

Abstract
Peroxidation of membranes and lipoproteins converts “inert” phospholipids into a plethora of oxidatively modified phospholipids (oxPL) that can act as signaling molecules. In this review, we will discuss four major classes of oxPL: mildly oxygenated phospholipids, phospholipids with oxidatively truncated acyl chains, phospholipids with cyclized acyl chains, and phospholipids that have been oxidatively N-modified on their headgroups by reactive lipid species. For each class of oxPL we will review the chemical mechanisms of their formation, the evidence for their formation in biological samples, the biological activities and signaling pathways associated with them, and the catabolic pathways for their elimination. We will end by briefly highlighting some of the critical questions that remain about the role of oxPL in physiology and disease.
Keywords: Lipid peroxidation, Oxidized phospholipids, Platelet-Activating Factor Acetyl Hydrolase, CD36, Toll-Like Receptors, Lipid aldehydes
1. Introduction
A number of excellent reviews on various aspects of oxidatively modified phospholipids (oxPL) have been previously published (Aldrovandi and O’Donnell, 2013; Bochkov et al., 2010; Salomon, 2012; Spickett and Dever, 2005). However, the field of oxPL continues to rapidly evolve and new signaling pathways and new mechanism of action for oxPL have been discovered. We will especially highlight the newest class of oxPL, the oxidatively N-modified phospholipids. Our review will primarily focus on studies where authentic synthetic compounds have been used to elucidate the biological effects of oxPL, rather than studies using uncharacterized mixtures of oxidized compounds such as oxidized 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphocholine (oxPAPC). Our feeling is that rigorous elucidation of signaling pathways requires authentic, synthetized oxPL and structure-activity relationship studies rather than undefined mixtures where overlapping activities confound the results.
1.1 Nomenclature for oxPL
Before proceeding, it may also be worthwhile to explain our use of nomenclature and abbreviations in this review. Use of the full IUPAC name for lipids is always unbearably cumbersome and different authors have used a wide variety of trivial names and abbreviations to label various oxPL in their work. Any label must convey sufficient structural detail about the oxPL for the reader to easily make comparisons with other oxPL while remaining as simple to remember as possible. In mammalian cells, glycerophospholipids including phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), and phosphatidylinositol (PI) generally carry polyunsaturated fatty acids (PUFA) esterified at the sn-2 position and saturated or monounsaturated fatty acids esterified at the the sn-1 position. Thus, because generally only the sn-2 groups is oxidatively modified, we will abbreviate oxPL products using only their modified sn-2 group hyphenated to the parent phospholipid from which it derives (e.g. 1-acyl-2-(15-hydroxyeicosatetraenoate)-sn-glycero-3-phosphoethanolamine will be abbreviated as 15-HETE-PE). For consistency, we will do this even in the case of oxPL commonly abbreviated with their sn-1 palmitate moiety (e.g. we will use OV-PC to designate 1-palmitoyl-2-(5-oxovaleroyl)- sn-glycero-3-phosphocholine rather than POVPC). We note that most structure activity relationship studies on oxPL have only utilized palmitate at the sn-1 position, yet those that have explored effects of changing the sn-1 group from palmitate to other common acyl chains such as oleate or stearate typically find little differences, so that this simplification seems justified. The exception to sn-1 moieties are alkylacyl phospholipids, so we will abbreviate alkylacyl PC as platelet-activating factor analogs (e.g. 1-O-alkyl-2- butanoyl -sn-glycero-phosphocholine will be referred to as butanoyl-PAF), and plasmalogens (alkenylacyl PE) as pPE (e.g. HETE-pPE). A significant part of this review focuses on the newest class of oxPL, the oxidatively N-modified phospholipids. To avoid confusion with sn-2 modifications, these will be abbreviated with the letter N proceeding the aldehyde modifying the phospholipid headgroup. Therefore, intact PE where its headgroup has been N-modified by isolevuglandin (IsoLG) will be designated as N-IsoLG-PE, while arachidonyl-PE where its sn-2 O-acyl chain has undergone oxygenation and cyclization to form IsoLG will be designated as IsoLG-PE.
1.2 Initiation of peroxidation and formation of four classes of oxPL
Peroxidation of PUFA can occur both enzymatically (e.g. by lipoxygenases) or non-enzymatically via reactive oxygen species (e.g. hydroxyl radicals) generated by oxidases such as myeloperoxidase, NADPH oxidases, or xanthine oxidases or by environmental sources such as cigarette smoke, pollution, radiation, or UV exposure. Whatever the source of initiating radical, lipid peroxidation occurs via abstraction of a bis-allylic hydrogen from a PUFA by a radical species and then subsequent reaction of molecular oxygen with the resulting carbon-centered lipid radical to form a lipid peroxyl radical (Fig 1). Enzyme-catalyzed peroxidation typically generates only a single enantiomer of one specific regioisomer of lipid peroxide. For instance, peroxidation of arachidonyl-PC by 15-lipoxygenase (15-LOX) generates 15S-hydroperoxyeicosatetraenoate-PC (15S-HPETE-PC). In contrast, non-enzymatic peroxidation generates racemic mixtures with multiple regio- and stereo-isomers. Thus, non-enzymatic peroxidation of arachidonyl-PC generates both the R- and S- enantiomers of 5-HPETE-PC, 8-HPETE-PC, 9-HPETE-PC, 11-HPETE-PC, 12-HPETE-PC, and 15-HPETE-PC (Fig 1). Non-enzymatic peroxidation of linoleoyl-PC generates both the R- and S- enantiomers of 9-HPODE-PE and 13-HPODE-PC (Fig 2).
Figure 1.

Peroxidation of arachidonyl-PC forms six phospholipid hydroperoxide regioisomers. Hydrogen abstraction is the first step of lipid peroxidation and this readily occurs at bis-allylic hydrogens of 1,4-pentadiene groups because the resulting radical is in resonance across all five carbons of the pentadiene group. There are three pairs of bis-allylic hydrogens in arachidonyl-PC (labeled a, b, c) and the regioisomer of hydroperoxy-eicosatetraenoyl-PC (HPETE-PC) formed depends on the position of the initial hydrogen abstraction. Molecular oxygen, which is a diradical, reacts with the lipid radical to form a phospholipid peroxyl radical. In the absence of local environmental factors, addition of oxygen is equally favored at either the 1 or 5 positions of the pentadiene group so that yields of the two regioisomers are similar. Subsequent hydrogen abstraction (often from an adjacent PUFA) gives the PL-OOH and propagates the radical reaction to neighboring phospholipids.
Figure 2.

Peroxidation of linoleoyl-PC. Linoleoyl-PC has only one pair of bis-allylic hydrogens so that hydrogen abstraction at this position (labeled a) yields two regioisomers of phospholipid hydroperoxides.
Each of these phospholipid hydroperoxides (PL-OOH) can go on to form a wide range of secondary products with the yield of each of these secondary products depending on variables within the local environment such as other lipid species present, the redox status of the cells or surrounding tissue, and the availability of hydrogen donors such as vitamin E. Given the number of oxPL species that can be generated, it is helpful to group them based on structural similarities. For the purposes of this review, we will group oxPL into four major classes: i) mildly oxygenated oxPL (e.g. 15-HETE-PE), ii) oxPL with oxidatively truncated sn-2 acyl chains (e.g. OV-PC), iii) oxPL with cyclized acyl chains (e.g. 15-F2-IsoP-PE), and the newly discovered class of oxidatively N-modified phospholipids (Fig 3). This fourth class of oxPL forms when reactive lipid species, including those from the first three classes of oxPL, react with the primary amines found in the head groups of PE, pPE, and PS. For each class of oxPL we will examine the biochemical pathway of their formation, evidence for their formation in biological samples, their biological activities and signaling pathways, and their metabolism. As will be seen, biological activities of one oxPL class can overlap with other classes. In some cases, sufficient structure-activity relationship studies have been carried out to understand how this occurs, but often these sorts of studies are still lacking.
Figure 3.

Peroxidation of membrane phospholipids generate four classes of oxidatively modified phospholipids (oxPL). Representative examples from peroxidation of 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphocholine (PAPC) and 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphoethanolamine (PAPE) are shown. Class I oxPL include all mildly oxygenated phospholipids such as 15-HETE-PE and 14,15-EET-PC. Class II oxPL include all truncated oxPL such as OV-PC and KOdiA-PC. Class III oxPL include cyclized oxPL such as 15-F2-IsoP-PE and EI-PC. Class IV include oxidatively N-modified PL such as N-IsoLG-PE and N-C4:0CA-
2. Class I: Mildly oxygenated oxPL
2.1 Chemical mechanisms for formation of mildly oxygenated oxPL
Addition of oxygen to PUFA generates peroxyl-, hydroxyl-, keto-, and epoxy-fatty acids. The most stable of these are the hydroxyl fatty acids, with hydroxylated arachidonate being designated as hydroxyeicosatetraenoate (HETE) and hydroxylated linoleate being designated as hydroxyoctadecaenoate (HODE). Formation of mildly oxygenated oxPL occurs via three mechanisms in vivo (Fig 4): i) direct enzymatic oxygenation of the phospholipid via lipoxygenases, ii) enzymatic oxygenation of a free fatty acid by lipoxygenases or cytochrome P450 enzymes with subsequent esterified into a lysophospholipids, and iii) non-enzymatic oxygenation of the phospholipid via reactive oxygen species. The first two mechanisms generate specific stereoisomers of mildly oxygenated phospholipids, while the third generates a series of stereo- and regio-isomers of these same mildly oxygenated phospholipids.
Figure 4.
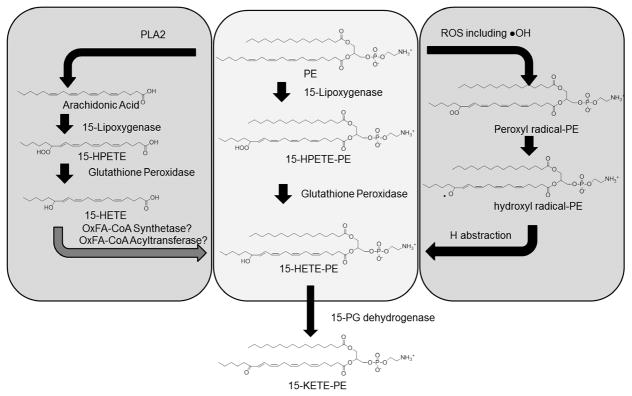
Mildly oxygenated phospholipids can form by three general mechanisms: direct enzymatic oxygenation of the phospholipid (center mechanism), indirect enzymatic oxygenation of PUFA with subsequent re-esterification (left panel), or non-enzymatic peroxidation via free radicals (right panel). Direct enzymatic peroxidation of phospholipid (PE is shown in this example) can be performed by lipoxygenases such as 15-lipoxygenase to generate HPETE-PL which are subsequent reduced by glutathione peroxidase to HETE-PL. The HETE-PL can then be converted to KETE-PL by 15-prostaglandin dehydrogenase. Alternatively, 15-lipoxygenase can act on unesterified fatty acid to generate HPETE, which is then reduced by glutathione peroxidase to its HETE, and this is subsequently esterified into phospholipid by currently unknown oxidized fatty acid synthetase and acyltransferase. These same reactions can take place non-enzymatically, with hydroxyl radicals generated as byproducts of oxidase being particularly good at peroxidation of phospholipids.
Many lipoxygenases (LOX), including human 15-LOX, its murine equivalent 12/15-LOX, and soybean 15- LOX can directly utilize phospholipids including PE and plasmalogens (pPE) as substrates, thus generating the phospholipid with hydroxylated fatty acids at the sn-2 position(PL-OOH) (Maskrey et al., 2007; Morgan et al., 2009). Glutathione peroxidase is then hypothesized to convert the peroxyl group to a hydroxyl group (PL-OH) forming HETE-PE and HODE-PE (Aldrovandi and O’Donnell, 2013). Treatment of IL-4 induced human monocytes with ionophore activates 15-LOX activity and generates four major species of 15S-HETE modified phospholipids: three 15-HETE pPE and one 15-HETE-PE (Maskrey et al., 2007). Less than 5% of the hydroxylated phospholipid was 15-HETE-PC, and no significant amounts of oxygenated PG, PS, or PI were found. Activation of human bronchial epithelial cells with IL-13 also leads to expression of 15-LOX and increased levels of 15-HETE-PE (Zhao et al., 2009).
How mammalian 15-LOX selectively oxygenates PE species within cells is unknown but this finding suggests an evolutionarily optimized signaling pathway, rather than non-specific oxygenation. That 15-HETE-PE forms in human monocytes via direct oxygenation of PE by 15-LOX was shown by the absence of [18O]15-HPETE-PE when monocytes were activated in the presence of H218O (Maskrey et al., 2007). If re-esterification of 15-HETE was a major pathway in these cells, 50% of the 15-HETE-PE formed would have been [18O] labeled, as free arachidonate formed by PLA2 in the presence of H218O has one of the two carboxyl oxygens [18O] labeled, so that subsequent oxygenation would generate [18O]15-HETE and then half of any re-esterified 15-HETE would still contain this [18O] label. Furthermore, adding exogenous [18O]15-HETE did not result in the formation of [18O]15-HETE-PE in monocytes. Of note, murine macrophages express 12/15-LOX and 12-HETE-PE was generated after activation of wild-type murine macrophages with calcium ionophore, but not after activation of macrophages derived from 12/15-LOX null mice (Morgan et al., 2009). The 12-HETE-PE was also shown not to contain [18O] label, demonstrating that the 12/15-LOX of mice also directly oxygenated PE.
After formation of HETE-PE, the enzyme 15-prostaglandin dehydrogenase converts these products to their keto-analogs (15-KETE-PE) (Hammond et al., 2012). Thus, ionophore stimulated human monocytes form the four 15-KETE-PE species analogous to their four 15-HETE-PE species and murine macrophages form the12-KETE-PE species analogous to their 12-HETE-PE species. KETE-PE readily react with nucleophiles such as thiol moiety of glutathione.
In contrast to the 15-HETE-PE and 12-HETE-PE formed in monocyte/macrophages by direct oxygenation of PE species, 12-HETE-PE and 12-HETE-PC formed in platelets activated by thrombin appear to form from the re-esterification of 12-HETE into these phospholipids (Thomas et al., 2010). Thus, activation of platelets in the presence of H218O leads to 40–50% of 12-HETE-PE and 12-HETE-PC carrying the 18O label. Furthermore, recombinant platelet 12-LOX is unable to oxidize PE. However, addition of exogenous 12-HETE to platelets does not result in esterification of the 12-HETE, suggesting there is very tight coupling between AA oxygenation and re-esterification. The enzyme(s) responsible for HETE re-esterification have not yet been identified, but presumably include a fatty acid CoA synthetase that generates HETE-CoA and then an acyltransferase that links the HETE-CoA to lysophospholipids.
Cytochrome P450 monoxygenases (e.g. CYP2C and CYP2J) can also add a single oxygen to specific double bonds of arachidonic acid to form four regioisomers of epoxy eicosatetraenoic acids (EET) (Spector and Norris, 2007) (Fig 5). Epoxy-fatty acid are rapidly converted to dihydroxy-fatty acids (e.g. diHETE) by the action of soluble epoxide hydrolase. 90% of EET circulating in plasma are esterified to phospholipids including PC, PE, and PI (Karara et al., 1992). Chiral chromatography of these phospholipid esterified EET demonstrated that 68–84% of each EET regioisomer was of a single enantiomer, suggesting enzymatic oxygenation. EET are preferentially incorporated into PE > PC > PI > PS (Bernstrom et al., 1992). Rat liver homogenates failed to directly oxygenate phospholipids, but rapidly incorporated radiolabeled free EET into CoA derivatives and then into lysophospholipids (Karara et al., 1992). The acyl CoA synthase and O-acyl transferase responsible for the esterification of EET has not yet been identified.
Figure 5.
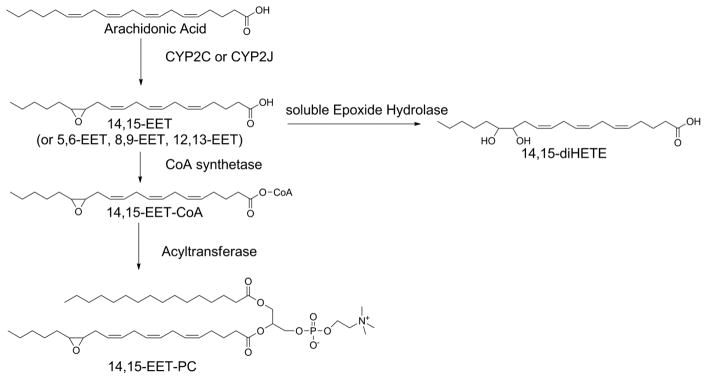
Formation of epoxyeicosanoid (EET) esterified phospholipids by the action of cytochrome P450s.
While PL-OOH are the primary products of lipid peroxidation by non-enzymatic mechanisms, PL-OOH are converted to alkoxyl radicals in the presence of reducing metals such as iron. These alkoxyl radicals then either abstract an hydrogen from an adjacent donor to form hydroxyl-fatty acids or react with their own double bonds to form epoxy-fatty acids. For instance, exposure of red blood cells to tert-butylhydroperoxide (tBuOOH) generates HETE-PE, HETE-PC, and HETE-PS, along with EET-PE, EET-PC, and EET-PS(Nakamura et al., 1997). Incubation of PC containing-liposome with xanthine oxidase and acetaldehyde (to generate hydrogen peroxide) leads to extensive formation of HPETE-PC and HETE-PC (measured as methyl esters after methanolic saponification) (Porter and Lehman, 1982). Interestingly, oxidation of red blood cells with xanthine oxidase/hypoxanthine/Fe3+ (which primarily generates •OH radicals) resulted in extensive fragmentation of the phospholipids rather than mildly oxygenated phospholipids (Kawai et al., 1999). Incubation of plasma with activated neutrophils (to release myeloperoxidase) leads to extensive formation of HETE- and HODE-containing phospholipids, while neutrophil from patients with defective myeloperoxidase does not (Zhang et al., 2002).
2.2 Formation of mildly oxygenated oxPL in biological samples
Although increased lipid peroxidation has been demonstrated in many disease states, there are very few studies that have specifically measure mildly oxygenated oxPL in vivo. The most commonly used methods such as xylenol orange for lipid peroxides or base hydrolysis prior to mass spectral analysis for HETE and HODE do not distinguish between oxygenated PUFA esterified to phospholipids versus those esterified to cholesterol or triglycerides. For this reason, while studies have shown increased levels plasma of HETE, HODE, or EET in non-alcoholic fatty liver disease (Feldstein et al., 2010), coronary artery disease (Shishehbor et al., 2006), and asthma (Chu et al., 2002; Zhao et al., 2009), and these increases are likely the result of increases in mildly oxygenated phospholipids, this cannot be stated with absolute certainty.
However, some studies have directly monitored specific oxygenated phospholipids by mass spectrometry or have separated lipids into specific phospholipid classes and then performed base hydrolysis to identify specific oxygenated fatty acids present. For instance, phospholipid esterified EET are found in endothelium, kidney, liver, and plasma lipoproteins (Spector and Norris, 2007) and these esterified EET are rapidly released upon exposure to agonist (Spector et al., 2004). More recently, thioglycollate challenge of mice showed that levels of HETE-PE levels in peritoneal exudates increased nearly 10-fold by 7 days after challenge, in contrast with 15-LOX null mice that had barely detectable levels of HETE-PE (Uderhardt et al., 2012). 15-KETE-PE levels are significantly increased in the bronchoalveolar lavage fluid of patients with cystic fibrosis (Hammond et al., 2012). Future studies using these mass spectrometry methods will be helpful in accessing the conditions where mildly oxygenated versus highly oxidized oxPL are formed, their relative concentrations, and the underlying processes that lead to changes in their levels.
2.3 Molecular targets, signaling pathways, and biological activities of mildly oxygenated oxPL
2.3.1 Regulation of immunogenic response to apoptotic cells
HETE-PE is a key regulator of immunogenic responses to apoptotic cells because it binds to a critically important cell adhesion protein, milk fat globule-EGF factor 8 protein (MFG-E8). MFG-E8 facilitates the interaction of phagocytes with apoptotic cells by interacting with these two different cell types via its two distinct binding domains and thereby acting as a bridge between these cells. One domain of MFG-E8 binds to PS (which is usually displayed on the outer leaflet of plasma membrane of cells only during apoptosis), while the other domain binds to αvβ3 and αvβ5 integrins on phagocytes (Aziz et al., 2011). Uderhardt et al showed that PE oxygenated by 15-LOX outcompetes PS for binding to MFG-E8 (Uderhardt et al., 2012). Because resident macrophages normally express 12/15-LOX and thus HETE-PE, they are able to sequester MFG-E8 away from non-resident, inflammatory monocytes. This ensures that phagocytosis of apoptotic cells is carried out by resident macrophages rather than by non-resident, inflammatory monocytes. This non-immunogenic disposal by resident macrophages maintains self-tolerance. Resident macrophages of mice lacking 12/15-LOX fail to take up apoptotic cells, which allows their uptake by inflammatory macrophage and leads to the development of autoimmune disease (Uderhardt et al., 2012).
Oxidized PS, consisting primarily of H(P)ETE-PS, was also reported to have better binding affinity for MFG-E8 than unoxygenated PS (Borisenko et al., 2004), suggesting that oxidized PS may also be an important MFG-E8 ligand. However, when Tyurin et al performed protein lipid overlay to identify receptors that interacted with oxidized PS, they found no enhancement of binding of MFG-E8 to oxidized PS compared to PS (Tyurin et al., 2014). Instead, they found that the receptors that bound with somewhat higher affinity to oxidized PS compared to PS included brain-specific angiogenesis inhibitor-1 and growth arrest specific-6. Of interest, they found that incubation of apoptotic cells with the secreted form of PAF-AH, which hydrolyzes a wide variety of oxPL species generated in apoptotic cells but not native PL species, suppressed phagocytosis of apoptotic cells by macrophages (Tyurin et al., 2014). This finding strongly implicates the participation of oxPL in the phagocytosis of apoptotic cells.
2.3.2 Activation of ERK signaling
The ERK signaling cascade is not active under basal conditions because the initiating kinase, Raf-1, is bound to PE binding protein 1 (PEBP1), also known as Raf-1 Kinase inhibitor protein. When released from PEBP1, Raf-1 can then phosphorylate its downstream substrates such as MEK-1 (Odabaei et al., 2004). 15-HETE-PE appears to activate ERK signaling by binding PEBP1, allowing the release of Raf-1 (Zhao et al., 2011). Activation of 15-LOX in human bronchial epithelial cells by treatment with IL-13 increases both 15-HETE-PE levels and ERK phosphorylation. Adding 15-HETE-PE is sufficient to displace Raf-1 from PEBP1 and to increase ERK phosphorylation. These findings suggest that activation of 15-LOX causes oxygenation of the PE bound to PEBP1, which leads to the release of Raf-1 from inhibition by PEBP1 (Zhao et al., 2011). The structure-activity relationship for oxPL binding to PEBP1 has not yet been fully explored, and merits further investigation. For instance, does activation of other lipoxygenases to form 12-HETE-PE or 5-HETE-PC also lead to PEBP1 binding and induction of ERK signaling or can other oxPL also bind to PEBP1?
The IL-13/LOX/PEBP1/ERK pathway may be relevant to IL-13 signaling in asthma. IL-13 induces the expression of mucins (MUC), particularly MUC5AC (Zhao et al., 2009). MUC5AC is one of the most abundant mucin expressed in airway epithelium, and is overexpressed in asthmatic epithelium (Morcillo and Cortijo, 2006; Thornton et al., 2008; Turner and Jones, 2009). 15-LOX expression levels in bronchial epithelium correlate with asthma severity (Zhao et al., 2009). Furthermore, inhibition of 15-LOX in human bronchial epithelial cells blocks synthesis of 15-HETE-PE in response to IL-13 and also the expression of MUC5AC. Addition of 15-HETE can partially restore MUC5AC expression during 15-LOX inhibition (Zhao et al., 2009). Whether 15-HETE-PE interactions with PEBP1 plays a direct role in IL-13 induced mucous expression and asthma needs further study.
2.3.3 Platelet aggregation
12-HETE-PE and 12-HETE-PC stimulate coagulation by facilitating the generation of thrombin. Cleavage of prothrombin requires formation of a negatively charged surface (previously thought to be primarily due to externalization of PS) that allows coagulation factors to bind. Interestingly, oxygenation of arachidonate by 12-LOX and esterification in PE to form 12-HETE-PE leads to its externalization in platelets (Thomas et al., 2010). Liposomes containing synthetic 12-HETE-PC (synthetic 12-HETE-PE was not available) dramatically increased thrombin generation when incubated with platelets. The mechanism underlying this increased production was not determined, nor whether 15-HETE-PC (or 15-HETE-PE) could also induce thrombin generation. Oxygenation of phospholipids might increase the total negative charge of the membrane because the presence of the hydroxyl group may force the acyl chain that is normally buried in the bilayer to rise to the aquaeous interface and in so doing, may force the negatively charged phosphate group to also be more exposed on the surface. More study on the effects of these mildly oxygenated phospholipids on membrane conformation and charge is clearly needed.
2.3.4. Inhibition of LPS signaling
At relatively high concentrations (IC50 ~30 uM), HETE-PC can inhibit LPS induced E-selectin expression. However, this activity appear to be far less potent than other oxPL generated by oxidation of arachidonyl-PE, -PC, -PG, or –PS, where less than 1 μM was required (von Schlieffen et al., 2009). Thus, it seems unlikely that this is an important activity of HETE-PC.
2.3.5. PPARγ
Both HETE-PE and its oxidized counterpart KETE-PE are weak agonists of PPARγ (Hammond et al., 2012), a property they share with many other lipids and lipid metabolites (Wahli and Michalik, 2012). Incubation with either 2.5 μM 15-KETE-PE or 15-HETE-PE induced PPRE reporter constructs and the expression of PPARγ regulated genes such as CD36 in monocyte/macrophage (Hammond et al., 2012). However, because 15-HETE is also a ligand for PPARγ, it is unclear if this is a direct effect of the oxygenated PE or if there is hydrolysis of the esterified HETE and KETE to generate the active unesterified form.
2.3.6. L-type calcium channel
Phospholipids containing EET potently inhibit the L-type calcium channel (Chen et al., 1999). The exact mechanism of this activity is not known, and all four EET regioisomers have similar effects, so that changes in membrane conformation have been posited to drive this inhibition (Spector and Norris, 2007).
2.4. Metabolism of mildly oxygenated oxPL
To some extent, the formation of HETE- and KETE-phospholipid by glutathione peroxidase and 15-prostaglandin dehydrogenase represents metabolism of HPETE-phospholipids. However, it is unclear what the final fates of these mildly oxygenated oxPL are. Presumably, they are cleaved from the phospholipid by PLA2, perhaps including platelet-activating factor acetylhydrolases (PAF-AH) and then undergo beta-oxidation either in the mitochondria or peroxisome. Given their apparent importance in regulating apoptosis and platelet activation, further elucidation of their metabolism seems critical.
3. Class II: Oxidatively truncated oxPL
3.1 Chemical mechanisms for formation of oxidatively truncated oxPL
Although PL-OOH are the primary products of lipid peroxidation, the generation of lipid peroxyl radicals readily allows a series of reactions that result in fragmentation of the acyl chain. This fragmentation typically generates two complementary products: a fragmented acyl chain that remains esterified to the phospholipid (a truncated oxPL) and a fragmented alkyl or alkenal chain that can diffuse (Fig 6). Both fragments are biologically important. This section will focus on the truncated phospholipid, while the section on N-oxidative modified PL will discuss what happens with the fragmented alkenal that modifies proteins, PE, and other primary amines.
Figure 6.
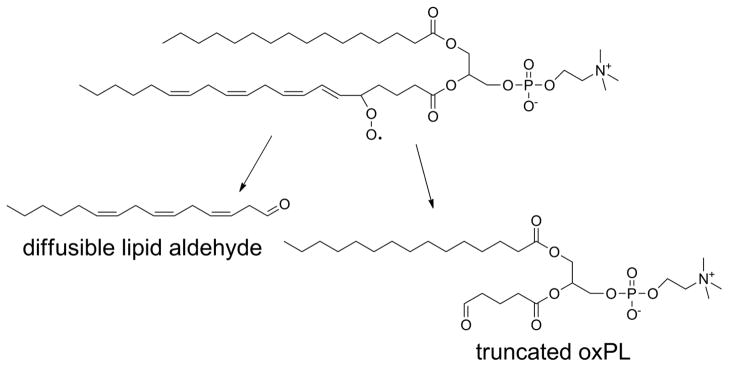
Oxidative fragmentation of PUFA esterified phospholipids generates two complementary fragments. The first fragment is a lipid aldehyde (or its oxidized or reduced analog), that is no longer attached to the phospholipid and is therefore free to diffuse into cytoplasm or extracellular space. The remaining fragment is a phospholipid now carrying a highly truncated sn-2 chain.
The most abundant forms of truncated oxPL generated by lipid peroxidation have shortened sn-2 acyl chains one carbon less in length than the first double bond in the parent, unoxidized lipid or shortened sn-2 acyl chain of the same length as the carbon of the first double bond and a ω-hydroxy, -aldehyde, or -carboxylate groups (Tanaka et al., 1994; Tanaka et al., 1993; Tanaka et al., 1995) (Fig 7). A number of different mechanisms have been invoked to rationalize fragmentation of lipid hydroperoxides to these products and their reciprocal alkanyl chains. These include β-scission, Hock rearrangement, formation of diepoxy carbinyls, and peroxyl dimers. Even with highly reductionist in vitro systems, there is still great uncertainty about the relative contribution of each of these particular chemical pathways.
Figure 7.
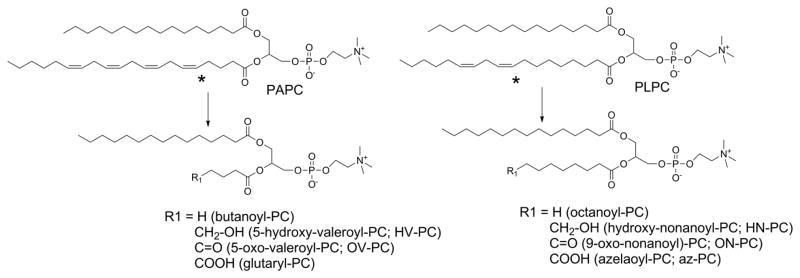
The most abundant oxidatively truncated oxPL are predicted by position of first double bond (marked by asterisk). Major products have O-acyl chains one carbon shorter than the position of this first double bond or O-acyl chain with one of three terminal ω-oxygen moieties on the carbon of this first double bond. The major products for arachidonyl-PC and linoleoyl-PC are listed, as they represent the most abundant products.
3.1.1. beta-scission
Formation of truncated oxPL with shortened acyl chains lacking omega;-oxo groups is most easily rationalized by β-scission. This requires formation of an alkoxyl radical from the PL-OOH as previously described for the formation of PL-OH. But in this case, a concerted radical reaction of the alkoxyl radical with its neighboring carbons forms a conjugated aldehyde while fragmenting the adjacent carbon-carbon bond, leaving a carbon centered alkyl radical (Fig 8). This radical then abstracts an hydrogen from another PUFA or other hydrogen donor to form a stable alkane. If the initial PL-OOH species is 5-HPETE-PC, then the β-scission products are hexadecatetraenal and butanoyl-PC. If 15-HPETE-PC is formed instead, then the β-scission products are pentane and 15-oxopentadecatetraenoate-PC. The formation of pentane from β-scission of arachidonate and linoleate peroxides and ethane from EPA and DHA peroxides is the basis of using exhaled pentane and ethane as biomarkers of in vivo lipid peroxidation (Dillard et al., 1977; Knutson et al., 2000).
Figure 8.

Beta-scission generates volatile alkanes and oxPL with very short O-acyl chains lacking ω-oxygens. Lipid peroxyl radicals form lipid alkoxyl radicals in the presence of iron. Reaction of the alkoxyl radical with the neighboring carbon-carbon bond fragments the acyl chain, leaving an alkyl radical and a lipid aldehyde. Abstraction of hydrogen from a nearby PUFA or other H donor by the alkyl radical generates an alkane. The major beta-scission products for each of the major PL-OOH are listed.
3.1.2. Hock rearrangement
The formation of shortened sn-2 acyl with omega;-oxo groups is most easily rationalized by “Hock rearrangement”. Here, the conjugated alkyl chain of the initial PL-OOH undergoes alkyl chain migration to form an intermediate hemiacetal and then hydrolysis, leaving two fragmented aldehydes as products (Fig 9). Under oxidizing conditions, the aldehydes are converted to carboxylates. Thus, Hock rearrangement of 5-HpETE-PC first generates PC with a 5-oxovaleroyl at the sn-2 position (OV-PC), which then oxidizes to generate a glutaryl group at the sn-2 position (glutaryl-PC). Alternatively, OV-PC can be reduced by aldose reductase to form 5-hydroxyvaleroyl-PC (HV-PC). Hock rearrangement of 9-HPODE-PC forms PC with azeaolyl group at the sn-2 position (az-PC).
Figure 9.
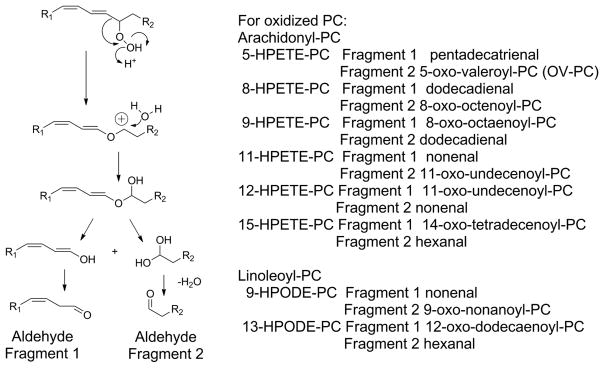
Hock rearrangement of PL-OOH fragments PUFA into two complimentary lipid aldehydes. Alkyl chain migration to the beta oxygen of the peroxide and subsequent addition of water to the resulting cation forms a hemiacetal which readily hydrolyzes to two aldehydic fragments. The predicted products for each HPETE-PC and HPODE-PC are listed. It is worth noting that for the two internal double bonds of arachidonyl-PC, the hydroperoxide located on either of the two original carbons in the double bond will generate the same two products (e.g. 8-HPETE-PC and 9-HPETE-PC give the same two products). Additionally some products such as hexanal and nonenal can form from peroxidation of either arachidonyl-PC or linoleoyl-PC.
By itself, the Hock rearrangement does not explain the formation of γ-ketoalkenoates and their reduced analogs such as γ-hydroxyalkenoates, γ-ketoalkenals, and γ-hydroxyalkenals that are commonly found with lipid peroxidation. However, the formation of these compounds can be rationalized by a second peroxidation step for aldehydic fragments with double bonds at the γ-position. Presumably, these behave very similarly to the 1,4-pentadienes of PUFA, so that a radical can abstract a hydrogen at the beta position, with molecular oxygen then rapidly reacting with the resulting carbon-center radical to generate γ-peroxy-alkenals that then form either γ-hydroxy-alkenals or γ-keto-alkenals (Fig 10). These aldehydes easily oxidize to form their analogous alkenoates. Thus, Hock rearrangement of 8-HPETE-PC could first give 8-oxo-octenoyl-PC, which then peroxidizes to form 5-keto-8-oxo-oct-6-enoate-PC (KOOA-PC) and 5-hydroxy-8-oxo-oct-6-enoate-PC (HOOA-PC) and then these aldehydes easily oxidize to form 5-keto-6-octendioate-PC (KOdiA-PC) and 5-hydroxy-6-octendioate-PC (HOdiA-PC). The analogous reactions for 13-HPODE-PC could give rise to 9-keto-12-oxo-10-dodecenoate-PC (KODA-PC) and 9-hydroxy-12-oxo-10-dodecenoate-PC (HODA-PC) that then are easily oxidized to 9-keto-10-dodecendioate-PC (KDdiA-PC) and 9-hydroxy-10-dodecendioate-PC (HDdiA-PC).
Figure 10.

Predicted secondary peroxidation of Hock rearrangement products to generate γ-ketoalkenoates and γ-hydroxyalkenoates. Hock rearrangement products with double bonds at γ-position relative to aldehyde (e.g. 8-oxo-oct-6-enoyl-PC) are predicted to be susceptible to H abstraction at the beta position. Addition of molecular oxygen to this lipid radical at the γ-position generates a peroxyl radical (e.g. 5-peroxy-8-oxo-oct-6-enoate-PC) that can subsequently react to form either γ-keto or γ-hydroxy moieties. Oxidation of the aldehyde generates the final γ-keto-or γ-hydroxy-alkenoate moieties (e.g. KOdiA-PC and HOdiA-PC).
3.1.3. Diepoxycarbinyl intermediates
As an alternative to formation of γ-hydroxyalkenoates by secondary peroxidation of Hock rearrangement products, Gu and Salomon proposed a mechanism where fragmentation occurs via formation of a diepoxy carbinyl intermediate (Fig 11). In this mechanism, alkoxyl radicals form then react with the adjacent double bond to give an epoxide in two successive rounds. The resulting diepoxy carbinyl can fragment in either direction to produce complementary aldehydic fragments, one of which would always be a γ-ketoalkenal or γ-hydroxyalkenal (Gu and Salomon, 2012).
Figure 11.
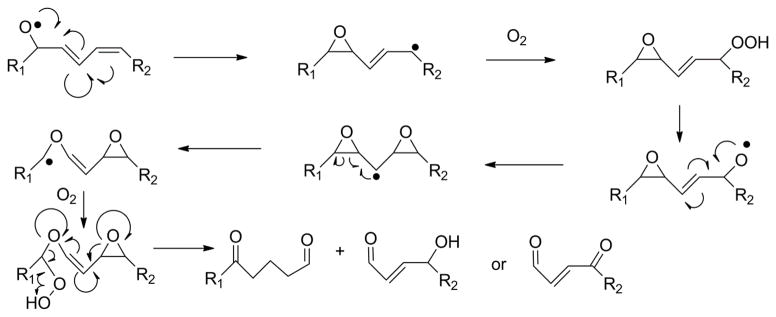
Alternative mechanism proposed for formation of γ-keto- or γ-hydroxy-alkenoate moieties via diepoxy carbinyl intermediate (Gu and Salomon, 2012). Formation of alkoxyl radical leads to monoepoxy carbinyl, which adds molecular oxygen to form epoxyperoxide. Subsequent scission of peroxide to form epoxy alkoxyl radical allows formation of diepoxy carbinyl radical which can undergo fragmentation at either epoxide. Fragmentation of one epoxide leads to formation of hydroxyl or keto groups by the other epoxide.
3.1.4. Other potential mechanisms
While both of the mechanisms above can be used to rationalize the formation of γ-hydroxyalkenals andγ-ketoalkenals like 4-hydroxynonenal (HNE) and 4-oxo-nonenal (ONE), respectively, it is important to note that several other mechanisms have been put forward for the formation of HNE. For instance, Schneider at al presented evidence for the formation of peroxy dimers with subsequent fragmentation that would account for both the formation of HNE and other oxidatively truncated PL such as OV-PC (Schneider et al., 2008) (Fig 12).
Figure 12.
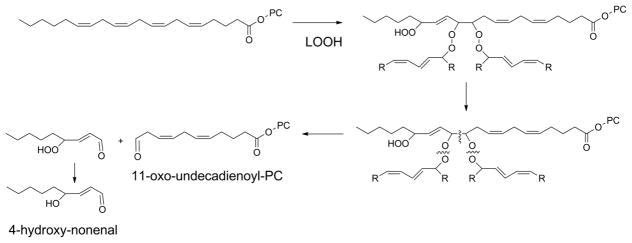
Another alternative mechanism for formation of γ-keto- or γ-hydroxy-alkenals and –alkenoates. Peroxidation of arachidonyl-PC can generate peroxide trimers. When these peroxides are on adjacent carbons, they readily fragment to form aldehydes, which would lead to the formation of γ-hydroxyalkenals.
3.2. Formation of oxidatively truncated oxPL in biological samples
Because the initial characterization of truncated oxPL was in oxLDL, characterization in vivo has focused on their formation in models of atherosclerosis. OV-PC and glutaryl-PC levels in aorta increase from about 20 ng/mg in rabbits fed a control diet to about 60 ng/mg tissue in those fed a cholesterol-rich Western diet (Watson et al., 1997). Truncated oxPL with ω-oxo groups (based on their ability to be derivatized by 9-(chloromethyl)anthracene, their retention time on HPLC, and their ability to stimulate the PAF receptor) are higher in plasma of elderly individuals compared to young adults, and in patients undergoing cardiopulmonary bypass (Frey et al., 2000). Levels of truncated oxPL derived from oxidation of arachidonyl-PC including HOOA-PC, KOOA-PC, HOdiA-PC, KOdiA-PC, OV-PC, and glutaryl-PC are about 5-fold higher (1–3 ng/mg arachidonyl-PC) in aorta isolated from aged WHHL rabbits with significant atherosclerotic lesion, compared to aorta from young WHHL rabbits (Podrez et al., 2002a). Similar increases are found for truncated oxPL from oxidized linoleoyl-PC such as HODA-PC, KODA-PC, HDdiA-PC, and KDdiA-PC, while ON-PC and ND-PC reach about 5–10 ng/mg linoleoyl-PC (Podrez et al., 2002a). In wild-type mice fed a normal chow diet, plasma concentrations of HOdiA-PC, KOdiA-PC, KOOA-PC, HDdiA-PC, KDdiA-PC, HODA-PC, and KODA-PC range from 60 to 280 nM. The plasma concentration of these oxPL are 3- to 7-fold higher in ApoE−/− mice fed a chow diet and 10- to 30-fold higher in ApoE−/− mice fed a cholesterol-rich Western diet, so that concentrations of KOdiA-PC and KODA-PC can reach 3.0 and 2.3μM, respectively (Podrez et al., 2007). Feeding LDLR−/− mice a Western diet increases levels of these same truncated oxPL by 4- to 6-fold compared those given a normal chow diet, so that levels of KOdiA-PC and KODA-PC reached 2.2 μM and 1.4 μM, respectively. In zebrafish larvae fed a high cholesterol diet, OV-PC and glutaryl-PC levels increased about 10-fold compared to standard diet (Fang et al., 2010). These results support the notion that biological activities which can be induced by low μM concentrations of these truncated oxPL are likely to be important during early pathogenesis of atherosclerosis.
A number of other inflammatory stimuli can induce the formation of oxidatively truncated oxPL, so that oxPL likely participates in feed-forward mechanism for inflammation. For instance, treatment of endothelial cells with IL-1β increases the levels of OV-PC and glutaryl-PC by 50% (Subbanagounder et al., 2002b). Exposure of human pneumocyte cell lines (A549) to influenza A virus induces 2-fold increases in OV-PC and glutaryl-PC (Van Lenten et al., 2004). Min mice are an animal model of human familial adenomatous polyposis (FAP) and analysis of intestinal polyps from 10 and 20 week old Min mice showed 2- to 6-fold increases in levels of ON-PC, Az-PC, OV-PC, and glutaryl-PC compared to wild-type mice (Ikeda et al., 2011). Treatment with TNFα increased levels of AZ-PC in Jurkat cells about 3-fold (Latchoumycandane et al., 2012).
Exposure to light also increases formation of truncated oxPL. For instance, exposure of human keratinocytes to UVB light increased levels of butanoyl-PAF and butenoyl-PAF nearly 100-fold (Marathe et al., 2005). UVB light also generated az-PAF (Zhang et al., 2005). Furthermore, UVB irradiation of fibroblast from mice deficient in xeroderma pigmentosum type A DNA repair gene led to 2-fold higher levels butanoyl-PAF, OV-PAF, and glutaryl-PAF (Yao et al., 2012). Exposure of dark adapted rats to physiological light for even as little as 4 hours increased retinal concentration of OV-PC and glutaryl-PC, AZ-PC, and ON-PC about 2-fold compared to rats without this light exposure (Sun et al., 2006). OV-PC, glutaryl-PC, AZ-PC and ON-PC have also been detected in vitreous fluid obtained from healthy elderly humans (Pollreisz et al., 2013).
γ-hydroxyalkenal-PC undergo spontaneous dehydration and cyclization to form furans. Increased levels of these more stable furans were detected after experimental cerebral infarction induced by ligation of a middle cerebral artery (Gao et al., 2006).
In addition to the mass spectrometry studies mentioned above, there are many other studies that have used non-specific means of detection such as anti-oxidized phospholipid antibody immunoreactivity to evaluate changes in truncated oxPL in various disease states. For instance, using E06 to non-specifically measure oxPL, increased E06 immunoreactivity has been reported in plasma of patients undergoing percutaneous coronary intervention (Tsimikas et al., 2004), in BAL fluid of acid-treated mice (Imai et al., 2008), in BAL fluid of H1N1 influenza A treated mice (Crowe et al., 2009), and with extent of coronary artery disease (Tsimikas et al., 2005). Increased immunoreactivity of DLH3, another anti-oxPC antibody, was found in liver specimens from humans with non-alcoholic steatohepatisis (Ikura et al., 2006) and in macula of patients with age-related macular degeneration (Suzuki et al., 2007). These measurements support the notion that lipid peroxidation occurs in these disease conditions, but do not provide definitive evidence for any specific species.
3.3. Molecular targets, signaling pathways, and biological activities of truncated oxPL
3.3.1. PAF-like activity
Perhaps the earliest and best characterized activities of oxidatively truncated oxPLs are as platelet-activating factor (PAF) receptor agonists. Binding of authentic PAF (1-alkyl-2-acetyl-sn-glycero-3-phosphocholine) to this Gq-protein coupled receptor activates PLC and PKC, eventually leading to cPLA2 activation and subsequent prostaglandin synthesis. While named for its ability to activate platelets, the PAF receptor is expressed in a wide range of leukocytes including polymorphonuclear leukocytes (PMN) and monocytes, and activation of PAF receptor also induces adhesion of these leukocytes to endothelial cells.
The first experiments that demonstrated oxPL could act via the PAF receptor were performed by Smiley et al, who showed that oxidized arachidonyl-PC induced PMN adhesion which could be blocked by PAF receptor antagonists (Smiley et al., 1991). Subsequently, Patel et al demonstrated that exposure of endothelial cells to hydrogen peroxide also resulted in the formation of membrane blebs that that potently activates the PAF receptor of PMN (Patel et al., 1992). Inhibition of PAF synthesis did not block formation of PAF receptor agonists in these blebs yet these agonists were susceptible to degradation by PAF-AH. Berliner et al had previously shown that a phospholipid component of oxidized LDL stimulated monocyte binding to endothelial cells (Berliner et al., 1990; Cushing et al., 1990). They later showed that this activity was susceptible to inhibition by PAF receptor antagonist and to degradation by PAF-AH (Leitinger et al., 1997; Watson et al., 1995). Importantly, the relevance of these in vitro findings was verified by studies using intravital fluorescence microscopy and skinfold-chamber model in hamsters. These studies showed that administration of PAF receptor inhibitors to hamsters blocked the ability of injected oxLDL to induce leukocyte rolling and adhesion (Lehr et al., 1993).
In addition to the effect of oxPC on endothelial cells, Heery et al showed that oxLDL induced smooth muscle proliferation, another key atherogenic action, as well as PMN adhesion to gelatin (Heery et al., 1995). Both of these activities could be blocked by PAF receptor antagonists and treatment with PAF-AH. In reversed phase HPLC, the activity of oxLDL eluded after the retention time for authentic PAF but prior to the retention time for unoxidized phospholipids, consistent with previous finding for the PAF-like activity of oxidized PAPC. These phospholipid fractions were shown to stimulate arachidonic acid release in CHO cells transfected with the PAF receptor, but not in non-transfected cells, demonstrating they were authentic PAF receptor ligands (Heery et al., 1995).
During the studies to elucidate the PAF like activity of oxPL, parallel studies were performed to isolate the active components. A key breakthrough were studies that showed that the PAF-like activity isolated from oxLDL could not be degraded by PLA1, suggesting that truncated alkylacyl phospholipids rather than diacyl phospholipids were responsible (Marathe et al., 1999). This was consistent with the findings that oxidation of arachidonyl-PAF generated PAF-like activity that was about 800-fold more potent than oxidation of arachidonyl-PC. Subsequent mass spectrometric analysis of oxidized arachidonyl-PAF showed that butanoyl-PAF and butenoyl-PAF are the most prominent phospholipids present in the HPLC fractions with PAF-like activity. Synthetic butanoyl-PAF and butenoyl-PAF are nearly as potent as authentic PAF (acetyl-PAF) at stimulating calcium release in HEK293 cells transfected with the PAF receptor. The PAF like activity of oxLDL co-eluted in HPLC with butanoyl-PAF and butenoyl-PAF, supporting the notion that these are major PAF-like lipids in oxLDL (Marathe et al., 1999).
The extent that other truncated oxPL besides butanoyl-PAF and butenoyl-PAF contribute to the PAF-like activity of oxidized lipoproteins and membranes is not fully resolved. The oxidation of arachidonyl-PAF present in LDL generates both OV-PAF via Hock rearrangement and butanoyl-PAF and butenoyl-PAF via β-scission. The original studies by the McIntyre lab demonstrated that synthetic OV-PC (generated by ozonolysis of PAPC) activated PMN adhesion to gelatin and that this adhesion is blocked by PAF receptor antagonists (Smiley et al., 1991). Ozonolysis of arachidonyl-PAF also generates potent PAF-like activity, so butanoyl-PAF and butenoyl-PAF seem unlikely to entirely account for the PAF-like activity of oxLDL.
Although relatively few studies have used the more active OV-PAF, a number of studies have used the commercially available OV-PC to examine the contribution of these species and activation of the PAF receptor to atherosclerosis. OV-PC induces monocyte adhesion to endothelial cells and this effect can be inhibited by PAF receptor antagonists (Subbanagounder et al., 1999). OV-PC binds to PAF receptor expressed on macrophages and induces calcium mobilization (Pegorier et al., 2006). OV-PC also induces inflammatory gene expression in monocytes, but authentic PAF can only induce expression of a subset of these genes suggesting that OV-PC also interacts with other receptors (Pegorier et al., 2006). OV-PC induces bone-marrow derived stem cell migration that is blocked by PAF receptor specific antagonists or RNAi depletion of PAF receptor (Shin et al., 2011). These effects on migration depend on induction of Kruppel-like factor 4.
During studies to identify the active species of PAF-like lipids in oxLDL, various alkylacyl precursors were oxidized to determine which of these could give rise to PAF receptor agonists (Marathe et al., 1999). While oxidation of arachidonyl-PAF gave robust PAF receptor agonist activity as expected, it was surprising to find that oxidation of linoleoyl-PAF also gave strong PAF agonist activity. This result raised that possibility that AZ-PAF might also be a PAF receptor agonist, because octanoyl-PAF, the other major fragmentation product of linoleoyl-PAF, does not agonize the PAF receptor. Subsequently, Gopfert et al showed that 10 μM OV-PC, glutaryl-PC, and AZ-PC could all stimulate shape-change in platelets, but that they reported that these effects were not blocked by the PAF receptor antagonist WEB2086 (Gopfert et al., 2005). However, when Chen et al examined the effects of submicromolar concentrations of AZ-PAF on platelet activation, they found that 200 nM AZ-PAF activated calcium in platelets and that 100 nM AZ-PAF synergistically activated platelets induced with suboptimal concentrations of thrombin, ADP, or collagen (Chen et al., 2009a). The activity of this submicromolar AZ-PAF was blocked by PAF receptor antagonists (Chen et al., 2009a). Thus, the PAF receptor activity of AZ-PL appear to absolutely require an O-alkyl chain at sn-1, unlike OV-PL where OV-PC can activate the PAF receptor, though with substantially less potency than OV-PAF.
3.3.2. PPARα and PPARγ
During studies of oxLDL activation of monocytes, it became apparent that authentic PAF could not mimic all of the effects of oxLDL. Because these effects were most strongly induced by oxidized linoleoyl-PAF, AZ-PAF was used to identify this additional receptor. A candidate receptor approach eventually identified PPARγ as the receptor, with AZ-PAF potently (100 nM) inducing PPRE-reporter expression when PPARγ is heterologously expressed (Davies et al., 2001). Furthermore, AZ-PAF induces expression of CD36, a PPARγ responsive genes. Direct binding of AZ-PAF to recombinant PPARγ potently (Kd 40 nM) displaces a radiolabeled PPARγ agonist (BRL49653). Interestingly, anti-CD36 antibodies block the uptake of AZ-PAF by monocytes, suggesting that AZ-PAF is a CD36 ligand and that induction of CD36 and PPARγ expression by AZ-PAF induce a feed-forward mechanism (Davies et al., 2001). Further studies found that both the PPARγ agonist rosiglitazone and AZ-PAF induce COX-2 expression in monocytes, which increased their ability to secrete PGE2 (Pontsler et al., 2002). It should be noted that this effect contrasts with the ability of various PPARγ agonists to inhibit inflammatory signaling in macrophages induced by IFNγ, phorbol ester, or LPS (Inoue et al., 2000; Jiang et al., 1998; Ricote et al., 1998). The reason for this paradox is not fully understand, but is in keeping with the observation that truncated oxPL are functionally partial agonists of inflammation, so that by themselves they induce modest inflammatory effects while they suppress inflammatory effects when combined with full agonists.
In contrast to AZ-PC which activates PPARγ, the shorter oxPL such as OV-PC and glutaryl-PC activate PPARα (Lee et al., 2000). For instance, treatment with 5 ug/mL OV-PC or 2.5 ug/mL glutaryl-PC induced about a 3-fold increase in PPRE reporter expression in endothelial cells, which primarily express PPARα, rather than PPARγ. Similarly, OV-PC and glutaryl-PC induce PPAR reporter expression in CV-1 cells transiently transfected with PPARα, but not PPARδ or PPARγ Finally, endothelial cells derived from PPARα−/− mice failed to synthesize IL-8 and MCP-1 in response to oxidized arachidonyl-PC or oxLDL (Lee et al., 2000). It is worth noting that others who have examined the PPARα dependent activation of endothelial cells by oxLDL and oxPL found that this activity was partially blocked by the PLA2 inhibitor (Delerive et al., 2000). This finding was interpreted to indicate that it was unesterified oxidized lipids rather than the oxPL that were the active PPARα ligands, although an alternative interpretation is that the required activation of PLA2 is downstream of the initial PPARα binding and activation.
3.3.3. Scavenger receptors CD36 and SR-BI
The scavenger receptors SR-BI and CD36 play important roles in the transfer of lipids from lipoproteins to peripheral cells, in macrophage cell signaling, and in formation of foam cells. Both SR-BI and CD36 bind oxLDL with high affinity (Endemann et al., 1993; Gillotte-Taylor et al., 2001). Because of the importance of CD36 for the uptake of oxLDL and formation of foam cells, Podrey et al performed a series of elegant studies to identify oxPL species bound by CD36. These studies revealed that γ-ketoalkenoate-PC are critically important CD36 ligands for generation of foam cells (Podrez et al., 2002a; Podrez et al., 2002b).
Myeloperoxidase driven oxidation of liposomes produced eight major species with high affinity to CD36 binding (Podrez et al., 2002b). Isolation and characterization of these oxPL showed each had an α,β unsaturated aldehyde or carboxylate at the ω-terminus of the fragmented chain and a γ-hydroxyl- or γ-keto- group (e.g. KOdiA-PC). They then performed structure activity relationship studies to access the structural requirements for CD36 binding using competition experiments with radiolabeled oxLDL (Table 1).
Table 1.
Potency of CD36 binding by γ-ketoalkenoate–PC and analogs
| |||||
|---|---|---|---|---|---|
| PC | Sn-2 group | n= | R1 | R2 | IC50 (μM) |
| KDdiA-PC | 9-keto-10-dodecendioate | 5 | =O | -OH | 2.0 |
| KOdiA-PC | 5-keto-6-octendioate | 1 | =O | -OH | 3.9 |
| KODA-PC | 9-keto-12-oxo-10-dodecenoate | 5 | =O | -H | 2.5 |
| KOOA-PC | 5-keto-8-oxo-6-octenoate | 1 | =O | -H | 6.2 |
| HDdiA-PC | 9-hydroxy-10-dodecendioate | 5 | -OH | -OH | 14.5 |
| HOdiA-PC | 5-hydroxy-6-octendioate | 1 | -OH | -OH | 15.5 |
| HODA-PC | 9-hydroxy-12-oxo-10-dodecenoate | 5 | -OH | -H | 75.4 |
| HOOA-PC | 5-hydroxy-8-oxo-6-octenoate | 1 | -OH | -H | 46.8 |
| OV-PC | 5-oxo-valeroate | n/a | n/a | -H | 138.3 |
Adapted from (Podrez et al., 2002b). IC50 is for inhibition of CD36 binding to radiolabeled oxLDL. All PC had sn-1 palmitoyl and sn-3 phosphocholine groups.
They found that the truncated oxPC with esterified γ-ketoalkenoates at sn-2 (e.g. KDdiA-PC and KOdiA-PC) were the most potent competitors for CD36 binding. Replacing the ω-carboxylate with an ω-aldehyde reduced potency 1.5-fold. Replacing the γ-keto group with a γ-hydroxy group reduced potency three to seven-fold (Podrez et al., 2002b). PC esterified with ω-aldehydes lacking both the γ-functional group and double bond (e.g. OV-PC) can compete for oxLDL binding but are even less potent (Boullier et al., 2005; Podrez et al., 2002b). Nevertheless, OV-PC signaling via CD36 was reported to play an important role in inflammatory corneal neovascularization (Mwaikambo et al., 2006). HPETE-PC and HPODE-PC and the non-esterifed γ-hydroxyalkenal failed to effectively compete for CD36 binding as did cholesterol esterified analogs (Podrez et al., 2002b).
Subsequent work showed that these γ-ketoalkenoate-PC and their analogs form “lipid whiskers” on the surface of membranes, with the truncated acyl chain sticking out of the membrane bilayer to be recognized by CD36 (Greenberg et al., 2008). CD36 was essential for uptake of liposomes with these truncated oxPC and addition of these oxPC to cholesterol containing lipoparticles drove accumulation of cholesterol and formation of foam cells in mouse perioteneal macrophages derived from wild-type, but not CD36−/− mice (Podrez et al., 2002a).
In addition to binding to CD36, γ-ketoalkenoate-PC and its reduced analogs also bind with high affinity to SR-BI (Ashraf et al., 2008). KOOA-PC markedly inhibited uptake of radiolabeled oxLDL by SR-BI expressing CHO cells. Furthermore, small unilamellar vesicles constituted with KDdiA-PC bound to these SR-BI expressing CHO cells, while vesicles lacking KDdiA-PC failed to bind. KDdiA-PC and KOdiA-PC bind with high affinity to recombinant SRBI and potently inhibited HDL uptake by SR-BI expressing cells including hepatocytes. In addition to γ-ketoalkenoate-PC, OV-PC also competes with oxLDL for binding to SR-BI (Gillotte-Taylor et al., 2001).
The ability of γ-ketoalkenoate-PC and other truncated oxPL to bind SR-BI has been interpreted as likely to promote atherogenesis, since SR-BI binding to HDL is critical to reversal cholesterol effux, so that competition for efflux would potentially lead to peripheral cholesterol accumulation. However, it is worth considering that SR-BI mediated clearance of oxidized lipoproteins may dramatically reduce the levels of these proinflammatory particles, which may be more important for protecting against the atherosclerosis.
3.3.4. Inflammatory responses including adhesion and cytokine secretion
One of the earliest identified bioactivities for truncated oxPL was the ability to activate endothelial cell interactions with leukocytes. OV-PC and glutaryl-PC both induce endothelial cell activation, expression of proinflammatory cytokines, and binding of monocytes, while only glutaryl-PC induces PMN adhesion to endothelium (Leitinger et al., 1999; Subbanagounder et al., 1999). These truncated oxPL are present in membrane vesicles released from activated cells and apoptotic blebs which stimulate endothelial cell activation (Huber et al., 2002). The mechanism by which OV-PC and glutaryl-PC induce endothelial cell activation and monocyte adhesion differs. Endothelial activation by OV-PC, but not glutaryl-PC, can be inhibited by PAF receptor antagonists (Subbanagounder et al., 1999). OV-PC induces monocyte adhesion by inducing surface expression of the fibronectin connecting segment-1 domain (Leitinger et al., 1999), while glutaryl-PC induces E-selectin and VCAM-1 expression. OV-PC induces cAMP-mediated pathways suggesting agonism of Gs coupled receptor(s), while glutaryl-PC induces chloride ion conductance. Subsequent experiments showed that OV-PC activates endothelial R-Ras downstream of cAMP activation, which in turn activates PI3-kinase (Cole et al., 2003).
Of great interest are recent studies that suggest that the OV-PC metabolite 5-hydroxyvaleroyl-PC (HV-PC) induces cytokine expression in macrophages, rather than OV-PC itself (Vladykovskaya et al., 2011). HV-PC is 100-fold more potent in inducing cytokine expression than OV-PC. HV-PC forms by aldose reductase-mediated reduction of the aldehyde group and OV-PC fails to induce cytokine expression in macrophages lacking aldose reductase. Treatment with the aldose reductase inhibitor tolrestat also blocks OV-PC induction of cytokine expression (Vladykovskaya et al., 2011).
Similar to OV-PC and glutaryl-PC, at least one γ-ketoalkenoate-PC analog (HOOA-PC) is known to activate monocyte binding to endothelial cells and endothelial expression of MCP-1 and IL-8 expression (Subbanagounder et al., 2002a). The ability of other γ-ketoalkenoate-PC analogs to activate endothelial cells in this manner has not been tested.
3.3.5. Binding to TLR4 co-activators and inhibition of LPS signaling
OV-PC and glutaryl-PC were the first oxidatively truncated oxPL shown to exert inhibitory effects on LPS signaling, but this is now recognized as a common feature of many oxPL. Because both oxPL and LPS activate TLR signaling, the inhibition of LPS signaling by oxPL initially appears paradoxical. However, this effect is consistent with a paradigm where oxPL are high affinity, partial agonists for components of the TLR signaling pathway, so that weak activation of TLR signaling by oxPL inhibits the far more robust activation of this signaling by LPS.
LPS invokes proinflammatory signaling by binding to soluble MD-2 and then forming a complex with the extracellular domain of the membrane spanning pattern recognition receptor TLR4 (Fig 13) (Kumar et al., 2011). These TLR4-MD-2 complexes can signal by both CD14 dependent and CD14-independent mechanisms, with LPS-binding protein (LBP) assisting in extracting LPS and presenting it to these complexes. Park et al showed that when LPS binds to MD-2 and TLR4, five of the six acyl chains of LPS are buried within the large hydrophobic pocket of MD-2, with the remaining acyl chain and the glucosamine backbone interacting with both the surface of MD-2 and the TLR4 dimer to form a signaling complex (Park et al., 2009). Bacterial lipopeptides can also interact with the hydrophobic pockets of TLR2/TLR1 and TLR2/TLR6 heterodimers to activate similar signaling cascades (Manavalan et al., 2011).
Figure 13.
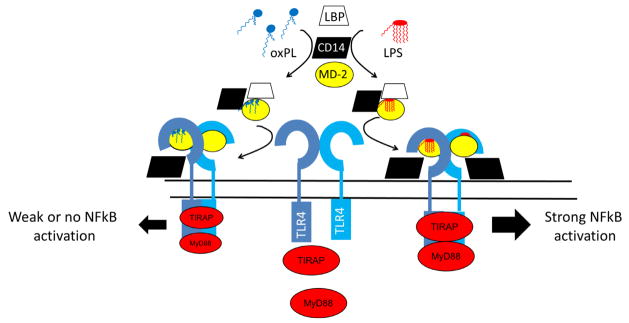
Oxidatively truncated oxPL compete with LPS for binding to MD-2, CD14, and LBP and thereby inhibit LPS signaling. MD-2 bound to LPS promotes formation of highly active TLR4 dimer that activates TIRAP and MyD88 and therefore NFkB. Truncated oxPL like OV-PC or KOdiA-PC also bind to MD-2, CD14, or LBP but only weakly activate TLR4 signaling, so that their net effect is to inhibit LPS induced signaling via TLR4. Adapted in part from (Kawai and Akira, 2011) and (Park et al., 2009).
The induction of cytokine expression by OV-PC and other oxPL can be blocked by ablation of TLR2 and TLR4 (Imai et al., 2008; Kadl et al., 2011; Miller et al., 2005; Walton et al., 2003) consistent with their being TLR agonists. The formation of oxPL and subsequent activation of TLR4 by these oxPL has been identified as a key pathway mediating acid- and viral-induced acute lung injury (Imai et al., 2008).
While OV-PC and other oxPL directly induce endothelial cell activation, they paradoxically also inhibit bacterial lipopolysaccharide (LPS) induced expression of E-selectin by endothelial cells, as well as subsequent PMN binding to this activated endothelium (Leitinger et al., 1999). A key aspect to understanding this effect are studies showing that high concentrations (30 μM and higher) of OV-PC and glutaryl-PC effectively compete with LPS for binding to LBP and CD14 (Bochkov et al., 2002; von Schlieffen et al., 2009) as well as MD-2 (Erridge et al., 2008). Unlike LPS, which is full agonist of TLR signaling, OV-PC and other oxPL are only partial agonists of TLR signaling, so that the net effect of coincubation of OV-PC and LPS is inhibition of LPS signaling (Bochkov et al., 2002; Erridge et al., 2008; Erridge et al., 2007) (Fig 12).
Walton et al found that γ-ketoalkenoate-PL including KOdiA-PC, KOdiA-PE, KHdiA-PE, KDdiA-PE, and HDdiA-PE were more potent than OV-PC or glutaryl-PC at inhibiting LPS induced IL-8 expression by endothelial cells (Walton et al., 2006). Kim et al found that KOdiA-PC competed with LPS for binding to MD-2, rather than CD14 or LBP (Kim et al., 2013). Additionally, they found that KOdiA-PC prevented LPS from inducing macrophages to express TNF, IFN-beta, and COX-2 and that an inhibitor of acid sphingomyelinase blocked KOdiA-PC’s effects.
3.3.6. SMC proliferation and migration
In atherogenesis, vascular smooth muscle cells (SMC) undergo phenotypic switching from differentiated SMC to proliferating SMC showing down-regulation of differentiation marker genes and enhanced capacity for migration. Exposing cultured SMC to oxLDL produces this phenotypic switching and proliferation, and this effect can be blocked by PAF receptor antagonists (Heery et al., 1995). Subsequent characterization of the active components in oxLDL revealed that OV-PC, but not glutaryl-PC, induces this proliferation of SMC (Chatterjee et al., 2004). OV-PC reduces connexin 43 (Cx43) levels, but both OV-PC and glutary-PC increase Cx43 phosphorylation, which is thought to be an important step in SMC proliferation (Johnstone et al., 2009). Treatment of SMC with OV-PC stimulates the activity of UDP-galactose:glucosylceramide galactosyltransferase (GalT-2) and the formation of lactosylceramide which leads to activation of p44 MAP kinase (Chatterjee et al., 2004). At high concentrations (~30 μM or higher), both OV-PC and glutary-PC inhibited expression of SMC differentiation markers such as actin and myosin heavy chain via their activation of Kruppel-like transcription factor 4 (KLF4) (Pidkovka et al., 2007). Induction of KLF4 was dependent on OV-PC induced phosphorylation of ERK1/2 and Elk-1, with Elk-1 physically interacting with KLF4 (Yoshida et al., 2008). KLF4 also interacts with histone deacetylase 5 (HDAC5) to recruit it to histone H4 of the actin promoter (Yoshida et al., 2008). OV-PC activation of KLF4 also induces expression of extracellular matrix proteins including type VIII collagen, Col8a1 (Cherepanova et al., 2009).
Both OV-PC and glutaryl-PC invoke calcium entry into proliferating SMC, leading to their migration, and this effect is inhibited by antibodies to transient receptor potential cation channel 5 (TRPC5) and TRPC1 (Al-Shawaf et al., 2010). OV-PC and glutaryl-PC could also invoke calcium currents in HEK293 cells overexpressing TRPC5 but not TRPM2 or TRPM3. The effects of glutaryl-PC and OV-PC on depended on G(i/o)proteins, which is interesting given that other effects of these lipids appear to result from agonism of Gq- and Gs-coupled receptors (Al-Shawaf et al., 2010)
3.3.7. ER stress response, ceramide formation, and apoptosis
When AZ-PAF is given at μM concentrations, rather than the nM concentration needed for PAF receptor or PPARγ activity, it displays strong apoptotic effects (Chen et al., 2007). The acyl analog AZ-PC is about half as potent as AZ-PAF. Exogenously administered AZ-PAF rapidly internalizes to the mitochondria and depolarizes their membrane through the action of Bid (Chen et al., 2009b; Chen et al., 2007). The transfer of exogenous AZ-PAF or AZ-PC to intracellular compartments is dependent on the action of TMEM30a and reducing levels of this transporter markedly reduces induction of apoptosis (Chen et al., 2011). Once in the mitochondria, AZ-PAF induces the release of cytochrome c and apoptosis-inducing factor, which activates caspase 3 and 9 and directly leads to apoptosis.(Chen et al., 2009b; Chen et al., 2007). The formation of AZ-PC appears to be part of the physiological pathways for inducing apoptosis, as TNFα strongly induces formation of AZ-PC in Jurkat cells, and overexpression of either gluthathione peroxidase-4 or PAF-AH2 suppresses both the formation of AZ-PC and markedly inhibits the ability to TNF to induce apoptosis (Latchoumycandane et al., 2012).
When given at extremely high concentrations (50 μM or greater) both OV-PC and glutaryl-PC begin to have significant effects on the viability of cultured cells. The effect of these two oxPL may be mediated by different pathways. Because OV-PC contains an aldehydic moiety, it can react with lysine residues of protein. Proteins modified by lipid aldehydes often partially unfold and aggregate and thereby activate the endoplasmic recticulum (ER) stress response. The ER stress response activates three downstream pathways (PERK, IRE1, and ATF6), which halts general protein synthesis and increases expression of the ER chaperone protein BiP/Grp78 and proteases so that unfolded and damaged protein can be cleared from the cell (Lai et al., 2007). Prolonged activation of these pathways initiates apoptosis, thereby preventing the damaged cells from altering function of adjacent cells. Exposure of endothelial cells to oxPAPC results in upregulation of genes represented of the ER stress response (Gargalovic et al., 2006a; Gargalovic et al., 2006b), but the exact lipids responsible for these effects have not yet been identified. What has been shown is that exposure of isolated mouse cardiac myocytes cells to 50 μM OV-PC increases ER stress response markers including phosphorylated PERK and eIF2alpha, as well as higher levels of ATF3 (Keith et al., 2009). In contrast, the reduced metabolite of OV-PC (HV-PC) fails to induce these effects, supporting the importance of the aldehyde moiety for this effect. Overexpression of aldose reductase, which metabolizes OV-PC to HV-PC, reduced markers of ER stress response in cardiac myocytes of mice subjected to ischemia-reperfusion. Structure-activity relationship studies examining the potency of OV-PC versus other aldehydic oxPL for induction of ER stress signaling have not been performed. These seem critical given the relatively weak potency of OV-PC and its relatively weak reactivity with primary amines. As will be discussed in the section on class III oxPL, EI-PC is far more potent than OV-PC at inducing markers of ER stress and is also more reactive, leading to speculation that potency may be a function of reactivity.
While glutaryl-PC cannot react with proteins in a similar manner as OV-PC, at 50 μM both OV-PC and glutaryl-PC induce apoptosis in cultured SMC (Fruhwirth et al., 2006; Loidl et al., 2003). These effects were abolished in the presence of high levels of serum, due to hydrolysis of OV-PC and glutaryl-PC. The apoptotic effects of OV-PC and glutaryl-PC in SMC appear to result from their ability to induce formation of ceramide via sphingomyelinases (Loidl et al., 2003). OV-PC and glutaryl-PC also induce apoptosis in macrophages, with OV-PAF and glutaryl-PAF showing no greater potency, so that these effects do not appear to be dependent on agonism of the PAF receptor (Stemmer et al., 2012). In macrophages, induction of ceramide formation by OV-PC and glutaryl-PC occurs by the activity of both acid sphingomyelinase and ceramide synthetase (Halasiddappa et al., 2013; Stemmer et al., 2012). In neurons, OV-PC induces apoptosis by producing ceramide via neutral sphingomyelinase rather than acid sphingomyelinase (Qin et al., 2009).
KOdiA-PC and KDdiA-PC both synergize with submaximal concentration of thapsigargin to induce ER stress responses and apoptosis in macrophages, and this was dependent on CD36 (Seimon et al., 2010).
3.3.8. Targets of HDL anti-inflammatory activity
High density lipoprotein (HDL) has at least three anti-atherogenic effects: its capacity for reverse cholesterol transport, its capacity to inhibit inflammatory responses induced by oxLDL and LPS, and its capacity to inhibit oxidation. ApoAI is the major protein of HDL and has been shown to contribute to all three of these properties. ApoAI consists of multiple repeats of amphipathic helices of similar sequence. Short therapeutic peptides based on the consensus sequence of these helices have been developed and have shown strong anti-atherogenic effects in animal models. These ApoAI mimetic peptides bind with high affinity to OV-PC and glutaryl-PC (Van Lenten et al., 2008). KOdiA-PC and KDdiA-PC bound with even higher affinity than AZ-PC or glutaryl-PC (Epand et al., 2009). While ApoAI is the major protein of HDL, there are many other minor components that have been shown to have anti-inflammatory effects including apoJ. Interestingly, ApoJ also adopts an amphipathic helical structure in liposomes and binds with high affinity to micelles with even a low percentage of KOdiA-PC (Mishra et al., 2011). These results suggest that the anti-atherogenic effects of HDL results in part from binding of these various amphipathic helical proteins to γ-ketoalkenoate-PC.
3.4. Metabolism
3.4.1. PAF-AH
Perhaps the most important mechanism for metabolism of oxidatively truncated oxPL is the hydrolysis of the oxidatively modified sn-2 chain by PAF acetylhydrolases. The initial PAF acetylhydrolase enzyme that was characterized was the activity secreted by macrophages that circulates on plasma lipoproteins (plasma PAF-AH) and is also referred to as lipoprotein-associated PLA2 (Hakkinen et al., 1999; Ostermann et al., 1989; Stafforini, 2009; Stafforini et al., 1989; Stafforini et al., 1990). Structure-activity relationship studies with plasma PAF-AH found that in addition to authentic PAF, it also degraded truncated oxPL generated by oxidation of arachidonyl-PC (Stremler et al., 1989). The efficiency of plasma PAF-AH for hydrolysis of 1-alkyl-2-acyl-PC dropped dramatically when the sn-2 acyl chain was 5 carbons or longer.
Addition of an ω-oxo group to the acyl chain allowed PAF-AH to efficiently hydrolyze acyl chains up to 9 carbons long. While other oxPL such as IsoP-PL and PL-OOH can be hydrolyzed by this enzyme, the catalytic efficiency for these longer oxPL is much lower (Davis et al., 2008; Kriska et al., 2007; Stafforini et al., 2006). Plasma PAF-AH destroys the PAF-like activity of oxPAPC (Stremler et al., 1989), as well as endothelial and smooth muscle cell activating activity of oxLDL (Heery et al., 1995; Watson et al., 1995).
Plasma PAF-AH expression is increased by inflammatory stimuli, presumably as a mechanism to counteract the resulting increase in oxPL (Shi et al., 2007; Wu et al., 2004). Because of this, increased levels of PAF-AH correlate with increased risk for coronary artery disease (Caslake and Packard, 2005; Dada et al., 2002; Packard et al., 2000; Zheng et al., 2012). Increased PAF-AH levels may be pathogenic because they increase lysoPC levels (Shi et al., 2007). On the other hand, humans with inactivating polymorphisms in PAF-AH have increased severity of a number of disease associated with oxPL including asthma, cardiovascular disease, and stroke (Stafforini, 2009). Furthermore, a number of animal studies that overexpressed human PAF-AH in vivo for therapeutic benefit showed efficacy including in atherosclerosis (Hase et al., 2002; Quarck et al., 2001), diabetes (Lee et al., 1999), paw edema and pleurisy (Tjoelker et al., 1995), necrotizing enterocolitis (Caplan et al., 1997), acute pancreatitis (Bedirli et al., 2004), anaphylactic shock (Fukuda et al., 2000), and sepsis (Gomes et al., 2006).
In addition to the plasma form of PAF-AH, there are two intracellular forms of PAF-AH. The intracellular type II PAF-AH (PAF-AH2) shows 41% sequence similarity to the plasma PAF-AH, while intracellular type I PAF-AH is a trimeric protein with no sequence similarity (Hattori et al., 1996). PAF-AH2 shows similar substrate specificity to plasma PAF-AH (Min et al., 2001). PAF-AH2 translocates from the cytoplasm to membranes after exposure to oxidants and overexpression of PAF-AH2 inhibits oxidant induced cell death (Matsuzawa et al., 1997). PAF-AH2−/− mice develop normally, however, they have a delayed recovery in response to carbon tetrachloride mediated hepatic injury (Kono et al., 2008). Fibroblasts derived from these mice are also much more sensitive to tert-butylhydroperoxide induce cytotoxicity (Kono et al., 2008). Worms where PAF-AH2 is deleted show morphogenesis defects and epithelial organization defects (Inoue et al., 2004).
3.4.2. Aldo-keto Reductases
A very important step in metabolism of aldehydic truncated oxPL is the reduction of the aldehyde moiety to the stable, non-reactive hydroxyl moiety. The enzymes that catalyze this reaction are aldo-keto reductases (AKR) (Srivastava et al., 2004). Structure-activity relationship studies with various aldo-reductase found that OV-PC is reduced by AKR1A and AKR1B family members, but not by AKR6 and AKR7 family members (Spite et al., 2007). AKR1B also reduces ON-PC. AKR1A4 most efficiently reduces γ-hydroxy-alkenal-PC. The importance of this reduction is demonstrated by the effects of inhibitors of AKR, which increase atherosclerotic lesion size (Srivastava et al., 2009).
3.4.3. Paraoxonase
ApoE−/− mice lacking paraoxonase-1 (PON1−/−) have increased levels of OV-PC and glutaryl-PC in their IDL and LDL fractions, along with greater lesion size (Shih et al., 2000). Furthermore, endothelial cells derived from PON1−/− mice are more susceptible to cell death induced by oxLDL than those derived from wild-type mice (Garcia-Heredia et al., 2013). Purified PON1 hydrolyzed ON-PC and OV-PC and inhibited their ability to induce monocyte binding to endothelial cells (Ahmed et al., 2003). However, the interpretation of these studies is somewhat clouded by the discovery that PON1 isolated from HDL typically co-purifies with PAF-AH and that recombinant PON1 lacks the ability to hydrolyze oxPL (Kriska et al., 2007). Nevertheless, in rabbits fed a high-fat diet and then subjected to balloon injury, virus-mediated transgenic overexpression of PON1 led to decreased oxidative stress, LOX-1 accumulation in aorta, and reduced intimal thickening (Miyoshi et al., 2007). Thus, the presence of PON1 appears to be important in metabolizing truncated oxPL, whether or not it directly participates in their hydrolysis.
3.4.4. Transport of truncated oxPL
Although hydrolysis of circulating oxPL by plasma PAF-AH helps to reduce plasma levels of oxidatively truncated oxPL, recent experiment have shown that cellular uptake of circulating oxPL plays the most important role in the short circulation time of these oxPL (t1/2 < 30 secs)(Liu et al., 2011). Because this transport mechanism can be saturated, too rapid accumulation of oxPL or inhibition of transport by other substrates may lead to marked increases in circulating levels of oxPL. The specific enzymes involved in uptake of circulating oxPL transport have not been determined. However, Liu et al showed that siRNA against TMEM30a significantly slowed uptake of labeled oxPL by endothelial cells in culture (Liu et al., 2011), suggesting that this transmembrane protein may be a key player in this activity.
4. Class III: oxPL with cyclized acyl chains
4.1. Chemical mechanisms for formation of cyclized oxPL
4.1.1. Isoprostanes and isolevuglandin-PL
Formation of a phospholipid peroxyl radical when the peroxidized fatty acid contains at least three double bonds (e.g. arachidonate) has the potential to form a bicyclic endoperoxide. The first step in the formation of the bicyclic endoperoxide is 5-exo-cyclization to form a 1,2-dioxolanyl radical (Fig 14). This cyclization helps bring the two halves of the acyl chain into juxtaposition and allows the radical to react in trans with a double bond to form a cyclopentane ring. The resulting carbon center radical undergoes delocalization and then rapidly reacts with any available molecular oxygen to form a peroxide. In the presence of iron this peroxide undergoes scission to form the hydroxyl radical and then a stable hydroxyl group. Alternatively, the peroxide can react with an adjacent double bond to form an epoxide.
Figure 14.

Products of lipid 1,2-dioxoanyl radical include bicyclic endoperoxides, diepoxyalcohols, isofurans, and serial cyclic endoperoxides. 5-exo-cyclization of initial peroxyl radical generates 1,2-dioxolanyl radical. If the radical reacts with double bond on opposite side of the endoperoxide, a bicyclic endoperoxide is formed that can then go on to form various prostaglandin-like moieties. Alternatively, 1,2-dioxoanyl the radical can react with the endoperoxide to form a diepoxy radical that with the addition of another molecule of oxygen leads to the generation of diepoxyalcohol. This diepoxyalcohol has been postulated to be a precursor for the isofurans. Finally, the 1,2-dioxoanyl radical can react with a molecule of oxygen and then, if another double bond is appropriately positioned, undergo another round of 5-exo-cyclization to form a serial cyclic endoperoxide.
Bicyclic endoperoxides with an additional hydroxyl group on the acyl chain are given the trivial name of H-type isoprostanes, based on their structural similarity to prostaglandin H2. A subscript after the H is added to designate the number of double bonds in the esterified isoprostane (IsoP). To designate the precise regioisomer, the carbon number of the hydroxyl group is added prior to ring designation. Thus, the oxidation of arachidonyl-PC generates four regioisomers of bicyclic endoperoxides: 5-H2-IsoP-PC, 8-H2-IsoP-PC, 12-H2-IsoP-PC and 15-H2-IsoP (Fig 15).
Figure 15.

Formation of a large family of prostaglandin like molecules (isoprostanes, IsoP) by peroxidation of arachidonyl-PC. Initial peroxidation forms four regioisomers of bicyclic endoperoxides (H2-IsoP), designated by the position of their hydroxyl group. Each of these regio-isomers can then form PC esterified with F2-IsoP, A2-isothromboxane (A2-IsoTx), D2-IsoP, E2-IsoP, epoxy IsoP (potentially including epoxy E2, D2, F2, A2, and J2-IsoP), and the acyclic isolevuglandin (IsoLG). Only F2-IsoP is chemically stable, with the remaining products undergoing further chemical reactions to form secondary products. In particular, IsoLG reacts with any nearby amines to form pyrrole adducts.
Bicyclic endoperoxides are quite unstable and the endoperoxide ring quickly opens to form more stable products (Fig 14). Fragmentation of the oxygen-oxygen bond of the endoperoxide ring forms F-, D-, and E- type IsoP, with F-type being the most stable (Fig 14). For this reason, F2-IsoP-PL are widely used as markers of lipid peroxidation in vivo. D- and E-type IsoP can rapidly dehydrate to form J- and A-type IsoP, respectively. These cyclopentone IsoP can react via Michael addition with cellular thiols like cysteine to form thiol adducts. The epoxy analog of each of these IsoP can also form, with 5,6-epoxy-E2-IsoP-PC (EI-PC) being the most commonly studied.
Fragmentation of the carbon-carbon bond of the endoperoxide ring, rather than its oxygen-oxygen bond, forms γ-ketoaldehydes given the trivial name of isolevuglandins (IsoLG) or isoketals. To correspond to the prostaglandin literature, IsoLG with the ketone on the same side as the carboxylate are designated as E-type IsoLG, while those with the ketone on the opposite side from the carboxylate are designated D-type IsoLG. Thus a total of 8 IsoLG regioisomers are formed by peroxidation of arachidonyl-PC. These γ-ketoaldehydes react nearly instantaneously with any nearby primary amines to form pyrrole adducts. The reaction of esterified IsoLG with the ethanolamine headgroup of PE will be discussed more extensively in section 5.1.3.
4.1.2. Diepoxyalchohol-PC, Isofuran-PC, and serial cyclic endoperoxide-PC
There are alternative chemical pathways for the 1,2-dioxolanyl radical besides formation of the bicyclic endoperoxide. For instance, rather than the carbon centered radical reacting in trans with a juxtaposed double bond, it can instead react with the endoperoxide to form two epoxide rings, and then react with molecular oxygen to form a diepoxyalcohol (Fig 14). These diepoxyalcohols have also been postulated to be the primary precursor of the isofurans (IsoF) (Jahn et al., 2008). This mechanism is as an alternative to the original mechanisms proposed for IsoF formation (Fessel et al., 2002). Another alternative pathway for the 1,2-dioxolanyl radical, if it includes appropriately positioned additional double bonds, is the addition of molecular oxygen to form serial cyclic endoperoxides (Fig 14).
4.2. Formation of oxPL with cyclized acyl chains in biological samples
Measurement of unesterified 15-F2-IsoP by GC/MS after derivatization is one of the most widely used methods to assess lipid peroxidation in vivo. Often this method also indirectly measures esterified 15-F2-IsoP because samples are treated with base or phospholipases to convert the esterified 15-F2-IsoP to unesterified 15-F2-IsoP. Other isomers, such as 5-F2-IsoP, have also occasionally been used for these measurements. Physiological and pathological conditions where esterified 15-F2-IsoP increase have been cataloged in numerous reviews and include cardiovascular diseases, neurodegenerative diseases, obesity, aging, cancer, exposure to industrial chemicals and radiation, and many other stressors (Basu, 2008; Davies and Roberts, 2011; Jahn et al., 2008; Milatovic and Aschner, 2009; Milne et al., 2005b; Milne et al., 2011; Roberts et al., 2005). While the standard base hydrolysis method cannot distinguish between PL-, cholesterol-, and triglyceride-esterified IsoP, what studies that have been done to elucidate the donor of the esterified suggest that phospholipids are the primary carriers of IsoP. Direct measurement of 15-F2-IsoP-PC and –PE have been performed in a limited number of cases (Kayganich-Harrison et al., 1993; Yin et al., 2009). It should also be noted that while plasma measurements of F2-IsoP typically measure only the unesterified 15-F2-IsoP, there is generally reasonable correlation between plasma esterified 15-F2-IsoP and plasma unesterified 15-F2-IsoP. In plasma, HDL is the major lipoprotein carrier of both free and esterified 15-F2-IsoP and this is likely to be due at least in part to the ability of ApoAI to facilitate transfer of oxPL from tissue to HDL.
Although it is anticipated that 5,6-epoxy-E2-IsoP (EI-PC) would be formed proportionately to F2-IsoP-PC, there is very little data on its formation in vivo. A biologically active oxPL with m/z 828.6 was originally identified in oxPAPC and the aortic levels of this unidentified oxPL was shown to increase in mice fed an atherogenic diet (Watson et al., 1997). Subsequently, the oxPL in oxPAPC with m/z 828.6 was characterized as EI-PC (Watson et al., 1999). The only other published report specifically measuring epoxy-isoprostane in vivo appears to be one showing that EI-PC accumulates in macrophages infected with the leprosy causing bacteria (Cruz et al., 2008). While the E06 antibody commonly used as marker of oxPL formation in vivo does recognize EI-PC (Watson et al., 1997), this is also true of many other oxPL, so increased levels of E06 binding in disease is not definitive evidence for increased levels of EI-PC.
Like other members of the IsoP family, levels of esterified IsoLG would be anticipated to rise under the same conditions as for esterified 15-F2-IsoP; however, because of their reactivity, IsoLG cannot be measured in their unreacted state in vivo. Instead, esterified IsoLG are measured as their pyrrole, lactam, and hydroxylactam adducts to proteins, PE, and DNA. As with 15-F2-IsoP, these adducts are almost always measured after base hydrolysis. Thus, while phospholipids are assumed to be the primary donors for esterified IsoLG adducts, this cannot be claimed with absolute certainty. Increased IsoLG-protein adducts have been found in atherosclerosis, myocardial infarction, end stage renal disease, industrial chemical exposure, hyperoxia, allergic inflammation, experimental sepsis, ethanol ingestion, glaucoma, and bright light exposure (Brame et al., 2004; Charvet et al., 2013b; Davies et al., 2004; Fukuda et al., 2005; Govindarajan et al., 2008; Poliakov et al., 2004; Roychowdhury et al., 2009; Salomon et al., 2000; Talati et al., 2006). Evidence for modification of PE by esterified IsoLG will be discussed in section 5.2.
Very little in vivo data is available about the formation of esterified diepoxy alcohols, IsoF, or serial cyclic endoperoxides. Esterified IsoF are increased in post-mortem substantia nigra taken from patients with Parkinson’s disease compared to aged-matched controls (Fessel et al., 2003). Esterified IsoF also form in the liver after carbon tetrachloride poisoning (Song et al., 2008). Plasma levels of unesterified IsoF, which likely reflect at least to some extent changes in esterified IsoF in tissue, are elevated in age-related macular degeneration, severe sepsis, mercury exposure, acute kidney injury, and preterm resuscitation (Alkazemi et al., 2013; Billings et al., 2011; Brantley et al., 2012; Montine et al., 2004; Vento et al., 2009; Ware et al., 2011).
4.3. Biological activities and signaling pathways of cyclized oxPL
Little is known about the activity of 15-F2-IsoP-PC or 15-F2-IsoP-PE because of the difficulty in synthesizing large amounts of these compounds. While 15-F2-IsoP-PL can obviously serve as a precursor to generate 15-F2-IsoP, it seems reasonable to speculate that 15-F2-IsoP-PL may also have unique activities not dependent on their hydrolysis. Nevertheless, there are currently no published studies that we are aware of that have used synthetic 15-F2-IsoP-PL (or any other F2-IsoP-PL regioisomer) to access such bioactivity. Ironically, although the synthesis of 5,6-epoxy-E2-IsoP-PC (EI-PC) is even more challenging than the synthesis of 15-F2-IsoP-PC, far more is known about the biological activity of EI-PC because the UCLA group that discovered it was willing and able to invest sufficient time and resources to develop robust methods for its synthesis (Jung et al., 2005; Jung et al., 2008).
Like 15-F2-IsoP-PC, the difficulty of synthesizing IsoLG-PC or IsoLG-PE isomers has made it difficult to directly ascertain the biological activity of the esterified forms of IsoLG. Unlike 15-F2-IsoP, however, much of the effect of unesterified IsoLG on protein function appears to be dependent on its ability to modify lysine residues, rather than the precise chemical composition of the side chains. This conclusion is drawn from experiments where IsoLG analogs lacking the side chains (e.g. 4-oxo-pentanal), or that are esterified (e.g. methyl-IsoLG) invoke similar effects as IsoLG itself. Thus, it seems reasonable to speculate that IsoLG-PC and IsoLG-PE may invoke at least some, if not most, of the same effects on proteins as unesterified IsoLG.
4.3.1. Thromboxane A2 receptor: smooth muscle cell contraction and platelet activation
Unesterified 15-F2-IsoP and 15-E2-IsoP are potent vasoconstrictors with 100 nM being sufficient to induce this effect, which can be blocked by thromboxane A2 (TxA2) receptor (TP) antagonists(Cracowski et al., 2001; Hoffman et al., 1997; Mohler et al., 1996; Morrow et al., 1994; Mueed et al., 2008; Wagner et al., 1997). Similarly, unesterified 15-F2-IsoP induces smooth muscle cell contraction and bronchoconstriction in the lung and this effect is blocked by TP antagonists (Bernareggi et al., 1998). In hepatic stellate cells, unesterified 15-F2-IsoP induces cell proliferation and collagen synthesis and these effects are also block by TP antagonists (Gardi et al., 2008). In these same cells, 15-F2-IsoP induces IP3 formation, decreases cAMP levels, and activates ERK, p38MAPK, and JNK (Acquaviva et al., 2013). Treatment of platelet rich plasma 15-F2-IsoP induces platelet expression of P-selectin and TxA2 synthesis (Spinelli et al., 2013).
Because 15-F2-IsoP and 15-E2-IsoP induce TxA2 synthesis, the ability of TP antagonists to block their effects does not imply they are direct TP agonists. Nevertheless, studies do indicate that 15-F2-IsoP directly binds TP (Kd 57 nM) and activates TP with the Phe196 residue of TP being critical for this activation by 15-F2-IsoP, but not for other agonists such as TxA2(Khasawneh et al., 2008). Interestingly, 15-F2-IsoP appears to activate TPα/TPβ heterodimers better than either homodimer (Wilson et al., 2007). Several other F2-IsoP regio- and stereo-isomers, including 12-F2-IsoP are able to evoke vasoconstriction, but 5-F2-IsoP stereoisomers are relatively poor activators (Hou et al., 2004).
Very low concentrations of 15-E2-IsoLG (1 nM) potentiates the activation of platelets and TxA2 synthesis induced by sub-threshold concentrations of collagen (Bernoud-Hubac et al., 2009). Whether this activity is mediated by TP has not been evaluated. 15-E2-IsoLG induced p38MAPK activation and cPLA2 activity. Platelet potentiation was blocked by the aldehyde scavenger pyridoxamine, indicating that 15-E2-IsoLG modification of either a protein target or of PE was responsible for this effect. Because N-IsoLG-PE activates p38MAPK in endothelial cells, a primary role for N-IsoLG-PE seems likely. Platelet potentiation was also blocked by the PLA2 inhibitor AACOCF3, consistent with arachidonic acid release and TxA2 synthesis participating in the potentiation by 15-E2-IsoLG.
4.3.2. EP2 receptor: activation of endothelial cells, cytokine secretion and monocyte adhesion
EI-PC activates endothelial cells to display adhesion molecules and secrete cytokines, facilitating the binding of monocytes (Watson et al., 1997; Watson et al., 1999). Interestingly, other regioisomers of EI-PC including 8,9-, 11,12-, and 14,15-EI-PC epoxy-E2-isoprostane-PC also induced IL-8 and MCP-1 secretion, as did their dehydrated epoxy-A2-isoprostane-PC analogs, and all of these analogs were more potent than OV-PC and glutaryl-PC (Subbanagounder et al., 2002b). EI-PC elicits this activation via cAMP mediated responses including R-Ras activation and PI3-Kinase activity (Cole et al., 2003). Therefore, Li et al screened EI-PC against candidate Gs-coupled receptors expressed in HEK293 cells. Among these candidate Gs-coupled receptors, 5,6-EI-PC was found to potently activate PGE2 receptor sub-type 2 (EP2) (EC50 109 nM), while OV-PC did not (Li et al., 2006). In comparison, the EC50for PGE2 itself is 31 nM. EI-PC also competes with PGE2 for binding to EP2 and the DP/EP1/EP2 antagonist AH6809 blocks the ability of EI-PC to induce IL-10 in monocytes. The selective EP2 agonist butaprost induces monocyte binding to endothelium in a similar manner as EI-PC. Thus EI-PC appears to be an authentic EP2 agonist and activation of EP2 likely accounts for many of its activities in endothelium and monocytes.
15-F2-IsoP may also act via EP2, but the evidence is more ambiguous. There have been a number of reports suggesting that there are additional receptors for 15-F2-IsoP besides TP (Fukunaga et al., 1993; Fukunaga et al., 1995; Fukunaga et al., 1997). In the pulmonary vasculature, 15-E2-IsoP was able to induce contraction even in the presence of TP antagonists, and this non-TP dependent signaling could be blocked by the DP/EP1/EP2 antagonist AH 6809 (Janssen and Tazzeo, 2002). 15-E2-IsoP induced airway smooth muscle contraction could also be inhibited by AH 6809 as well as by EP4 antagonists (Clarke et al., 2005). Similarly, at high concentrations (30 μM) 15-F2-IsoP inhibits potassium-depolarization-evoked release of aspartate from retinae and again this effect is attenuated by AH 6809 (Opere et al., 2005). 5-F2-IsoP also attenuates this release, but this is blocked by EP1 and EP4 antagonists (Jamil et al., 2012). Examination of high affinity binding sites for 15-F2-IsoP in platelets identified a second binding site which signals through increasing cAMP, but this binding site did not appear to be any of the known prostanoid receptors such as EP, DP, or IP (Khasawneh et al., 2008).
15-E2-IsoLG also induces monocyte binding to endothelial cells (EC50 ~5 μM) (Guo et al., 2011). As will be discussed in subsequent section on N-modified phospholipid, this effect appears to be mediated via reaction of 15-E2-IsoLG with PE to form N-IsoLG-PE, rather than directly by 15-E2-IsoLG itself or by modification of protein. Currently, there is no direct evidence that N-IsoLG-PE acts via EP2 or other prostanoid receptors. A very early report on the effects of the dehydration product of 15-E2-IsoLG suggested that it competed with PGE2 for binding to a receptor that induced uterine contraction (Foreman and Solomon, 1992; Foreman et al., 1987), but uterine contractions appear to be mediated by EP1 and EP3, rather than EP2(Lebel et al., 2004).
4.3.3. Endothelial barrier function and ER stress response
Physiologically relevant concentrations (~1 μM) of EI-PC enhance endothelial barrier function and can reverse the effects of OV-PC (Birukova et al., 2013). EI-PC markedly increases VE-cadherin expression, which likely accounts for its effects on barrier function. Treatment of endothelial cells with modest concentrations of EI-PC (1.2 μM) induces ER stress responses such as increased ATF3 mRNA (Gharavi et al., 2007). EI-PC is one of the components of oxPAPC (among many other oxPL) and treatment of endothelial cells with oxPAPC alters expression of >1000 genes, including those of the ER stress response such as ATF4 and XBP1 (Gargalovic et al., 2006b). The relationships between EI-PC, ER stress response induced by oxPAPC, and endothelial cell activation need to be more rigorously defined.
The effect of unesterified 15-E2-IsoLG on endothelial cell barrier function has been examined by intrahemispherical injection and measurement of blood brain barrier function by Evans Blue dye. 15-E2-IsoLG markedly worsens barrier function, being more potent than PGE2 at inducing leak of Evans blue dye. 100 nmol 15-E2-IsoLG was sufficient to induce visible necrosis (Schmidley et al., 1992).
4.3.4. Inhibition of dendritic cell function and T cell activation
Appropriate antigen presentation to T cells and the ensuing immunogenic responses requires differentiation of dendritic cells, which is marked by increased expression of CD1. Exposure to oxLDL suppresses CD1 expression, so that the accumulation of EI-PC in the dendritic cells of leprous individuals led Cruz et al to examine the effects of EI-PC on dendritic cell differentiation and function (Cruz et al., 2008). They found that EI-PC inhibited expression of CD1 but not MHC class II. Pretreatment of dendritic cells with 600 nM EI-PC almost entirely blocked their ability to induce T-cell activation, as measured by IFNγ secretion. EI-PC also blocked TLR1/2 ligand induced IL-12 secretion and CYP27b1 expression but enhanced IL-10 secretion (Cruz et al., 2008).
4.3.5. PPARα activation
Modest levels (1 to 2 μM) of both epoxy-E2-IsoP-PC and epoxy-A2-IsoP-PC induced 5 to 15 fold increases in PPARα activity, suggesting these were potent ligands for the PPARα (Subbanagounder et al., 2002b).
4.3.6. Other receptors and signaling mechanisms for IsoP
12-F2-IsoP activates the FP receptor of human cilary bodies, which may be important in intraocular pressure (Kunapuli et al., 1997). 15-F2-IsoP induces expression of lectinlike oxidized LDL receptor-1 in trophoblast cells, along with subsequent uptake of LDL by these cells (Halvorsen et al., 2001). 15-A2-IsoP invokes anti-inflammatory effects at high concentrations, which may be due to its ability to modify IKK leading to NFκB inhibition (Lappas et al., 2007).
ApoAI binds and extracts oxPL from tissue, which may account for the finding that HDL is the major carrier of IsoP-PC in circulation (Proudfoot et al., 2009). The ApoAI mimetic peptide L-4F binds to EI-PC with far greater affinity (Kd 10 pM) than it does to OV-PC (Kd 3.6 nM) or KOdiA-PC (Kd 1.6 nM)(Van Lenten et al., 2008). Gharavi et al found that HDL inhibits the ability of EI-PC to induce IL-8 and LDL receptor mRNA expression by endothelium (Gharavi et al., 2007). HDL also inhibits the ability of EI-PC to induce ER stress response. Interestingly, HDL fails to inhibit the ability of EI-PC to induce redox genes such as heme oxygenase-1 and heat shock protein A1A and this appears to be because HDL fails to inhibit NRF-2 activation by EI-PC (Gharavi et al., 2007). The mechanisms accounting for this differential effect is not currently understood.
4.3.7. Induction of cell death: inhibition of proteasome function and mitochondrial respiration
One of the earliest observations about the effects of IsoLG was their ability to induce cell death. As mentioned previously, even low concentrations of 15-E2-IsoLG induced necrosis in the brain (Schmidley et al., 1992). 15-E2-IsoLG also potently induces cell death in fibroblasts (LC50 = 230 nM) and neurons (LC50=670 nM) (Davies et al., 2002; Murthi et al., 1993). Interestingly, IsoLG modification of neurotoxic peptides, such as amyloid beta1-42, further increases their toxicity (Boutaud et al., 2006). A number of mechanisms may underlie this cytotoxicity including inhibition of proteasomal degradation (Davies et al., 2002), endoplasmic recticulum stress (Guo et al., 2011), and inhibition of mitochondrial respiration and subsequent induction of mitochondrial swelling and cytochrome c release (Stavrovskaya et al., 2010).
Inhibition of proteasomal degradation induces accumulation of protein aggregates and cell death. The ability of IsoLG to induce crosslinking and aggregation of proteins, led to exploration of their effects on the ability of the proteasome to degrade modified proteins. Modification of a model unfolded protein with equimolar IsoLG led to significantly reduced hydrolysis by the 20S proteasome, while modification of this same protein with 10 molar equivalents of IsoLG led to complete inhibition of its degradation by the 20 S proteasome (Davies et al., 2002). IsoLG modification of other proteolytic enzymes, such as calpain-1 also inhibits their activity and renders them resistant to proteasomal degradation so that they accumulate in cells (Govindarajan et al., 2008). IsoLG-modified protein inhibits degradation of unmodified protein as well (Davies et al., 2002). Presumably this inhibition results from the non-degradable IsoLG-modified protein remaining within the barrel of the proteasome, blocking access by other target proteins to the protease sites. Accumulation of various proteins would then trigger ER stress and apoptotic cell death.
The physiological relevance of IsoLG mediated inhibition of proteasomal degradation is currently unclear. IsoLG-modified proteins accumulate in glaucomatous trabecular meshwork, particularly in astrocytes, and the inhibition of proteasomal degradation by IsoLG has therefore been postulated to contribute to glaucoma pathology (Govindarajan et al., 2009; Govindarajan et al., 2008). Proteasome activity is markedly decreased in Alzheimer’s disease and appears to contribute to neuronal loss in the disease (Cecarini et al., 2007; Keller et al., 2000; Lopez Salon et al., 2003). IsoLG and their analogous docosahexaenoic acid analogs, neuroketals, accumulate in the brains of Alzheimer’s patients and in animal models of neurodegeneration, so that they may contribute to this loss of proteasomal activity (Davies et al., 2011; Zagol-Ikapitte et al., 2005). Currently, one of the best methods to assess the contribution of IsoLG and other amine-directed reactive aldehydes to disease is the use of selective aldehyde scavengers such as pyridoxamine, pentylpyridoxamine, and salicylamine that prevent IsoLG modification of proteins (Amarnath et al., 2004; Davies et al., 2006a; Zagol-Ikapitte et al., 2010a; Zagol-Ikapitte et al., 2010b). It is therefore interesting to note that treatment with salicylamine blocked the age-related loss of working memory in a mouse model of Alzheimer’s disease (Davies et al., 2011). The effects of the aldehyde scavenger on proteasomal activity was not assessed in these mice, so the exact relationship between accumulation of IsoLG-modified proteins, changes in proteasomal activity, neurodegeneration, and loss of working memory needs to be more fully elucidated.
In addition to proteasomal inhibition, another mechanism whereby IsoLG may induce cell death is by their effects on mitochondrial respiration. 15-E2-IsoLG dose dependently (EC50 1.6 μM) potentiates mitochondrial swelling induced by calcium (Stavrovskaya et al., 2010). 15-E2-IsoLG activates other aspects of mitochondrial driven cell death pathway, such as inhibition of respiration, induction of the mitochondrial permeability transition, and cytochrome c release. Inhibition of the permeability transition by cyclosporin A blocked 15-E2-IsoLG induced mitochondrial swelling but not cytochrome c release. Cytochrome c release appears to be driven by direct IsoLG modification, as IsoLG modified cytochrome c bound to mitochondria with much poorer affinity than unmodified cytochrome c (Stavrovskaya et al., 2010). Further studies are needed to assess the contribution of IsoLG induced changes in mitochondrial function and to more fully elucidate the relationship between mitochondrial function and IsoLG mediated cell death. Studies examining the effect of aldehyde scavengers should be particularly helpful in this regard.
4.3.8. Inhibition of ion channels
Modification of ion channels by IsoLG and IsoLG-PL may be important in the proarrhythmic effects of oxidative injury. Proteins modified by IsoLG accumulate in the epicardial border zone of the heart after infarction, in the same region where alterations in Na+ and K+ channel activity lead to development of ventricular arrhythmia (Boyden et al., 2007; Fukuda et al., 2005). 15-E2-IsoLG inhibited inward K+ rectifying current in an atrial tumor myocyte cell line, AT-1 (IC50 2.2 μM), (Brame et al., 2004). Interestingly, maximal inhibition only occurred after 60 minutes, which is consistent with the time course of crosslink formation, suggesting that crosslinking of the K+ channel was required for its inhibition. Furthermore, both the activating and deactivating currents were inhibited, suggesting total loss of K+ channel function, rather than selective inhibition.
15-E2-IsoLG modification also potentiated inactivation of sodium current in myocytes (HL-1) and in HEK293 heterologously expressing Nav1.5 (Fukuda et al., 2005). In contrast to the K+ channel, IsoLG modification had no effect on the activation phase of Nav1.5. Treating Nav1.5 expressing cells with oxidants also potentiated inactivation of sodium current and several lines of evidence implicate IsoLG as the primary active agent in oxidative stress induced Nav1.5 inactivation. First, while oxidant treatment of Nav1.5 expressing cells generates many oxPL products including IsoLG protein adducts, HNE, and 15-F2-IsoP, neither 15-F2-IsoP nor HNE could mimic the effect of synthetic IsoLG on Nav1.5 (Fukuda et al., 2005). Additionally, both IsoLG and oxidant induced Nav1.5 inactivation could be blocked by salicylamine and pentylpyridoxamine, which readily scavenge IsoLG but not HNE (Nakajima et al., 2010). Oxidation of Nav1.5 expressing cells leads to direct lipid modification of Nav1.5 and salicylamine blocks this modification (Nakajima et al., 2010). These studies support the proposed working model where myocardial infarction generates IsoLG that subsequently modifies Nav1.5, causing it to more readily inactivate and therefore resulting in arrhythmia (Boyden et al., 2007). Current studies are underway to examine the effect of aldehyde scavengers such as salicylamine and pentylpyridoxamine on infarction induced ventricular arrhythmias.
4.3.9. Inhibition of sterol metabolism: CYP27A1 modification
Cholesterol homeostasis in an important aspect of many cells, and the mitochondrial cytochrome P450 enzyme CYP27A1 functions to eliminate cytotoxic 7-ketocholesterol from cells including the retina. Deficiencies in CYP27A1 cause cerebrotendinous xanthomatosis and mice lacking CYP27A1 have focal cholesterol deposits in their retina and faulty vascularization (Omarova et al., 2012). Exposure to light leads to oxidative post-translational modification of CYP27A1 in retina, but not brain, which is likely due to light induced peroxidation of PUFA. These observations led Charvet et al to characterize post-translational modification of CYP27A1 in the retina, and they demonstrated that specific lysine residues of CYP27A1 are modified by IsoLG (Charvet et al., 2011). Furthermore, they demonstrated that modification of CYP27A1 by 12-E2-IsoLG markedly inhibits its activity and pinpointed the exact Lys residues responsible. In particular, they showed that substituting Arg for Lys-358 protected against IsoLG mediated loss of function (Charvet et al., 2013a). Most recently, they demonstrated that the aldehyde scavenger pyridoxamine blocked bright light induced formation of IsoLG-protein adducts in the retina and the development of abnormal mitochondrial morphology and autofluorescence in these retina (Charvet et al., 2013b).
4.4. Metabolism
To date, the primary known metabolic enzyme for cyclized oxPL is PAF-acetylhydrolase (PAF-AH). Although these cyclized oxPL are not as good of substrates for hydrolysis as oxidatively truncated oxPL, they are still readily hydrolyzed by recombinant plasma PAF-AH (Stafforini et al., 2006). In mice lacking the intracellular type II PAF-AH (PAF-AH2−/− mice), carbon tetrachloride treatment markedly elevated PC esterified levels of 15-F2-IsoP, suggesting that this enzyme is also critical for their hydrolysis (Kono et al., 2008). It is not clear if esterified IsoLG that covalently modify proteins are substrates for PAF-AHs. It is of interest that the initial imine adduct formed during carbon tetrachloride exposure is almost entirely the esterified form, while later evolving adducts are unesterified, raising the possiblility that they are hydrolyzed by PAF-AH to some extent (Brame et al., 2004).
After formation of unesterified IsoP, the metabolism of IsoP is similar to that of prostaglandins with dehydrogenation and beta-oxidation except that dinor-metabolites, rather than tetranor-metabolites are formed (Awad et al., 1993; Chiabrando et al., 1999; Milne et al., 2005a). These dinor-metabolites are readily detected in the urine by GC/MS and LC/MS methodologies (Davies et al., 2006b; Morales et al., 2001; Morrow et al., 1999).
5. Class IV: Oxidatively N-modified phospholipids (PL)
5.1. Chemical mechanisms of N-modification of phospholipids
The first three classes of oxPL all include some species of reactive lipids. For instance, class I oxPL includes15-KETE-PE, class II oxPL includes OV-PC and KOdiA-PC as well as the complementary diffusable hydroxyalkenals formed during fragmentation. Class III oxPL include the extremely reactive IsoLG-phospholipids as well as EI-PC. Additional sources of lipid aldehydes include long-chain unsaturated aldehydes formed during oxidation of plasmalogens and malondialdehyde (MDA) formed both by lipid peroxidation and by thromboxane synthesis.
Lipid aldehydes (and ketones) react with primary amines to form imine adducts or in the case of α,β-unsaturated lipid aldehydes, to form both imine adducts and Michael adducts (Fig 16). Two groups of glycerophospholipids, PE and PS, are ready targets for reaction with lipid aldehydes because their headgroups sport primary amines. In contrast, PC with its quaternary amine does not react with lipid aldehydes. PI lacks primary amines, so it is also not a ready target for lipid aldehyde modification.
Figure 16.

Reaction of lipid aldehydes with amines generates imine or Michael adducts. Targeted amines include the lysyl residues of proteins, PE, PS, nucleic acids, and small molecular cytosolic amines such as spermidines. Recent studies suggest that some aldehydes like IsoLG modify PE to a greater extent than other targets.
5.1.1. PE modified by 4-hydroxyalkenals: N-HNE-PE, N-HHE-PE, and N-HDDE-PE
4-hydroxy-2E-nonenal (HNE) is perhaps the most intensively studied of the reactive lipid aldehydes. Increased levels of HNE protein adduct have been detected in many degenerative diseases. While HNE preferentially undergoes Michael addition with the cysteine residues of protein, it also reacts with the lysine residues of proteins via Michael addition and imine formation (Nadkarni and Sayre, 1995). The ability of HNE to react with lysine in vivo raised the possibility it would also react with other primary amines in vivo such as PE and PS.
In 1986, Esterbauer et al found that HNE formed fluorescent chromophores with phospholipids (Esterbauer et al., 1986). Later, Guichardant et al chemically characterized the reaction of HNE with PE and PS. Under their experimental conditions, they found that the major product of reaction with PE was the Michael adduct, while the imine and pyrrole adducts were formed to a lesser extent (Guichardant et al., 1998). When reacting HNE with PS, the only product they detected was the Michael adduct, probably due to the poor accessibility of the amine in PS (Fig 17).
Figure 17.

Characterized products of HNE reaction with PE and PS. Reaction of PE with HNE generated primarily the N-HNE-PE Michael adduct, but also the N-HNE-PE pyrrole adducts. Reaction of HNE with PS generated only very low yield of N-HNE-PS Michael adduct (Guichardant et al., 1998).
Besides HNE, other hydroxyalkenals derived from n-3 fatty acids including 4-hydroxy-2E-hexenal HHE) and 4-hydroxydodeca-(2E,6Z)-dienal (HDDE) also modify PE, generating both Michael adducts and Schiff bases. The reactivity of hydroxyalkenal to PE is proportional to the hydrophobicity of the alkenal, with HHDE > HNE> HHE. Interestingly, all these hydroxyalkenals favor modification of plasmalogen PE over other PEs (Bacot et al., 2003).
5.1.2. Acrolein modified PE
Although not as reactive as hydroxyalkenals and ketoalkenals, acrolein and other α,β-unsaturated lipid aldehyde without these γ-functional groups still react with primary amines via Michael addition and imine bonds (Esterbauer et al., 1991). Characterization of acrolein-modified lysine demonstrated that lysine formed Schiff base or 1,4-Michael adducts with one acrolein molecule first, followed by a 1,4-Michael addition of a second acrolein. The compound was further cyclized under aldol condensation to form Nα-acetyl-Nε-(3-methylpyridinium)lysine N-MP-Lys or N ε-(3-formyl-3,4-dehydropiperidino)lysine (N-FDP-Lys) (Furuhata et al., 2003; Uchida et al., 1998) (Fig 18). Berry and Murphy investigated acrolein reactions with PE and they found only N-FDP-PE and not the imine adduct or its secondary product N-MP-PE (Zemski Berry and Murphy, 2007). Of course, this does not exclude the possibility that such adducts might form under different conditions.
Figure 18.
Proposed products of PE reaction with acrolein. Based on the reaction of acrolein with lysine, it was expected that the reaction of acrolein with PE would generate both N-(3-methylpyridinium)-PE (N-MP-PE) and N-(3-formyl-3,4-dehydropiperidino)-PE (N-FDP-PE) (Furuhata et al., 2003; Uchida et al., 1998). However, only formation N-FDP-PE has been detected in vitro (Zemski Berry and Murphy, 2007).
5.1.3. IsoLG modified PE
Bernoud-Hubac et al characterized the reaction of 15-E2-IsoLG with PE and found that IsoLG covalently modifies PE with greater reactivity than HNE (Bernoud-Hubac et al., 2004). Using LC-MS/MS, they detected an initial imine adduct that rapidly evolved into more stable pyrrole species including lactam and hydroxylactam. Formation of the hydroxylactam species predominates during t-butylhydroperoxide catalyzed peroxidation of PL (Guo et al., 2012). These findings are consistent with lysine modification by IsoLG where reaction of IsoLG first forms a hemiaminal adduct, which is either directly dehydrated to Schiff base or first cyclized to pyrrolidine and then dehydrated to pyrrole (Fig 19). Like HNE, IsoLG can also modify PS, but does so with extremely poor yield. Of great importance was the recent discovery that exogenous addition of IsoLG to cells results in more PE modification than protein modification (Sullivan et al., 2010), despite the high reactivity of IsoLG for lysines (Brame et al., 1999). This suggests that many of the effects of IsoLG previously attributed to modification of protein may in fact be due to its ability to modify PE.
Figure 19.

Characterized products of IsoLG reaction with PE. Reaction of IsoLG with PE initially forms hemiaminal, which then either forms the highly irreversible imine adduct (minor product) or the highly stable N-IsoLG-PE pyrrole adduct (major product). Under oxidizing conditions, the pyrrole adduct generally rapidly evolves to form the N-IsoLG-PE hydroxylactam adduct (Bernoud-Hubac et al., 2004; Li et al., 2009; Sullivan et al., 2010).
5.1.4. Malondialdehyde (MDA) modified PE
MDA is one of the most abundant lipid fragments formed in lipid peroxidation. It covalently reacts with amines including lysine, PE, and PS to form N-propenal-, N-1,4-dihydropyridine-3,5-dicarbeldehyde- and N-propenal-crosslinks (including protein-phospholipid crosslinks) as well as other uncharacterized adducts (Fig 20) (Bhuyan et al., 1996; Borchman et al., 1989; Burcham and Kuhan, 1996; Draper et al., 1988; Kikugawa et al., 1984; Uchida et al., 1997). More recently, Guo et al analyzed the reaction of MDA with PE and confirmed the formation of N-1,4-dihydropyridine-3,5-dicarbeldehyde-PE (N-DHP-PE) and N-propenal-PE, although other products were also present (Guo et al., 2012).
Figure 20.
Characterized products of malondialdehyde (MDA) reaction with PE. Reaction of MDA with PE initially forms imine adduct, which undergoes double bond rearrangement to form the more stable N-propenal-PE. This can then undergo further reaction to yield crosslinked products (either with PE or proteins) or undergo reaction with two additional MDA to form N-1,4-dihydropyridine-3,5-dicarbeldehyde-PE (N-DHP-PE) (Guo et al., 2012).
5.1.5. Amide-linked PE (N-Acyl PE and N-Carboxyacyl PE)
Recently, an entire family of oxidatively N-modified PE with amide linkages has been discovered. The initial member of this family, N-hexanoyl-PE, was found byTsuji et al when they examined the reaction of 13-HPODE with PE (Tsuji et al., 2003). Guo et al subsequently systematically characterized the most abundant aldehyde-modified PE generated by the oxidation of arachidonate in the presence of dipalmitoyl-PE. These included several N-acyl-PE with short chains such as N-formyl-PE, N-acetyl-PE, N-hexanoyl-PE, and N-4-hydroxy-nanenoyl-PE (Guo et al., 2012) (Fig 21). Even more interesting was the finding of a number of N-carboxyacyl-PE (N-CA-PE) including N-C1:0CA-PE, N-C4:0CA-PE, N-C7:1CA-PE, and N-C9:2CA-PE.
Figure 21.
Major species of oxidatively N-modified PE with amide linkage found during in vitro peroxidation of arachidonyl-PC in presence of PE (Guo et al., 2012).
Currently no general mechanism for the formation of an amide bond from the reaction of lipid aldehydes with primary amines has been described. However, t-butylhydroperoxide is used in synthesis reactions to peroxidize imine bonds, which then lose water to form the amides, so we speculate that similar reaction would occur with lipid peroxides. A similar type of reaction from imine to amide has been proposed during the reaction of 4-hydroperoxy-nonenal with lysine to form N-4-hydroxy-nanenoyl-lysine (Shimozu et al., 2011). Thus the aldehydes formed during fragmentation such as hexanal and 8-oxo-octaenoate-PC could react with PE to initially form an imine bond, and then peroxidation of this bond by LOOH would rationalize the formation of N-hexanoyl-PE and N-C7:1CA-PE, respectively (Fig 22). If this reaction mechanism is correct, then these products should arise from the γ-ketoalkenals and alkenals species generated during formation of class II oxPL, so that at least some of the ω-carboxyl groups will be esterified at the sn-2 group of a PC or PE to form a phospholipid-phospholipid crosslink. Subsequent hydrolysis by PLA2 would then release the cross-linked phospholipids to generate N-CA-PE and lysoPC.
Figure 22.

Proposed mechanisms for formation of N-modified PE with amide linkage. Lipid aldehydes formed by Hock rearrangement (e.g. hexanal or 8-oxo-oct-6-enoyl-PC) react with PE to give imine adducts. Neighboring lipid peroxides transfer their peroxides to the imine adducts to yield peroxyheminals that then lose water to form the highly stable amide bond (e.g. N-hexanoyl-PE and N-C7:1CA-PE).
5.1.6. PL modified by oxidized plasmalogen (N-alkyl-PL)
Oxidation of plasmalogen produced long chain aldehydes (such as pentadecanal, heptadecanal and heptadecenal) and α-hydroxy aldehydes (such as α-hydroxyhexadecanal and α-hydroxyoctadecanal) (Stadelmann-Ingrand et al., 2001). These lipid aldehydes can also covalently modify PE (Stadelmann-Ingrand et al., 2004), leading to formation of Schiff bases detected as N-alkyl-PE after reduction (Fig 23).
Figure 23.
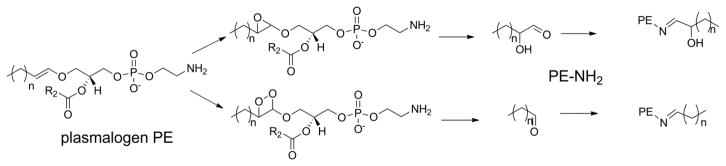
Formation of long-chained aldehydes from oxidation of plasmalogen with subsequent formation of N-alkyl-PE (Felde and Spiteller, 1995; Jira and Spiteller, 1996; Morand et al., 1988; Stadelmann-Ingrand et al., 2004).
5.1.7. N-glycated PL
The open chain form of glucose is an aldehyde that can modify the amine group of PE via Maillard reaction. Glycation of PE was detected in LC-MS as an imine adduct that subsequently rearranges to form N-Amadori-PE (Fountain et al., 1999; Ravandi et al., 1995) (Fig 24). Additional studies characterizing the structures of PE modified by glucose found that a major product was N-(5-hydroxymethyl-1-H-pyrrole-2-carbaldehyde)-PE and proposed that this product formed from N-Amadori-PE through a series of dehydration and rearrangements steps (Lederer and Baumann, 2000). Glycoxidation of PE may also be the origin of N-carboxymethyl-PE (Requena et al., 1997). The imine adduct of N-glycated PS has also been identified in vitro by TLC and LC-MS, although no such detection in vivo has been reported yet (Ravandi et al., 1995).
Figure 24.
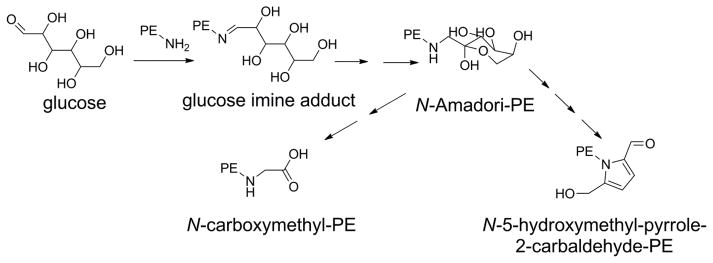
Characterized products of glucose reaction with PE. The open form of glucose reacts with PE to form an imine adduct which rearranges to form the stable N-Amadori-PE. A series of dehydration and rearrangement steps has been proposed to lead from this N-Amadori-PE to N-(5-hydroxmethyl-1-pyrrole-2 –carbaldehyde)-PE. N-Amadori-PE also likely decomposes to form N-carboxymethyl-PE.
5.2. Formation of oxidatively N-modified PL in biological samples
Increased levels of protein/peptides modified by lipid aldehydes have been measured in many disease conditions, but until recently very few studies had examined whether corresponding changes in oxidatively N-modified PL also occur. We will therefore briefly summarize these studies below.
Retinal levels of N-hydroxyalkenal-PE including N-HHE-PE, N-HNE-PE and N-HDDE-PE increased after induction of diabetes in rats using streptozotocin (Bacot et al., 2007). The species of N-hydroxyalkenal-PE produced by oxidation of platelets appears to be dependent on the oxidative stress applied. For example, diamide-treated platelets produced moderate increases in only N-HNE-PE, while aspirin and mercaptosuccinic acid treated platelets produced both N-HNE-PE and N-HDDE-PE, but not N-HHE-PE (Bacot et al., 2007).
The hydroxylactam species of N-IsoLG-PE and the propenal species of N-MDA-PE increase when HDL is incubated with myeloperoxidase or t-butylhydroperoxide (Guo et al., 2012). Liver N-IsoLG-PE levels increase when mice chronically consume ethanol and plasma (Li et al., 2009). N-IsoLG-PE levels also increase in subjects with age-related macular degeneration (Li et al., 2009). N-MDA-PE has not yet been detected in vivo.
N-hexanoyl-PE has been found in several types of biological samples upon oxidation, including oxidized erythrocyte ghosts and HDL, 13-HPODE-treated RBC ghost membrane, and livers from rats injected with CCl4 (Guo et al., 2012; Hisaka et al., 2010; Tsuji et al., 2003).
N-alkyl-PE formation was observed when brain homogenate was subjected to UV irradiation or Fe2+/ascobate oxidation. The time course of this N-alkyl-PE formation correlated with the release of α-hydroxy aldehydes from oxidized plasmalogens (Stadelmann-Ingrand et al., 2004).
The presence of N-Amadori-PE has been reported in a variety of tissues, including rat plasma (Bucala et al., 1993; Lertsiri et al., 1998; Oak et al., 2002; Pamplona et al., 1995; Ravandi et al., 2000), liver from streptozotocin-induced diabetic rats (Bucala et al., 1993; Lertsiri et al., 1998; Oak et al., 2002; Pamplona et al., 1995; Ravandi et al., 2000), LDL from diabetic subjects (Bucala et al., 1993; Lertsiri et al., 1998; Oak et al., 2002; Pamplona et al., 1995; Ravandi et al., 2000), human plasma/RBC (Bucala et al., 1993; Lertsiri et al., 1998; Oak et al., 2002; Pamplona et al., 1995; Ravandi et al., 2000), and atherosclerotic tissue (Bucala et al., 1993; Lertsiri et al., 1998; Oak et al., 2002; Pamplona et al., 1995; Ravandi et al., 2000).
5.3. Biological activities and signaling pathways of oxidatively N-modified PL
5.3.1. Stimulation of platelet prothrombinase activity
The observation that high levels of thrombin are often found around the sites of atherosclerotic plaques, along with the important roles oxidized LDL plays in the pathophysiology of atherosclerosis, prompted the studies of physiological effects of oxidized LDL in the process of coagulation. Not surprisingly, oxidized LDL was found to be a potent stimulant for platelet-dependent thrombin formation. More importantly, such stimulation is attributed mainly to the actions of diacyl-PE modified by oxidized unsaturated PC on platelet prothrombinase complexes (Zieseniss et al., 2001). Treatment of platelets with synthetic N-HNE-PE or N-HNE-lysoPE increased thrombin formation. Chemical reduction of these compounds inactivated their prothrombinase activity suggesting that the imine adduct was the key bioactive species.
As discussed in the previous section on cyclized oxPL, very low concentrations of 15-E2-IsoLG (1 nM) potentiate platelet activation and TxA2 synthesis induced by sub-threshold concentrations of collagen (Bernoud-Hubac et al., 2009). While there are no published studies indicating whether formation of N-IsoLG-PE is responsible for this effect, this remains a distinct possibility. For instance, N-IsoLG-PE induces p38MAPK activation in endothelial cells, and IsoLG treated platelets also have markedly elevated phosphorylation of p38MAPK.
5.3.2. Induction of endothelial cell activation and leukocyte adhesion
The proinflammatory activity of oxidatively N-modified PE has been most rigorously explored with N-IsoLG-PE. N-IsoLG-PE induces surface expression of the adhesion molecules ICAM-1, VCAM-1 and E-selectin in human umbilical vein endothelial cells, with 1 μM being sufficient to induce near maximal expression (Guo et al., 2011). Similar concentrations of N-IsoLG-PE induced expression of mRNA for IL-8 and MCP-1 and binding of monocytes to the endothelial cells. N-IsoLG-PE was more potent than IsoLG itself at inducing monocyte binding to endothelium. This finding, along with previous data showing that IsoLG modifies PE to a greater extent than protein, suggests that formation of N-IsoLG-PE is the primary mechanism whereby IsoLG induces its proinflammatory response. Structure activity relationship studies found that the N-IsoLG-lysoPE and N-IsoLG-ethanolamine (N-IsoLG-Etn) were about 5-fold and 10-fold less potent, respectively, than N-IsoLG-PE at inducing monocyte binding to endothelium. N-4-oxo-pentanal modified PE, which forms a pyrrole adduct similar to N-IsoLG-PE but without the sidechains, induces adhesion nearly to the same extent as N-IsoLG-PE, suggesting that these sidechains are dispensable for this activity.
N-HNE-PE and N-ONE-PE both activate endothelial cells so that they facilitate the binding of monocytes (Guo et al., 2012). N-HNE-PE was somewhat more potent than N-ONE-PE, being able to induce near maximal adhesion at 1 μM rather than 5 μM. This result suggests that the Michael adduct form is somewhat more potent than the pyrrole form.
The dihydropyridine (DHP) species of N-MDA-PE adduct was able to induce monocyte binding to endothelial cells (Guo et al., 2012). In contrast, N-acrolein-PE was only a weak partial agonist. One species of amide-linked oxidatively N-modified PE, N-C11:0CA-PE was able to induce endothelial cell activation, while N-C4:0CA-PE and various N-acyl-PE did not. This result suggests that both an ω-caboxylate group and an acyl chain of sufficient length are required to induce endothelial cell activation.
Treatment of monocytes and dendritic cells with N-glycated PE also enhances their expression of cytokines such as TNFα, MIP-1b, IL-6, IL-8 and IL-1. (Simoes et al., 2013). Interestingly, oxidation of the sn-2 side chain of the N-glycated PE further enhanced cytokine expression in some cases.
5.3.3. Induction of ER stress responses and cytotoxicity
Addition of exogenous N-IsoLG-PE to endothelial cells leads to its rapid internalization into ER and perinuclear membranes (Guo et al., 2011). Furthermore, N-IsoLG-PE markedly alters membrane curvature. This raised the possibility that it induces ER stress response. Indeed, N-IsoLG-PE induces expression of CHOP and BiP, along with phosphorylation of p38MARK. Additionally, known inhibitors of the ER stress response, such as the chemical chaperone (TUDCA) and the serine protease inhibitor AEBSF, significantly reduced inflammatory effects of N-IsoLG-PE (Guo et al., 2011). Consistent with the relationship between prolonged ER stress and cell death, prolonged exposures to N-IsoLG-PE induces cell death (Sullivan et al., 2010). These results support the conclusion that N-IsoLG-PE exerts its biological effects in part via ER stress. It is also important to note that N-IsoLG-PE is far more potent than OV-PC and shares similar potency as EI-PC for induction of ER stress. Given the potential reactivity of EI-PC, it may be important to determine to what extent PE modification by EI-PC might also account for its ability to induce ER stress.
5.3.4. Macrophage uptake of LDL
Ravandi et al. showed that N-glycated PE induces THP-1 macrophage uptake of LDL and accumulation of cholesteryl esters and triacylglycerols (Ravandi et al., 1999). Specifically, they found that incubation of THP-1 macrophages with N-glycated PE enriched LDL promotes a more rapid increase in cell-associated LDL than incubation with control PE-enriched LDL. Furthermore, the incubation with N-glycated PE enriched LDL led to significantly greater levels of total cholesteryl esters, total cholesterol, and triacylglycerols in the THP-1 macrophages, compared with control PE enriched LDL. Of note, glycation of LDL results in accumulation of cholesteryl esters and apolipoprotein B-100 protein in primary human monocyte-derived macrophages (Brown et al., 2007). Because enrichment of LDL with N-glycated PE mimics these biological effects of glycated LDL, it raises the possibility that modification of PE mediates all or most of the effect of LDL glycation.
5.3.5. Angiogenesis
Increased angiogenesis plays an important role in diabetic microangiopathy. Because N-glycated-PE including N-Amadori-PE are increased during diabetes, the effect of N-Amadori-PE on endothelial cells has been examined. Exposure of endothelial cells to N-Amadori-PE (1 to 5 μM) led to significant increases in the rate of their proliferation (Oak et al., 2003). Furthermore, treatment with N-Amadori-PE increased migration and tube formation (Nakagawa et al., 2005; Oak et al., 2003). Interestingly, treatment of N-Amadori-PE induced expression of matrix metalloproteinase-2 (MMP-2), but not MMP-1, MMP-9, or vascular endothelial growth factor (VEGF).
5.3.6 Telomerase activity
In addition to its effects on angiogenesis, recent studies have raised the possibility that N-Amadori-PE may contribute to development of cancer through an effect on telomerase. For instance, in PANC-1 human pancreatic carcinoma cells, exposure to N-Amadori-PE up-regulates hTERT mRNA expression though induction of c-myc expression. (Eitsuka et al., 2012)
5.3.7 Promotion of lipid peroxidation
Glycation of PE increases membrane lipid order and lipid hydration, suggesting that formation of N-glycated PE also leads to formation of lipid fragments by oxidation (Obsil et al., 1999). Ravandi et al showed that that increased levels of N-glycated PE in LDL correlated with increases in both hydroperoxides and lipid aldehydes (Ravandi et al., 2000). Importantly, incorporation of N-glycated PE into LDL induced a rapid oxidation of polyunsaturated cholesteryl esters. Moreover, incubation of synthetic N-Amadori-PE with unsaturated fatty acids remarkably increased formation of lipid fragments and lipid hydroperoxides (Breitling-Utzmann et al., 2001; Oak et al., 2000). N-glycated PE appears to induce lipid peroxidation by formation of free radicals as this effect can by blocked by superoxide dismutase, mannitol, catalase and metal chelator (Oak et al., 2000).
5.3.8. Ca2+−-ATPase
Addition of N-glycated PE decreases the affinity of phospholipids for plasma-membrane Ca2+-ATPase (PMCA), which triggers a structural rearrangement of PMCA and decreases its thermal stability (Levi et al., 2008).
5.4. Metabolism
Very little is known about the metabolism of oxidatively N-modified PE and PS. Because of the proinflammatory effects of these oxPL, reduced metabolism of these compounds might promote development of inflammatory diseases including atherosclerosis. Despite the fact that oxidatively N-modified PE have bulky headgroups, they are still substrates for many of the promiscuous phospholipases used for in vitro chemical analysis. Thus N-IsoLG-PE can be hydrolyzed by S. chromofuscus PLD and bee venom PLA2 (Li et al., 2009; Sullivan et al., 2010), N-glycated-PE can be hydrolyzed by PLA2 from cobra venom, PLC from Clostridium perfringens, and type III PLD from peanut (Lertsiri et al., 1998), N-hexanoyl-PE can be hydrolyzed by S. chromofuscus PLD (Hisaka et al., 2010), and N-HNE-PE can be hydrolyzed by the secreted PLA2 from pancreas, but not the PLD from cabbage (Guichardant et al., 2002).
To characterize the metabolism of N-IsoLG-PE in cultured cells, Guo et al treated HEK293 cells with synthetic IsoLG to generate N-IsoLG-PE in their membranes and then followed the degradation of this N-IsoLG-PE over time. Levels rapidly declined over six hours, supporting the existence of phospholipases that catabolize N-modified PE (Guo et al., 2013). Treatment of HEK293 cells with siRNA specific for N-acyl PE hydrolyzing phospholipase D (NAPE-PLD) elevated levels of N-IsoLG-PE compared to cells treated with control siRNA, suggesting that NAPE-PLD is one of the principal enzymes that hydrolyzes N-IsoLG-PE. The catalytic efficiency (Vmax/Km) of recombinant mouse NAPE-PLD for N-IsoLG-PE is 3.85 mL min−1 mg−1, which is close to its catalytic efficiency for C16:0N-acyl-PE of 12.9 mL min−1 mg−1. The product of NAPE-PLD hydrolysis of N-IsoLG-PE is N-IsoLG-ethanolamine, which is ~10-fold less potent than N-IsoLG-PE (Guo et al., 2011), so that loss of NAPE-PLD activity is likely to increase the potency of N-IsoLG-PE for inducing inflammation. These studies also showed that N-IsoLG-ethanolamine is further degraded by activities found in HEK293 lysates, which would presumably lead to further inactivation. Whether NAPE-PLD participates in the hydrolysis of other oxidatively N-modified PL is unknown, but seems likely.
6. Summary and the future of oxPL research
The past twenty years of research on oxPL have led to some remarkable discoveries. Not only have a larger number of bioactive oxPL been characterized, but the basic outlines of their participation in physiological and pathological processes have emerged. These new discoveries have led to several paradigm shifts. The first paradigm shift came when the notion that peroxidation of lipids was merely a marker of the collateral damage wrought by the immune system’s war on microbial invaders was replaced with the notion that oxPL directly contributed to the pathological inflammatory responses leading to atherosclerosis and other diseases. A second paradigm shift has come as it has now become apparent that in addition to contributing to pathophysiology, the formation of some oxPL is an important part of physiological responses.
A major reason for these paradigm shifts has been our evolving understanding of the pervasiveness of oxPL formation and of the complexity of the cellular responses to their formation. The simplistic notion that oxPL invoke effects merely by acting as detergents in the cell membrane is no longer tenable. Instead, it is clear that oxPL invoke precise signaling mechanisms and can fine tune responses in a manner similar to that described for other well-accepted lipid mediators like prostaglandins. For instance, the finding that resident macrophages use 12/15-LOX mediated HETE-PE expression to sequester MFG-E8 and thereby ensure that they and not inflammatory monocytes take up apoptotic cells strongly suggest that mildly oxygenated oxPL are physiological lipid mediators, not merely unintended byproducts. Similarly, the finding that several classes of oxPL modestly induce inflammatory responses, but limit the inflammatory response to LPS by competing for co-activators of TLR4 suggests that formation of oxPL may be useful feedback mechanism to attenuate overly exuberant inflammatory responses to microbial invasion.
With this in mind, one of the major challenges for future studies will be to determine which oxPL are indeed physiological mediators, with well-regulated biosynthesis, catabolism, and signaling pathways. The previous characterization of the major oxPL species and the development of mass spectrometric lipidomics approaches should facilitate these future studies. While current shotgun lipidomics approaches are usually not sensitive enough for measurement of oxPL, their general approach of measuring multiple lipid classes and species at once, rather than only one or a few species at a time is a critical step forward. Now is the time for studies quantifying changes in as many of the major species of oxPL as possible during well-defined physiological responses. It would be particularly helpful to focus on cellular systems where activation of lipoxygenases and oxidases occurs as part of physiological responses.
In parallel with studies defining the regulation of oxPL concentrations, similarly systematic studies on the signaling pathways activated by physiological rather than pharmacological concentrations of these oxPL need to be performed. One of the most helpful advances in the past twenty years has been the identification of specific receptors such as the PAF receptor, PPARs, various prostaglandin receptors, CD36, and SR-BI that mediate some of the responses to oxPL. With the sequencing of the genome and the wholesale cloning of G-protein coupled receptors, nuclear hormone receptors, and other orphan receptors, it should now be possible to move from a somewhat haphazard candidate approach for identifying oxPL receptors to a more rigorous, structure-activity relationship based analysis. At the same time, the emergence of RNA-seq platforms should facilitate systematic characterization of downstream cellular responses to each major species of oxPL. Thus previous gene array studies performed for oxPAPC can and need to be expanded to specific oxPL and to a wide range of additional cellular systems.
One of the major challenges that oxPL research faces is the flagging interest from governmental agencies and pharmaceutical companies funding health research due to a number of failed clinical trials for therapeutics aimed at oxPL. All too frequently, therapeutics that have shown great promise in animal studies have shown no efficacy in human clinical trials. Included in this list of apparent failures are dietary antioxidants like vitamin C and E, PAF receptor antagonists, recombinant PAF acetylhydrolase, and ApoAI mimetic peptides. These failures should remind us of the importance of systematic investigations into the role of oxPL in physiological responses and especially how these responses vary between species. For instance, one reason for the failure of vitamin E trials may have been that the doses used in most trials, which were based on animal studies, actually turn out to be too low to reduce formation of oxPL in humans (at least as measured by F2-IsoP levels). In fact, a number of studies now indicate that lowering circulating F2-IsoP levels in humans is difficult. The reasons for this resistance are unknown, but probably worth serious investigation. Thus, the failure of a few promising therapeutics, while certainly disappointing to patients, scientists, and investors alike, should not be allowed to invalidate a long line of evidence supporting the potential role of oxPL in physiological and pathophysiological processes. Instead, the failure of these first-line therapeutics must serve as a catalyst for more systematic investigations that can unravel the complex biology surrounding oxPL.
Highlights.
Oxidatively modified phospholipids can be grouped into four broad categories base on their structures.
Elevated levels found in a variety of physiological and pathophysiological conditions.
Identified molecular targets include G-protein coupled receptors, scavenger receptors, and nuclear hormone receptors.
Biological effects include platelet activation, modulation of immune cell function, cell migration, and cytotoxicity.
Identified catabolizing enzymes include platelet-activating factor hydrolase, aldo-keto reductases, and paraoxonase.
Acknowledgments
This work was supported by funds from the Vanderbilt Department of Pharmacology and the National Institutes of Health: NIH P30 ES000267 (Vanderbilt Center in Molecular Toxicology).
List of Abbreviations
- ApoAI
apolipoprotein AI
- ApoE
apolipoprotein E
- AKR
aldo-keto reductases
- arachidonyl-PAF
1-O-alkyl-2-arachidonoyl-sn-glycero-3-phosphocholine
- arachidonyl-PC
1-acyl-2-arachidonoyl-sn-glycero-3-phosphocholine
- ATF
activating transcription factor
- AZ-PAF
1-O-alkyl-2-azeaolyl-sn-glycero-3-phosphocholine
- AZ-PC
1-acyl-2-azeaolyl- sn-glycero-3-phosphocholine
- BAL
bronchoalveolar lavage
- BiP/Grp78
binding immunoglobin protein/glucose-regulated protein 78
- butanoyl-PAF
1-O-alkyl-2- butanoyl -sn-glycero-3-phosphocholine
- butanoyl-PC
1-acyl-2- butanoyl -sn-glycero-3-phosphocholine
- CD14
Cluster of Differentiation 14
- CD36
Cluster of Differentiation 36, fatty acid translocase
- CHO
Chinese Hamster Ovary cells
- CoA
coenzyme A
- COX
cyclooxygenase
- Cx43
connexin 43
- CYP
cytochrome P450
- diHETE
dihydroxyeicosatetraenoate
- DP
prostanoid receptor for prostaglandin D2
- EET
epoxyeicosatetraenoate
- EET-PC
1-acyl-2-epoxyeicosatetraenoate-sn-glycero-3-phosphocholine
- EET-PE
1-acyl-2-epoxyeicosatetraenoate-sn-glycero-3-phosphoethanolamine
- EET-PS
1-acyl-2-epoxyeicosatetraenoate-sn-glycero-3-phosphoserine
- EI-PC
1-acyl-2-(5,6-epoxy-E2-isoprostane)-sn-glycero-3-phosphocholine
- EP
prostanoid receptor for prostaglandin E2
- ER
endoplasmic recticulum
- ERK
extracellular-signal regulated kinase
- Etn
ethanolamine
- GalT-2
UDP-galactose-glucosylceramide galactosyltransferase
- glutaryl-PC
1-acyl-2-glutaryl-sn-glycero-3-phosphocholine
- HDdiA-PC
1-acyl-2-(9-hydroxy-10-dodecendioate)-sn-glycero-3-phosphocholine
- HDAC5
histone deacetylase 5
- HDL
high density lipoprotein
- HETE
hydroxyeicosatetraenoate
- HETE-PC
1-acyl-2-hydroeicosatetraenoate-sn-glycero-3-phosphocholine
- HETE-PE
1-acyl-2-hydroeicosatetraenoate-sn-glycero-3-phosphoethanolamine
- HNE
4-hydroxynonenal
- HOOA-PC
1-acyl-2-(5-hydroxy-8-oxo-oct-6-enoate)-sn-glycero-3-phosphocholine
- HODE
hydroxyoctadecaenoate
- HODE-PE
1-acyl-2-hydroxyeicosatetraenoate-sn-glycero-3-phosphoethanolamine
- HPETE
hydroperoxyeicosatetraenoate
- HPETE-PC
1-acyl-2-hydroperoxyeicosatetraenoate-sn-glycero-3-phosphocholine
- HPETE-PL
hydroperoxyeicosatetraenoate containing phospholipid
- HPLC
high performance liquid chromatography
- HV-PC
1-acyl-2-(5-hydroxyvaleroyl)-sn-glycero-3-phosphocholine
- IL-8
interleukin 8
- IL-13
interleukin 13
- IRE1
inositol-requiring enzyme-1
- IsoF
isofuran
- IsoLG
isolevuglandin, isoketal
- IsoLG-PC
1-acyl-2-isolevuglandin-sn-glycero-3-phospocholine
- IsoLG-PE
1-acyl-2-isolevuglandin-sn-glycero-3-phospoethanolamine
- IsoP
isoprostane
- IsoP-PC
1-acyl-2-isoprostanyl-sn-glycero-3-phospoethanolamine
- IsoP-PL
phospholipid containing isoprostane
- JNK
c-Jun N-terminal kinase
- KDdiA-PC
1-acyl-2-(9-keto-10-dodecendioate)-sn-glycero-3-phosphocholine
- KETE
ketoeicosatetraenoate
- KETE-PL
ketoeicosatetraenoate containing phospholipid
- KLF4
Kruppel-like transcription factor 4
- KODA-PC
1-acyl-2-(9-keto-12-oxo-10-dodecenoate)-sn-glycero-3-phosphocholine
- KOdiA-PC
1-acyl-2-(5-keto-6-octendioate)-sn-glycero-3-phosphocholine
- KOOA-PC
1-acyl-2-(5-keto-8-oxo-oct-6-enoate)-sn-glycero-3-phosphocholine
- linoleoyl-PAF
1-O-alkyl-2-linoleoyl-sn-glycero-3-phosphocholine
- LOX
lipoxygenase
- LBP
lipopolysaccharide binding protein
- LPS
lipopolysaccharide
- MCP-1
monocyte chemotactic protein-1
- MD-2
myb-regulated gene clone MD-1 family member-2
- MDA
malondialdehyde
- MFG-E8
milk fat globule-EGF factor 8 protein
- MMP-2
matrix metalloproteinase-2
- MUC5AC
mucin protein type 5AC
- NAPE-PLD
N-acyl-phosphatidylethanolamine hydrolyzing phospholipase D
- N-Amadori-PE
phosphatidylethanolamine N-modified with Amadori group
- N-acrolein-PE
phosphatidylethanolamine N-modified by reaction with acrolein
- N-(Cx:y)CA-PE
phosphatidylethanolamine N-modified with carboxylacyl group with x carbons and y double bonds
- N-DHP-PE
phosphatidylethanolamine N-modified with1,4-dihydropyridine-3,5-dicarbeldehyde group
- N-glycated-PE
phosphatidylethanolamine N-modified by reaction with glucose
- N-HDDE-PE
phosphatidylethanolamine N-modified by reaction with 4-hydroxydodecadienal
- N-hexanoyl-PE
phosphatidylethanolamine N-modified with hexanoyl group
- N-HHE-PE
phosphatidylethanolamine N-modified by reaction with 4-hydroxyhexanal
- N-HNE-PE
phosphatidylethanolamine N-modified by reaction with 4-hydroxynonenal
- N-FDP-PE
phosphatidylethanolamine N-modified with 3-formyl-3,4-dehydropiperidine group
- N-IsoLG-PE
phosphatidylethanolamine N-modified by reaction with isolevuglandin
- N-propenal-PE
phosphatidylethanolamine N-modified with propenal group
- N-MDA-PE
phosphatidylethanolamine N-modified by reaction with malondialdehyde
- N-MP-PE
phosphatidylethanolamine N-modified with 3-methylpyridinium group
- N-ONE-PE
phosphatidylethanolamine N-modified by reaction with 4-oxo-nonenal
- octanoyl-PAF
1-O-alkyl-2-octanoyl-sn-glycero-3-phosphocholine
- ONE
4-oxo-nonenal
- ON-PC
1-acyl-2-(9-oxo-nonanoyl)- sn-glycero-3-phosphocholine
- OV-PC
1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphocholine
- OV-PAF
1-O-alkyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphocholine
- oxLDL
oxidized low density lipoprotein
- oxPAPC
oxidized 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphocholine
- oxPL
Oxidatively modified phospholipids
- PAF
Platelet-activating factor; 1-O-alkyl-2-acetyl-sn-glycero-phosphocholine
- PAF-AH
platelet-activating factor acetylhydrolase
- PAPC
1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphocholine
- PAPE
1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphoethanolamine
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PEBP1
phosphatidylethanolamine binding protein 1
- PERK
protein kinase R-like endoplasmic recticulum kinase
- PGE2
prostaglandin E2
- PI
phosphatidylinositol
- PL
phospholipid
- PLA2
phospholipase A2
- PLD
phospholipase D
- PL-OH
hydroxylated phospholipid
- PL-OOH
phospholipid hydroperoxides
- PMN
polymorphonuclear leukocytes, neutrophils
- PON
paraoxonase
- PPAR
peroxisomal proliferator activated receptor
- pPE
plasmalogen; alkenylacyl phosphatidylethanolamine
- PS
phosphatidylserine
- PUFA
polyunsaturated fatty acids
- RNAi
RNA interference
- siRNA
small interfering RNA
- SMC
smooth muscle cells
- SR-BI
scavenger receptor-BI
- TLR
Toll-like receptor
- TMEM30a
Transmembrane protein 30a
- TNF
tumor necrosis factor
- TP
prostanoid receptor for prostaglandin H2/thromboxane A2
- TRPC
transient receptor potential cation channel
- TRPM
transient receptor potential cation channel, subfamily M
- TxA2
thromboxane A2
- VEGF
vascular endothelial growth factor
- VCAM-1
vascular cell adhesion molecule-1
- WHHL
Watanable heritable hyperlipidemic rabbit
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acquaviva A, Vecchio D, Arezzini B, Comporti M, Gardi C. Signaling pathways involved in isoprostane-mediated fibrogenic effects in rat hepatic stellate cells. Free Radic Biol Med. 2013;65:201–207. doi: 10.1016/j.freeradbiomed.2013.06.023. [DOI] [PubMed] [Google Scholar]
- Ahmed Z, Babaei S, Maguire GF, Draganov D, Kuksis A, La Du BN, Connelly PW. Paraoxonase-1 reduces monocyte chemotaxis and adhesion to endothelial cells due to oxidation of palmitoyl, linoleoyl glycerophosphorylcholine. Cardiovasc Res. 2003;57:225–231. doi: 10.1016/s0008-6363(02)00659-4. [DOI] [PubMed] [Google Scholar]
- Al-Shawaf E, Naylor J, Taylor H, Riches K, Milligan CJ, O’Regan D, Porter KE, Li J, Beech DJ. Short-term stimulation of calcium-permeable transient receptor potential canonical 5-containing channels by oxidized phospholipids. Arterioscler Thromb Vasc Biol. 2010;30:1453–1459. doi: 10.1161/ATVBAHA.110.205666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrovandi M, O’Donnell VB. Oxidized PLs and vascular inflammation. Curr Atheroscler Rep. 2013;15:323. doi: 10.1007/s11883-013-0323-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkazemi D, Egeland GM, Roberts LJ, 2nd, Chan HM, Kubow S. New insights regarding tissue Se and Hg interactions on oxidative stress from plasma IsoP and IsoF measures in the Canadian Inuit population. J Lipid Res. 2013;54:1972–1979. doi: 10.1194/jlr.M033068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amarnath V, Amarnath K, Amarnath K, Davies S, Roberts LJ., 2nd Pyridoxamine: an extremely potent scavenger of 1,4-dicarbonyls. Chem Res Toxicol. 2004;17:410–415. doi: 10.1021/tx0300535. [DOI] [PubMed] [Google Scholar]
- Ashraf MZ, Kar NS, Chen X, Choi J, Salomon RG, Febbraio M, Podrez EA. Specific oxidized phospholipids inhibit scavenger receptor bi-mediated selective uptake of cholesteryl esters. J Biol Chem. 2008;283:10408–10414. doi: 10.1074/jbc.M710474200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awad JA, Morrow JD, Takahashi K, Roberts LJ., 2nd Identification of non-cyclooxygenase-derived prostanoid (F2-isoprostane) metabolites in human urine and plasma. J Biol Chem. 1993;268:4161–4169. [PubMed] [Google Scholar]
- Aziz M, Jacob A, Matsuda A, Wang P. Review: milk fat globule-EGF factor 8 expression, function and plausible signal transduction in resolving inflammation. Apoptosis. 2011;16:1077–1086. doi: 10.1007/s10495-011-0630-0. [DOI] [PubMed] [Google Scholar]
- Bacot S, Bernoud-Hubac N, Baddas N, Chantegrel B, Deshayes C, Doutheau A, Lagarde M, Guichardant M. Covalent binding of hydroxy-alkenals 4-HDDE, 4-HHE, and 4-HNE to ethanolamine phospholipid subclasses. J Lipid Res. 2003;44:917–926. doi: 10.1194/jlr.M200450-JLR200. [DOI] [PubMed] [Google Scholar]
- Bacot S, Bernoud-Hubac N, Chantegrel B, Deshayes C, Doutheau A, Ponsin G, Lagarde M, Guichardant M. Evidence for in situ ethanolamine phospholipid adducts with hydroxy-alkenals. J Lipid Res. 2007;48:816–825. doi: 10.1194/jlr.M600340-JLR200. [DOI] [PubMed] [Google Scholar]
- Basu S. F2-isoprostanes in human health and diseases: from molecular mechanisms to clinical implications. Antioxid Redox Signal. 2008;10:1405–1434. doi: 10.1089/ars.2007.1956. [DOI] [PubMed] [Google Scholar]
- Bedirli A, Gokahmetoglu S, Sakrak O, Soyuer I, Ince O, Sozuer E. Beneficial effects of recombinant platelet-activating factor acetylhydrolase and BN 52021 on bacterial translocation in cerulein-induced pancreatitis. Eur Surg Res. 2004;36:136–141. doi: 10.1159/000077254. [DOI] [PubMed] [Google Scholar]
- Berliner JA, Territo MC, Sevanian A, Ramin S, Kim JA, Bamshad B, Esterson M, Fogelman AM. Minimally modified low density lipoprotein stimulates monocyte endothelial interactions. J Clin Invest. 1990;85:1260–1266. doi: 10.1172/JCI114562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernareggi M, Rossoni G, Berti F. Bronchopulmonary effects of 8-epi-PGF2A in anaesthetised guinea pigs. Pharmacological research: the official journal of the Italian Pharmacological Society. 1998;37:75–80. doi: 10.1006/phrs.1997.0266. [DOI] [PubMed] [Google Scholar]
- Bernoud-Hubac N, Alam DA, Lefils J, Davies SS, Amarnath V, Guichardant M, Roberts LJ, II, Lagarde M. Low concentrations of reactive [gamma]-ketoaldehydes prime thromboxane-dependent human platelet aggregation via p38-MAPK activation. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 2009;1791:307–313. doi: 10.1016/j.bbalip.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Bernoud-Hubac N, Fay LB, Armarnath V, Guichardant M, Bacot S, Davies SS, Roberts LJ, 2nd, Lagarde M. Covalent binding of isoketals to ethanolamine phospholipids. Free Radic Biol Med. 2004;37:1604–1611. doi: 10.1016/j.freeradbiomed.2004.07.031. [DOI] [PubMed] [Google Scholar]
- Bernstrom K, Kayganich K, Murphy RC, Fitzpatrick FA. Incorporation and distribution of epoxyeicosatrienoic acids into cellular phospholipids. J Biol Chem. 1992;267:3686–3690. [PubMed] [Google Scholar]
- Bhuyan DK, Master RW, Bhuyan KC. Crosslinking of aminophospholipids in cellular membranes of lens by oxidative stress in vitro. Biochim Biophys Acta. 1996;1285:21–28. doi: 10.1016/s0005-2736(96)00142-3. [DOI] [PubMed] [Google Scholar]
- Billings FTt, Ball SK, Roberts LJ, 2nd, Pretorius M. Postoperative acute kidney injury is associated with hemoglobinemia and an enhanced oxidative stress response. Free Radic Biol Med. 2011;50:1480–1487. doi: 10.1016/j.freeradbiomed.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova AA, Starosta V, Tian X, Higginbotham K, Koroniak L, Berliner JA, Birukov KG. Fragmented oxidation products define barrier disruptive endothelial cell response to OxPAPC. Translational research: the journal of laboratory and clinical medicine. 2013;161:495–504. doi: 10.1016/j.trsl.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochkov VN, Kadl A, Huber J, Gruber F, Binder BR, Leitinger N. Protective role of phospholipid oxidation products in endotoxin-induced tissue damage. Nature. 2002;419:77–81. doi: 10.1038/nature01023. [DOI] [PubMed] [Google Scholar]
- Bochkov VN, Oskolkova OV, Birukov KG, Levonen AL, Binder CJ, Stockl J. Generation and biological activities of oxidized phospholipids. Antioxid Redox Signal. 2010;12:1009–1059. doi: 10.1089/ars.2009.2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchman D, Yappert MC, Rubini RQ, Paterson CA. Distribution of phospholipid-malondialdehyde-adduct in the human lens. Curr Eye Res. 1989;8:939–946. [PubMed] [Google Scholar]
- Borisenko GG, Iverson SL, Ahlberg S, Kagan VE, Fadeel B. Milk fat globule epidermal growth factor 8 (MFG-E8) binds to oxidized phosphatidylserine: implications for macrophage clearance of apoptotic cells. Cell Death Differ. 2004;11:943–945. doi: 10.1038/sj.cdd.4401421. [DOI] [PubMed] [Google Scholar]
- Boullier A, Friedman P, Harkewicz R, Hartvigsen K, Green SR, Almazan F, Dennis EA, Steinberg D, Witztum JL, Quehenberger O. Phosphocholine as a pattern recognition ligand for CD36. J Lipid Res. 2005;46:969–976. doi: 10.1194/jlr.M400496-JLR200. [DOI] [PubMed] [Google Scholar]
- Boutaud O, Montine TJ, Chang L, Klein WL, Oates JA. PGH2-derived levuglandin adducts increase the neurotoxicity of amyloid beta1–42. J Neurochem. 2006;96:917–923. doi: 10.1111/j.1471-4159.2005.03586.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden PA, Davies SS, Viswanathan PC, Amarnath V, Balser JR, Roberts LJ., 2nd Potential role of isoketals formed via the isoprostane pathway of lipid peroxidation in ischemic arrhythmias. J Cardiovasc Pharmacol. 2007;50:480–486. doi: 10.1097/FJC.0b013e31815a0564. [DOI] [PubMed] [Google Scholar]
- Brame CJ, Boutaud O, Davies SS, Yang T, Oates JA, Roden D, Roberts LJ., 2nd Modification of proteins by isoketal-containing oxidized phospholipids. J Biol Chem. 2004;279:13447–13451. doi: 10.1074/jbc.M313349200. [DOI] [PubMed] [Google Scholar]
- Brame CJ, Salomon RG, Morrow JD, Roberts LJ., 2nd Identification of extremely reactive gamma-ketoaldehydes (isolevuglandins) as products of the isoprostane pathway and characterization of their lysyl protein adducts. J Biol Chem. 1999;274:13139–13146. doi: 10.1074/jbc.274.19.13139. [DOI] [PubMed] [Google Scholar]
- Brantley MA, Jr, Osborn MP, Sanders BJ, Rezaei KA, Lu P, Li C, Milne GL, Cai J, Sternberg P., Jr Plasma biomarkers of oxidative stress and genetic variants in age-related macular degeneration. American journal of ophthalmology. 2012;153:460–467. e461. doi: 10.1016/j.ajo.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitling-Utzmann CM, Unger A, Friedl DA, Lederer MO. Identification and quantification of phosphatidylethanolamine-derived glucosylamines and aminoketoses from human erythrocytes--influence of glycation products on lipid peroxidation. Arch Biochem Biophys. 2001;391:245–254. doi: 10.1006/abbi.2001.2406. [DOI] [PubMed] [Google Scholar]
- Brown BE, Rashid I, van Reyk DM, Davies MJ. Glycation of low-density lipoprotein results in the time-dependent accumulation of cholesteryl esters and apolipoprotein B-100 protein in primary human monocyte-derived macrophages. Febs J. 2007;274:1530–1541. doi: 10.1111/j.1742-4658.2007.05699.x. [DOI] [PubMed] [Google Scholar]
- Bucala R, Makita Z, Koschinsky T, Cerami A, Vlassara H. Lipid advanced glycosylation: pathway for lipid oxidation in vivo. Proc Natl Acad Sci U S A. 1993;90:6434–6438. doi: 10.1073/pnas.90.14.6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burcham PC, Kuhan YT. Introduction of carbonyl groups into proteins by the lipid peroxidation product, malondialdehyde. Biochem Biophys Res Commun. 1996;220:996–1001. doi: 10.1006/bbrc.1996.0521. [DOI] [PubMed] [Google Scholar]
- Caplan MS, Lickerman M, Adler L, Dietsch GN, Yu A. The role of recombinant platelet-activating factor acetylhydrolase in a neonatal rat model of necrotizing enterocolitis. Pediatr Res. 1997;42:779–783. doi: 10.1203/00006450-199712000-00010. [DOI] [PubMed] [Google Scholar]
- Caslake MJ, Packard CJ. Lipoprotein-associated phospholipase A2 as a biomarker for coronary disease and stroke. Nature clinical practice. Cardiovascular medicine. 2005;2:529–535. doi: 10.1038/ncpcardio0321. [DOI] [PubMed] [Google Scholar]
- Cecarini V, Ding Q, Keller JN. Oxidative inactivation of the proteasome in Alzheimer’s disease. Free Radic Res. 2007;41:673–680. doi: 10.1080/10715760701286159. [DOI] [PubMed] [Google Scholar]
- Charvet C, Liao WL, Heo GY, Laird J, Salomon RG, Turko IV, Pikuleva IA. Isolevuglandins and mitochondrial enzymes in the retina: mass spectrometry detection of post-translational modification of sterol-metabolizing CYP27A1. J Biol Chem. 2011;286:20413–20422. doi: 10.1074/jbc.M111.232546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charvet CD, Laird J, Xu Y, Salomon RG, Pikuleva IA. Posttranslational modification by an isolevuglandin diminishes activity of the mitochondrial cytochrome P450 27A1. J Lipid Res. 2013a;54:1421–1429. doi: 10.1194/jlr.M035790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charvet CD, Saadane A, Wang M, Salomon RG, Brunengraber H, Turko IV, Pikuleva IA. Pretreatment with pyridoxamine mitigates isolevuglandin-associated retinal effects in mice exposed to bright light. J Biol Chem. 2013b;288:29267–29280. doi: 10.1074/jbc.M113.498832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Berliner JA, Subbanagounder GG, Bhunia AK, Koh S. Identification of a biologically active component in minimally oxidized low density lipoprotein (MM-LDL) responsible for aortic smooth muscle cell proliferation. Glycoconjugate journal. 2004;20:331–338. doi: 10.1023/B:GLYC.0000033629.54962.68. [DOI] [PubMed] [Google Scholar]
- Chen J, Capdevila JH, Zeldin DC, Rosenberg RL. Inhibition of cardiac L-type calcium channels by epoxyeicosatrienoic acids. Mol Pharmacol. 1999;55:288–295. doi: 10.1124/mol.55.2.288. [DOI] [PubMed] [Google Scholar]
- Chen R, Brady E, McIntyre TM. Human TMEM30a promotes uptake of antitumor and bioactive choline phospholipids into mammalian cells. J Immunol. 2011;186:3215–3225. doi: 10.4049/jimmunol.1002710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Chen X, Salomon RG, McIntyre TM. Platelet activation by low concentrations of intact oxidized LDL particles involves the PAF receptor. Arterioscler Thromb Vasc Biol. 2009a;29:363–371. doi: 10.1161/ATVBAHA.108.178731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Feldstein AE, McIntyre TM. Suppression of mitochondrial function by oxidatively truncated phospholipids is reversible, aided by bid, and suppressed by Bcl-XL. J Biol Chem. 2009b;284:26297–26308. doi: 10.1074/jbc.M109.018978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Yang L, McIntyre TM. Cytotoxic phospholipid oxidation products. Cell death from mitochondrial damage and the intrinsic caspase cascade. J Biol Chem. 2007;282:24842–24850. doi: 10.1074/jbc.M702865200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanova OA, Pidkovka NA, Sarmento OF, Yoshida T, Gan Q, Adiguzel E, Bendeck MP, Berliner J, Leitinger N, Owens GK. Oxidized phospholipids induce type VIII collagen expression and vascular smooth muscle cell migration. Circ Res. 2009;104:609–618. doi: 10.1161/CIRCRESAHA.108.186064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiabrando C, Valagussa A, Rivalta C, Durand T, Guy A, Zuccato E, Villa P, Rossi JC, Fanelli R. Identification and measurement of endogenous beta-oxidation metabolites of 8-epi-Prostaglandin F2alpha. J Biol Chem. 1999;274:1313–1319. doi: 10.1074/jbc.274.3.1313. [DOI] [PubMed] [Google Scholar]
- Chu HW, Balzar S, Westcott JY, Trudeau JB, Sun Y, Conrad DJ, Wenzel SE. Expression and activation of 15-lipoxygenase pathway in severe asthma: relationship to eosinophilic phenotype and collagen deposition. Clinical and experimental allergy: journal of the British Society for Allergy and Clinical Immunology. 2002;32:1558–1565. doi: 10.1046/j.1365-2222.2002.01477.x. [DOI] [PubMed] [Google Scholar]
- Clarke DL, Belvisi MG, Hardaker E, Newton R, Giembycz MA. E-ring 8-isoprostanes are agonists at EP2- and EP4-prostanoid receptors on human airway smooth muscle cells and regulate the release of colony-stimulating factors by activating cAMP-dependent protein kinase. Mol Pharmacol. 2005;67:383–393. doi: 10.1124/mol.104.006486. [DOI] [PubMed] [Google Scholar]
- Cole AL, Subbanagounder G, Mukhopadhyay S, Berliner JA, Vora DK. Oxidized phospholipid-induced endothelial cell/monocyte interaction is mediated by a cAMP-dependent R-Ras/PI3-kinase pathway. Arterioscler Thromb Vasc Biol. 2003;23:1384–1390. doi: 10.1161/01.ATV.0000081215.45714.71. [DOI] [PubMed] [Google Scholar]
- Cracowski JL, Devillier P, Chavanon O, Sietchiping-Nzepa FA, Stanke-Labesque F, Bessard G. Isoprostaglandin E2 type-III (8-iso-prostaglandin E2) evoked contractions in the human internal mammary artery. Life Sci. 2001;68:2405–2413. doi: 10.1016/s0024-3205(01)01032-3. [DOI] [PubMed] [Google Scholar]
- Crowe CR, Chen K, Pociask DA, Alcorn JF, Krivich C, Enelow RI, Ross TM, Witztum JL, Kolls JK. Critical role of IL-17RA in immunopathology of influenza infection. J Immunol. 2009;183:5301–5310. doi: 10.4049/jimmunol.0900995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz D, Watson AD, Miller CS, Montoya D, Ochoa MT, Sieling PA, Gutierrez MA, Navab M, Reddy ST, Witztum JL, Fogelman AM, Rea TH, Eisenberg D, Berliner J, Modlin RL. Host-derived oxidized phospholipids and HDL regulate innate immunity in human leprosy. J Clin Invest. 2008;118:2917–2928. doi: 10.1172/JCI34189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushing S, Berliner J, Valente A, Territo M, Navab M, Parhami F, Gerrity RG, Schwartz C, Fogelman A. Minimally modified low density lipoprotein induces monocyte chemotactic protein (MCP-1) in human endothelial and smooth muscle cells. Proc Natl Acad Sci USA. 1990;87:5134–5138. doi: 10.1073/pnas.87.13.5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dada N, Kim NW, Wolfert RL. Lp-PLA2: an emerging biomarker of coronary heart disease. Expert review of molecular diagnostics. 2002;2:17–22. doi: 10.1586/14737159.2.1.17. [DOI] [PubMed] [Google Scholar]
- Davies SS, Amarnath V, Montine KS, Bernoud-Hubac N, Boutaud O, Montine TJ, Roberts LJ., 2nd Effects of reactive gamma-ketoaldehydes formed by the isoprostane pathway (isoketals) and cyclooxygenase pathway (levuglandins) on proteasome function. Faseb J. 2002;16:715–717. doi: 10.1096/fj.01-0696fje. [DOI] [PubMed] [Google Scholar]
- Davies SS, Bodine C, Matafonova E, Pantazides BG, Bernoud-Hubac N, Harrison FE, Olson SJ, Montine TJ, Amarnath V, Roberts LJ., II Treatment with a gamma-ketoaldehyde scavenger prevents working memory deficits in hApoE4 mice. J Alzheimers Dis. 2011;27:49–59. doi: 10.3233/JAD-2011-102118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SS, Brantley EJ, Voziyan PA, Amarnath V, Zagol-Ikapitte I, Boutaud O, Hudson BG, Oates JA, Roberts LJ., II Pyridoxamine Analogues Scavenge Lipid-Derived gamma-Ketoaldehydes and Protect against H(2)O(2)-Mediated Cytotoxicity. Biochemistry. 2006a;45:15756–15767. doi: 10.1021/bi061860g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SS, Pontsler AV, Marathe GK, Harrison KA, Murphy RC, Hinshaw JC, Prestwich GD, Hilaire AS, Prescott SM, Zimmerman GA, McIntyre TM. Oxidized Alkyl Phospholipids Are Specific, High Affinity Peroxisome Proliferator-activated Receptor γ Ligands and Agonists. Journal of Biological Chemistry. 2001;276:16015–16023. doi: 10.1074/jbc.M100878200. [DOI] [PubMed] [Google Scholar]
- Davies SS, Roberts LJ., 2nd F2-isoprostanes as an indicator and risk factor for coronary heart disease. Free Radic Biol Med. 2011;50:559–566. doi: 10.1016/j.freeradbiomed.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SS, Talati M, Wang X, Mernaugh RL, Amarnath V, Fessel J, Meyrick BO, Sheller J, Roberts LJ., 2nd Localization of isoketal adducts in vivo using a single-chain antibody. Free Radic Biol Med. 2004;36:1163–1174. doi: 10.1016/j.freeradbiomed.2004.02.014. [DOI] [PubMed] [Google Scholar]
- Davies SS, Zackert W, Luo Y, Cunningham CC, Frisard M, Roberts LJ., 2nd Quantification of dinor, dihydro metabolites of F2-isoprostanes in urine by liquid chromatography/tandem mass spectrometry. Anal Biochem. 2006b;348:185–191. doi: 10.1016/j.ab.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Davis B, Koster G, Douet LJ, Scigelova M, Woffendin G, Ward JM, Smith A, Humphries J, Burnand KG, Macphee CH, Postle AD. Electrospray ionization mass spectrometry identifies substrates and products of lipoprotein-associated phospholipase A2 in oxidized human low density lipoprotein. J Biol Chem. 2008;283:6428–6437. doi: 10.1074/jbc.M709970200. [DOI] [PubMed] [Google Scholar]
- Delerive P, Furman C, Teissier E, Fruchart J, Duriez P, Staels B. Oxidized phospholipids activate PPARalpha in a phospholipase A2-dependent manner. FEBS Lett. 2000;471:34–38. doi: 10.1016/s0014-5793(00)01364-8. [DOI] [PubMed] [Google Scholar]
- Dillard CJ, Dumelin EE, Tappel AL. Effect of dietary vitamin E on expiration of pentane and ethane by the rat. Lipids. 1977;12:109–114. doi: 10.1007/BF02532981. [DOI] [PubMed] [Google Scholar]
- Draper HH, Hadley M, Lissemore L, Laing NM, Cole PD. Identification of N-epsilon-(2-propenal)lysine as a major urinary metabolite of malondialdehyde. Lipids. 1988;23:626–628. doi: 10.1007/BF02535610. [DOI] [PubMed] [Google Scholar]
- Eitsuka T, Nakagawa K, Ono Y, Tatewaki N, Nishida H, Kurata T, Shoji N, Miyazawa T. Amadori-glycated phosphatidylethanolamine up-regulates telomerase activity in PANC-1 human pancreatic carcinoma cells. FEBS Lett. 2012;586:2542–2547. doi: 10.1016/j.febslet.2012.06.027. [DOI] [PubMed] [Google Scholar]
- Endemann G, Stanton LW, Madden KS, Bryant CM, White RT, Protter AA. CD36 is a receptor for oxidized low density lipoprotein. J Biol Chem. 1993;268:11811–11816. [PubMed] [Google Scholar]
- Epand RF, Mishra VK, Palgunachari MN, Anantharamaiah GM, Epand RM. Anti-inflammatory peptides grab on to the whiskers of atherogenic oxidized lipids. Biochim Biophys Acta. 2009;1788:1967–1975. doi: 10.1016/j.bbamem.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erridge C, Kennedy S, Spickett CM, Webb DJ. Oxidized phospholipid inhibition of toll-like receptor (TLR) signaling is restricted to TLR2 and TLR4: roles for CD14, LPS-binding protein, and MD2 as targets for specificity of inhibition. J Biol Chem. 2008;283:24748–24759. doi: 10.1074/jbc.M800352200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erridge C, Webb DJ, Spickett CM. Toll-like receptor 4 signalling is neither sufficient nor required for oxidised phospholipid mediated induction of interleukin-8 expression. Atherosclerosis. 2007;193:77–85. doi: 10.1016/j.atherosclerosis.2006.08.032. [DOI] [PubMed] [Google Scholar]
- Esterbauer H, Benedetti A, Lang J, Fulceri R, Fauler G, Comporti M. Studies on the mechanism of formation of 4-hydroxynonenal during microsomal lipid peroxidation. Biochim Biophys Acta. 1986;876:154–166. doi: 10.1016/0005-2760(86)90329-2. [DOI] [PubMed] [Google Scholar]
- Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- Fang L, Harkewicz R, Hartvigsen K, Wiesner P, Choi SH, Almazan F, Pattison J, Deer E, Sayaphupha T, Dennis EA, Witztum JL, Tsimikas S, Miller YI. Oxidized cholesteryl esters and phospholipids in zebrafish larvae fed a high cholesterol diet: macrophage binding and activation. J Biol Chem. 2010;285:32343–32351. doi: 10.1074/jbc.M110.137257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldstein AE, Lopez R, Tamimi TA, Yerian L, Chung YM, Berk M, Zhang R, McIntyre TM, Hazen SL. Mass spectrometric profiling of oxidized lipid products in human nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. J Lipid Res. 2010;51:3046–3054. doi: 10.1194/jlr.M007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fessel JP, Hulette C, Powell S, Roberts LJ, 2nd, Zhang J. Isofurans, but not F2-isoprostanes, are increased in the substantia nigra of patients with Parkinson’s disease and with dementia with Lewy body disease. J Neurochem. 2003;85:645–650. doi: 10.1046/j.1471-4159.2003.01709.x. [DOI] [PubMed] [Google Scholar]
- Fessel JP, Porter NA, Moore KP, Sheller JR, Roberts LJ., 2nd Discovery of lipid peroxidation products formed in vivo with a substituted tetrahydrofuran ring (isofurans) that are favored by increased oxygen tension. Proc Natl Acad Sci USA. 2002;99:16713–16718. doi: 10.1073/pnas.252649099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foreman D, Solomon RG. Evidence that the prostaglandin E2 receptor and the anhydrolevuglandin E2 receptor in the rat uterus are the same receptor. Prostaglandins. 1992;43:263–270. doi: 10.1016/0090-6980(92)90094-a. [DOI] [PubMed] [Google Scholar]
- Foreman D, Zuk L, Miller DB, Salomon RG. Effects of E2 levuglandins on the contractile activity of the rat uterus. Prostaglandins. 1987;34:91–98. doi: 10.1016/0090-6980(87)90266-8. [DOI] [PubMed] [Google Scholar]
- Fountain WC, Requena JR, Jenkins AJ, Lyons TJ, Smyth B, Baynes JW, Thorpe SR. Quantification of N-(glucitol)ethanolamine and N-(carboxymethyl)serine: two products of nonenzymatic modification of aminophospholipids formed in vivo. Anal Biochem. 1999;272:48–55. doi: 10.1006/abio.1999.4147. [DOI] [PubMed] [Google Scholar]
- Frey B, Haupt R, Alms S, Holzmann G, Konig T, Kern H, Kox W, Rustow B, Schlame M. Increase in fragmented phosphatidylcholine in blood plasma by oxidative stress. J Lipid Res. 2000;41:1145–1153. [PubMed] [Google Scholar]
- Fruhwirth GO, Moumtzi A, Loidl A, Ingolic E, Hermetter A. The oxidized phospholipids POVPC and PGPC inhibit growth and induce apoptosis in vascular smooth muscle cells. Biochim Biophys Acta. 2006;1761:1060–1069. doi: 10.1016/j.bbalip.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Fukuda K, Davies SS, Nakajima T, Ong BH, Kupershmidt S, Fessel J, Amarnath V, Anderson ME, Boyden PA, Viswanathan PC, Roberts LJ, 2nd, Balser JR. Oxidative mediated lipid peroxidation recapitulates proarrhythmic effects on cardiac sodium channels. Circ Res. 2005;97:1262–1269. doi: 10.1161/01.RES.0000195844.31466.e9. [DOI] [PubMed] [Google Scholar]
- Fukuda Y, Kawashima H, Saito K, Inomata N, Matsui M, Nakanishi T. Effect of human plasma-type platelet-activating factor acetylhydrolase in two anaphylactic shock models. Eur J Pharmacol. 2000;390:203–207. doi: 10.1016/s0014-2999(99)00920-6. [DOI] [PubMed] [Google Scholar]
- Fukunaga M, Makita N, Roberts LJ, 2nd, Morrow JD, Takahashi K, Badr KF. Evidence for the existence of F2-isoprostane receptors on rat vascular smooth muscle cells. Am J Physiol. 1993;264:C1619–1624. doi: 10.1152/ajpcell.1993.264.6.C1619. [DOI] [PubMed] [Google Scholar]
- Fukunaga M, Yura T, Badr KF. Stimulatory effect of 8-Epi-PGF2 alpha, an F2-isoprostane, on endothelin-1 release. J Cardiovasc Pharmacol. 1995;26(Suppl 3):S51–52. [PubMed] [Google Scholar]
- Fukunaga M, Yura T, Grygorczyk R, Badr KF. Evidence for the distinct nature of F2-isoprostane receptors from those of thromboxane A2. Am J Physiol. 1997;272:F477–483. doi: 10.1152/ajprenal.1997.272.4.F477. [DOI] [PubMed] [Google Scholar]
- Furuhata A, Ishii T, Kumazawa S, Yamada T, Nakayama T, Uchida K. N(epsilon)-(3-methylpyridinium)lysine, a major antigenic adduct generated in acrolein-modified protein. J Biol Chem. 2003;278:48658–48665. doi: 10.1074/jbc.M309401200. [DOI] [PubMed] [Google Scholar]
- Gao S, Zhang R, Greenberg ME, Sun M, Chen X, Levison BS, Salomon RG, Hazen SL. Phospholipid hydroxyalkenals, a subset of recently discovered endogenous CD36 ligands, spontaneously generate novel furan-containing phospholipids lacking CD36 binding activity in vivo. J Biol Chem. 2006;281:31298–31308. doi: 10.1074/jbc.M604039200. [DOI] [PubMed] [Google Scholar]
- Garcia-Heredia A, Marsillach J, Rull A, Triguero I, Fort I, Mackness B, Mackness M, Shih DM, Joven J, Camps J. Paraoxonase-1 inhibits oxidized low-density lipoprotein-induced metabolic alterations and apoptosis in endothelial cells: a nondirected metabolomic study. Mediators Inflamm. 2013;2013:156053. doi: 10.1155/2013/156053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardi C, Arezzini B, Monaco B, De Montis MG, Vecchio D, Comporti M. F2-isoprostane receptors on hepatic stellate cells. Lab Invest. 2008;88:124–131. doi: 10.1038/labinvest.3700712. [DOI] [PubMed] [Google Scholar]
- Gargalovic PS, Gharavi NM, Clark MJ, Pagnon J, Yang WP, He A, Truong A, Baruch-Oren T, Berliner JA, Kirchgessner TG, Lusis AJ. The Unfolded Protein Response Is an Important Regulator of Inflammatory Genes in Endothelial Cells. Arterioscler Thromb Vasc Biol. 2006a;26:2490–2496. doi: 10.1161/01.ATV.0000242903.41158.a1. [DOI] [PubMed] [Google Scholar]
- Gargalovic PS, Imura M, Zhang B, Gharavi NM, Clark MJ, Pagnon J, Yang WP, He A, Truong A, Patel S, Nelson SF, Horvath S, Berliner JA, Kirchgessner TG, Lusis AJ. Identification of inflammatory gene modules based on variations of human endothelial cell responses to oxidized lipids. Proceedings of the National Academy of Sciences. 2006b;103:12741–12746. doi: 10.1073/pnas.0605457103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharavi NM, Gargalovic PS, Chang I, Araujo JA, Clark MJ, Szeto WL, Watson AD, Lusis AJ, Berliner JA. High-density lipoprotein modulates oxidized phospholipid signaling in human endothelial cells from proinflammatory to anti-inflammatory. Arterioscler Thromb Vasc Biol. 2007;27:1346–1353. doi: 10.1161/ATVBAHA.107.141283. [DOI] [PubMed] [Google Scholar]
- Gillotte-Taylor K, Boullier A, Witztum JL, Steinberg D, Quehenberger O. Scavenger receptor class B type I as a receptor for oxidized low density lipoprotein. Journal of Lipid Research. 2001;42:1474–1482. [PubMed] [Google Scholar]
- Gomes RN, Bozza FA, Amancio RT, Japiassu AM, Vianna RC, Larangeira AP, Gouvea JM, Bastos MS, Zimmerman GA, Stafforini DM, Prescott SM, Bozza PT, Castro-Faria-Neto HC. Exogenous platelet-activating factor acetylhydrolase reduces mortality in mice with systemic inflammatory response syndrome and sepsis. Shock. 2006;26:41–49. doi: 10.1097/01.shk.0000209562.00070.1a. [DOI] [PubMed] [Google Scholar]
- Gopfert MS, Siedler F, Siess W, Sellmayer A. Structural identification of oxidized acyl-phosphatidylcholines that induce platelet activation. J Vasc Res. 2005;42:120–132. doi: 10.1159/000083461. [DOI] [PubMed] [Google Scholar]
- Govindarajan B, Junk A, Algeciras M, Salomon RG, Bhattacharya SK. Increased isolevuglandin-modified proteins in glaucomatous astrocytes. Molecular vision. 2009;15:1079–1091. [PMC free article] [PubMed] [Google Scholar]
- Govindarajan B, Laird J, Salomon RG, Bhattacharya SK. Isolevuglandin-Modified Proteins, Including Elevated Levels of Inactive Calpain-1, Accumulate in Glaucomatous Trabecular Meshwork. Biochemistry. 2008 doi: 10.1021/bi701517m. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg ME, Li XM, Gugiu BG, Gu X, Qin J, Salomon RG, Hazen SL. The lipid whisker model of the structure of oxidized cell membranes. J Biol Chem. 2008;283:2385–2396. doi: 10.1074/jbc.M707348200. [DOI] [PubMed] [Google Scholar]
- Gu X, Salomon RG. Fragmentation of a linoleate-derived gamma-hydroperoxy-alpha,beta-unsaturated epoxide to gamma-hydroxy- and gamma-oxo-alkenals involves a unique pseudo-symmetrical diepoxycarbinyl radical. Free Radic Biol Med. 2012;52:601–606. doi: 10.1016/j.freeradbiomed.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guichardant M, Bernoud-Hubac N, Chantegrel B, Deshayes C, Lagarde M. Aldehydes from n-6 fatty acid peroxidation. Effects on aminophospholipids. Prostaglandins Leukot Essent Fatty Acids. 2002;67:147–149. doi: 10.1054/plef.2002.0412. [DOI] [PubMed] [Google Scholar]
- Guichardant M, Taibi-Tronche P, Fay LB, Lagarde M. Covalent modifications of aminophospholipids by 4-hydroxynonenal. Free Radic Biol Med. 1998;25:1049–1056. doi: 10.1016/s0891-5849(98)00149-x. [DOI] [PubMed] [Google Scholar]
- Guo L, Chen Z, Amarnath V, Davies SS. Identification of novel bioactive aldehyde-modified phosphatidylethanolamines formed by lipid peroxidation. Free Radic Biol Med. 2012;53:1226–1238. doi: 10.1016/j.freeradbiomed.2012.07.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Chen Z, Cox BE, Amarnath V, Epand RF, Epand RM, Davies SS. Phosphatidylethanolamines modified by gamma-ketoaldehyde (gammaKA) induce endoplasmic reticulum stress and endothelial activation. J Biol Chem. 2011;286:18170–18180. doi: 10.1074/jbc.M110.213470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Gragg SD, Chen Z, Zhang Y, Amarnath V, Davies SS. Isolevuglandin-modified phosphatidylethanolamine is metabolized by NAPE-hydrolyzing phospholipase D. Journal of Lipid Research. 2013 doi: 10.1194/jlr.M042556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakkinen T, Luoma JS, Hiltunen MO, Macphee CH, Milliner KJ, Patel L, Rice SQ, Tew DG, Karkola K, Yla-Herttuala S. Lipoprotein-associated phospholipase A(2), platelet-activating factor acetylhydrolase, is expressed by macrophages in human and rabbit atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 1999;19:2909–2917. doi: 10.1161/01.atv.19.12.2909. [DOI] [PubMed] [Google Scholar]
- Halasiddappa LM, Koefeler H, Futerman AH, Hermetter A. Oxidized phospholipids induce ceramide accumulation in RAW 264.7 macrophages: role of ceramide synthases. PLoS One. 2013;8:e70002. doi: 10.1371/journal.pone.0070002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halvorsen B, Staff AC, Henriksen T, Sawamura T, Ranheim T. 8-iso-prostaglandin F(2alpha) increases expression of LOX-1 in JAR cells. Hypertension. 2001;37:1184–1190. doi: 10.1161/01.hyp.37.4.1184. [DOI] [PubMed] [Google Scholar]
- Hammond VJ, Morgan AH, Lauder S, Thomas CP, Brown S, Freeman BA, Lloyd CM, Davies J, Bush A, Levonen AL, Kansanen E, Villacorta L, Chen YE, Porter N, Garcia-Diaz YM, Schopfer FJ, O’Donnell VB. Novel keto-phospholipids are generated by monocytes and macrophages, detected in cystic fibrosis, and activate peroxisome proliferator-activated receptor-gamma. J Biol Chem. 2012;287:41651–41666. doi: 10.1074/jbc.M112.405407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hase M, Tanaka M, Yokota M, Yamada Y. Reduction in the extent of atherosclerosis in apolipoprotein E-deficient mice induced by electroporation-mediated transfer of the human plasma platelet-activating factor acetylhydrolase gene into skeletal muscle. Prostaglandins Other Lipid Mediat. 2002;70:107–118. doi: 10.1016/s0090-6980(02)00015-1. [DOI] [PubMed] [Google Scholar]
- Hattori K, Adachi H, Matsuzawa A, Yamamoto K, Tsujimoto M, Aoki J, Hattori M, Arai H, Inoue K. cDNA cloning and expression of intracellular platelet-activating factor (PAF) acetylhydrolase II. Its homology with plasma PAF acetylhydrolase. J Biol Chem. 1996;271:33032–33038. doi: 10.1074/jbc.271.51.33032. [DOI] [PubMed] [Google Scholar]
- Heery JM, Kozak M, Stafforini DM, Jones DA, Zimmerman GA, McIntyre TM, Prescott SM. Oxidatively modified LDL contains phospholipids with platelet-activating factor-like activity and stimulates the growth of smooth muscle cells. J Clin Invest. 1995;96:2322–2330. doi: 10.1172/JCI118288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisaka S, Yamada N, Naito K, Osawa T. The immunological and chemical detection of N-(hexanoyl)phosphatidylethanolamine and N-(hexanoyl)phosphatidylserine in an oxidative model induced by carbon tetrachloride. Biochem Biophys Res Commun. 2010;393:631–636. doi: 10.1016/j.bbrc.2010.02.043. [DOI] [PubMed] [Google Scholar]
- Hoffman SW, Moore S, Ellis EF. Isoprostanes: free radical-generated prostaglandins with constrictor effects on cerebral arterioles. Stroke. 1997;28:844–849. doi: 10.1161/01.str.28.4.844. [DOI] [PubMed] [Google Scholar]
- Hou X, Roberts LJ, 2nd, Gobeil F, Jr, Taber D, Kanai K, Abran D, Brault S, Checchin D, Sennlaub F, Lachapelle P, Varma D, Chemtob S. Isomer-specific contractile effects of a series of synthetic f2-isoprostanes on retinal and cerebral microvasculature. Free Radic Biol Med. 2004;36:163–172. doi: 10.1016/j.freeradbiomed.2003.10.024. [DOI] [PubMed] [Google Scholar]
- Huber J, Vales A, Mitulovic G, Blumer M, Schmid R, Witztum JL, Binder BR, Leitinger N. Oxidized membrane vesicles and blebs from apoptotic cells contain biologically active oxidized phospholipids that induce monocyte-endothelial interactions. Arterioscler Thromb Vasc Biol. 2002;22:101–107. doi: 10.1161/hq0102.101525. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Mutoh M, Teraoka N, Nakanishi H, Wakabayashi K, Taguchi R. Increase of oxidant-related triglycerides and phosphatidylcholines in serum and small intestinal mucosa during development of intestinal polyp formation in Min mice. Cancer science. 2011;102:79–87. doi: 10.1111/j.1349-7006.2010.01754.x. [DOI] [PubMed] [Google Scholar]
- Ikura Y, Ohsawa M, Suekane T, Fukushima H, Itabe H, Jomura H, Nishiguchi S, Inoue T, Naruko T, Ehara S, Kawada N, Arakawa T, Ueda M. Localization of oxidized phosphatidylcholine in nonalcoholic fatty liver disease: impact on disease progression. Hepatology. 2006;43:506–514. doi: 10.1002/hep.21070. [DOI] [PubMed] [Google Scholar]
- Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, Ermolaeva M, Veldhuizen R, Leung YH, Wang H, Liu H, Sun Y, Pasparakis M, Kopf M, Mech C, Bavari S, Peiris JS, Slutsky AS, Akira S, Hultqvist M, Holmdahl R, Nicholls J, Jiang C, Binder CJ, Penninger JM. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133:235–249. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H, Tanabe T, Umesono K. Feedback control of cyclooxygenase-2 expression through PPARgamma. Journal of Biological Chemistry. 2000;275:28028–28032. doi: 10.1074/jbc.M001387200. [DOI] [PubMed] [Google Scholar]
- Inoue T, Sugimoto A, Suzuki Y, Yamamoto M, Tsujimoto M, Inoue K, Aoki J, Arai H. Type II platelet-activating factor-acetylhydrolase is essential for epithelial morphogenesis in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2004;101:13233–13238. doi: 10.1073/pnas.0405507101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn U, Galano JM, Durand T. Beyond prostaglandins--chemistry and biology of cyclic oxygenated metabolites formed by free-radical pathways from polyunsaturated fatty acids. Angew Chem Int Ed Engl. 2008;47:5894–5955. doi: 10.1002/anie.200705122. [DOI] [PubMed] [Google Scholar]
- Jamil J, Wright A, Harrison N, Kegey E, Flowers AF, Flyod NJ, Kotera C, Guy A, Galano JM, Durand T, Njie-Mbye YF, Ohia SE, Opere CA. Regulation of [(3)H]d-aspartate release by the 5-F(2t)-isoprostane and its 5-epimer in isolated bovine retina. Neurochem Res. 2012;37:574–582. doi: 10.1007/s11064-011-0645-5. [DOI] [PubMed] [Google Scholar]
- Janssen LJ, Tazzeo T. Involvement of TP and EP3 Receptors in Vasoconstrictor Responses to Isoprostanes in Pulmonary Vasculature. Journal of Pharmacology and Experimental Therapeutics. 2002;301:1060–1066. doi: 10.1124/jpet.301.3.1060. [DOI] [PubMed] [Google Scholar]
- Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- Johnstone SR, Ross J, Rizzo MJ, Straub AC, Lampe PD, Leitinger N, Isakson BE. Oxidized phospholipid species promote in vivo differential cx43 phosphorylation and vascular smooth muscle cell proliferation. Am J Pathol. 2009;175:916–924. doi: 10.2353/ajpath.2009.090160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung ME, Berliner JA, Angst D, Yue D, Koroniak L, Watson AD, Li R. Total synthesis of the epoxy isoprostane phospholipids PEIPC and PECPC. Org Lett. 2005;7:3933–3935. doi: 10.1021/ol051415y. [DOI] [PubMed] [Google Scholar]
- Jung ME, Berliner JA, Koroniak L, Gugiu BG, Watson AD. Improved synthesis of the epoxy isoprostane phospholipid PEIPC and its reactivity with amines. Org Lett. 2008;10:4207–4209. doi: 10.1021/ol8014804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadl A, Sharma PR, Chen W, Agrawal R, Meher AK, Rudraiah S, Grubbs N, Sharma R, Leitinger N. Oxidized phospholipid-induced inflammation is mediated by Toll-like receptor 2. Free Radic Biol Med. 2011;51:1903–1909. doi: 10.1016/j.freeradbiomed.2011.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karara A, Wei S, Spady D, Swift L, Capdevila JH, Falck JR. Arachidonic acid epoxygenase: structural characterization and quantification of epoxyeicosatrienoates in plasma. Biochem Biophys Res Commun. 1992;182:1320–1325. doi: 10.1016/0006-291x(92)91877-s. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Kawai Y, Ogamo A, Nakagawa Y. Formation of the aldehydic choline glycerophospholipids in human red blood cell membrane peroxidized with an azo initiator. J Biochem. 1999;126:115–120. doi: 10.1093/oxfordjournals.jbchem.a022411. [DOI] [PubMed] [Google Scholar]
- Kayganich-Harrison KA, Rose DM, Murphy RC, Morrow JD, Roberts LJ., 2nd Collision-induced dissociation of F2-isoprostane-containing phospholipids. J Lipid Res. 1993;34:1229–1235. [PubMed] [Google Scholar]
- Keith RJ, Haberzettl P, Vladykovskaya E, Hill BG, Kaiserova K, Srivastava S, Barski O, Bhatnagar A. Aldose reductase decreases endoplasmic reticulum stress in ischemic hearts. Chemico-biological interactions. 2009;178:242–249. doi: 10.1016/j.cbi.2008.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller JN, Hanni KB, Markesbery WR. Impaired proteasome function in Alzheimer’s disease. J Neurochem. 2000;75:436–439. doi: 10.1046/j.1471-4159.2000.0750436.x. [DOI] [PubMed] [Google Scholar]
- Khasawneh FT, Huang JS, Mir F, Srinivasan S, Tiruppathi C, Le Breton GC. Characterization of isoprostane signaling: Evidence for a unique coordination profile of 8-iso-PGF2α with the thromboxane A2 receptor, and activation of a separate cAMP-dependent inhibitory pathway in human platelets. Biochemical Pharmacology. 2008;75:2301–2315. doi: 10.1016/j.bcp.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikugawa K, Kosugi H, Asakura T. Effect of malondialdehyde, a product of lipid peroxidation, on the function and stability of hemoglobin. Arch Biochem Biophys. 1984;229:7–14. doi: 10.1016/0003-9861(84)90124-3. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Choi NY, Koo JE, Kim SY, Joung SM, Jeong E, Lee JY. Suppression of Toll-like receptor 4 activation by endogenous oxidized phosphatidylcholine, KOdiA-PC by inhibiting LPS binding to MD2. Inflamm Res. 2013;62:571–580. doi: 10.1007/s00011-013-0609-0. [DOI] [PubMed] [Google Scholar]
- Knutson MD, Handelman GJ, Viteri FE. Methods for measuring ethane and pentane in expired air from rats and humans. Free Radical Biology and Medicine. 2000;28:514–519. doi: 10.1016/s0891-5849(99)00230-0. [DOI] [PubMed] [Google Scholar]
- Kono N, Inoue T, Yoshida Y, Sato H, Matsusue T, Itabe H, Niki E, Aoki J, Arai H. Protection against oxidative stress-induced hepatic injury by intracellular type II platelet-activating factor acetylhydrolase by metabolism of oxidized phospholipids in vivo. J Biol Chem. 2008;283:1628–1636. doi: 10.1074/jbc.M708622200. [DOI] [PubMed] [Google Scholar]
- Kriska T, Marathe GK, Schmidt JC, McIntyre TM, Girotti AW. Phospholipase action of platelet-activating factor acetylhydrolase, but not paraoxonase-1, on long fatty acyl chain phospholipid hydroperoxides. J Biol Chem. 2007;282:100–108. doi: 10.1074/jbc.M608135200. [DOI] [PubMed] [Google Scholar]
- Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. International reviews of immunology. 2011;30:16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- Kunapuli P, Lawson JA, Rokach J, FitzGerald GA. Functional characterization of the ocular prostaglandin f2alpha (PGF2alpha) receptor. Activation by the isoprostane, 12-iso-PGF2alpha. J Biol Chem. 1997;272:27147–27154. doi: 10.1074/jbc.272.43.27147. [DOI] [PubMed] [Google Scholar]
- Lai E, Teodoro T, Volchuk A. Endoplasmic Reticulum Stress: Signaling the Unfolded Protein Response. Physiology. 2007;22:193–201. doi: 10.1152/physiol.00050.2006. [DOI] [PubMed] [Google Scholar]
- Lappas M, Permezel M, Holdsworth SJ, Zanoni G, Porta A, Rice GE. Antiinflammatory effects of the cyclopentenone isoprostane 15-A(2)-IsoP in human gestational tissues. Free Radic Biol Med. 2007;42:1791–1796. doi: 10.1016/j.freeradbiomed.2007.03.009. [DOI] [PubMed] [Google Scholar]
- Latchoumycandane C, Marathe GK, Zhang R, McIntyre TM. Oxidatively truncated phospholipids are required agents of tumor necrosis factor alpha (TNFalpha)-induced apoptosis. J Biol Chem. 2012;287:17693–17705. doi: 10.1074/jbc.M111.300012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebel W, Riccardi K, Grasser WA, Terry K, Thompson D, Paralkar VM. Prostaglandin E2 receptor subtype EP-2 is not involved in the induction of non-pregnant guinea pig uterine contractions associated with terminal pregnancy. Prostaglandins, leukotrienes, and essential fatty acids. 2004;71:399–404. doi: 10.1016/j.plefa.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Lederer MO, Baumann M. Formation of a phospholipid-linked pyrrolecarbaldehyde from model reactions of D-glucose and 3-deoxyglucosone with phosphatidyl ethanolamine. Bioorg Med Chem. 2000;8:115–121. doi: 10.1016/s0968-0896(99)00264-3. [DOI] [PubMed] [Google Scholar]
- Lee ES, Jiang J, Sund GC, Simonson WT, Graham J, Dietsch G, Schimpf B, Bieg S, Peterman G, Lernmark A. Recombinant human platelet-activating factor acetylhydrolase reduces the frequency of diabetes in the diabetes-prone BB rat. Diabetes. 1999;48:43–49. doi: 10.2337/diabetes.48.1.43. [DOI] [PubMed] [Google Scholar]
- Lee H, Shi W, Tontonoz P, Wang S, Subbanagounder G, Hedrick CC, Hama S, Borromeo C, Evans RM, Berliner JA, Nagy L. Role for peroxisome proliferator-activated receptor alpha in oxidized phospholipid-induced synthesis of monocyte chemotactic protein-1 and interleukin-8 by endothelial cells. Circ Res. 2000;87:516–521. doi: 10.1161/01.res.87.6.516. [DOI] [PubMed] [Google Scholar]
- Lehr HA, Seemuller J, Hubner C, Menger MD, Messmer K. Oxidized LDL-induced leukocyte/endothelium interaction in vivo involves the receptor for platelet-activating factor. Arterioscler Thromb. 1993;13:1013–1018. doi: 10.1161/01.atv.13.7.1013. [DOI] [PubMed] [Google Scholar]
- Leitinger N, Tyner TR, Oslund L, Rizza C, Subbanagounder G, Lee H, Shih PT, Mackman N, Tigyi G, Territo MC, Berliner JA, Vora DK. Structurally similar oxidized phospholipids differentially regulate endothelial binding of monocytes and neutrophils. Proc Natl Acad Sci U S A. 1999;96:12010–12015. doi: 10.1073/pnas.96.21.12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitinger N, Watson AD, Faull KF, Fogelman AM, Berliner JA. Monocyte binding to endothelial cells induced by oxidized phospholipids present in minimally oxidized low density lipoprotein is inhibited by a platelet activating factor receptor antagonist. Adv Exp Med Biol. 1997;433:379–382. doi: 10.1007/978-1-4899-1810-9_82. [DOI] [PubMed] [Google Scholar]
- Lertsiri S, Shiraishi M, Miyazawa T. Identification of deoxy-D-fructosyl phosphatidylethanolamine as a non-enzymic glycation product of phosphatidylethanolamine and its occurrence in human blood plasma and red blood cells. Biosci Biotechnol Biochem. 1998;62:893–901. doi: 10.1271/bbb.62.893. [DOI] [PubMed] [Google Scholar]
- Levi V, Villamil Giraldo AM, Castello PR, Rossi JP, Gonzalez Flecha FL. Effects of phosphatidylethanolamine glycation on lipid-protein interactions and membrane protein thermal stability. Biochem J. 2008;416:145–152. doi: 10.1042/BJ20080618. [DOI] [PubMed] [Google Scholar]
- Li R, Mouillesseaux KP, Montoya D, Cruz D, Gharavi N, Dun M, Koroniak L, Berliner JA. Identification of prostaglandin E2 receptor subtype 2 as a receptor activated by OxPAPC. Circ Res. 2006;98:642–650. doi: 10.1161/01.RES.0000207394.39249.fc. [DOI] [PubMed] [Google Scholar]
- Li W, Laird JM, Lu L, Roychowdhury S, Nagy LE, Zhou R, Crabb JW, Salomon RG. Isolevuglandins covalently modify phosphatidylethanolamines in vivo: Detection and quantitative analysis of hydroxylactam adducts. Free Radic Biol Med. 2009;47:1539–1552. doi: 10.1016/j.freeradbiomed.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Chen R, Marathe GK, Febbraio M, Zou W, McIntyre TM. Circulating platelet-activating factor is primarily cleared by transport, not intravascular hydrolysis by lipoprotein-associated phospholipase A2/PAF acetylhydrolase. Circ Res. 2011;108:469–477. doi: 10.1161/CIRCRESAHA.110.228742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loidl A, Sevcsik E, Riesenhuber G, Deigner HP, Hermetter A. Oxidized phospholipids in minimally modified low density lipoprotein induce apoptotic signaling via activation of acid sphingomyelinase in arterial smooth muscle cells. J Biol Chem. 2003;278:32921–32928. doi: 10.1074/jbc.M306088200. [DOI] [PubMed] [Google Scholar]
- Lopez Salon M, Pasquini L, Besio Moreno M, Pasquini JM, Soto E. Relationship between beta-amyloid degradation and the 26S proteasome in neural cells. Exp Neurol. 2003;180:131–143. doi: 10.1016/s0014-4886(02)00060-2. [DOI] [PubMed] [Google Scholar]
- Manavalan B, Basith S, Choi S. Similar Structures but Different Roles - An Updated Perspective on TLR Structures. Frontiers in physiology. 2011;2:41. doi: 10.3389/fphys.2011.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marathe GK, Davies SS, Harrison KA, Silva AR, Murphy RC, Castro-Faria-Neto H, Prescott SM, Zimmerman GA, McIntyre TM. Inflammatory platelet-activating factor-like phospholipids in oxidized low density lipoproteins are fragmented alkyl phosphatidylcholines. J Biol Chem. 1999;274:28395–28404. doi: 10.1074/jbc.274.40.28395. [DOI] [PubMed] [Google Scholar]
- Marathe GK, Johnson C, Billings SD, Southall MD, Pei Y, Spandau D, Murphy RC, Zimmerman GA, McIntyre TM, Travers JB. Ultraviolet B radiation generates platelet-activating factor-like phospholipids underlying cutaneous damage. J Biol Chem. 2005;280:35448–35457. doi: 10.1074/jbc.M503811200. [DOI] [PubMed] [Google Scholar]
- Maskrey BH, Bermudez-Fajardo A, Morgan AH, Stewart-Jones E, Dioszeghy V, Taylor GW, Baker PR, Coles B, Coffey MJ, Kuhn H, O’Donnell VB. Activated platelets and monocytes generate four hydroxyphosphatidylethanolamines via lipoxygenase. J Biol Chem. 2007;282:20151–20163. doi: 10.1074/jbc.M611776200. [DOI] [PubMed] [Google Scholar]
- Matsuzawa A, Hattori K, Aoki J, Arai H, Inoue K. Protection against oxidative stress-induced cell death by intracellular platelet-activating factor-acetylhydrolase II. J Biol Chem. 1997;272:32315–32320. doi: 10.1074/jbc.272.51.32315. [DOI] [PubMed] [Google Scholar]
- Milatovic D, Aschner M. Measurement of isoprostanes as markers of oxidative stress in neuronal tissue. Current protocols in toxicology/editorial board, Mahin D Maines. 2009;Chapter 12(Unit12):14. doi: 10.1002/0471140856.tx1214s39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller YI, Viriyakosol S, Worrall DS, Boullier A, Butler S, Witztum JL. Toll-like receptor 4-dependent and -independent cytokine secretion induced by minimally oxidized low-density lipoprotein in macrophages. Arterioscler Thromb Vasc Biol. 2005;25:1213–1219. doi: 10.1161/01.ATV.0000159891.73193.31. [DOI] [PubMed] [Google Scholar]
- Milne GL, Gao L, Porta A, Zanoni G, Vidari G, Morrow JD. Identification of the major urinary metabolite of the highly reactive cyclopentenone isoprostane 15-A(2t)-isoprostane in vivo. J Biol Chem. 2005a;280:25178–25184. doi: 10.1074/jbc.M502891200. [DOI] [PubMed] [Google Scholar]
- Milne GL, Musiek ES, Morrow JD. F2-isoprostanes as markers of oxidative stress in vivo: an overview. Biomarkers: biochemical indicators of exposure, response, and susceptibility to chemicals. 2005b;10(Suppl 1):S10–23. doi: 10.1080/13547500500216546. [DOI] [PubMed] [Google Scholar]
- Milne GL, Yin H, Hardy KD, Davies SS, Roberts LJ., 2nd Isoprostane generation and function. Chemical reviews. 2011;111:5973–5996. doi: 10.1021/cr200160h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min JH, Wilder C, Aoki J, Arai H, Inoue K, Paul L, Gelb MH. Platelet-activating factor acetylhydrolases: broad substrate specificity and lipoprotein binding does not modulate the catalytic properties of the plasma enzyme. Biochemistry. 2001;40:4539–4549. doi: 10.1021/bi002600g. [DOI] [PubMed] [Google Scholar]
- Mishra VK, Palgunachari MN, Hudson JS, Shin R, Keenum TD, Krishna NR, Anantharamaiah GM. Structure and lipid interactions of an anti-inflammatory and anti-atherogenic 10-residue class G(*) apolipoprotein J peptide using solution NMR. Biochim Biophys Acta. 2011;1808:498–507. doi: 10.1016/j.bbamem.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi M, Nakano Y, Sakaguchi T, Ogi H, Oda N, Suenari K, Kiyotani K, Ozono R, Oshima T, Yoshida T, Chayama K. Gene delivery of paraoxonase-1 inhibits neointimal hyperplasia after arterial balloon-injury in rabbits fed a high-fat diet. Hypertens Res. 2007;30:85–91. doi: 10.1291/hypres.30.85. [DOI] [PubMed] [Google Scholar]
- Mohler ER, Franklin MT, Adam LP. Intracellular signaling by 8-epi-prostaglandin F2 alpha is mediated by thromboxane A2/prostaglandin endoperoxide receptors in porcine carotid arteries. Biochem Biophys Res Commun. 1996;225:915–923. doi: 10.1006/bbrc.1996.1272. [DOI] [PubMed] [Google Scholar]
- Montine KS, Quinn JF, Zhang J, Fessel JP, Roberts LJ, 2nd, Morrow JD, Montine TJ. Isoprostanes and related products of lipid peroxidation in neurodegenerative diseases. Chem Phys Lipids. 2004;128:117–124. doi: 10.1016/j.chemphyslip.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Morales CR, Terry ES, Zackert WE, Montine TJ, Morrow JD. Improved assay for the quantification of the major urinary metabolite of the isoprostane 15-F(2t)-Isoprostane (8-iso-PGF(2alpha)) by a stable isotope dilution mass spectrometric assay. Clin Chim Acta. 2001;314:93–99. doi: 10.1016/s0009-8981(01)00637-4. [DOI] [PubMed] [Google Scholar]
- Morcillo EJ, Cortijo J. Mucus and MUC in asthma. Curr Opin Pulm Med. 2006;12:1–6. doi: 10.1097/01.mcp.0000198064.27586.37. [DOI] [PubMed] [Google Scholar]
- Morgan AH, Dioszeghy V, Maskrey BH, Thomas CP, Clark SR, Mathie SA, Lloyd CM, Kuhn H, Topley N, Coles BC, Taylor PR, Jones SA, O’Donnell VB. Phosphatidylethanolamine-esterified eicosanoids in the mouse: tissue localization and inflammation-dependent formation in Th-2 disease. J Biol Chem. 2009;284:21185–21191. doi: 10.1074/jbc.M109.021634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow JD, Minton TA, Mukundan CR, Campbell MD, Zackert WE, Daniel VC, Badr KF, Blair IA, Roberts LJ., 2nd Free radical-induced generation of isoprostanes in vivo. Evidence for the formation of D-ring and E-ring isoprostanes. J Biol Chem. 1994;269:4317–4326. [PubMed] [Google Scholar]
- Morrow JD, Zackert WE, Yang JP, Kurhts EH, Callewaert D, Dworski R, Kanai K, Taber D, Moore K, Oates JA, Roberts LJ. Quantification of the major urinary metabolite of 15-F2t-isoprostane (8-iso-PGF2alpha) by a stable isotope dilution mass spectrometric assay. Anal Biochem. 1999;269:326–331. doi: 10.1006/abio.1999.4008. [DOI] [PubMed] [Google Scholar]
- Mueed I, Tazzeo T, Liu C, Pertens E, Zhang Y, Cybulski I, Semelhago L, Noora J, Lamy A, Teoh K, Chu V, Janssen LJ. Isoprostanes constrict human radial artery by stimulation of thromboxane receptors, Ca2+ release, and RhoA activation. The Journal of Thoracic and Cardiovascular Surgery. 2008;135:131–138. doi: 10.1016/j.jtcvs.2007.06.023. [DOI] [PubMed] [Google Scholar]
- Murthi KK, Friedman LR, Oleinick NL, Salomon RG. Formation of DNA-protein cross-links in mammalian cells by levuglandin E2. Biochemistry. 1993;32:4090–4097. doi: 10.1021/bi00066a034. [DOI] [PubMed] [Google Scholar]
- Mwaikambo BR, Sennlaub F, Ong H, Chemtob S, Hardy P. Activation of CD36 inhibits and induces regression of inflammatory corneal neovascularization. Invest Ophthalmol Vis Sci. 2006;47:4356–4364. doi: 10.1167/iovs.05-1656. [DOI] [PubMed] [Google Scholar]
- Nadkarni DV, Sayre LM. Structural definition of early lysine and histidine adduction chemistry of 4-hydroxynonenal. Chem Res Toxicol. 1995;8:284–291. doi: 10.1021/tx00044a014. [DOI] [PubMed] [Google Scholar]
- Nakagawa K, Oak JH, Miyazawa T. Angiogenic potency of Amadori-glycated phosphatidylethanolamine. Ann N Y Acad Sci. 2005;1043:413–416. doi: 10.1196/annals.1333.048. [DOI] [PubMed] [Google Scholar]
- Nakajima T, Davies SS, Matafonova E, Potet F, Amarnath V, Tallman KA, Serwa RA, Porter NA, Balser JR, Kupershmidt S, Roberts LJ., 3rd Selective gamma-ketoaldehyde scavengers protect Nav1.5 from oxidant-induced inactivation. J Mol Cell Cardiol. 2010;48:352–359. doi: 10.1016/j.yjmcc.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Bratton DL, Murphy RC. Analysis of epoxyeicosatrienoic and monohydroxyeicosatetraenoic acids esterified to phospholipids in human red blood cells by electrospray tandem mass spectrometry. Journal of mass spectrometry: JMS. 1997;32:888–896. doi: 10.1002/(SICI)1096-9888(199708)32:8<888::AID-JMS548>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Oak J, Nakagawa K, Miyazawa T. Synthetically prepared Aamadori-glycated phosphatidylethanolaminecan trigger lipid peroxidation via free radical reactions. FEBS Lett. 2000;481:26–30. doi: 10.1016/s0014-5793(00)01966-9. [DOI] [PubMed] [Google Scholar]
- Oak JH, Nakagawa K, Miyazawa T. UV analysis of Amadori-glycated phosphatidylethanolamine in foods and biological samples. J Lipid Res. 2002;43:523–529. [PubMed] [Google Scholar]
- Oak JH, Nakagawa K, Oikawa S, Miyazawa T. Amadori-glycated phosphatidylethanolamine induces angiogenic differentiations in cultured human umbilical vein endothelial cells. FEBS Lett. 2003;555:419–423. doi: 10.1016/s0014-5793(03)01237-7. [DOI] [PubMed] [Google Scholar]
- Obsil T, Amler E, Obsilova V, Pavlicek Z. Effect of aminophospholipid glycation on order parameter and hydration of phospholipid bilayer. Biophys Chem. 1999;80:165–177. doi: 10.1016/s0301-4622(99)00067-8. [DOI] [PubMed] [Google Scholar]
- Odabaei G, Chatterjee D, Jazirehi AR, Goodglick L, Yeung K, Bonavida B. Raf-1 kinase inhibitor protein: structure, function, regulation of cell signaling, and pivotal role in apoptosis. Advances in cancer research. 2004;91:169–200. doi: 10.1016/S0065-230X(04)91005-6. [DOI] [PubMed] [Google Scholar]
- Omarova S, Charvet CD, Reem RE, Mast N, Zheng W, Huang S, Peachey NS, Pikuleva IA. Abnormal vascularization in mouse retina with dysregulated retinal cholesterol homeostasis. J Clin Invest. 2012;122:3012–3023. doi: 10.1172/JCI63816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opere CA, Zheng WD, Huang J, Adewale A, Kruglet M, Ohia SE. Dual effect of isoprostanes on the release of [3H]D-aspartate from isolated bovine retinae: role of arachidonic acid metabolites. Neurochem Res. 2005;30:129–137. doi: 10.1007/s11064-004-9694-3. [DOI] [PubMed] [Google Scholar]
- Ostermann G, Kostner GM, Gries A, Malle E, Till U. The contribution of individual lipoproteins to the degradation of platelet-activating factor in human serum. Haemostasis. 1989;19:160–168. doi: 10.1159/000215910. [DOI] [PubMed] [Google Scholar]
- Packard CJ, O’Reilly DS, Caslake MJ, McMahon AD, Ford I, Cooney J, Macphee CH, Suckling KE, Krishna M, Wilkinson FE, Rumley A, Lowe GD. Lipoprotein-associated phospholipase A2 as an independent predictor of coronary heart disease. West of Scotland Coronary Prevention Study Group. N Engl J Med. 2000;343:1148–1155. doi: 10.1056/NEJM200010193431603. [DOI] [PubMed] [Google Scholar]
- Pamplona R, Bellmunt MJ, Portero M, Riba D, Prat J. Chromatographic evidence for Amadori product formation in rat liver aminophospholipids. Life Sci. 1995;57:873–879. doi: 10.1016/0024-3205(95)02020-j. [DOI] [PubMed] [Google Scholar]
- Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–1195. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- Patel KD, Zimmerman GA, Prescott SM, McIntyre TM. Novel leukocyte agonists are released by endothelial cells exposed to peroxide. J Biol Chem. 1992;267:15168–15175. [PubMed] [Google Scholar]
- Pegorier S, Stengel D, Durand H, Croset M, Ninio E. Oxidized phospholipid: POVPC binds to platelet-activating-factor receptor on human macrophages. Implications in atherosclerosis. Atherosclerosis. 2006;188:433–443. doi: 10.1016/j.atherosclerosis.2005.11.015. [DOI] [PubMed] [Google Scholar]
- Pidkovka NA, Cherepanova OA, Yoshida T, Alexander MR, Deaton RA, Thomas JA, Leitinger N, Owens GK. Oxidized phospholipids induce phenotypic switching of vascular smooth muscle cells in vivo and in vitro. Circ Res. 2007;101:792–801. doi: 10.1161/CIRCRESAHA.107.152736. [DOI] [PubMed] [Google Scholar]
- Podrez EA, Byzova TV, Febbraio M, Salomon RG, Ma Y, Valiyaveettil M, Poliakov E, Sun M, Finton PJ, Curtis BR, Chen J, Zhang R, Silverstein RL, Hazen SL. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med. 2007;13:1086–1095. doi: 10.1038/nm1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podrez EA, Poliakov E, Shen Z, Zhang R, Deng Y, Sun M, Finton PJ, Shan L, Febbraio M, Hajjar DP, Silverstein RL, Hoff HF, Salomon RG, Hazen SL. A novel family of atherogenic oxidized phospholipids promotes macrophage foam cell formation via the scavenger receptor CD36 and is enriched in atherosclerotic lesions. J Biol Chem. 2002a;277:38517–38523. doi: 10.1074/jbc.M205924200. [DOI] [PubMed] [Google Scholar]
- Podrez EA, Poliakov E, Shen Z, Zhang R, Deng Y, Sun M, Finton PJ, Shan L, Gugiu B, Fox PL, Hoff HF, Salomon RG, Hazen SL. Identification of a novel family of oxidized phospholipids that serve as ligands for the macrophage scavenger receptor CD36. J Biol Chem. 2002b;277:38503–38516. doi: 10.1074/jbc.M203318200. [DOI] [PubMed] [Google Scholar]
- Poliakov E, Meer SG, Roy SC, Mesaros C, Salomon RG. Iso[7]LGD2-protein adducts are abundant in vivo and free radical-induced oxidation of an arachidonyl phospholipid generates this D series isolevuglandin in vitro. Chem Res Toxicol. 2004;17:613–622. doi: 10.1021/tx034185+. [DOI] [PubMed] [Google Scholar]
- Pollreisz A, Afonyushkin T, Oskolkova OV, Gruber F, Bochkov VN, Schmidt-Erfurth U. Retinal pigment epithelium cells produce VEGF in response to oxidized phospholipids through mechanisms involving ATF4 and protein kinase CK2. Experimental eye research. 2013;116:177–184. doi: 10.1016/j.exer.2013.08.021. [DOI] [PubMed] [Google Scholar]
- Pontsler AV, St Hilaire A, Marathe GK, Zimmerman GA, McIntyre TM. Cyclooxygenase-2 is induced in monocytes by peroxisome proliferator activated receptor gamma and oxidized alkyl phospholipids from oxidized low density lipoprotein. J Biol Chem. 2002;277:13029–13036. doi: 10.1074/jbc.M109546200. [DOI] [PubMed] [Google Scholar]
- Porter NA, Lehman LS. Xanthine-Oxidase Initiated Oxidation of Model Membranes -Effect of Position of Abstractable Hydrogen-Atoms in the Bilayer on the Distribution of Products. Journal of the American Chemical Society. 1982;104:4731–4732. [Google Scholar]
- Proudfoot JM, Barden AE, Loke WM, Croft KD, Puddey IB, Mori TA. HDL is the major lipoprotein carrier of plasma F2-isoprostanes. J Lipid Res. 2009;50:716–722. doi: 10.1194/jlr.M800607-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Testai FD, Dawson S, Kilkus J, Dawson G. Oxidized phosphatidylcholine formation and action in oligodendrocytes. J Neurochem. 2009;110:1388–1399. doi: 10.1111/j.1471-4159.2009.06231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarck R, De Geest B, Stengel D, Mertens A, Lox M, Theilmeier G, Michiels C, Raes M, Bult H, Collen D, Van Veldhoven P, Ninio E, Holvoet P. Adenovirus-mediated gene transfer of human platelet-activating factor-acetylhydrolase prevents injury-induced neointima formation and reduces spontaneous atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2001;103:2495–2500. doi: 10.1161/01.cir.103.20.2495. [DOI] [PubMed] [Google Scholar]
- Ravandi A, Kuksis A, Marai L, Myher JJ. Preparation and characterization of glucosylated aminoglycerophospholipids. Lipids. 1995;30:885–891. doi: 10.1007/BF02537478. [DOI] [PubMed] [Google Scholar]
- Ravandi A, Kuksis A, Shaikh NA. Glycated Phosphatidylethanolamine Promotes Macrophage Uptake of Low Density Lipoprotein and Accumulation of Cholesteryl Esters and Triacylglycerols. Journal of Biological Chemistry. 1999;274:16494–16500. doi: 10.1074/jbc.274.23.16494. [DOI] [PubMed] [Google Scholar]
- Ravandi A, Kuksis A, Shaikh NA. Glucosylated glycerophosphoethanolamines are the major LDL glycation products and increase LDL susceptibility to oxidation: evidence of their presence in atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20:467–477. doi: 10.1161/01.atv.20.2.467. [DOI] [PubMed] [Google Scholar]
- Requena JR, Ahmed MU, Fountain CW, Degenhardt TP, Reddy S, Perez C, Lyons TJ, Jenkins AJ, Baynes JW, Thorpe SR. Carboxymethylethanolamine, a biomarker of phospholipid modification during the maillard reaction in vivo. J Biol Chem. 1997;272:17473–17479. doi: 10.1074/jbc.272.28.17473. [DOI] [PubMed] [Google Scholar]
- Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- Roberts LJ, 2nd, Fessel JP, Davies SS. The biochemistry of the isoprostane, neuroprostane, and isofuran Pathways of lipid peroxidation. Brain Pathol. 2005;15:143–148. doi: 10.1111/j.1750-3639.2005.tb00511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roychowdhury S, McMullen MR, Pritchard MT, Li W, Salomon RG, Nagy LE. Formation of gamma-ketoaldehyde-protein adducts during ethanol-induced liver injury in mice. Free Radic Biol Med. 2009;47:1526–1538. doi: 10.1016/j.freeradbiomed.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon RG. Structural identification and cardiovascular activities of oxidized phospholipids. Circ Res. 2012;111:930–946. doi: 10.1161/CIRCRESAHA.112.275388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon RG, Batyreva E, Kaur K, Sprecher DL, Schreiber MJ, Crabb JW, Penn MS, DiCorletoe AM, Hazen SL, Podrez EA. Isolevuglandin-protein adducts in humans: products of free radical-induced lipid oxidation through the isoprostane pathway. Biochim Biophys Acta. 2000;1485:225–235. doi: 10.1016/s1388-1981(00)00038-x. [DOI] [PubMed] [Google Scholar]
- Schmidley JW, Dadson J, Iyer RS, Salomon RG. Brain tissue injury and blood-brain barrier opening induced by injection of LGE2 or PGE2. Prostaglandins, leukotrienes, and essential fatty acids. 1992;47:105–110. doi: 10.1016/0952-3278(92)90145-9. [DOI] [PubMed] [Google Scholar]
- Schneider C, Boeglin WE, Yin H, Porter NA, Brash AR. Intermolecular peroxyl radical reactions during autoxidation of hydroxy and hydroperoxy arachidonic acids generate a novel series of epoxidized products. Chem Res Toxicol. 2008;21:895–903. doi: 10.1021/tx700357u. [DOI] [PubMed] [Google Scholar]
- Seimon TA, Nadolski MJ, Liao X, Magallon J, Nguyen M, Feric NT, Koschinsky ML, Harkewicz R, Witztum JL, Tsimikas S, Golenbock D, Moore KJ, Tabas I. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab. 2010;12:467–482. doi: 10.1016/j.cmet.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Zhang P, Zhang L, Osman H, Mohler ER, 3rd, Macphee C, Zalewski A, Postle A, Wilensky RL. Role of lipoprotein-associated phospholipase A2 in leukocyte activation and inflammatory responses. Atherosclerosis. 2007;191:54–62. doi: 10.1016/j.atherosclerosis.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Shih DM, Xia YR, Wang XP, Miller E, Castellani LW, Subbanagounder G, Cheroutre H, Faull KF, Berliner JA, Witztum JL, Lusis AJ. Combined serum paraoxonase knockout/apolipoprotein E knockout mice exhibit increased lipoprotein oxidation and atherosclerosis. J Biol Chem. 2000;275:17527–17535. doi: 10.1074/jbc.M910376199. [DOI] [PubMed] [Google Scholar]
- Shimozu Y, Hirano K, Shibata T, Shibata N, Uchida K. 4-Hydroperoxy-2-nonenal is not just an intermediate but a reactive molecule that covalently modifies proteins to generate unique intramolecular oxidation products. J Biol Chem. 2011;286:29313–29324. doi: 10.1074/jbc.M111.255737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SH, Song HY, Kim MY, Do EK, Lee JS, Kim JH. Oxidized phosphatidylcholine induces migration of bone marrow-derived mesenchymal stem cells through Kruppel-like factor 4-dependent mechanism. Mol Cell Biochem. 2011;352:109–115. doi: 10.1007/s11010-011-0745-1. [DOI] [PubMed] [Google Scholar]
- Shishehbor MH, Zhang R, Medina H, Brennan ML, Brennan DM, Ellis SG, Topol EJ, Hazen SL. Systemic elevations of free radical oxidation products of arachidonic acid are associated with angiographic evidence of coronary artery disease. Free Radic Biol Med. 2006;41:1678–1683. doi: 10.1016/j.freeradbiomed.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoes C, Silva AC, Domingues P, Laranjeira P, Paiva A, Domingues MR. Modified phosphatidylethanolamines induce different levels of cytokine expression in monocytes and dendritic cells. Chem Phys Lipids. 2013;175–176:57–64. doi: 10.1016/j.chemphyslip.2013.07.008. [DOI] [PubMed] [Google Scholar]
- Smiley PL, Stremler KE, Prescott SM, Zimmerman GA, McIntyre TM. Oxidatively fragmented phosphatidylcholines activate human neutrophils through the receptor for platelet-activating factor. J Biol Chem. 1991;266:11104–11110. [PubMed] [Google Scholar]
- Song WL, Lawson JA, Reilly D, Rokach J, Chang CT, Giasson B, FitzGerald GA. Neurofurans, Novel Indices of Oxidant Stress Derived from Docosahexaenoic Acid. J Biol Chem. 2008;283:6–16. doi: 10.1074/jbc.M706124200. [DOI] [PubMed] [Google Scholar]
- Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Progress in Lipid Research. 2004;43:55–90. doi: 10.1016/s0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- Spector AA, Norris AW. Action of epoxyeicosatrienoic acids on cellular function. Am J Physiol Cell Physiol. 2007;292:C996–1012. doi: 10.1152/ajpcell.00402.2006. [DOI] [PubMed] [Google Scholar]
- Spickett CM, Dever G. Studies of phospholipid oxidation by electrospray mass spectrometry: from analysis in cells to biological effects. BioFactors. 2005;24:17–31. doi: 10.1002/biof.5520240103. [DOI] [PubMed] [Google Scholar]
- Spinelli SL, Lannan KL, Casey AE, Croasdell A, Curran TM, Henrichs KF, Pollock SJ, Milne GA, Refaai MA, Francis CW, Phipps RP, Blumberg N. Isoprostane and isofuran lipid mediators accumulate in stored red blood cells and influence platelet function in vitro. Transfusion. 2013 doi: 10.1111/trf.12485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spite M, Baba SP, Ahmed Y, Barski OA, Nijhawan K, Petrash JM, Bhatnagar A, Srivastava S. Substrate specificity and catalytic efficiency of aldo-keto reductases with phospholipid aldehydes. Biochem J. 2007;405:95–105. doi: 10.1042/BJ20061743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Spite M, Trent JO, West MB, Ahmed Y, Bhatnagar A. Aldose reductase-catalyzed reduction of aldehyde phospholipids. J Biol Chem. 2004;279:53395–53406. doi: 10.1074/jbc.M403416200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Vladykovskaya E, Barski OA, Spite M, Kaiserova K, Petrash JM, Chung SS, Hunt G, Dawn B, Bhatnagar A. Aldose reductase protects against early atherosclerotic lesion formation in apolipoprotein E-null mice. Circ Res. 2009;105:793–802. doi: 10.1161/CIRCRESAHA.109.200568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadelmann-Ingrand S, Favreliere S, Fauconneau B, Mauco G, Tallineau C. Plasmalogen degradation by oxidative stress: production and disappearance of specific fatty aldehydes and fatty alpha-hydroxyaldehydes. Free Radic Biol Med. 2001;31:1263–1271. doi: 10.1016/s0891-5849(01)00720-1. [DOI] [PubMed] [Google Scholar]
- Stadelmann-Ingrand S, Pontcharraud R, Fauconneau B. Evidence for the reactivity of fatty aldehydes released from oxidized plasmalogens with phosphatidylethanolamine to form Schiff base adducts in rat brain homogenates. Chem Phys Lipids. 2004;131:93–105. doi: 10.1016/j.chemphyslip.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Stafforini DM. Biology of platelet-activating factor acetylhydrolase (PAF-AH, lipoprotein associated phospholipase A2) Cardiovascular drugs and therapy/sponsored by the International Society of Cardiovascular Pharmacotherapy. 2009;23:73–83. doi: 10.1007/s10557-008-6133-8. [DOI] [PubMed] [Google Scholar]
- Stafforini DM, Carter ME, Zimmerman GA, McIntyre TM, Prescott SM. Lipoproteins alter the catalytic behavior of the platelet-activating factor acetylhydrolase in human plasma. Proc Natl Acad Sci U S A. 1989;86:2393–2397. doi: 10.1073/pnas.86.7.2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafforini DM, Elstad MR, McIntyre TM, Zimmerman GA, Prescott SM. Human macrophages secret platelet-activating factor acetylhydrolase. J Biol Chem. 1990;265:9682–9687. [PubMed] [Google Scholar]
- Stafforini DM, Sheller JR, Blackwell TS, Sapirstein A, Yull FE, McIntyre TM, Bonventre JV, Prescott SM, Roberts LJ., 2nd Release of free F2-isoprostanes from esterified phospholipids is catalyzed by intracellular and plasma platelet-activating factor acetylhydrolases. J Biol Chem. 2006;281:4616–4623. doi: 10.1074/jbc.M507340200. [DOI] [PubMed] [Google Scholar]
- Stavrovskaya IG, Baranov SV, Guo X, Davies SS, Roberts LJ, 2nd, Kristal BS. Reactive gamma-ketoaldehydes formed via the isoprostane pathway disrupt mitochondrial respiration and calcium homeostasis. Free Radic Biol Med. 2010 doi: 10.1016/j.freeradbiomed.2010.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmer U, Dunai ZA, Koller D, Purstinger G, Zenzmaier E, Deigner HP, Aflaki E, Kratky D, Hermetter A. Toxicity of oxidized phospholipids in cultured macrophages. Lipids in health and disease. 2012;11:110. doi: 10.1186/1476-511X-11-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stremler KE, Stafforini DM, Prescott SM, Zimmerman GA, McIntyre TM. An oxidized derivative of phosphatidylcholine is a substrate for the platelet-activating factor acetylhydrolase from human plasma. J Biol Chem. 1989;264:5331–5334. [PubMed] [Google Scholar]
- Subbanagounder G, Deng Y, Borromeo C, Dooley AN, Berliner JA, Salomon RG. Hydroxy alkenal phospholipids regulate inflammatory functions of endothelial cells. Vascular pharmacology. 2002a;38:201–209. doi: 10.1016/s1537-1891(02)00170-2. [DOI] [PubMed] [Google Scholar]
- Subbanagounder G, Leitinger N, Shih PT, Faull KF, Berliner JA. Evidence that phospholipid oxidation products and/or platelet-activating factor play an important role in early atherogenesis: in vitro and In vivo inhibition by WEB 2086. Circ Res. 1999;85:311–318. doi: 10.1161/01.res.85.4.311. [DOI] [PubMed] [Google Scholar]
- Subbanagounder G, Wong JW, Lee H, Faull KF, Miller E, Witztum JL, Berliner JA. Epoxyisoprostane and epoxycyclopentenone phospholipids regulate monocyte chemotactic protein-1 and interleukin-8 synthesis. Formation of these oxidized phospholipids in response to interleukin-1beta. J Biol Chem. 2002b;277:7271–7281. doi: 10.1074/jbc.M107602200. [DOI] [PubMed] [Google Scholar]
- Sullivan CB, Matafonova E, Roberts LJ, II, Amarnath V, Davies SS. Isoketals form cytotoxic phosphatidylethanolamine adducts in cells. J Lipid Res. 2010;51:999–1009. doi: 10.1194/jlr.M001040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Finnemann SC, Febbraio M, Shan L, Annangudi SP, Podrez EA, Hoppe G, Darrow R, Organisciak DT, Salomon RG, Silverstein RL, Hazen SL. Light-induced oxidation of photoreceptor outer segment phospholipids generates ligands for CD36-mediated phagocytosis by retinal pigment epithelium: a potential mechanism for modulating outer segment phagocytosis under oxidant stress conditions. J Biol Chem. 2006;281:4222–4230. doi: 10.1074/jbc.M509769200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Kamei M, Itabe H, Yoneda K, Bando H, Kume N, Tano Y. Oxidized phospholipids in the macula increase with age and in eyes with age-related macular degeneration. Molecular vision. 2007;13:772–778. [PMC free article] [PubMed] [Google Scholar]
- Talati M, Meyrick B, Peebles RS, Jr, Davies SS, Dworski R, Mernaugh R, Mitchell D, Boothby M, Roberts LJ, 2nd, Sheller JR. Oxidant stress modulates murine allergic airway responses. Free Radic Biol Med. 2006;40:1210–1219. doi: 10.1016/j.freeradbiomed.2005.11.012. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Iimori M, Tsukatani H, Tokumura A. Platelet-aggregating effects of platelet-activating factor-like phospholipids formed by oxidation of phosphatidylcholines containing an sn-2-polyunsaturated fatty acyl group. Biochim Biophys Acta. 1994;1210:202–208. doi: 10.1016/0005-2760(94)90122-8. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Minamino H, Unezaki S, Tsukatani H, Tokumura A. Formation of platelet-activating factor-like phospholipids by Fe2+/ascorbate/EDTA-induced lipid peroxidation. Biochim Biophys Acta. 1993;1166:264–274. doi: 10.1016/0005-2760(93)90107-k. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Tokumura A, Tsukatani H. Platelet-activating factor (PAF)-like phospholipids formed during peroxidation of phosphatidylcholines from different foodstuffs. Biosci Biotechnol Biochem. 1995;59:1389–1393. doi: 10.1271/bbb.59.1389. [DOI] [PubMed] [Google Scholar]
- Thomas CP, Morgan LT, Maskrey BH, Murphy RC, Kuhn H, Hazen SL, Goodall AH, Hamali HA, Collins PW, O’Donnell VB. Phospholipid-esterified eicosanoids are generated in agonist-activated human platelets and enhance tissue factor-dependent thrombin generation. J Biol Chem. 2010;285:6891–6903. doi: 10.1074/jbc.M109.078428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton DJ, Rousseau K, McGuckin MA. Structure and function of the polymeric mucins in airways mucus. Annu Rev Physiol. 2008;70:459–486. doi: 10.1146/annurev.physiol.70.113006.100702. [DOI] [PubMed] [Google Scholar]
- Tjoelker LW, Wilder C, Eberhardt C, Stafforini DM, Dietsch G, Schimpf B, Hooper S, Le Trong H, Cousens LS, Zimmerman GA, Yamada Y, McIntyre TM, Prescott SM, Gray PW. Anti-inflammatory properties of a platelet-activating factor acetylhydrolase. Nature. 1995;374:549–553. doi: 10.1038/374549a0. [DOI] [PubMed] [Google Scholar]
- Tsimikas S, Brilakis ES, Miller ER, McConnell JP, Lennon RJ, Kornman KS, Witztum JL, Berger PB. Oxidized phospholipids, Lp(a) lipoprotein, and coronary artery disease. N Engl J Med. 2005;353:46–57. doi: 10.1056/NEJMoa043175. [DOI] [PubMed] [Google Scholar]
- Tsimikas S, Lau HK, Han KR, Shortal B, Miller ER, Segev A, Curtiss LK, Witztum JL, Strauss BH. Percutaneous coronary intervention results in acute increases in oxidized phospholipids and lipoprotein(a): short-term and long-term immunologic responses to oxidized low-density lipoprotein. Circulation. 2004;109:3164–3170. doi: 10.1161/01.CIR.0000130844.01174.55. [DOI] [PubMed] [Google Scholar]
- Tsuji K, Kawai Y, Kato Y, Osawa T. Formation of N-(hexanoyl)ethanolamine, a novel phosphatidylethanolamine adduct, during the oxidation of erythrocyte membrane and low-density lipoprotein. Biochem Biophys Res Commun. 2003;306:706–711. doi: 10.1016/s0006-291x(03)01038-6. [DOI] [PubMed] [Google Scholar]
- Turner J, Jones CE. Regulation of mucin expression in respiratory diseases. Biochem Soc Trans. 2009;37:877–881. doi: 10.1042/BST0370877. [DOI] [PubMed] [Google Scholar]
- Tyurin VA, Balasubramanian K, Winnica D, Tyurina YY, Vikulina AS, He RR, Kapralov AA, Macphee CH, Kagan VE. Oxidatively modified phosphatidylserines on the surface of apoptotic cells are essential phagocytic ‘eat-me’ signals: cleavage and inhibition of phagocytosis by Lp-PLA. Cell Death Differ. 2014 doi: 10.1038/cdd.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida K, Kanematsu M, Morimitsu Y, Osawa T, Noguchi N, Niki E. Acrolein is a product of lipid peroxidation reaction. Formation of free acrolein and its conjugate with lysine residues in oxidized low density lipoproteins. J Biol Chem. 1998;273:16058–16066. doi: 10.1074/jbc.273.26.16058. [DOI] [PubMed] [Google Scholar]
- Uchida K, Sakai K, Itakura K, Osawa T, Toyokuni S. Protein modification by lipid peroxidation products: formation of malondialdehyde-derived N(epsilon)-(2-propenol)lysine in proteins. Arch Biochem Biophys. 1997;346:45–52. doi: 10.1006/abbi.1997.0266. [DOI] [PubMed] [Google Scholar]
- Uderhardt S, Herrmann M, Oskolkova OV, Aschermann S, Bicker W, Ipseiz N, Sarter K, Frey B, Rothe T, Voll R, Nimmerjahn F, Bochkov VN, Schett G, Kronke G. 12/15-lipoxygenase orchestrates the clearance of apoptotic cells and maintains immunologic tolerance. Immunity. 2012;36:834–846. doi: 10.1016/j.immuni.2012.03.010. [DOI] [PubMed] [Google Scholar]
- Van Lenten BJ, Wagner AC, Jung CL, Ruchala P, Waring AJ, Lehrer RI, Watson AD, Hama S, Navab M, Anantharamaiah GM, Fogelman AM. Anti-inflammatory apoA-I-mimetic peptides bind oxidized lipids with much higher affinity than human apoA-I. J Lipid Res. 2008;49:2302–2311. doi: 10.1194/jlr.M800075-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lenten BJ, Wagner AC, Navab M, Anantharamaiah GM, Hui EK, Nayak DP, Fogelman AM. D-4F, an apolipoprotein A-I mimetic peptide, inhibits the inflammatory response induced by influenza A infection of human type II pneumocytes. Circulation. 2004;110:3252–3258. doi: 10.1161/01.CIR.0000147232.75456.B3. [DOI] [PubMed] [Google Scholar]
- Vento M, Moro M, Escrig R, Arruza L, Villar G, Izquierdo I, Roberts LJ, 2nd, Arduini A, Escobar JJ, Sastre J, Asensi MA. Preterm resuscitation with low oxygen causes less oxidative stress, inflammation, and chronic lung disease. Pediatrics. 2009;124:e439–449. doi: 10.1542/peds.2009-0434. [DOI] [PubMed] [Google Scholar]
- Vladykovskaya E, Ozhegov E, Hoetker JD, Xie Z, Ahmed Y, Suttles J, Srivastava S, Bhatnagar A, Barski OA. Reductive metabolism increases the proinflammatory activity of aldehyde phospholipids. J Lipid Res. 2011;52:2209–2225. doi: 10.1194/jlr.M013854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Schlieffen E, Oskolkova OV, Schabbauer G, Gruber F, Bluml S, Genest M, Kadl A, Marsik C, Knapp S, Chow J, Leitinger N, Binder BR, Bochkov VN. Multi-hit inhibition of circulating and cell-associated components of the toll-like receptor 4 pathway by oxidized phospholipids. Arterioscler Thromb Vasc Biol. 2009;29:356–362. doi: 10.1161/ATVBAHA.108.173799. [DOI] [PubMed] [Google Scholar]
- Wagner RS, Weare C, Jin N, Mohler ER, Rhoades RA. Characterization of signal transduction events stimulated by 8-epi-prostaglandin(PG)F2 alpha in rat aortic rings. Prostaglandins. 1997;54:581–599. doi: 10.1016/s0090-6980(97)00127-5. [DOI] [PubMed] [Google Scholar]
- Wahli W, Michalik L. PPARs at the crossroads of lipid signaling and inflammation. Trends in endocrinology and metabolism: TEM. 2012;23:351–363. doi: 10.1016/j.tem.2012.05.001. [DOI] [PubMed] [Google Scholar]
- Walton KA, Gugiu BG, Thomas M, Basseri RJ, Eliav DR, Salomon RG, Berliner JA. A role for neutral sphingomyelinase activation in the inhibition of LPS action by phospholipid oxidation products. J Lipid Res. 2006;47:1967–1974. doi: 10.1194/jlr.M600060-JLR200. [DOI] [PubMed] [Google Scholar]
- Walton KA, Hsieh X, Gharavi N, Wang S, Wang G, Yeh M, Cole AL, Berliner JA. Receptors involved in the oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine-mediated synthesis of interleukin-8. A role for Toll-like receptor 4 and a glycosylphosphatidylinositol-anchored protein. J Biol Chem. 2003;278:29661–29666. doi: 10.1074/jbc.M300738200. [DOI] [PubMed] [Google Scholar]
- Ware LB, Fessel JP, May AK, Roberts LJ., 2nd Plasma biomarkers of oxidant stress and development of organ failure in severe sepsis. Shock. 2011;36:12–17. doi: 10.1097/SHK.0b013e318217025a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson A, Leitinger M, Navab M, Faull K, Horkko S, Witztum J, Palinski W, Schwenke DC, Salomon R, Sha W, Subbanagounder G, Fogelman A, Berliner J. Structural identification by mass spectrometry of oxidized phospholipids in minimally oxidized low density lipoprotein that induce monocyte/endothelial interactions and evidence for their presence in vivo. J Biol Chem. 1997;272:13597–13607. doi: 10.1074/jbc.272.21.13597. [DOI] [PubMed] [Google Scholar]
- Watson AD, Navab M, Hama SY, Sevanian A, Prescott SM, Stafforini DM, McIntyre TM, Du BN, Fogelman AM, Berliner JA. Effect of platelet activating factor-acetylhydrolase on the formation and action of minimally oxidized low density lipoprotein. J Clin Invest. 1995;95:774–782. doi: 10.1172/JCI117726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson AD, Subbanagounder G, Welsbie DS, Faull KF, Navab M, Jung ME, Fogelman AM, Berliner JA. Structural identification of a novel pro-inflammatory epoxyisoprostane phospholipid in mildly oxidized low density lipoprotein. J Biol Chem. 1999;274:24787–24798. doi: 10.1074/jbc.274.35.24787. [DOI] [PubMed] [Google Scholar]
- Wilson SJ, McGinley K, Huang AJ, Smyth EM. Heterodimerization of the α and β isoforms of the human thromboxane receptor enhances isoprostane signaling. Biochemical and Biophysical Research Communications. 2007;352:397–403. doi: 10.1016/j.bbrc.2006.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Zimmerman GA, Prescott SM, Stafforini DM. The p38 MAPK pathway mediates transcriptional activation of the plasma platelet-activating factor acetylhydrolase gene in macrophages stimulated with lipopolysaccharide. J Biol Chem. 2004;279:36158–36165. doi: 10.1074/jbc.M402454200. [DOI] [PubMed] [Google Scholar]
- Yao Y, Harrison KA, Al-Hassani M, Murphy RC, Rezania S, Konger RL, Travers JB. Platelet-activating factor receptor agonists mediate xeroderma pigmentosum A photosensitivity. J Biol Chem. 2012;287:9311–9321. doi: 10.1074/jbc.M111.332395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Cox BE, Liu W, Porter NA, Morrow JD, Milne GL. Identification of intact oxidation products of glycerophospholipids in vitro and in vivo using negative ion electrospray iontrap mass spectrometry. Journal of mass spectrometry: JMS. 2009;44:672–680. doi: 10.1002/jms.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Gan Q, Owens GK. Kruppel-like factor 4, Elk-1, and histone deacetylases cooperatively suppress smooth muscle cell differentiation markers in response to oxidized phospholipids. Am J Physiol Cell Physiol. 2008;295:C1175–1182. doi: 10.1152/ajpcell.00288.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagol-Ikapitte I, Amarnath V, Bala M, Roberts LJ, 2nd, Oates JA, Boutaud O. Characterization of scavengers of gamma-ketoaldehydes that do not inhibit prostaglandin biosynthesis. Chem Res Toxicol. 2010a;23:240–250. doi: 10.1021/tx900407a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagol-Ikapitte I, Masterson TS, Amarnath V, Montine TJ, Andreasson KI, Boutaud O, Oates JA. Prostaglandin H(2)-derived adducts of proteins correlate with Alzheimer’s disease severity. J Neurochem. 2005;94:1140–1145. doi: 10.1111/j.1471-4159.2005.03264.x. [DOI] [PubMed] [Google Scholar]
- Zagol-Ikapitte I, Matafonova E, Amarnath V, Bodine CL, Boutaud O, Tirona RG, Oates JA, Roberts LJ, II, Davies SS. Determination of the Pharmacokinetics and Oral Bioavailability of Salicylamine, a Potent gamma-Ketoaldehyde Scavenger, by LC/MS/MS. Pharmaceutics. 2010b;2:18–29. doi: 10.3390/pharmaceutics2010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemski Berry KA, Murphy RC. Characterization of acrolein-glycerophosphoethanolamine lipid adducts using electrospray mass spectrometry. Chem Res Toxicol. 2007;20:1342–1351. doi: 10.1021/tx700102n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Southall MD, Mezsick SM, Johnson C, Murphy RC, Konger RL, Travers JB. Epidermal peroxisome proliferator-activated receptor gamma as a target for ultraviolet B radiation. J Biol Chem. 2005;280:73–79. doi: 10.1074/jbc.M409795200. [DOI] [PubMed] [Google Scholar]
- Zhang R, Shen Z, Nauseef WM, Hazen SL. Defects in leukocyte-mediated initiation of lipid peroxidation in plasma as studied in myeloperoxidase-deficient subjects: systematic identification of multiple endogenous diffusible substrates for myeloperoxidase in plasma. Blood. 2002;99:1802–1810. [PubMed] [Google Scholar]
- Zhao J, Maskrey B, Balzar S, Chibana K, Mustovich A, Hu H, Trudeau JB, O’Donnell V, Wenzel SE. Interleukin-13-induced MUC5AC is regulated by 15-lipoxygenase 1 pathway in human bronchial epithelial cells. Am J Respir Crit Care Med. 2009;179:782–790. doi: 10.1164/rccm.200811-1744OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, O’Donnell VB, Balzar S, St Croix CM, Trudeau JB, Wenzel SE. 15-Lipoxygenase 1 interacts with phosphatidylethanolamine-binding protein to regulate MAPK signaling in human airway epithelial cells. Proc Natl Acad Sci U S A. 2011;108:14246–14251. doi: 10.1073/pnas.1018075108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GH, Xiong SQ, Mei LJ, Chen HY, Wang T, Chu JF. Elevated plasma platelet activating factor, platelet activating factor acetylhydrolase levels and risk of coronary heart disease or blood stasis syndrome of coronary heart disease in Chinese: a case control study: a case-control study. Inflammation. 2012;35:1419–1428. doi: 10.1007/s10753-012-9455-4. [DOI] [PubMed] [Google Scholar]
- Zieseniss S, Zahler S, Muller I, Hermetter A, Engelmann B. Modified phosphatidylethanolamine as the active component of oxidized low density lipoprotein promoting platelet prothrombinase activity. J Biol Chem. 2001;276:19828–19835. doi: 10.1074/jbc.M007506200. [DOI] [PubMed] [Google Scholar]