with clodronate liposomes prior to the induction of nerve injury.33 In contrast, Liu et al showed that i.v. injections of clodronate liposomes immediately after nerve injury and 2 days later alleviated ongoing nerve injury–induced allodynia and reduced the number of macrophages in the injured nerve.17 Development of diabetic neuropathy was delayed after macrophage depletion by repeated i.v. injections with clodronate liposomes.22 In contrast, persistent hyperalgesia after intraplantar injection with Freund’s adjuvant was not affected by clodronate liposome–mediated monocyte/macrophage depletion locally in the hindpaw.1 It should be noted that in all studies described above, the role of monocytes/macrophages in models of chronic pain was investigated, because these studies aimed at testing the hypothesis that monocytes/macrophages contribute to the generation or maintenance of chronic pain. This is in sharp contrast to our present study, in which we tested the hypothesis that monocytes/macrophages contribute to the resolution of pain and therefore employed a model of transient inflammatory hyperalgesia. The results of our study indicate that in the context of transient hyperalgesia induced by a single injection of a mild inflammatory stimulus such as IL-1β or carrageenan, systemic monocyte/macrophage depletion markedly prolongs hyperalgesia. We therefore suggest that monocytes/macrophages originating in the periphery are key to the resolution of transient hyperalgesia through regulation at the level of the DRG. In previous studies, we already showed that there is no increase in the expression of CX3CR1 mRNA or Iba-1+ cells in the paw of WT and LysM-GRK2+/− mice after intraplantar IL-1β.42 Moreover, we show here that there was no decrease in CX3CR1 expression in the paw after CCR2-treatment. These observations further support our hypothesis that the monocytes/macrophages regulate resolution of the pain response at the level of the DRG and/or spinal cord, and not via an effect in the paw.
In search for the pathway via which peripheral monocytes/macrophages promote resolution of transient inflammatory hyperalgesia, we focused on the antiinflammatory cytokine IL-10. Our data show that i.t. injection of anti-IL-10 in WT mice prolongs the duration of IL-1β and carrageenan hyperalgesia. In addition, local anti-IL-10 treatment in the paw did not influence IL-1-induced hyperalgesia, indicating that IL-10 signaling in the spinal cord or DRG is required for spontaneous resolution of hyperalgesia in these models of acute inflammatory pain. It has been shown before that i.t. treatment with IL-10 or local overexpression of IL-10 alleviates hyperalgesia in mouse and rat models of neuropathic pain.24 However, until now it was not known that IL-10 signaling, likely downstream of IL-10 production by peripheral monocytes/macrophages, is required for the spontaneous resolution of hyperalgesia in response to a short-lasting peripheral inflammatory stimulus.
We reported earlier that the transition from acute to persistent IL-1β-hyperalgesia in LysM-GRK2+/− mice is associated with persistent microglia/macrophage activation in the spinal cord with an M1 or proinflammatory phenotype. In addition, after intraplantar IL-1β injection, microglia/macrophages isolated from the spinal cord of LysM-GRK2+/− mice express significantly more proinflammatory (M1) cytokines and less anti-inflammatory (M2) cytokines compared to cells from WT mice.43 Here we show that in vitro, GRK2-deficient monocytes/macrophages produce less of the anti-inflammatory M2-type cytokine IL-10 and increased levels of the proinflammatory M1-type cytokines IL-1β and TNF-α. Moreover, we show that transfer of WT, but not IL-10-deficient, BMDM promotes resolution of hyperalgesia and reduces the expression of CD16/32, a marker of proinflammatory M1 microglia/macrophage polarization in LysM-GRK2+/− spinal cord. Collectively these findings indicate that the transition from acute to persistent hyperalgesia in LysM-GRK2+/− mice is caused by reduced capacity of GRK2-deficient peripheral macrophages to produce IL-10, which inhibits the M1-type spinal cord microglia/macrophage activity.
Transfer of GRK2-deficient BMDM to LysM-GRK2+/− mice transiently increased hyperalgesia, indicating that a further increase in the number of peripheral blood monocytes per se does not attenuate hyperalgesia. These findings further support the hypothesis that the cytokine profile of peripheral monocytes/macrophages determines whether these cells are capable of inhibiting the pain response.
In our previous studies, we showed that GRK2 levels of primary microglia cultures from LysM-GRK2+/− mice were reduced by approximately 50% compared to WT microglia.29,42 However, in the present study, we show that the level of GRK2 protein or mRNA in freshly isolated naive microglia or in microglia isolated from the spinal cord of LysM-GRK2+/− mice at 24 hours after intraplantar IL-1β was not different from the level of GRK2 protein or mRNA in WT microglia. This finding is in line with the notion that the LysM promoter is not active in naive freshly isolated microglia but becomes active in these cells after in vitro stimulation.2,21 The fact that GRK2 is reduced in macrophages, but not in microglia, from naive LysM-GRK2+/− mice or from LysM-GRK2+/− mice after intraplantar IL-1β further supports the hypothesis that the peripheral monocytes/macrophage components are key for regulating the transition from acute to persistent pain.
Our current findings that IL-10-producing BMDM suppress spinal cord inflammatory activity and promote resolution of transient inflammatory hyperalgesia are reminiscent of earlier findings regarding the contribution of BMDM to injury and repair of the damaged spinal cord. In a model of spinal cord injury, Shechter et al demonstrated that peripheral monocytes infiltrate into the damaged central nervous system to regulate recovery. They also showed that adoptive transfer of WT BMDM, but not of IL-10-deficient BMDM, promoted recovery from spinal cord injury.36 The model of spinal cord injury is associated with damage to the blood-brain barrier, which will facilitate the infiltration of BMDM into the spinal cord. Notably, it is unlikely that the blood-brain barrier is damaged or more permeable in response to a single intraplantar injection of IL-1β. Yet, i.v. adoptive transfer of monocytes promotes resolution of hyperalgesia, and this effect can be mimicked by i.t. delivery of a much lower number of BMDM. However, even though the transfer of BMDM reduced the expression of proinflammatory microglia marker CD16/32 in the spinal cord, we did not detect infiltration of donor BMDM into the spinal cord, but only in the DRGs. Based on these results, we propose that the control of resolution of hyperalgesia by BMDM involves IL-10 signaling in the DRG resulting in suppression of nociceptor sensitization and reduction of microglial activation in the spinal cord. However, we cannot exclude that other differences between WT and IL10−/− BMDM, for example, in the capacity to produce resolvins11,12 or other anti-inflammatory mediators, may contribute as well. In addition, we cannot exclude that i.v. WT BMDM promotes resolution of hyperalgesia via an effect of transferred cells on the peripheral nerve.
Earlier studies have shown that chronic constriction injury in rats leads to a decrease in IL-10 protein in the DRG.15 In addition, overexpression of IL-10 in the spinal cord alleviates chronic hyperalgesia in models of chronic neuropathic pain.25,26 Successful treatment of neuropathic pain in rats is associated with an increase in IL-10 in the spinal cord.41 In line with these observations in animal models, an inverse correlation between pain intensity and plasma IL-10 has been reported in patients with chronic pain receiving opioid therapy. Moreover, plasma IL-10 levels were 2-fold higher in patients with painless compared to painful neuropathy.46 It remains to be determined whether a decrease in IL-10 production in patients with chronic pain is associated with reduced expression of GRK2 in peripheral monocytes/macrophages. Notably, we showed earlier that the painful autoimmune diseases rheumatoid arthritis and multiple sclerosis are associated with a decrease in the level of GRK2 in peripheral blood mononuclear cells as compared to healthy individuals. 18,38 Moreover, analysis of cytokine profiles in these patients showed increased proinflammatory cytokines such as TNF-α and decreased levels of antiinflammatory cytokines such as IL-10 and transforming growth factor beta.4,28,44
In conclusion, we propose that IL-10-producing peripheral monocytes/macrophages are key to the resolution of transient hyperalgesia and thereby may contribute to preventing the transition from acute to persistent inflammatory pain. We also propose that GRK2 deficiency in peripheral monocytes/macrophages is sufficient to promote transition from acute to persistent pain because of the reduced capacity of these cells to produce IL-10. Future studies should elucidate whether low GRK2 expression and/or impaired IL-10 production by peripheral monocytes/macrophages represent biomarkers for the risk of developing persistent pain after inflammation.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This study was supported by NIH grants RO1 NS 073939 and RO1 NS 074999 and by a STARS award from the University of Texas System.
Footnotes
The authors declare no competing financial interests.
Supplementary data related with this article can be found in the online version, at http://dx.doi.org/10.1016/j.jpain.2014.01.491.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brack A, Labuz D, Schiltz A, Rittner HL, Machelska H, Schafer M, Reszka R, Stein C. Tissue monocytes/macrophages in inflammation: Hyperalgesia versus opioid-mediated peripheral antinociception. Anesthesiology. 2004;101:204–211. doi: 10.1097/00000542-200407000-00031. [DOI] [PubMed] [Google Scholar]
- 2.Cho IH, Hong J, Suh EC, Kim JH, Lee H, Lee JE, Lee S, Kim CH, Kim DW, Jo EK, Lee KE, Karin M, Lee SJ. Role of microglial IKKbeta in kainic acid-induced hippocampal neuronal cell death. Brain. 2008;131(Pt 11):3019–3033. doi: 10.1093/brain/awn230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clark AK, Grist J, Al-Kashi A, Perretti M, Malcangio M. Spinal cathepsin S and fractalkine contribute to chronic pain in collagen induced arthritis. Arthritis Rheum. 2012;64:2038–2047. doi: 10.1002/art.34351. [DOI] [PubMed] [Google Scholar]
- 4.Correale J, Farez MF, Ysrraelit MC. Increase in multiple sclerosis activity after assisted reproduction technology. Ann Neurol. 2012;72:682–694. doi: 10.1002/ana.23745. [DOI] [PubMed] [Google Scholar]
- 5.De Leo JA, Tawfik VL, LaCroix-Fralish ML. The tetrapartite synapse: Path to CNS sensitization and chronic pain. Pain. 2006;122:17–21. doi: 10.1016/j.pain.2006.02.034. [DOI] [PubMed] [Google Scholar]
- 6.DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90:1–6. doi: 10.1016/s0304-3959(00)00490-5. [DOI] [PubMed] [Google Scholar]
- 7.Echeverry S, Shi XQ, Rivest S, Zhang J. Peripheral nerve injury alters blood-spinal cord barrier functional and molecular integrity through a selective inflammatory pathway. J Neurosci. 2011;31:10819–10828. doi: 10.1523/JNEUROSCI.1642-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eijkelkamp N, Heijnen CJ, Willemen HL, Deumens R, Joosten EA, Kleibeuker W, den HI, van Velthoven CT, Nijboer C, Nassar MA, Dorn GW, Wood JN, Kavelaars A. GRK2: A novel cell-specific regulator of severity and duration of inflammatory pain. J Neurosci. 2010;30:2138–2149. doi: 10.1523/JNEUROSCI.5752-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fukuda S, Nagano M, Yamashita T, Kimura K, Tsuboi I, Salazar G, Ueno S, Kondo M, Kunath T, Oshika T, Ohneda O. Functional endothelial progenitor cells selectively recruit neurovascular protective monocyte-derived F4/80(+)/Ly6c(+) macrophages in a mouse model of retinal degeneration. Stem Cells. 2013;31:2149–2161. doi: 10.1002/stem.1469. [DOI] [PubMed] [Google Scholar]
- 10.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 11.Hong S, Lu Y. Omega-3 fatty acid-derived resolvins and protectins in inflammation resolution and leukocyte functions: Targeting novel lipid mediator pathways in mitigation of acute kidney injury. Front Immunol. 2013;4:13. doi: 10.3389/fimmu.2013.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsiao HM, Sapinoro RE, Thatcher TH, Croasdell A, Levy EP, Fulton RA, Olsen KC, Pollock SJ, Serhan CN, Phipps RP, Sime PJ. A novel anti-inflammatory and pro-resolving role for resolvin D1 in acute cigarette smoke-induced lung inflammation. PLoS One. 2013;8:e58258. doi: 10.1371/journal.pone.0058258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, Divino CM, Chen SH. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66:1123–1131. doi: 10.1158/0008-5472.CAN-05-1299. [DOI] [PubMed] [Google Scholar]
- 14.Institute of Medicine. [Accessed March 11, 2014];Relieving pain in America: A blueprint for transforming prevention, care, education, and research. Available at: http://www.iom.edu/relievingpain.
- 15.Jancalek R, Svizenska I, Klusakova I, Dubovy P. Bilateral changes of IL-10 protein in lumbar and cervical dorsal root ganglia following proximal and distal chronic constriction injury of peripheral nerve. Neurosci Lett. 2011;501:86–91. doi: 10.1016/j.neulet.2011.06.052. [DOI] [PubMed] [Google Scholar]
- 16.Ledeboer A, Jekich BM, Sloane EM, Mahoney JH, Langer SJ, Milligan ED, Martin D, Maier SF, Johnson KW, Leinwand LA, Chavez RA, Watkins LR. Intrathecal interleukin-10 gene therapy attenuates paclitaxel-induced mechanical allodynia and proinflammatory cytokine expression in dorsal root ganglia in rats. Brain Behav Immun. 2007;21:686–698. doi: 10.1016/j.bbi.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu T, van RN, Tracey DJ. Depletion of macrophages reduces axonal degeneration and hyperalgesia following nerve injury. Pain. 2000;86:25–32. doi: 10.1016/s0304-3959(99)00306-1. [DOI] [PubMed] [Google Scholar]
- 18.Lombardi MS, Kavelaars A, Schedlowski M, Bijlsma JW, Okihara KL, Van de PM, Ochsmann S, Pawlak C, Schmidt RE, Heijnen CJ. Decreased expression and activity of G-protein-coupled receptor kinases in peripheral blood mononuclear cells of patients with rheumatoid arthritis. FASEB J. 1999;13:715–725. doi: 10.1096/fasebj.13.6.715. [DOI] [PubMed] [Google Scholar]
- 19.Loram LC, Harrison JA, Sloane EM, Hutchinson MR, Sholar P, Taylor FR, Berkelhammer D, Coats BD, Poole S, Milligan ED, Maier SF, Rieger J, Watkins LR. Enduring reversal of neuropathic pain by a single intrathecal injection of adenosine 2A receptor agonists: A novel therapy for neuropathic pain. J Neurosci. 2009;29:14015–14025. doi: 10.1523/JNEUROSCI.3447-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mack M, Cihak J, Simonis C, Luckow B, Proudfoot AE, Plachy J, Bruhl H, Frink M, Anders HJ, Vielhauer V, Pfirstinger J, Stangassinger M, Schlondorff D. Expression and characterization of the chemokine receptors CCR2 and CCR5 in mice. J Immunol. 2001;166:4697–4704. doi: 10.4049/jimmunol.166.7.4697. [DOI] [PubMed] [Google Scholar]
- 21.Mawhinney LA, Thawer SG, Lu WY, Rooijen N, Weaver LC, Brown A, Dekaban GA. Differential detection and distribution of microglial and hematogenous macrophage populations in the injured spinal cord of lys-EGFP-ki transgenic mice. J Neuropathol Exp Neurol. 2012;71:180–197. doi: 10.1097/NEN.0b013e3182479b41. [DOI] [PubMed] [Google Scholar]
- 22.Mert T, Gunay I, Ocal I, Guzel AI, Inal TC, Sencar L, Polat S. Macrophage depletion delays progression of neuropathic pain in diabetic animals. Naunyn Schmiedebergs Arch Pharmacol. 2009;379:445–452. doi: 10.1007/s00210-008-0387-3. [DOI] [PubMed] [Google Scholar]
- 23.Mildner A, Mack M, Schmidt H, Bruck W, Djukic M, Zabel MD, Hille A, Priller J, Prinz M. CCR2+Ly-6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain. 2009;132(Pt 9):2487–2500. doi: 10.1093/brain/awp144. [DOI] [PubMed] [Google Scholar]
- 24.Milligan ED, Langer SJ, Sloane EM, He L, Wieseler-Frank J, O’Connor K, Martin D, Forsayeth JR, Maier SF, Johnson K, Chavez RA, Leinwand LA, Watkins LR. Controlling pathological pain by adenovirally driven spinal production of the anti-inflammatory cytokine, interleukin-10. Eur J Neurosci. 2005;21:2136–2148. doi: 10.1111/j.1460-9568.2005.04057.x. [DOI] [PubMed] [Google Scholar]
- 25.Milligan ED, Penzkover KR, Soderquist RG, Mahoney MJ. Spinal interleukin-10 therapy to treat peripheral neuropathic pain. Neuromodulation. 2012;15:520–526. doi: 10.1111/j.1525-1403.2012.00462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Milligan ED, Soderquist RG, Malone SM, Mahoney JH, Hughes TS, Langer SJ, Sloane EM, Maier SF, Leinwand LA, Watkins LR, Mahoney MJ. Intrathecal polymer-based interleukin- 10 gene delivery for neuropathic pain. Neuron Glia Biol. 2006;2:293–308. doi: 10.1017/S1740925X07000488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009;10:23–36. doi: 10.1038/nrn2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moelants EA, Mortier A, Grauwen K, Ronsse I, Van DJ, Proost P. Citrullination of TNF-alpha by peptidylarginine deiminases reduces its capacity to stimulate the production of inflammatory chemokines. Cytokine. 2013;61:161–167. doi: 10.1016/j.cyto.2012.09.011. [DOI] [PubMed] [Google Scholar]
- 29.Nijboer CH, Heijnen CJ, Willemen HL, Groenendaal F, Dorn GW, van BF, Kavelaars A. Cell-specific roles of GRK2 in onset and severity of hypoxic-ischemic brain damage in neonatal mice. Brain Behav Immun. 2010;24:420–426. doi: 10.1016/j.bbi.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nikolic T, Geutskens SB, van RN, Drexhage HA, Leenen PJ. Dendritic cells and macrophages are essential for the retention of lymphocytes in (peri)-insulitis of the nonobese diabetic mouse: A phagocyte depletion study. Lab Invest. 2005;85:487–501. doi: 10.1038/labinvest.3700238. [DOI] [PubMed] [Google Scholar]
- 31.Peregrin S, Jurado-Pueyo M, Campos PM, Sanz-Moreno V, Ruiz-Gomez A, Crespo P, Mayor F, Jr, Murga C. Phosphorylation of p38 by GRK2 at the docking groove unveils a novel mechanism for inactivating p38MAPK. Curr Biol. 2006;16:2042–2047. doi: 10.1016/j.cub.2006.08.083. [DOI] [PubMed] [Google Scholar]
- 32.Ren K, Dubner R. Interactions between the immune and nervous systems in pain. Nat Med. 2010;16:1267–1276. doi: 10.1038/nm.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rutkowski MD, Pahl JL, Sweitzer S, van RN, DeLeo JA. Limited role of macrophages in generation of nerve injury-induced mechanical allodynia. Physiol Behav. 2000;71:225–235. doi: 10.1016/s0031-9384(00)00333-4. [DOI] [PubMed] [Google Scholar]
- 34.Saeij JP, Wiegertjes GF, Stet RJ. Identification and characterization of a fish natural resistance-associated macrophage protein (NRAMP) cDNA. Immunogenetics. 1999;50:60–66. doi: 10.1007/s002510050686. [DOI] [PubMed] [Google Scholar]
- 35.Scholz J, Woolf CJ. The neuropathic pain triad: Neurons, immune cells and glia. Nat Neurosci. 2007;10:1361–1368. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- 36.Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G, Rolls A, Mack M, Pluchino S, Martino G, Jung S, Schwartz M. Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med. 2009;6:e1000113. doi: 10.1371/journal.pmed.1000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: Mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174:83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 38.Vroon A, Kavelaars A, Limmroth V, Lombardi MS, Goebel MU, Van Dam AM, Caron MG, Schedlowski M, Heijnen CJ. G protein-coupled receptor kinase 2 in multiple sclerosis and experimental autoimmune encephalomyelitis. J Immunol. 2005;174:4400–4406. doi: 10.4049/jimmunol.174.7.4400. [DOI] [PubMed] [Google Scholar]
- 39.Waddell A, Ahrens R, Steinbrecher K, Donovan B, Rothenberg ME, Munitz A, Hogan SP. Colonic eosinophilic inflammation in experimental colitis is mediated by Ly6C(high) CCR2(+) inflammatory monocyte/macrophage-derived CCL11. J Immunol. 2011;186:5993–6003. doi: 10.4049/jimmunol.1003844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watkins LR, Hutchinson MR, Ledeboer A, Wieseler- Frank J, Milligan ED, Maier SF. Norman Cousins Lecture. Glia as the “bad guys”: Implications for improving clinical pain control and the clinical utility of opioids. Brain Behav Immun. 2007;21:131–146. doi: 10.1016/j.bbi.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilkerson JL, Gentry KR, Dengler EC, Wallace JA, Kerwin AA, Armijo LM, Kuhn MN, Thakur GA, Makriyannis A, Milligan ED. Intrathecal cannabilactone CB(2)R agonist, AM1710, controls pathological pain and restores basal cytokine levels. Pain. 2012;153:1091–1106. doi: 10.1016/j.pain.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Willemen HL, Eijkelkamp N, Wang H, Dantzer R, Dorn GW, Kelley KW, Heijnen CJ, Kavelaars A. Microglial/macrophage GRK2 determines duration of peripheral IL-1beta-induced hyperalgesia: Contribution of spinal cord CX3CR1, p38 and IL-1 signaling. Pain. 2010;150:550–560. doi: 10.1016/j.pain.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Willemen HL, Huo XJ, Mao-Ying QL, Zijlstra J, Heijnen CJ, Kavelaars A. MicroRNA-124 as a novel treatment for persistent hyperalgesia. J Neuroinflammation. 2012;9:143. doi: 10.1186/1742-2094-9-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yudoh K, Matsuno H, Nakazawa F, Yonezawa T, Kimura T. Reduced expression of the regulatory CD4+ T cell subset is related to Th1/Th2 balance and disease severity in rheumatoid arthritis. Arthritis Rheum. 2000;43:617–627. doi: 10.1002/1529-0131(200003)43:3<617::AID-ANR19>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 45.Zheng W, Ouyang H, Zheng X, Liu S, Mata M, Fink DJ, Hao S. Glial TNFalpha in the spinal cord regulates neuropathic pain induced by HIV gp120 application in rats. Mol Pain. 2011;7:40. doi: 10.1186/1744-8069-7-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zin CS, Nissen LM, O’Callaghan JP, Moore BJ, Smith MT. Preliminary study of the plasma and cerebrospinal fluid concentrations of IL-6 and IL-10 in patients with chronic pain receiving intrathecal opioid infusions by chronically implanted pump for pain management. Pain Med. 2010;11:550–561. doi: 10.1111/j.1526-4637.2010.00821.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.