

Abstract
Pirh2 (p53-induced RING-H2 domain protein, also known as Rchy1), is an E3 ubiquitin ligase involved in a negative-feedback loop with p53. Using NMR spectroscopy we show that Pirh2 is a unique cysteine-rich protein comprising three modular domains. The protein binds nine zinc ions using a variety of zinc coordination schemes including a RING domain and a novel left-handed β-spiral in which three zinc ions align three consecutive small β-sheets in an interleaved fashion. We demonstrate that Pirh2-p53 interaction is dependent on the C-terminal Zn-binding module of Pirh2 which binds to the tetramerization domain (TET) of p53. As a result, Pirh2 ubiquitylates preferably the tetrameric form of p53 in vitro and in vivo, suggesting that Pirh2 regulates protein turnover of the transcriptionally active form of p53 in the cell.
Tumor suppressor p53 is known for its prominent role in safeguarding genome integrity and preventing tumorigenesis. It induces cell cycle arrest or apoptosis in response to diverse cellular stresses, thus stopping damaged cells from proliferating1,2. The cellular levels of p53 are tightly-regulated by several ubiquitin E3 ligases that promote ubiquitylation of p53 and its subsequent targeting for 26S proteosome-dependent protein degradation3. Mdm2 is the prototypic p53 E3 ligase and a key negative regulator of p534. Mdm2 is overexpressed in a variety of human malignancies and is an attractive target for development of therapeutic compounds that could inhibit its activity and promote p53-dependent apoptosis in cancers overexpressing mdm25. The recent identification of several new p53 E3 ligases, namely Pirh2, COP1, TOPORS and ARF-BP1, adds more complexity to the p53 ubiquitylation pathway6–9 and may expand the potential targets for p53-dependent cancer therapies. However, the structural and catalytic properties of these new ligases and their physiological significance remain to be determined.
Pirh2 is a p53 inducible E3 ligase with a RING-H2 domain, and has been shown to ubiquitylate p53 in vivo and in vitro6. Like Mdm2, Pirh2 functions as a negative regulator of p53. Overexpression of Pirh2 decreases p53 levels in the cell and represses p53-dependent transactivation and growth inhibition. Abrogation of Pirh2 results in a steady increase in p53 cellular levels. Indeed, Pirh2 was shown to be overexpressed both in a mouse lung tumor model and human lung cancers10. The overexpression of Pirh2 was concomitant with decreased p53 levels in malignant tissues and was independent of Mdm2, suggesting a role for Pirh2 in tumorgenesis through regulation of p53 stability10.
To understand the molecular mechanism of Pirh2-mediated regulation of p53 stability, we have determined the solution structures of the three individual domains of human Pirh2 using NMR spectroscopy. Pirh2 contains a total of nine zinc-binding sites with six located at the N-terminal domain (NTD), two in the RING domain and one in the C-terminal domain (CTD). Nine zinc binding sites comprise three different zinc coordination schemes, ie. RING type cross-brace zinc coordination, C4 zinc finger and a novel left-handed β-spiral zinc-binding motif formed by three recurrent CCHC sequence motifs. We also report that Pirh2 interacts with p53 via the CTD, which targets the p53 TET domain, and with possible enhancement from a weak interaction between the NTD and p53 DNA binding domain. We find that Pirh2 preferably ubiquitylates the tetrameric form of p53 in vitro and in vivo, implying a role for Pirh2 in downregulating the transcriptionally active form of p53 in the cell. Our study provides a structural basis and a molecular mechanism for the function of Pirh2 in regulation of the p53 ubiquitylation pathway.
RESULTS
Human Pirh2 is a 261-residue protein containing three domains, the NTD (residues 1–137), the central RING domain (residues 138–189) and the CTD (residues 190–261) (Fig. 1a). There are 29 cysteine residues throughout the protein with 19 clustered in the NTD, 6 in the RING and 4 in the CTD (Fig. 1b). Full length Pirh2 protein could only be purified and studied at low concentrations (< 50 uM) and therefore was not amenable to structural studies under a variety of conditions. However, we were able to prepare suitable NMR samples for the three individual domains of human Pirh2 and solve their solution structures using three-dimensional heteronuclear NMR spectroscopy. The zinc coordination and binding stoichiometry of Pirh2 were determined based on the ICP-AES analysis and NMR restraints. The ensembles of the twenty lowest-energy conformers for these three domains are shown in Supplementary Fig. 1 online. Figure 2 demonstrates the overall structure and the zinc ion coordination scheme within each domain.
Figure 1.

The sequence and domain organization of Pirh2. (a) Schematic diagram illustrating the boundaries for the three domains of Pirh2 with the NTD in gray, the RING in blue and the CTD in brown. (b) The multiple sequence alignment of Pirh2 from eukaryotic organisms with the secondary structure elements of human Pirh2 are indicated above the sequence. Arrows designate β strands, solenoids for α-helices and asterisks indicating zinc coordinating cysteins and histidines. The secondary structure elements are colored in gray, blue and brown, representing NTD, RING and CTD, respectively. The alignment was generated by CLUSTAL W and was printed using the ESPript 2.2 software package.35
Figure 2.

Ribbon representations of the three individual Pirh2 domains and the zinc coordination schemes of Pirh2 (a) the Pirh2 NTD. The N lobe of the Pirh2 NTD showing the interleaved Zn1 and Zn2 sites coordinated by a sequence motif CHC4H2 and the Zn3 site coordinated by four cysteines. The C lobe of the Pirh2 NTD showing the left-handed β spiral structure with the three CCHC motifs coordinating Zn4, Zn5 and Zn6. (b) The Pirh2 RING domain showing the interleaved Zn7 and Zn8 sites coordinated by the sequence motif C3H2C3. (c) The Pirh2 CTD showing the Zn9 site coordinated by four cysteines. The nine zinc atoms are shown as spheres.
Solution structure of the Pirh2 NTD
The cysteine-rich Pirh2 NTD folds into two lobes shaped as a check mark with the N-terminal lobe (residues 1–82) forming the short arm and the elongated C lobe (residues 83–137) extending into the long arm. The N-terminal lobe consists of a four-stranded twisted antiparallel β-sheet and an α helix located on one side of the β-sheet. The 18 N-terminal residues are less ordered as reflected by random-coil chemical shifts, and the absence of medium or long range NOEs. The fold is largely defined by three zinc ions. The ligands for the first two zinc ions are interleaved with Cys20, His22, Cys40 and Cys43 coordinating the first zinc ion, and Cys33, Cys34, His44 and His50 binding the second zinc. The third zinc ion is chelated by four cysteine residues (Cys62, Cys65, Cys75 and Cys78), located on a β hairpin between β3 and β4 and on a rubredoxin knuckle type β turn between β4 and β5 (Fig. 2a).
The C lobe consists of six short antiparallel strands (residues 84–87, 92–96, 99–102, 107–111, 116–119 and 124–128), wrapped into a unique left-handed β-spiral by three zinc ions (4th – 6th zinc ions). Each zinc ion is bound by a CCHC sequence motif. The zinc chelating residues include Cys87, Cys90, His101 and Cys108 for the 4th zinc; Cys102, Cys105, His118 and Cys125 for the 5th zinc; and Cys119, Cys122, His134 and Cys136 for the 6th zinc (Fig. 2a). The overall structure of human pirh2 NTD is very similar to that of mouse pirh2 NTD (PDB:2DKT) with an r.m.s.d value of 1.274 Å (Cα atoms of the residues 18–119), in agreement with their highly conserved sequence (80% identity). Comparison with known protein structures via the DALI 11 server did not reveal any other homologous fold.
Solution structure of the Pirh2 RING domain
The Pirh2 RING domain folds into a globular structure which binds two zinc ions in a typical cross-brace arrangement. Two interleaved zinc-binding sites are situated at opposite sides of the domain, with the 7th zinc ion coordinated by Cys145, Cys148, His169 and Cys172 and the 8th zinc ion bound by Cys164, His166, Cys183 and Cys186 (Fig. 2b). The RING domain structure of Pirh2 is similar to that of other proteins. Notably, the distance between two zinc ions is 14.9 Å, a value close to the distances (approximately 14 Å) reported for the RING domain structures of PML (PDB:1BOR) and IEEHV (PDB:1CHC), reflecting conservation of the zinc-binding motif in the RING domain protein family12. Structurally homologous proteins to the Pirh2 RING domain include the RING domain of the Cul1-Rbx1-Skp1-F box-Skp2 SCF ubiquitin ligase complex (PDB:1LDJ-B) with RMSD value of 2.2 Å over 44 aligned Cα atoms, the BRCA1 RING-domain (PDB: 1JM7-A) with RMSD value of 1.9 Å over 43 aligned Cα atoms and the rice transcription regulator EL5 RING finger structure (PDB:1IYM-A) with RMSD value of 3.2 Å over 47 aligned Cα atoms. Superposition of the four RING domain structures shows substantial structural similarity in the zinc-binding motif, the central β-sheet (β13 and β14) and the helix (α2), despite limited sequence identity (15–20%) between the RING domains of these proteins. Residues P146, I147, L149, L168, Y173, M176, Y181, P184, L185 and M187 form a hydrophobic patch along a shallow depression on the molecular surface (see Supplementary Fig. 2 online), a feature shared with the E2 binding site of other RING domains such as c-Cbl13.
Solution structure of the Pirh2 CTD
The structure of the Pirh2 CTD consists of a 30-residue disordered loop (residues 187–215) followed by a compact, globular fold that incorporates a single zinc ion with four cysteine thiol groups (residues Cys222, Cys225, Cys240 and Cys243) forming a tetrahedral zinc coordination (Fig. 2c). Residues 198 to 205 within the unstructured region showed some α-helical propensity based on their backbone NMR resonances. However, the NOE pattern in this region does not support the existence of any rigid structure, suggesting that this region is natively disordered and may become an ordered α-helix under certain conditions such as binding. The globular core comprises a twisted three-stranded β sheet (residues 216 to 223, 227 to 233 and 247 to 251) with three strands arranged in the order of β2β1β3. Comparison with known protein structures using the DALI server11 did not reveal significant homologous folds (z score > 2.0). However, the topology resembles that of the zinc motif in domain B of the mini-chromosome maintenance (MCM) complex (PDB:1LTL, with RMSD value of 4.2Å for 51 Cα atoms) and that of the zinc ribbon in NOP10 of the H/ACA RNP complexes (PDB: 2EY4, with RMSD of 2.2Å for 30 Cα atoms). As both zinc motifs are involved in mediating complex formation, the C4 zinc finger of the Pirh2 CTD may also function as a protein interaction module.
In order to better understand the structure of full length Pirh2 we expressed the protein as an N-terminal GST fusion protein with a thrombin cleavage site. This protein could be purified in soluble and the GST removed form to yield a stable full length Pirh2 that was soluble and active (see below) at concentrations less than 50 μM. Full length Pirh2 eluted from a size exclusion column as a single peak of approximately 35 KDa, indicating that Pirh2 is a monomer in solution. In order to assess whether full length protein contained inter-domain interactions we titrated individual 15N-labeled domains with an unlabeled domain while monitoring the 15N NMR resonances. No changes in resonance frequencies were observed for any of the three domains when mixed with a two fold excess of either of the other two domains, implying that the full length protein contains three relatively independent domains (data not shown).
P53 interacts with both N- and C- terminal domains of Pirh2
To elucidate the molecular basis for the Pirh2-p53 interaction, protein-protein interaction studies were performed in which full length 6xHis tagged p53 was assayed against full length Pirh2 and its three individual domains, each fused to GST (GSTPirh2fl, GST-NTD, GST-RING and GST-CTD). The results showed that p53 interacted with full length GST-Pirh2, GST-NTD and GST-CTD, but not GST-RING. Based on the amount captured by the GST fusions, p53 appeared to bind to full length Pirh2 slightly better than either the Pirh2 NTD or Pirh2 CTD (Fig. 3a). To identify which region of p53 is responsible for binding to Pirh2, the interactions were tested between two GST-Pirh2 proteins (GST-NTD and GST-CTD) and four p53 fragments; the N-terminal 70 residues of p53, p53 DNA binding domain (p53DBD), p53 tetramerization domain (p53TET) and p53 C-terminal residues 355–393. Pirh2 NTD interacted with p53DBD while Pirh2 CTD bound to p53TET (Fig. 3b). This result suggests that Pirh2 may target p53 by binding to two different sites of p53 with its NTD binding to p53 DBD and its CTD interacting with p53TET.
Figure 3.

Pirh2 interacts with p53 via a two-site binding mode. (a) GST pull-down assays of GST-Pirh2 fusion proteins with the full-length p53. (b) GST pull-down assays of GST-Pirh2 NTD and GST-Pirh2 CTD fusions with purified p53 domains as indicated. The labeled lanes reflect loaded material (L), column flow-through after wash (W) and elutate (E). (c) Surface representations of the Pirh2 NTD (c) and CTD (d) showing the p53 binding interface, as determined by NMR chemical shift perturbation experiments. The residues whose resonances show substantial shifts upon addition of unlabeled p53 into 15N labeled Pirh2 samples are indicated (see Supplementary Fig. 3 and 4 online).
To confirm these observations and to map the p53 interaction sites on the NTD and CTD of Pirh2, a series of two dimensional 1H-15N Heteronuclear Single Quantum Coherence (HSQC) NMR spectra of Pirh2 NTD and CTD were collected and compared in the absence and the presence of the appropriate domain of p53. Upon addition of an increasing amount of p53DBD to 15N labeled Pirh2 NTD, perturbations were observed for the backbone amide resonances of His70, Ala71 and Glu76 and for the sidechain NE2 of the Gln72 (see Supplementary Fig. 3 online). These residues are all located close to the loop that binds the 3rd zinc in the hinge region of Pirh2 NTD and are conserved in mammals (Fig. 3c). However, complex formation was not saturated under the conditions of our NMR experiments (0.6 mM p53 DBD) and further titration data points could not be generated due to solubility limitations. Thus, p53 DBD interaction with Pirh2 NTD is very weak, estimated Kd >0.6 mM by comparison with the Kd value of Pirh2 CTD-p53TET (see below), consistent with the less-than (or sub-) stoichiometric binding observed in the protein-protein interaction experiments described above.
To characterize the interaction between p53 TET and the Pirh2 CTD, HSQC spectra of 15N-Pirh2 CTD were collected in the absence and the presence of p53TET. Upon addition of p53TET, resonance perturbations were observed for Pirh2 CTD, giving an estimated Kd of 0.6mM, confirming a weak interaction between these two domains. The affected residues of Pirh2 CTD include the backbone amide groups Ala249, Ala251, Arg254, Arg255 and I256 (see Supplementary Fig. 4a online). Additionally, the line width for residues Gly252 and Gly253 were broadened beyond the detection limit, suggesting the involvement of these two residues in the complex formation. These residues define a molecular surface on the Pirh2 CTD for interaction with p53TET (Fig. 3d).
P53 is transcriptionally active as a tetramer which is thought to form upon increased p53 protein concentrations in response to various stress stimuli14. To investigate whether other oligomeric forms of p53 affect its interaction with Pirh2, a dimeric TET domain mutant, M340Q/L344R (DM), and a monomeric mutant, L344P (MM), were tested under the same condition as the wild type p53 TET domain15. No changes were observed for Pirh2 CTD NMR resonances upon mixing with a two-fold excess of p53DM or p53MM, suggesting Pirh2 does not bind the dimeric or the monomeric oligomerization domains of p53 (see Supplementary Fig. 4b,c online).
Pirh2 RING interacts with UBE2D2/UbcH5b
To map the interactions between the ubiquitin conjugating enzyme E2 and Pirh2 we monitored the NMR resonances of the 15N-labeled Pirh2 RING domain upon addition of UBE2D2/UbcH5b (Fig. 4). The most perturbed region was seen in two zinc coordination sites including I147 and C148 from zinc-binding site I, and L185 and M187 from zinc-binding site II. The residues from the central β-sheet and the helix including L167, L168, T171, E174, M176, and Y181 were also greatly perturbed. From this NMR titration data, the interaction between the Pirh2 RING and UBE2D2/UbcH5b is estimated to have a Kd of 0.2 mM. Mutagenesis data of the most perturbed residues suggest that the hydrophobic patch on the RING domain surface mediates the RING-E2 interaction (data not shown). Notably, two RING domain mutants resulted in Pirh2 null activity in the ubiquitylation assay (see below). The mutant M176E disrupts the surface hydrophobicity and the RING-E2 interaction despite having an intact folded structure, while C186A affects zinc coordination thereby disrupting the global structure of the RING domain, as evidenced by 15N-HSQC spectra of the mutants.
Figure 4.
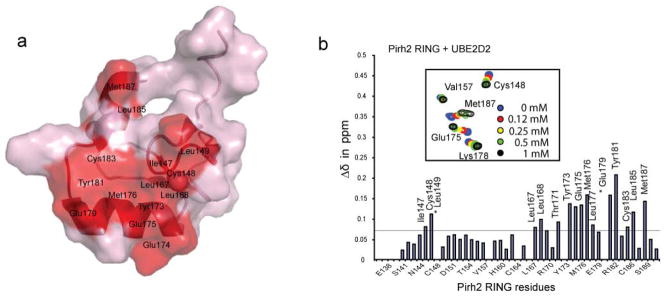
Mapping UBE2D2/UbcH5b binding interface. (a) Surface representation of the Pirh2 RING domain showing the UBE2D2/UbcH5b binding interface, as determined by NMR chemical shift perturbation experiments. Pro146 and Pro184, which lack amide resonances, are also likely to be involved (see Supplementary Fig. 2 online). (b) Composite chemical shift changes versus residue number for Pirh2 RING domain upon binding to UBE2D2/UbcH5b. The values shown were calculated by using the equation Δcomp = [ΔδHN2 + (ΔδN/5)2]1/2. The inset shows the chemical shift changes of the representative residues upon titration with UBE2D2/UbcH5b.
Pirh2 catalyzes ubiquitylation of tetrameric p53
To characterize the E3 ligase activity of Pirh2, we performed an in vitro ubiquitylation assay using recombinant GST-tagged full length Pirh2 (GST-Pirh2) in a reaction containing E1, E2 (UBE2D2/UbcH5b) and His-tagged ubiquitin with and without recombinant p53 protein. Ubiquitylation of Pirh2 and p53 was readily detected and was dependent on the presence of E1, E2, Ub and Pirh2 (see Supplementary Fig. 5 online). This result is consistent with the data reported by Leng et al. showing that Pirh2 is able to catalyze autoubiquitylation and ubiquitylation of p536.
To investigate the contribution of individual p53 residues on ubiquitylation activity, a series of p53 mutants were tested in in vitro ubiquitylation assays, including three p53 DNA binding domain (DBD) hot-spot mutants R273H, R175H and E285A, two tetramerization domain (TET) mutants DM and MM, and four deletion mutants; residues 1–360, residues 1–320, Δ325–355 (deletion of TET domain) and Δ70–311 (DBD deletion mutant) (Fig. 5a). In assays for Pirh2-mediated p53 ubiquitylation, the p53 DBD mutants displayed similar ubiquitylation levels and patterns as wild type p53. The p53 deletion mutants 1–360 and Δ70–311 were also readily ubiquitylated by Pirh2. However, all p53 point and deletion mutants involving the TET domain had substantially diminished ubiquitylation levels, strongly suggesting that a properly folded tetrameric TET domain is required for ubiquitylation of p53 by Pirh2 (Fig. 5b).
Figure 5.

Pirh2-mediated ubiquitilyation of both itself and p53. (a) Schematic diagram of the p53 mutants used in panel c. (b) Pirh2 mediated in vitro ubiquitylation assays of both wild type and mutant p53 constructs in the presence or absence of ubiquitin (UB). The reaction products were resolved on a 7.5% SDS-PAGE gel for lanes 1–10 and a 10% gel for lanes 11–20. Detection of p53 ubquitylation was performed as described in Methods. (c) Pirh2 expression results in reduced levels of tetrameric p53 in vivo. H1299 cells were transfected with either full length hPIRH2 or an empty vector control (pcDNA3(neo)), along with either tetrameric (p53 WT), dimeric (p53 DM) or monomeric (p53 MM) p53. Equal amounts of cell lysate were analyzed by SDS-PAGE and the amounts of p53, Pirh2 and β-actin were detected by immunoblot. The p53 (WT, DM and MM) levels were quantified as the ratio of p53 intensity to β-actin intensity. For each empty vector control experiment the p53 level was normalized to 1 in order to reflect the background level of endogenous Pirh2 activity against each type of transfected p53. The p53 levels (WT, DM and MM) in Pirh2 co-transfected cells are intensity ratio relative to the appropriate control experiment.
The Pirh2 CTD is required for p53 ubiquitylation
To investigate the sequence determinants for Pirh2 autoubiquitylation and ubiquitylation of p53, we created a series deletion-mutants with the N- and C- terminus of Pirh2 systematically truncated (Fig. 6a). The truncation mutants were designed to test not only the contribution of the three globular regions observed in the NMR structures, but also the disordered regions of the CTD and residues 120–138 that were previously reported to be responsible for interaction with p536,16. All deletion mutants were expressed in E. coli, purified as GST-fusions and tested for their ubiquitylation activity in the same way as the wild type Pirh2. In Pirh2 autoubiquitylation assays, the mutants 120–261 and 1–195 showed activity similar to that of wild type Pirh2 while no autoubiquitylation was detected in other fragments (Fig. 6b lane1–10). When the film was overexposed, faint ubiquitylated species could be observed for those fragments containing the intact RING domain (data not shown). No autoubiquitylation was detected for the fragment 1–137 under any conditions, consistent with the requirement for the RING domain. On the other hand, Pirh2-mediated ubiquitylation of p53 was only observed for wild type and Pirh2 mutants 120–261, 138–261 and 138–253 (Fig. 6c-e). The CTD deletion mutants and the RING domain mutants C186A and M176E showed no p53 ubiquitylation activity, indicating that the RING and most of the CTD are required for p53 ubiquitylation, and that the NTD is not required. The Pirh2 mutant, 138–253, retains the overall structural fold of the C-terminal Zn binding module but lacks residues 254–256 which are involved in p53 contacts. This mutant was considerably less active than wild type and Pirh2 120–261, 138–261, further supporting the role of the C-terminus of Pirh2 in mediating p53 interaction. While the NTD is clearly not required for p53 ubiquitylation, the weaker activity of Pirh2 120–261 and 138–261 mutants relative to wild type suggests that the NTD may contribute to optimal Pirh2 activity through its weak interaction with the p53 DBD.
Figure 6.

Assessment of the ubiquitylation activity of Pirh2 deletion mutants. (a) Schematic drawing of the Pirh2 deletion mutants and their activities in auto- and p53-ubiquitylation. (b) Autoubiquitylation (lanes 1–10, left panel) and ubiquitylation of p53 (lanes 11–20, right panel) using different Pirh2 deletion mutants as indicated. The degradation product of the GST-Pirh2 fusions to GST alone is noted (GST). (c) Ubiquitylation of p53 using various concentrations of the wild type Pirh2, the mutants 138–261 and 1–195 as indicated. (d) Ubiquitylation of p53 using the wild type Pirh2, and the mutants 138–261, 138–253, C186A and M176E. All Pirh2 mutants were purified as GST fusions and subjected to the in vitro ubiquitylation assays in the absence (lanes 1–10) and the presence of p53 (lanes 11–20). Autoubiquitylation of the Pirh2 mutants was evaluated by blotting with an antibody against GST. Ubiquitylation of p53 mediated by the Pirh2 mutants was evaluated by blotting with an antibody against p53 (Pab1801).
Pirh2 downregulates tetrameric p53 in vivo
The observation that Pirh2 selectively targets tetrameric p53 in vitro prompted us to test the function of Pirh2 on different oligomeric forms of p53 in vivo. Pirh2 or empty vector (pCDNA3(neo)) were co-transfected with wild type p53, p53DM or p53MM into p53-null H1299 cells. Cells were harvested after 40 hours. The levels of p53 (WT, DM or MM) were detected by western blotting with the p53 antibody DO-1 directed against the N-terminus of p53, and evaluated by pairwise comparison of Pirh2 cotransfected vs empty vector cultures. As expected, ectopic expression of Pirh2 decreased the levels of wild type p53 to about 20% of those without Pirh2 co-expression. This is consistent with the role of Pirh2 as an E3 ligase and a negative regulator of p53. However, the p53 DM and MM levels were only decreased to 60% and 80% respectively, suggesting that dimeric p53 is less sensitive to Pirh2 levels, with monomeric p53 having the least sensitivity (Fig. 5c). These data clearly indicate that Pirh2 efficiently targets and degrades tetrameric p53, the transcriptionally active form of p53 in the cell.
DISCUSSION
The structures of Pirh2 domains reveal several novel features in protein architecture and zinc coordination. An unusually high content of cysteine (11%) and histidine (8%) enables the coordination of nine zinc ions within this medium sized protein. The N-terminal lobe of Pirh2 NTD is a member of the CHY zinc finger family (PF05495) by virtue of its N-terminal CxHY sequence motif and is found only in eukaryotes. The structure of the N-terminal lobe shows a unique zinc coordination which combines a cross-brace zinc binding motif with a C4 zinc finger. The cross-brace zinc binding motif uses the consensus sequence CxHx(10)CCx(5)Cx(2)CHx(5)H to chelate two zinc ions. Though the topology of this cross-brace zinc binding motif in the NTD appears the same as the RING domain, the permutation and spacing of the cysteine and histidine residues for the zinc coordination are very different from the RING domain. The typical RING finger (pfam zf-C3HC4, PF00097) uses the consensus sequence Cx2Cx(9–39)Cx(1–3)Hx(2–3)Cx2Cx(4–48)Cx2C to bind two zinc ions and inevitably contains a hydrophobic patch for the recruitment of ubiquitin E2 ligases12,17. Both of these features are lacking in the Pirh2 NTD, explaining the lack of E3 ligase activity in the N-terminal lobe, despite its structural similarity to the RING domain.
The C lobe of the Pirh2 NTD folds into a three-turn β spiral held together by three zinc ions. The structure is novel in two respects. First, this left-handed solenoid structure is made of three successive strand-turn-strand motifs. These motifs are packed together by a novel combination of zinc ion coordination and hydrophobic interactions between sidechains from the strands. Second, the three zinc ions are interlocked by the sequence motif: Cx(2)Cx(10)HCx(2)Cx(2)Cx(9)HCx(2)Cx(2)Cx(8)HxC, in which the 1st histidine and 4th cysteine residues of each HCCC repeat are paired with the N-terminal cysteine pair to coordinate a zinc ion whereas the 2nd and 3rd cysteine residues are paired with the C-terminal His and Cys to bind another zinc ion (Fig. 2b). This type of zinc coordination has not been previously reported and is found to be conserved in eukaryotes. Protein sequences alignment reveals that many eukaryotic proteins contain similar sequence motif (see Supplementary Fig. 6 online). All these proteins have the HCCC repeats and the spacings within the HCCC repeat are conserved, suggesting that these proteins may also hold the β solenoid zinc finger structure.
Our data indicate that the three domains of Pirh2 are independently folded and do not interact with one another, suggesting that the full length protein has a modular arrangement of domains. The linker between the Pirh2 NTD and the RING domain is relatively short (~ 10 residues), suggesting that the NTD and the RING may form a more rigid connection. In contrast, the 30 amino-acid linker between the RING and the CTD (~30 residues) gives Pirh2 great flexibility to bring together the ubiquitin-charged E2 enzyme with substrates.
P53 is known to be a substrate for Pirh2. Here we find that Pirh2 interacts with p53 primarily via CTD residues 249–256 which recognize the p53 TET domain with a Kd on the order of 600 uM. The NTD of Pirh2 binds more weakly to the DBD of p53 and while not required for activity may contribute to optimal activity. The region between the N-terminal CHY zinc-finger and the RING domain of Pirh2 (residues 120–137) was reported to be required for p53 recognition because a mutant Pirh2 lacking residues 120–137 was unable to bind p536. In view of the present Pirh2 domain structures, this deletion disrupts both the 5th Zn and 6th Zn coordinating residues, and will lead to a drastically disturbed Pirh2 fold. Synthetic Pirh2 peptides derived from the same region (residues 120–130 and 126–137) bind very weakly (Kd > 1 mM) under physiological ionic strength conditions, implicating a weak and transient interaction between this Pirh2 peptide motif alone and p5316. Notably, we did not observe a direct interaction between Pirh2 120–137 in the context of the full-length protein or folded individual domains, suggesting that prior reports of this interaction may have been due to improper folding of the NTD.
The binding site on Pirh2 for the E2 ligase, UBE2D2/UbcH5b, was mapped to the hydrophobic depression on the surface of the RING domain using NMR titration and mutatgenesis. Based on the NMR titration data, Pirh2 RING binds UBE2D2/UbcH5b with an affinity of ~ 200 μM, consistent with values observed for the other E2-E3 complexes18. These data confirmed the central role of the RING domain in recruiting the E2 and mediating p53 ubiquitylation, in a manner similar to known RING-E2 complexes19,20.
Using an in vitro ubiquitylation assay, we found that the interaction of the Pirh2 CTD and p53 TET is essential for Pirh2 mediated p53 ubiquitylation. The Pirh2 fragment spanning residues 138–261, containing the RING domain and intact CTD but no NTD, was sufficient to ubiquitylate p53 while the Pirh2 fragments with C-terminal truncations had greatly reduced or no ubiquitylation activity. Importantly, the fragment 1–195 which lacks the entire C-terminus is still able to mediate autoubiquitylation, but not p53 ubiquitylation, suggesting an intact Pirh2 C-terminus is necessary for p53 ubiquitylation. Thus, the interaction of p53 via its TET domain with the CTD of Pirh2 appears to be necessary and sufficient for p53 ubiquitylation in vitro. The additional interaction of p53 DBD with the Pirh2 NTD, although not required for ubiquitylation activity, appears to augment Pirh2-mediated p53 ubiquitylation. Further characterization of p53 mutants for their ability to be ubiquitylated by Pirh2 in vitro, supported the importance of the tetrameric state of p53 in this activity; Pirh2 ubiquitylation of dimeric and monomeric mutants of p53 was severely curtailed. Mutations in the DBD of p53 had no effect on activity, again indicating a lesser role for this domain. Finally, we tested the p53 TET domain mutants for stability in H1299 cells when co-transfected with Pirh2. Consistent with the in vitro data, the levels of p53 oligomerization mutants were much higher than that of the wild type protein, reflecting defects in protein degradation for the DM and MM p53 proteins in the presence of Pirh2.
Together these data suggest a working model for Pirh2 mediated p53 ubiquitylation in which the CTD-TET domain interaction provides the majority of the binding energy and the NTD may further interact transiently with one or more of the p53 DNA binding domains (Fig. 7). Indeed, the combination of a primary and a secondary binding site together with the fact that WT p53 is tetrameric, may contribute to increased avidity and activity of the full length proteins as opposed to the individual domains studied here. This mode of interaction likely fixes p53 with respect to the ubiquitin-charged E2 which docks at the central RING domain of Pirh2. While the relative orientation of p53 domains is not likely to be rigid21, and that of the Pirh2 domains is not yet known, this model is conceivable in that it places the C-terminal Lysines of p53 in close proximity to the E2 active site. P53 ubiquitylation is thought to be the central cellular mechanism to downregulate p53 for proliferation in unstressed cells and to upregulate p53 in response to various stresses. As the main regulators of p53 stability, p53 E3 ligases have attracted enormous interest. They are often overexpressed in a variety of human malignancies and may represent a class of targets for the development of novel therapeutic compounds aimed at promoting p53-mediated growth inhibition and apoptosis in cancer cells. Indeed, Mdm2 has been an attractive therapeutic target and the subject of drug discovery activities in both academia and industry 5. We note here that Pirh2 binds to domains distinct from the transactivation domain recognized by Mdm2, possibly offering an alternative (and/or additional) strategy for p53 regulation. The structures and interaction data presented here provide insight into the molecular mechanism of Pirh2-mediated p53 ubiquitylation and point to the disruption of the p53 TET - Pirh2 CTD interaction as potential strategy for development of therapeutics.
Figure 7.

A model of a potential ternary complex between Pirh2, p53 and the E2 enzyme UBE2D2/UbcH5b. The top panel illustrates schematically how domains of tetrameric p53 interact with those of Pirh2. The UBE2D2-Pirh2 ring domain orientations are based on the crystal structure of the homologous complex formed between UBCH7 and the c-Cbl ring domain20. The active site cysteine of E2D2 which is charged with ubiquitin is shown in red.
EXPERIMENTAL PROCEDURES
Protein preparation
The full length Pirh2 and three individual domains NTD (residues 1–137), RING domain (residues 138–189) and CTD (residues 137–261) were PCR amplified and cloned between NdeI and BamHI of pGEX2TK vector (GE Healthcare). Proteins were expressed in Escherichia coli strain BL21 (DE3) (Stratagene) for five hours at room temperature after induction with 1 mM IPTG. The bacteria were grown in Luria Bertani (LB) media for non-labeled proteins and in M9 minimal media supplemented with 15N ammonium chloride (0.8 g/L), 13C glucose (2 g/L) and l50 μM ZnSO4 for uniformly labeled 15N/13C protein. All Pirh2 proteins were purified by standard GST-affinity chromatography. The Pirh2 proteins was further treated with thrombin and purified by size exclusion chromatography using a HiLoad 26/60 Superdex-75 column (GE Healthcare) to remove the GST-tag. The final NMR samples were prepared in buffer D containing 50 mM sodium phosphate (pH 7.0), 150 mM KCl, 10 μM ZnCl2 and 2 mM DTT at a concentration of 0.8 mM with 10% v/v D2O.
Full-length human p53 and p53 fragments including three p53 DBD mutants R273H, R175H and E285A, two tetramerization domain mutants M340Q/L344R (DM) and L344P (MM), and four deletion mutants, 1–360, 1–320, Δ325–355 and Δ70–311, were cloned into the pET15b vector (Novagen), expressed as 6XHis tagged fusion proteins and purified using standard metal affinity purification procedures. UBE2D2/UbcH5b was cloned into the pET15b vector, expressed and purified as described for p53 proteins.
NMR spectroscopy and data analysis
NMR spectra were recorded at 25 °C on Varian INOVA 500 MHz or 600 MHz spectrometers equipped with triple-resonance cold probes and Bruker Avance 500 MHz or 800 MHz spectrometer equipped with cryo probes. The assignments of 1H, 15N and 13C resonances of the RING domain were performed by a conventional manual procedure based on the following experiments: HNCO, HNCACB, CC(CO)NH-TOCSY, HC(CO)NH-TOCSY and HCCH-TOCSY spectra22,23. For NT and CT domains, the assignments of 1H, 15N and 13C resonances were obtained by an ABACUS24 approach using the following experiments: HNCO, CC(CO)NH-TOCSY, HC(CO)NH-TOCSY, (H)CCH-TOCSY and H(C)CH-TOCSY. NOE peaks were picked using manual procedures. The restraints for backbone ϕ and ψ torsion angles were derived from chemical shifts of backbone atoms using TALOS25. No hydrogen bond restraints were used. The initial structures calculated using only dihedral angle and NOE distance restrains were used to determine the coordinating residues to each zinc ion. Additional distance restraints between zinc atoms and their ligands were employed in the calculations of the final structure. These non-experimental distance restraints were developed based on the analysis of the geometrical properties of zinc binding sites observed in a dataset of high quality protein crystal structures26. These include: restraints of 2.25–2.40 Ǻ between Zn and Sγ of Cys residues, restraints of 2.00–2.20 Ǻ between Zn and Nε/Nδ of His residues, restraints of 3.28–3.38 Ǻ between Zn and Cβ of Cys residues. The distances between Sγ and Nε/Nδ atoms that coordinate the same zinc atom were also restrained within bounds of 3.70–4.00 Ǻ for Sγ-Sγ, 3.27–3.87 Ǻ for Sγ-Nε/Nδ, and 3.27–3.78 Ǻ for Nε-Nε, respectively. Automated NOE assignment27 and structure calculations28 were performed using CYANA (version 2.1) using its standard protocol. A total of 92%, 90%, and 88% of NOESY peaks were assigned for the Pirh2 NTD, RING domain, and CTD, respectively, after seven iterative cycles of automated structure calculation and NOE assignment. The final 20 lowest-energy structures were refined within the CNS package29 by performing a short constrained molecular dynamics simulation in explicit solvent30. After refinement, 99.7 % residues in Pirh2 NTD, 99.5% in RING and 99.6% in CTD are in favorable or allowed regions of Ramachandran plot. Resulting structures were analyzed using MOLMOL31, PROCHEK-NMR32, and PSVS validation software33. Figures were prepared using MOLMOL31 and Pymol (DeLano Scientific LLC).
Zinc stoichiometry analysis
The independent determinations of the zinc binding stoichiometry of the three Pirh2 domains were carried out by ICP-AES in the ANALEST laboratory, University of Toronto.
Chemical shift perturbation experiments
The chemical shift mapping on the Pirh2 domains were performed by monitoring the 1H-15N HSQC spectra of the uniformly 15N-labeled Pirh2 protein alone (0.2 mM) and with excess of the unlabeled interacting proteins until no further changes in chemical shifts were observed in the 1H-15N HSQC spectra. The HSQC spectra were recorded at 25 °C in a buffer containing 25 mM sodium phosphate (pH 7.0), 100 mM NaCl, 2 mM DTT and 10% v/v D2O on a Varian INOVA600 MHz spectrometer. The values shown were calculated by using the equation Δcomp = [ΔδHN2 + (ΔδN/5)2]1/2.
GST pull-down assays
Purified full-length p53 and p53 fragments were incubated with GST alone or a GST-Pirh2 fusion protein in an assay buffer containing 20 mM Tris (pH8.0), 100 mM NaCl, 2 mM DTT and 5 % v/v glycerol in a 1:1 molar ratio at 4 °C overnight in the presence of 50 μl glutathione-Sepharose beads (GE Healthcare). The mixture were then transferred to a microcolumn, and after entenxive washing with the assay buffer, bound proteins were eluted with 20 mM reduced glutathione and detected by SDS-PAGE and Coomassie staining.
In vitro ubiquitylation assay
Human ubiquitin activating enzyme E1 and 6xHis tagged ubiquitin were purchased from Boston Biochem. The ubiquitylation reaction was performed in a volume of 20 μL in a buffer of 50 mM Tris pH 7.6, 5 mM MgCl2, 2 mM ATP, 2 mM DTT. The reaction mixture typically contained E1 50 ng (Calbiochem), UBE2D2/UbcH5b (100ng), Ubiquitin 5 μg (Sigma), Pirh2 1 μg for detection of autoubiquitylation on E3s and with additional 0.5 ug of full-length p53 for detection of p53 ubiquitylation. After incubation at 30 °C for 90 mins, the reactions were stopped by addition of SDS-PAGE sample buffer and resolved on 7.5–10% SDS-PAGE gels. Ubiquitylated proteins were visualized and evaluated by western blot using monoclonal antibodies against GST (GE Healthcare) for Pirh2 autoubiquitylation, antibody against p53 (PAb1801)34 for p53 ubiquitylation.
Cell culture and DNA transfection
p53 null H1299 cells were cultured in alpha MEM with 10% FBS and antibiotics and transfected overnight by calcium phosphate with 10 μg of either pcDNA3 (neo), or pcDNA3 (neo) hPirh2 and 0.1μg of pcDNA3 (neo) p53 construct (wild-type, MM or DM). Cells were harvested 40 hours post-transfection using 1% v/v NP-40 lysis buffer. Westerns were performed using the following antibodies: p53 DO-1 (Santa Cruz), Pirh2 (Bethyl Laboratories), β-actin (Sigma). ImageQuant (5.2) was used to quantitate the expression levels from the western blots for p53 and B-actin.
Supplementary Material
Table 1.
NMR and refinement statistics for protein structures
| NMR distance and dihedral constraints | NTD | RING | CTD |
|---|---|---|---|
| Distance constraints | |||
| Total NOE | 2569 | 1036 | 909 |
| Intra-residue | 681 | 240 | 280 |
| Inter-residue | |||
| Sequential (|i − j| = 1) | 612 | 297 | 281 |
| Medium-range (|i − j| < 4) | 368 | 197 | 77 |
| Long-range (|i − j| > 5) | 908 | 302 | 271 |
| Intermolecular | 0 | 0 | 0 |
| Hydrogen bonds | 0 | 0 | 4 |
| Total dihedral angle restraints | 126 | 53 | 56 |
| φ | 63 | 27 | 28 |
| ψ | 63 | 26 | 28 |
| Zinc-ligand distance restraints | 66 | 22 | 11 |
| Structure statisticsa | |||
| Violationsb(mean ± s.d.) | |||
| Distance constraints (Å) | 0.15±0.36 | 0 | 0.05±0.22 |
| Dihedral angle constraints (°) | 0.67±0.70 | 0 | 0.15±0.36 |
| Max. dihedral angle violation (°) | 6.5 | 4.3 | 5.5 |
| Max. distance constraint violation (Å) | 0.83 | 0.27 | 0.59 |
| Deviations from idealized geometry | |||
| Bond lengths (Å) | 0.0048±0.0001 | 0.0046±0.0001 | 0.0066±0.0018 |
| Bond angles (°) | 0.64±0.01 | 0.55±0.01 | 0.78±0.05 |
| Impropers (°) | 2.25±0.10 | 1.39±0.09 | 1.73±0.12 |
| Average pairwise r.m.s. deviationc (Å) | |||
| Heavy | 1.30±0.13 | 1.11±0.14 | 1.05±0.14 |
| Backbone | 0.85±0.16 | 0.51±0.10 | 0.54±0.13 |
Ensemble of 20 lowest-energy structures out of 100 calculated.
Violations of more than 0.5 Å and 5° for distance and dihedral angles constraints, respectively.
NTD, residues 18–128; RING, 143–188; CTD, 216–251.
Acknowledgments
We thank Ayeda Ayed for helpful comments on the manuscript and members of the Arrowsmith lab for technical advice and discussion. This work was funded by the Canadian Cancer Society through grants from the National Cancer Institute of Canada (NCIC), Protein Structure Initiative of the National Institutes of Health (P50-GM62413-01) through the Northeast Structural Genomics Consortium, the Canada Research Chairs program (CHA). Y.S is supported by a fellowship from Leukaemia and Lymphoma Research Society of Canada.
Footnotes
Accession codes. The atomic coordinates for the Pirh2 NTD, RING and CTD domain have been deposited in Protein Data Bank with accession code 2K2C, 2JRJ and 2K2D, respectively.
AUTHOR CONTRIBUTIONS
Y.S, R.L. and E.T. designed and performed experiments; Y.S., A.L, B.W and M.S, determined the structures; R.L, J.L., S.S. and M.K. collected NMR data; S.D. provided constructs and reagents. Y.S. and C.H.A wrote the paper with input from all other authors.
References
- 1.Vousden KH, Prives C. P53 and prognosis: new insights and further complexity. Cell. 2005;120:7–10. doi: 10.1016/j.cell.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 2.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–31. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 3.Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol. 2003;13:49–58. doi: 10.1016/s1044-579x(02)00099-8. [DOI] [PubMed] [Google Scholar]
- 4.Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–7. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- 5.Chene P. Inhibiting the p53-MDM2 interaction: an important target for cancer therapy. Nat Rev Cancer. 2003;3:102–9. doi: 10.1038/nrc991. [DOI] [PubMed] [Google Scholar]
- 6.Leng RP, et al. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell. 2003;112:779–91. doi: 10.1016/s0092-8674(03)00193-4. [DOI] [PubMed] [Google Scholar]
- 7.Dornan D, et al. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature. 2004;429:86–92. doi: 10.1038/nature02514. [DOI] [PubMed] [Google Scholar]
- 8.Chen D, et al. ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell. 2005;121:1071–83. doi: 10.1016/j.cell.2005.03.037. [DOI] [PubMed] [Google Scholar]
- 9.Rajendra R, et al. Topors functions as an E3 ubiquitin ligase with specific E2 enzymes and ubiquitinates p53. J Biol Chem. 2004;279:36440–4. doi: 10.1074/jbc.C400300200. [DOI] [PubMed] [Google Scholar]
- 10.Duan W, et al. Expression of Pirh2, a newly identified ubiquitin protein ligase, in lung cancer. J Natl Cancer Inst. 2004;96:1718–21. doi: 10.1093/jnci/djh292. [DOI] [PubMed] [Google Scholar]
- 11.Holm L, Sander C. Dali: a network tool for protein structure comparison. Trends Biochem Sci. 1995;20:478–80. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- 12.Borden KL, Freemont PS. The RING finger domain: a recent example of a sequence-structure family. Curr Opin Struct Biol. 1996;6:395–401. doi: 10.1016/s0959-440x(96)80060-1. [DOI] [PubMed] [Google Scholar]
- 13.Fang S, Weissman AM. A field guide to ubiquitylation. Cell Mol Life Sci. 2004;61:1546–61. doi: 10.1007/s00018-004-4129-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laine A, et al. Regulation of p53 localization and activity by Ubc13. Mol Cell Biol. 2006;26:8901–13. doi: 10.1128/MCB.01156-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davison TS, et al. Structure and functionality of a designed p53 dimer. J Mol Biol. 2001;307:605–17. doi: 10.1006/jmbi.2001.4450. [DOI] [PubMed] [Google Scholar]
- 16.Friedler A, Veprintsev DB, Rutherford T, von Glos KI, Fersht AR. Binding of Rad51 and other peptide sequences to a promiscuous, highly electrostatic binding site in p53. J Biol Chem. 2005;280:8051–9. doi: 10.1074/jbc.M411176200. [DOI] [PubMed] [Google Scholar]
- 17.Joazeiro CA, Weissman AM. RING finger proteins: mediators of ubiquitin ligase activity. Cell. 2000;102:549–52. doi: 10.1016/s0092-8674(00)00077-5. [DOI] [PubMed] [Google Scholar]
- 18.Eletr ZM, Kuhlman B. Sequence determinants of E2-E6AP binding affinity and specificity. J Mol Biol. 2007;369:419–28. doi: 10.1016/j.jmb.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dominguez C, et al. Structural model of the UbcH5B/CNOT4 complex revealed by combining NMR, mutagenesis, and docking approaches. Structure. 2004;12:633–44. doi: 10.1016/j.str.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 20.Zheng N, Wang P, Jeffrey PD, Pavletich NP. Structure of a c-Cbl-UbcH7 complex: RING domain function in ubiquitin-protein ligases. Cell. 2000;102:533–9. doi: 10.1016/s0092-8674(00)00057-x. [DOI] [PubMed] [Google Scholar]
- 21.Ayed A, et al. Latent and active p53 are identical in conformation. Nat Struct Biol. 2001;8:756–60. doi: 10.1038/nsb0901-756. [DOI] [PubMed] [Google Scholar]
- 22.Kay LE. NMR methods for the study of protein structure and dynamics. Biochem Cell Biol. 1997;75:1–15. doi: 10.1139/o97-023. [DOI] [PubMed] [Google Scholar]
- 23.Bax A, Vuister GW, Grzesiek S, Delaglio F, Wang AC, Tschudin R, Zhu G. Measurement of homo- and heteronuclear J couplings from quantitative J correlation. Methods Enzymol. 1994;239:79–105. doi: 10.1016/s0076-6879(94)39004-5. [DOI] [PubMed] [Google Scholar]
- 24.Lemak A, Steren CA, Arrowsmith CH, Llinas M. Sequence specific resonance assignment via Multicanonical Monte Carlo search using an ABACUS approach. J Biomol NMR. 2008;41:29–41. doi: 10.1007/s10858-008-9238-2. [DOI] [PubMed] [Google Scholar]
- 25.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 26.Alberts IL, Nadassy K, Wodak SJ. Analysis of zinc binding sites in protein crystal structures. Protein Sci. 1998;7:1700–16. doi: 10.1002/pro.5560070805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guntert P, Mumenthaler C, Wuthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol. 1997;273:283–98. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 28.Grishaev A, et al. ABACUS, a direct method for protein NMR structure computation via assembly of fragments. Proteins. 2005;61:36–43. doi: 10.1002/prot.20457. [DOI] [PubMed] [Google Scholar]
- 29.Brunger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 30.Linge JP, Williams MA, Spronk CA, Bonvin AM, Nilges M. Refinement of protein structures in explicit solvent. Proteins. 2003;50:496–506. doi: 10.1002/prot.10299. [DOI] [PubMed] [Google Scholar]
- 31.Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–5. 29–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 32.Laskowski RA, Rullmann JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of proteins structures solved by NMR. J Biomol NMR. 1996;8:477–86. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 33.Bhattacharya A, Tejero R, Montelione GT. Evaluating protein structures determined by structural genomics consortia. Proteins. 2007;66:778–95. doi: 10.1002/prot.21165. [DOI] [PubMed] [Google Scholar]
- 34.Banks L, Matlashewski G, Crawford L. Isolation of human-p53 specific monoclonal antibodies and their use in the studies of human p53 expression. Eur J Biochem. 1986;159:529–534. doi: 10.1111/j.1432-1033.1986.tb09919.x. [DOI] [PubMed] [Google Scholar]
- 35.Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–8. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.