

Abstract
A practical, rapid, and highly regioselective Cu-catalyzed radiofluorination of (mesityl)(aryl)iodonium salts is described. This protocol utilizes [18F]KF to access 18F-labeled electron-rich, -neutral, and -deficient aryl fluorides under a single set of mild conditions. This methodology is applied to the synthesis of protected versions of two important radiotracers: 4-[18F]fluorophenylalanine and 6-[18F]fluoroDOPA.
Positron emission tomography (PET) is a powerful and minimally invasive medical imaging technique that provides kinetic physiochemical information.1 The most commonly used radioisotope for PET is 18F, which offers the advantages of high-resolution imaging, a relatively long lifetime (t1/2 = 110 min), and minimal perturbation of radioligand binding. Despite these advantages, the development of novel 18F radiotracers is currently impeded by a paucity of general and effective radiofluorination methods. There are currently few robust synthetic procedures for the incorporation of 18F into organic molecules with sufficient speed, selectivity, yield, radiochemical purity, and reproducibility to provide clinical imaging materials. Direct methods for the late stage nucleophilic [18F]fluorination of electron-rich aromatic substrates remains an especially long-standing challenge.2 A target of particular interest in this regard is 6-[18F]fluoroDOPA.3,4
The classical radiofluorination methods for electron-rich aryl rings utilize electrophilic fluorinating reagents derived from [18F]F2.2 However, [18F]F2 production typically requires 19F2 as a carrier gas, which leads to low specific activity (SA) radiotracers (typically <1.0 Ci/mmol) and requires specialized facilities. The development of [18F]KF production from [18O]water has provided the means to synthesize high SA radiotracers (>1000 Ci/mmol) through nucleophilic substitution (typically SN2 or SNAr).1 However, the use of [18F]KF is generally limited to the formation of primary sp3-C–F bonds or sp2-C–F bonds on electron-deficient (hetero)aromatics.
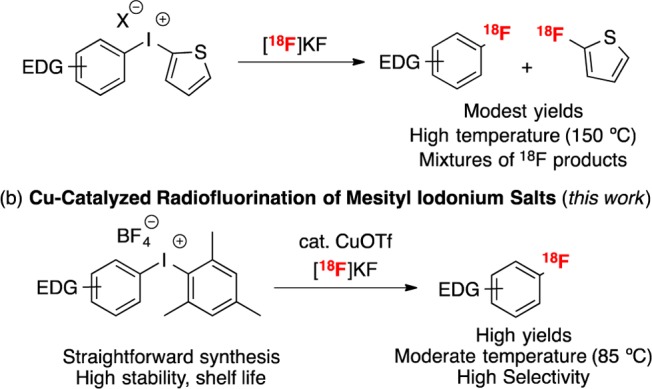
Two main strategies have been used to address these limitations. The first involves radiofluorination of powerful electrophiles such as diaryliodonium salts.5 Diaryliodonium salts bearing the 2-thienyl group have been shown to react with [18F]KF at elevated temperatures (often ≥150 °C) to afford [18F]fluororarenes (Scheme 1a).6 In these systems, the 2-thienyl group directs radiofluorination to the other aromatic ligand on iodine with moderate to good selectivity.6 However, the [(thienyl)(aryl)I+] starting materials are often challenging to prepare, suffer from low stability, and have a limited shelf life.7 Furthermore, with electron-neutral or -rich substrates, these transformations frequently require high temperatures, exhibit modest regioselectivity, demonstrate limited functional group tolerance, and provide low radiochemical yields.6 With the exception of recent work by DiMagno,8 it has proven challenging to access many important radiotracers using this method.
Scheme 1. Radiofluorination of Diaryliodonium Salts.
A second strategy applies transition metal catalysts and/or reagents to achieve radiofluorination with [18F]KF.9,10 Transition-metal catalysis offers opportunities for accelerating radiofluorination reaction rates as well as enhancing selectivity and reactivity. For instance, Hooker and Ritter have made progress in the radiofluorination of arenes using Pd9a,9b and Ni9c complexes.
We sought to develop a general, mild, high-yielding, and user-friendly procedure for the radiofluorination of diverse aromatic substrates by merging transition-metal catalysis with the fluorination of diaryliodonium reagents (Scheme 1b). We have recently demonstrated that Cu salts catalyze the fluorination of stable and synthetically accessible mesityl-substituted diaryliodonium salts.11 Here, the mesityl group directs oxidative addition and fluorination to the smaller aryl group on iodine, regardless of its electronic properties. In this paper, we demonstrate that this Cu-catalyzed fluorination method can be translated to a high specific activity radiofluorination of electron-rich, -neutral, and -deficient arene substrates. Furtheremore, this protocol enables the radiofluorination of protected versions of 4-[18F]fluorophenylalanine, and 6-[18F]fluoroDOPA.
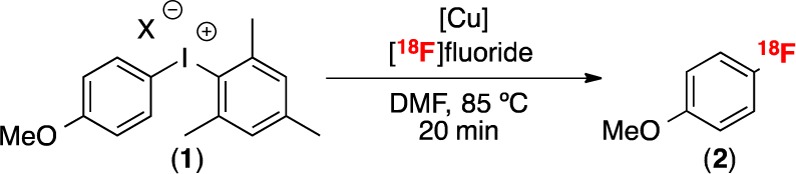
Our initial studies focused on the Cu-catalyzed radiofluorination of [4-OMePh-I-Mes]BF4 (1). This substrate was selected because the radiosynthesis of electron-rich fluoroarenes such as [18F]fluoroanisole remains a challenge.12 Gratifyingly, the use of Cu(OTf)2 as the catalyst, DMF as the solvent, and [18F]KF-18-crown-6 as the fluoride source resulted in 36% radiochemical conversion (RCC) to 4-[18F]fluoroanisole in 20 min (Table 1, entry 1). High selectivity was observed for 4-[18F]fluoroanisole, and <1% of [18F]fluoromesitylene was detected by radio-TLC or radio-HPLC. While the radiochemical conversion was reasonable, the reaction showed modest reproducibility (±19% yield over n = 15). To address this issue, we sought an alternate Cu precursor to catalyze the radiofluorination. A variety of CuI and CuII complexes were examined (for examples, see entries 2–4). Commercially available and bench stable (CH3CN)4CuOTf proved optimal, providing high radiochemical conversion and improved reproducibility (70 ± 11% RCC over n = 11, entry 4). A control experiment in the absence of Cu provided no detectable 4-[18F]fluoroanisole and only 6% RCC to [18F]fluoromesitylene (entry 5). A number of other variables were examined to optimize the radiofluorination including the ratio of [Cu] to 1, concentration of 1, temperature, counterion, and reaction time (Tables S1–S4, Supporting Information). Optimal conditions were as follows: 6 μmol loading of 1, 1:1 molar ratio of CuOTf to 1, 85 °C, 20 min, in a total volume of 750 μL of DMF. Under these conditions, 1 was obtained in 79 ± 8% radiochemical conversion (n = 38, entry 6). Notably, >50% RCC was obtained under most of the conditions examined, indicating that this transformation is remarkably insensitive to the reaction conditions (a particularly attractive attribute when translating this chemistry to use by nonexperts).
Table 1. Optimization of Radiofluorination of 1 To Form 4-[18F]Fluoroanisole.
| entry | [Cu] | X | RCC 2a (%) |
|---|---|---|---|
| 1b | Cu(OTf)2 | BF4 | 36 ± 19 (n = 15) |
| 2b | CuCO3·Cu(OH)2 | BF4 | 10 ± 6 (n = 3) |
| 3b | CuOTf·toluene | BF4 | 43 ± 15 (n = 3) |
| 4b | (CH3CN)4CuOTf | BF4 | 70 ± 11 (n = 11) |
| 5b | none | BF4 | <1 |
| 6c | (CH3CN)4CuOTf | BF4 | 79 ± 8 (n = 38) |
| 7c | (CH3CN)4CuOTf | PF6 | 53 ± 7 (n = 3) |
| 8c | (CH3CN)4CuOTf | OTs | 45 ± 26 (n = 3) |
| 9c | (CH3CN)4CuOTf | OTf | 15 ± 12 (n = 3) |
Radiochemical conversion was determined by radio-TLC (average of n runs). The identity of 4-[18F]fluoroanisole was confirmed by HPLC.
Conditions: [4-OMePh-I-Mes]BF4 (6 μmol), [Cu] (3 μmol), [18F]KF·18-crown-6·K2CO3 complex in DMF (250 μL, 300–700 μCi), total volume 750 μL.
Conditions: [4-OMePh-I-Mes]X (6 μmol), [Cu] (6 μmol). [18F]KF·18-crown-6·K2CO3 complex in DMF (250 μL, 300–700 μCi), total volume 750 μL.
One concern in the radiofluorination of 1 is the possibility for isotopic dilution via 18F/19F exchange between the BF4 counterion and the [18F]KF.13 In principle, this issue can be addressed by changing the counterion;14 however, an evaluation of different [4-OMePh-I-Mes]X salts (X = TsO, PF6, TfO) showed that the best conversion was obtained with BF4 (Table 1, entries 6–9). To test whether isotopic dilution from the BF4 counterion is, indeed, a problem, we compared the specific activity (SA) of the 4-[18F]fluoroanisole product obtained from [4-OMePh-I-Mes]BF4 to that from [4-OMePh-I-Mes]OTs. Automated syntheses were conducted in a standard module with 1500 mCi initial activity of 18F. Under automated conditions, [4-OMePh-I-Mes]BF4 afforded a RCC of 40 ± 10% and a SA of 1800 ± 800 Ci/mmol (n = 3), while [4-OMePh-I-Mes]OTs afforded 10 ± 2% RCC with a comparable SA of 3000 ± 1000 Ci/mmol (n = 3). These results indicate that isotopic dilution is not a significant problem under these reaction conditions.15
Importantly for the user, this transformation is performed under ambient conditions without the requirement for a drybox or extensive drying of reagents and glassware. The reaction mixture is homogeneous, and the remainder of 18F appears to be sequestered as inorganic fluoride salts. The copper catalyst is commercially available and is stable in solution over at least 4 h; for instance, a 77% RCC was obtained using a solution of (CH3CN)4CuOTf in DMF that was allowed to stand for 4 h under ambient conditions prior to use. Finally, 1 and the other (mesityl)(aryl)iodonium salts described herein are colorless, free-flowing solids that are shelf stable for months under ambient conditions when protected from intense light. Thus, this method is highly practical and amenable to routine automated synthesis.
These radiofluorination conditions were next applied to a diverse series of (mesityl)(aryl)iodonium tetrafluoroborate salts (Table 2). In all cases, modest to excellent radiochemical conversion was obtained without further optimization of the reaction conditions. All of the reactions in Table 2 were highly selective for a single 18F-containing product, with ≤2% fluoromesitylene detected. Furthermore, for all of the substrates examined, ≤2% of the corresponding fluoroarene product was observed in the absence of Cu catalyst, confirming that Cu is vital for accelerating the reaction rate as well as controlling selectivity.
Table 2. [18F]Fluorination of (Mesityl)(aryl)iodonium Saltsb.


Radiochemical conversion were determined by radio-TLC (average of n runs). The identity of each product was confirmed by HPLC.
Reaction conditions: precursor (6 μmol), Cu catalyst (6 μmol). [18F]KF·18-crown-6·K2CO3 complex in DMF (250 μL, 300–700 μCi). Total reaction volume 750 μL.
This protocol can be used for the radiofluorination of arenes containing multiple electron-donating methoxy substituents (Table 2, entries 2 and 3). Remarkably, even the highly electron-rich product 1-[18F]fluoro-3,4,5-trimethoxybenzene (4) was obtained in 14% RCC using this nucleophilic fluorination protocol. Steric factors do impact the radiochemical conversion; for instance, while 4-[18F]fluoroanisole was obtained in 79% RCC, the corresponding 2-[18F]fluoroanisole was formed in 30% RCC.
Electron-neutral and electron-deficient aryl rings also underwent radiofluorination in high yield and selectivity using this method (entries 6–10). A variety of functional groups, including amide NH bonds (6), ketones (8), aryl iodides (9), esters (10), and aldehydes (11), were well tolerated. This enabled the preparation of several known PET prosthetic groups, including 4-[18F]fluoroiodobenzene (entry 8),16 methyl [18F]fluorobenzoate (entry 9),17 and [18F]fluorobenzaldehyde (entry 10).18
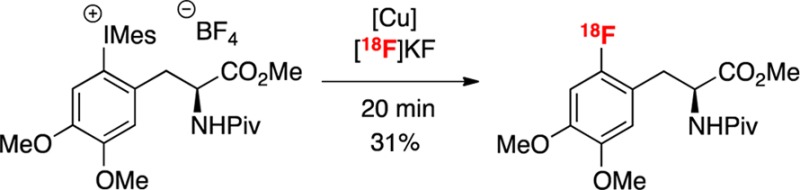
Having demonstrated that a diverse range of [18F]arylfluorides can be accessed via this methodology, the radiofluorination of several molecules of clinical relevance was investigated. Two (mesityl)(aryl)iodonium salts derived from aromatic amino acids (12 and 14) were prepared and subjected to the radiofluorination protocol (Scheme 2). Without any substrate-specific optimization, the radiolabeled products 13 and 15 were obtained in acceptable yields. Importantly, 13 is a protected version of 4-[18F]fluorophenylalanine (F-PHE), a radiotracer originally developed in the 1970s as a probe of pancreatic and cerebral protein synthesis.19 However, clinical applications of F-PHE to tumor imaging have not been realized partially due to the lack of an acceptable radio-synthesis. The original doses of F-PHE were prepared in low specific activity (<0.01 Ci/mmol) and required a dose “approaching toxic levels in order to obtain adequate sample count rates.”21b,20 The current protocol affords protected F-PHE (13) in 23% RCC (eq 1).
Scheme 2. Amino Acid Derived Substrates.
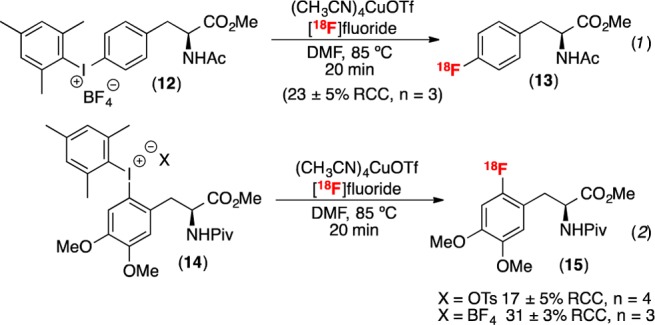
Finally, protected 6-[18F]fluoroDOPA 15 was prepared in 17 ± 6% RCC with a tosylate counterion and 30 ± 3% RCC with a tetrafluoroborate counterion (eq 2). While there has been much activity in the radiofluorination community aimed at accessing 6-[18F]fluoro-L-DOPA,21,22 current methods suffer from drawbacks that limit routine production of this material. This molecule has been of great interest to the PET community since the 1970s due to its numerous clinical applications.3,4However, despite decades of research, there is no routine automated synthesis of18F-DOPA in clinical use.5,6 To further demonstrate the utility of this method, we have performed the automated synthesis of 15 from the shelf stable salt 14-OTs. Under our standard conditions, 14-OTs afforded 17 ± 2% RCC of 15 with a SA of 4000 ± 2000 Ci/mmol (n = 2).23,24
In summary, this paper demonstrates a general, practical, and rapid Cu-catalyzed radiofluorination of diaryliodonium salts using [18F]KF. This method is compatible with a wide range of functional groups, is competent for electronically diverse aryl groups, and uniformly affords good selectivity for a single 18F-containing product. The synthesis of protected versions of clinically relevant tracers, 4-[18F]fluorophenylalanine and 6-[18F]fluoroDOPA, has been demonstrated. Application of this methodology to other molecules of clinical interest is ongoing and will be disclosed in due course.
Acknowledgments
We acknowledge the NIH (GM073836 (MSS) and T32-EB005172 (PJHS)) for financial support.
Supporting Information Available
Experimental procedures, optimization details, radio-TLC traces, HPLC traces, and spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Cai L.; Lu S.; Pike V. W. Eur. J. Org. Chem. 2008, 2853. [Google Scholar]; b Ametamey S. M.; Honer M.; Schubiger P. A. Chem. Rev. 2008, 108, 1501. [DOI] [PubMed] [Google Scholar]; c Miller P. W.; Long N. J.; Vilar R.; Gee A. D. Angew. Chem., Int. Ed. 2008, 47, 8998. [DOI] [PubMed] [Google Scholar]; d Littich R.; Scott P. J. H. Angew. Chem., Int. Ed. 2012, 51, 1106. [DOI] [PubMed] [Google Scholar]
- For a recent review on [18F]fluoroaryl synthesis, see:Tredwell M.; Gouverneur V. Angew. Chem., Int. Ed. 2012, 51, 11426. [DOI] [PubMed] [Google Scholar]
- a Garnett E. S.; Firnau G.; Chan P. K. H.; Sood S.; Belbeck L. W. Proc. Natl. Acad. Sci. U.S.A. 1978, 75, 464. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Garnett E. S.; Firnau G.; Nahmias C. Nature 1983, 305, 137. [DOI] [PubMed] [Google Scholar]; c Luxen A.; Guillaume M.; Melega W. P.; Pike V. W.; Solin O.; Wagner R. Int. J. Nucl. Med. Biol. 1992, 19, 149. [DOI] [PubMed] [Google Scholar]; d Rakshi J. S.; Uema T.; Ito K.; Bailey D. L.; Morrish P. K.; Ashburner J.; Dagher A.; Jenkins I. H.; Friston K. J.; Brooks D. J. Brain 1999, 122, 1637. [DOI] [PubMed] [Google Scholar]
- For the application of 6-[18F]F -DOPA to tumor imaging, see:; a Hoegerle S.; Altehoefer C.; Ghanem N.; Koehler G.; Waller C. F.; Scheruebl H.; Moser E.; Nitzsche E. Radiology 2001, 220, 373. [DOI] [PubMed] [Google Scholar]; b Becherer A.; Szabó M.; Karanikas G.; Wunderbaldinger P.; Angelberger P.; Raderer M.; Kurtaran A.; Dudczak R.; Kletter K. J. Nucl. Med. 2004, 45, 1161. [PubMed] [Google Scholar]; c Chen W.; Silverman D. H. S.; Delaloye S.; Czernin J.; Kamdar N.; Pope W.; Satyamurthy N.; Schiepers C.; Cloughesy T. J. Nucl. Med. 2006, 47, 904. [PubMed] [Google Scholar]
- a Grushin V. V.; Kantor M. M.; Tolstoya T. P.; Shcherbina T. M. Russ. Chem. Bull. 1984, 33, 2130. [Google Scholar]; b Shah A.; Pike V. W.; Widdowson D. A. J. Chem. Soc., Perkin Trans. 1 1998, 2043. [Google Scholar]; c Chun J.; Telu S.; Lu S.; Pike V. W. Org. Biomol. Chem. 2013, 11, 5094. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Chun J.; Pike V. W. Org. Biomol. Chem. 2013, 11, 6300. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Yusubov M. S.; Svitich D. Y.; Larkina M. S.; Zhdankin V. V. ARKIVOC 2013, 364. [Google Scholar]
- a Martín-Santamaría S.; Carroll M. A.; Carroll C. M.; Carter C. D.; Rzepa H. S.; Widdowson D. A.; Pike V. W. Chem. Commun. 2000, 649. [Google Scholar]; b Ross T. L.; Ermert J.; Hocke C.; Coenen H. H. J. Am. Chem. Soc. 2007, 129, 8018. [DOI] [PubMed] [Google Scholar]; c Carroll M. A.; Jones C.; Tang S.-L. J. Labelled Compd. Radiopharm. 2007, 50, 450. [Google Scholar]; d Jang K. S.; Jung Y.-W.; Gu G.; Koeppe R. A.; Sherman P. S.; Quesada C. A.; Raffel D. M. J. Med. Chem. 2013, 56, 7312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- In our hands, (2-thienyl)(aryl)iodonium derivatives exhibit modest stability; for example, [(4-OMePh)(2-thienyl)I]Br was found to decompose within 2 weeks under ambient conditions.
- Only one report in the patent literature has described accessing F-DOPA from diaryliodonium salts:DiMagno S.Fluorination of Aromatic Ring Systems. WO2010/048170A2, April 29, 2010.
- For transition metal-catalyzed aryl radiofluorination, see:; a Lee E.; Kamlet A. S.; Powers D. C.; Neumann C. N.; Boursalian G. B.; Furuya T.; Choi D. C.; Hooker J. M.; Ritter T. Science 2011, 334, 639. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kamlet A. S.; Neumann C. N.; Lee E.; Carlin S. M.; Moseley C. K.; Stephenson N.; Hooker J. M.; Ritter T. PLoS One 2013, 8, e59187. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lee E.; Hooker J. M.; Ritter T. J. Am. Chem. Soc. 2012, 134, 17456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For metal catalyzed sp3 radiofluorination, see:; a Topczewski J. J.; Tewson T. J.; Nguyen H. M. J. Am. Chem. Soc. 2011, 133, 19318. [DOI] [PubMed] [Google Scholar]; b Benedetto E.; Tredwell M.; Hollingworth C.; Khotavivattana T.; Brown J. M.; Gouverneur V. Chem. Sci. 2013, 4, 89. [Google Scholar]; c Graham T. J. A.; Lambert R. F.; Ploessl K.; Kung H. F.; Doyle A. G. J. Am. Chem. Soc. 2014, 136, 5291. [DOI] [PubMed] [Google Scholar]; d Huang X.; Liu W.; Ren H.; Neelamegam R.; Hooker J. M.; Groves J. T. J. Am. Chem. Soc. 2014, 136, 6842. [DOI] [PubMed] [Google Scholar]
- Ichiishi N.; Canty A. J.; Yates B. F.; Sanford M. S. Org. Lett. 2013, 15, 5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To achieve an acceptable yield, bis(p-methoxyphenyl)iodonium salts are required, which produces iodoanisole as a byproduct:Pike V. W.; Aigbirhio F. I. J. Chem. Soc., Chem. Commun. 1995, 2215. [Google Scholar]
- Jauregui-Osoro M.; Sunassee K.; Weeks A. J.; Berry D. J.; Paul R. L.; Cleij M.; Banga J. P.; O’Doherty M. J.; Marsden P. K.; Clarke S. E. M.; Ballinger J. R.; Szanda I.; Cheng S.-Y.; Blower P. J. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J.-H.; Lu S.; Pike V. W. Eur. J. Org. Chem. 2011, 4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- If 18F/19F exchange were rapid, the expected specific activity of 4-[18F]fluoroanisole would be <1 Ci/mmol.
- a Gail R.; Coenen H. H. Appl. Radiat. Isot. 1994, 45, 105. [Google Scholar]; b Steiniger B.; Wuest F. R. J. Labelled Compd. Radiopharm. 2006, 49, 817. [Google Scholar]; c Gao Z.; Gouverneur V.; Davis B. G. J. Am. Chem. Soc. 2013, 135, 13612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Basuli F.; Wu H.; Griffiths G. L. J. Labelled Compd. Radiopharm. 2011, 54, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Vaidyanathan G.; Zalutsky M. R. Nucl. Med. Biol. 1992, 19, 275. [Google Scholar]; c Hoehne A.; Behera D.; Parsons W. H.; James M. L.; Shen B.; Borgohain P.; Bodapati D.; Prabhakar A.; Gambhir S. S.; Yeomans D. C.; Biswal S.; Chin F. T.; Bois J. D. J. Am. Chem. Soc. 2013, 135, 18102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lemaire C.; Guillaume M.; Christiaens L.; Palmer A. J.; Cantineau R. Appl. Radiat. Isot. 1987, 38, 1033. [DOI] [PubMed] [Google Scholar]; b Haka M. S.; Kilbourn M. R.; Watkins G. L.; Toorongian S. A. J. Labelled Compd. Radiopharm. 1989, 27, 823. [Google Scholar]; c Poethko T.; Schottelius M.; Thumshirn G.; Hersel U.; Herz M.; Henriksen G.; Kessler H.; Schwaiger M.; Wester H.-J. J. Nucl. Med. 2004, 45, 892. [PubMed] [Google Scholar]
- a Goulding R. W.; Palmer A. J.; Thakur M. L. Radioisotopy 1971, 12, 1045. [Google Scholar]; b Cottrall M. F.; Taylor D. M.; McElwain T. J. Br. J. Radiol. 1973, 46, 277. [DOI] [PubMed] [Google Scholar]; c Bodsch W.; Coenen H. H.; Stöcklin G.; Takahashi K.; Hossmann K. A. J. Neurochem. 1988, 50, 979. [DOI] [PubMed] [Google Scholar]
- Since then, O-(2-[18F]fluoroethyl)-l-tyrosine has been utilized as a less than ideal but accessible surrogate for phenylalanine. See:; a Wester H. J.; Herz M.; Weber W.; Heiss P.; Senekowitsch-Schmidtke R.; Schwaiger M.; Stöcklin G. J. Nucl. Med. 1999, 40, 205. [PubMed] [Google Scholar]; b Langen K.-J.; Hamacher K.; Weckesser M.; Floeth F.; Stoffels G.; Bauer D.; Coenen H. H.; Pauleit D. Nucl. Med. Biol. 2006, 33, 287. [DOI] [PubMed] [Google Scholar]
- For 6-[18F]F-DOPA syntheses using [18F]F2 or a derivative thereof, see:; a Firnau G.; Chirakal R.; Garnett E. S. J. Nucl. Med. 1984, 25, 1228. [PubMed] [Google Scholar]; b Chirakal R.; Firnau G.; Couse J.; Garnett E. S. Appl. Radiat. Isot. 1984, 35, 651. [Google Scholar]; c Coenen H. H.; Franken K.; Kling P.; Stöcklin G. Appl. Radiat. Isot. 1988, 39, 1243. [Google Scholar]; d Namavari M.; Bishop A.; Satyamurthy N.; Bida G.; Barrio J. R. Appl. Radiat. Isot. 1992, 43, 989. [DOI] [PubMed] [Google Scholar]; e Stenhagen I. S. R.; Kirjavainen A. K.; Forsback S. J.; Jørgensen C. G.; Robins E. G.; Luthra S. K.; Solin O.; Gouverneur V. Chem. Commun. 2013, 49, 1386. [DOI] [PubMed] [Google Scholar]
- For syntheses of 6-[18F]F-DOPA from 18F–, see:; a Ding Y. S.; Shiue C. Y.; Fowler J. S.; Wolf A. P.; Plenevaux A. J. Fluorine Chem. 1990, 48, 189. [Google Scholar]; b Lemaire C.; Damhaut P.; Plenevaux A.; Comar D. J. Nucl. Med. 1994, 35, 1996. [PubMed] [Google Scholar]; c Lemaire C.; Gillet S.; Guillouet S.; Plenevaux A.; Aerts J.; Luxen A. Eur. J. Org. Chem. 2004, 2899. [Google Scholar]; d Wagner F. M.; Ermert J.; Coenen H. H. J. Nucl. Med. 2009, 50, 1724. [DOI] [PubMed] [Google Scholar]
- An unoptimized and uncorrected isolated RCY of 1.1% of 15 (16.5 mCi) was obtained via automated synthesis using 14-BF4 as starting material. This material had a specific activity of 290 Ci/mmol.
- Salts 12 and 14 were prepared from L-PHE and L-DOPA respectively. Optimization of the radiosynthesis is ongoing and to date ee’s have not yet been determined.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.