

Abstract
Whole-genome sequencing is becoming commonplace, but the accuracy and completeness of variant calling by the most widely used platforms from Illumina and Complete Genomics have not been reported. Here we sequenced the genome of an individual with both technologies to a high average coverage of ~76×, and compared their performance with respect to sequence coverage and calling of single-nucleotide variants (SNVs), insertions and deletions (indels). Although 88.1% of the ~3.7 million unique SNVs were concordant between platforms, there were tens of thousands of platform-specific calls located in genes and other genomic regions. In contrast, 26.5% of indels were concordant between platforms. Target enrichment validated 92.7% of the concordant SNVs, whereas validation by genotyping array revealed a sensitivity of 99.3%. The validation experiments also suggested that >60% of the platform-specific variants were indeed present in the genome. Our results have important implications for understanding the accuracy and completeness of the genome sequencing platforms.
The ability to sequence entire human genomes has the potential to provide enormous insights into human diversity and genetic disease, and is likely to transform medicine1,2. Several platforms for whole-genome sequencing have emerged3–7. Each uses relatively short reads (up to 450 bp) and through high-coverage DNA sequencing, variants are called relative to a reference genome. The platforms of two companies, Illumina and Complete Genomics (CG), have become particularly commonplace, and >90% of the complete human genome sequences reported thus far have been sequenced using these platforms5,8–11. Each of these platforms uses different technologies, and despite their increasingly common use, a detailed comparison of their performance has not been reported previously. Such a comparison is crucial for understanding accuracy and completeness of variant calling by each platform so that robust conclusions can be drawn from their genome sequencing data.
RESULTS
Sequence data generation
To examine the performance of Illumina and CG whole-genome sequencing technologies, we used each platform to sequence two sources of DNA, peripheral blood mononuclear cells (PBMCs) and saliva, from a single individual to high coverage. An Illumina HiSeq 2000 was used to generate 101-bp paired-end reads, and CG generated 35-bp paired-end reads. The average sequence coverage for each sample was ~76× (Table 1), which resulted in a total coverage equivalent to 300 haploid human genomes.
Table 1.
Whole-genome sequencing using CG and Illumina platforms
| CG | Illumina | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Sample | Bases (Gb) | Coverage (×) | Bases (Gb) | Coverage (×) | Reads | Mapped | After duplicate removal | ||
| Blood | 233.2 | 78 | 151.4 | 50 | 1,499,021,500 | 1,367,988,241 | 91% | 1,233,937,084 | 82% |
| Saliva | 218.6 | 73 | 307.1 | 102 | 3,040,306,840 | 2,614,663,882 | 86% | 2,354,594,740 | 77% |
| Total | 451.8 | 151 | 458.5 | 153 | 4,539,328,340 | 3,982,652,123 | 88% | 3,588,531,824 | 79% |
We aligned reads from both platforms to the human reference genome (NCBI build 37/HG19)12 and called SNVs. For Illumina, a total of 4,539,328,340 sequence reads, comprising 1,499,021,500 reads (151.4 Gb) from PBMCs and 3,040,306,840 reads (307.1 Gb) from saliva, were mapped to the reference genome using the Burrows-Wheeler Aligner13. About 88% mapped successfully. Duplicate reads were removed using the Picard software tool, resulting in 3,588,531,824 (79%, 362 Gb) mapped, nonduplicate reads (Table 1). Targeted realignment and base recalibration was performed using the Genome Analysis ToolKit (GATK)14. We used GATK to detect a total of 3,640,123 SNVs (3,570,658 from PBMCs and 3,528,194 from saliva) with a quality filter as defined by the 1000 Genomes Project11. CG generated a gross mapping yield of 233.2 Gb for the PBMC sample and 218.6 Gb for the saliva sample for a total of 451.8 Gb of sequence (Table 1). We analyzed these data using the CG Analysis pipeline to identify 3,394,601 SNVs (3,277,339 from PBMCs and 3,286,645 from saliva). A detailed comparison of PBMCs versus saliva differences has revealed that few of the tissue-specific calls could be validated by independent methods, and these results will be published elsewhere.
To examine the completeness of sequencing, we analyzed the depth and breadth of genomic coverage by each platform with the PBMC genome sequences. Both platforms covered the majority of the genome, and >95% of the genome was covered by 17 or more reads (Fig. 1a). The Illumina curve drops to zero coverage at much lower read depth than the CG curve because there are substantially fewer reads in the Illumina data set. We also noticed that CG generally is less uniform in coverage (Fig. 1b). This suggests that to achieve a certain level of coverage for most of the genome, CG requires more overall sequencing than Illumina.
Figure 1. Genome coverage at different read depths.
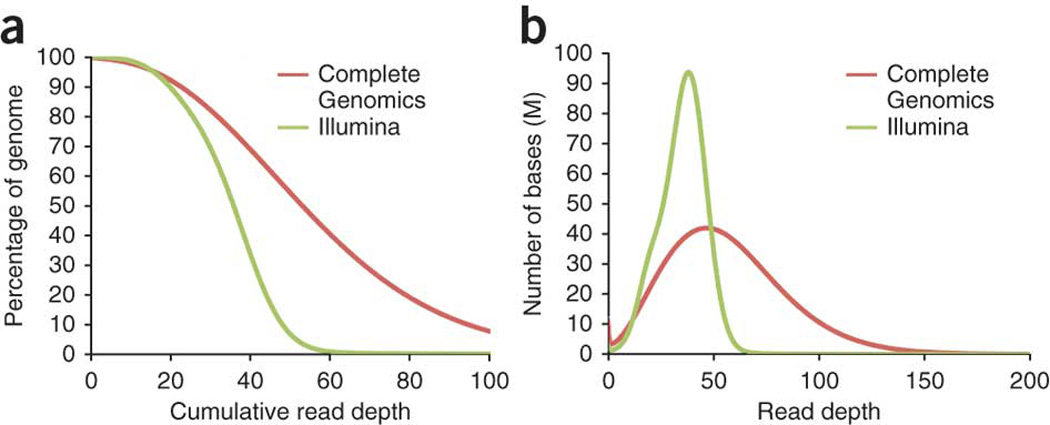
(a) Percentage of genome covered by different read depths in different platforms. (b) Histogram of genome coverage at different read depths.
Extensive differences in variant calling
We sought to compare the sensitivity and accuracy of each platform for SNV calling. In total, 88.1% (3,295,023 out of 3,739,701) of the unique SNVs were concordant—that is, either a homozygous or heterozygous SNV was detected at the same locus by the two platforms in at least one sample (Fig. 2a). We detected 444,678 SNVs by only one platform or the other but not both, of which 345,100 were specific to Illumina (10.5% of the Illumina combined SNVs) and 99,578 were CG-specific (3.0% of the CG combined SNVs). Among the Illumina-specific SNVs, 67% were ‘no-calls’ (that is, not a reference or variant call), 11% were reference calls and 22% were other types of calls (that is, complex and substitution calls) in CG. Similarly, 75% of the CG-specific SNVs were no-calls in Illumina, and 25% were reference calls (Fig. 2b). The higher percentage of no-calls in Illumina is likely because GATK does not make the complex and substitution calls as does the CG pipeline.
Figure 2. SNV detection and intersection.
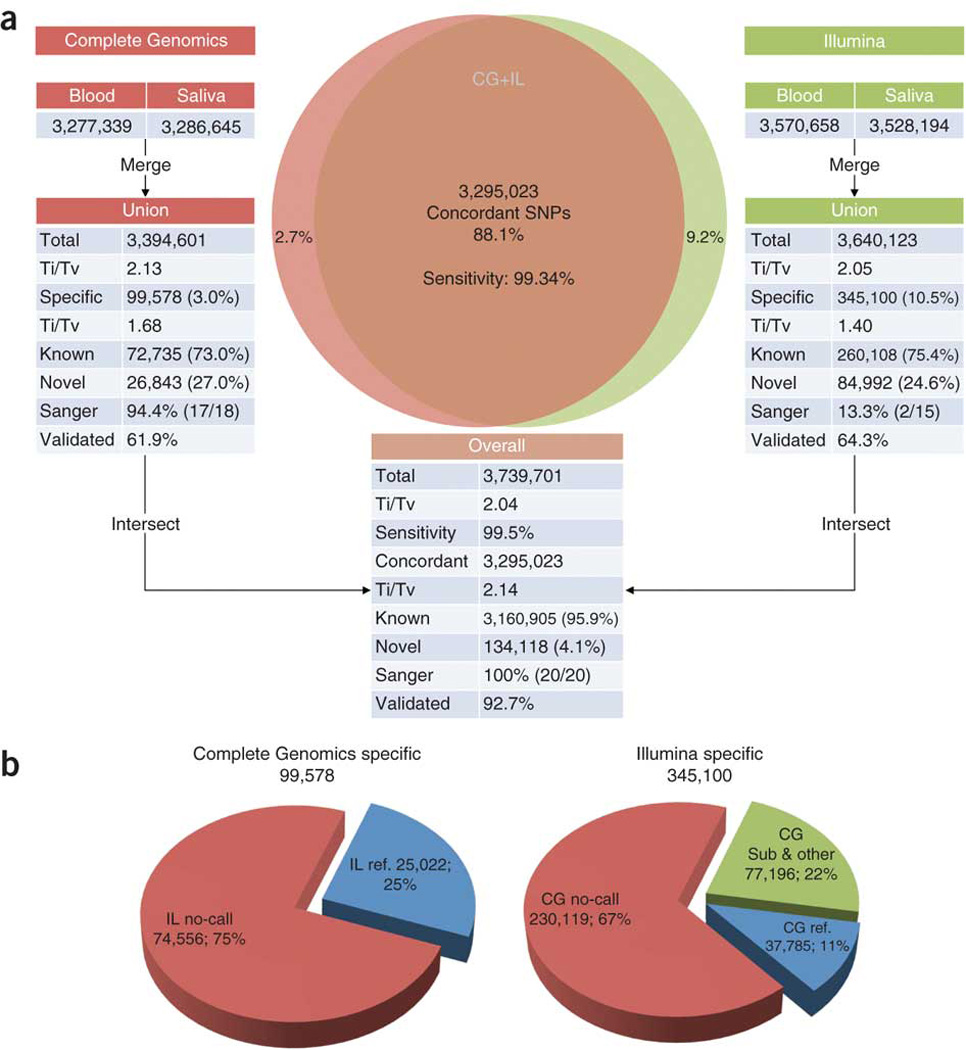
(a) SNVs detected from the PBMC and saliva samples in each platform were combined. The unions of SNVs in each platform were then intersected. Sensitivity was measured against the Illumina Omni array. Ti/Tv is the transition-to-transversion ratio. The known and novel counts were based on dbSNP. ‘Sanger’ and ‘validated’ represent validation by Sanger sequencing and Illumina sequencing (with Agilent target enrichment capture), respectively. (b) Comparing platform-specific SNVs to non-SNV calls in another platform. IL, Illumina; CG, Complete Genomics.
To assess the quality of the calls, we used four criteria: the transition/transversion ratio (ti/tv), quality scores, the heterozygous/homozygous call ratio and novel, platform-specific SNVs. The ti/tv ratio of 2.1 for SNVs in humans has been described in several previous studies, including the 1000 Genomes Project11. The ti/tv ratio for all of the SNVs detected in these genomes was 2.04, but in our data the ti/tv of SNVs concordant between the two platforms was 2.14. For all SNVs detected by the Illumina platform, ti/tv was 2.05, but for SNVs specific to Illumina it was only 1.40. Similarly, for SNVs detected by CG, ti/tv was 2.13, but for CG-specific SNVs, it was 1.68. Thus, the ti/tv of concordant SNVs was very close to that expected, whereas the platform-specific ti/tv was much lower, suggesting that the platform-specific calls were of lower accuracy. Inspection of the quality scores of the platform-specific SNVs showed that they were indeed lower than those for the concordant calls (Supplementary Fig. 1). Furthermore, the heterozygous/homozygous call ratio was 1.48 for the concordant calls, whereas the platform-specific ratios were indeed higher: 2.48 for Illumina-specific calls and 1.98 for CG-specific calls.
To examine the fraction of novel platform-specific SNVs, we noted that 3,160,905 (96.0%) of the concordant SNVs were present in dbSNP131 (ref. 15). In contrast, only 260,108 (75.4%) of the SNVs in the Illumina-specific set, and 72,735 (73.0%) of the SNVs in the CG-specific set were present in dbSNP131. Thus, the platform-specific call sets were enriched for novel SNVs, suggesting that they likely contain more errors. In addition, the overall genotype concordance rate (that is, the proportion of concordant calls having a consistent genotype—heterozygous or homozygous—across both platforms) for the concordant SNVs was 98.9%. The high genotype concordance rate and percentage of known SNVs indicate that the concordant SNVs were of high quality and accuracy.
To further assess the accuracy of the variant calling, we sought to validate our SNVs by using Omni Quad 1M Genotyping arrays, traditional Sanger sequencing and Agilent SureSelect target enrichment capture followed by sequencing on an Illumina HiSeq for both samples. Of the 260,112 heterozygous calls detected with the Omni array, 99.5% were present in the entire SNV data set, 99.34% were concordant calls and only 0.16% were platform-specific SNVs. This demonstrates that both platforms are sensitive to known SNVs and that few known single-nucleotide polymorphisms (SNPs) are detected by only one platform.
To directly determine accuracy, we sequenced randomly selected concordant and platform-specific regions for Sanger sequencing. We found that 20 of 20 concordant SNVs could be validated, whereas 2 of 15 (13.3%) Illumina-specific and 17 of 18 (94.4%) CG-specific SNVs could be validated. This suggests CG has higher accuracy than Illumina and that almost all the concordant calls are correct.
To attempt to examine accuracy on a larger scale, we used Agilent SureSelect target enrichment capture technology to capture 33,084 (9.6%) Illumina-specific, 3,015 (3.0%) CG-specific and 24,247 (0.7%) concordant SNVs for sequencing on an Illumina Hi-Seq instrument (Table 2). We found that the validation rate for the concordant SNVs was 92.7%, whereas the validation rate was 61.9% and 64.3% for the CG-specific and Illumina-specific SNVs. These results indicate that the platform-specific calls have a very high false-positive rate of at least 35%. We also found that 12.6–21.4% of the targeted SNVs were not called in the validation, possibly owing to nonunique regions that are difficult to map precisely. Because the capture validation was performed using Illumina DNA sequencing technology, it is difficult to directly compare the Illumina versus CG SNV rates with this approach. Nonetheless, these overall results indicate that concordant SNVs have high accuracy and platform-specific SNVs have a high false-positive rate.
Table 2.
Agilent SureSelect target enrichment capture with Illumina sequencing
| CG specific | Illumina specific | Concordant | ||||
|---|---|---|---|---|---|---|
| Total | 99,578 | — | 345,100 | — | 3,295,023 | — |
| Targeted | 3,015 | 3.0% | 33,084 | 9.6% | 24,247 | 0.7% |
| Not validated | 388 | 12.9% | 7,088 | 21.4% | 3,053 | 12.6% |
| Invalidated | 1,001 | 33.2% | 9,280 | 28.0% | 1,543 | 6.4% |
| Validated | 1,626 | 53.9% | 16,716 | 50.5% | 19,651 | 81.0% |
| Validation rate | — | 61.9% | — | 64.3% | — | 92.7% |
Association of genes with variant calling differences
To better understand the platform-specific calls, we investigated the association of SNVs from each platform with different genomic elements. We annotated both the platform-specific SNVs and concordant SNVs with gene and repeat annotations using Annovar16. In general, we did not find a significant difference between the associations of the platform-specific SNVs and the concordant SNVs with gene elements, such as exons and introns (Fig. 3a,b). For example, 1% and 32–38% of the platform-specific SNVs were associated with exonic and intronic regions, respectively, regardless of the platform. This correlates well with the portions of exons (~1.3%) and introns (~37%) in the whole human genome. Nonetheless, the CG-specific SNVs had a slightly stronger association (14%) with noncoding RNA than the Illumina-specific SNVs (12%) and concordant SNVs (11%). Overall, many platform-specific SNVs lie in RNA coding regions of the human genome, and thus deducing their accuracy is of high importance.
Figure 3. SNV association with different genomic elements.
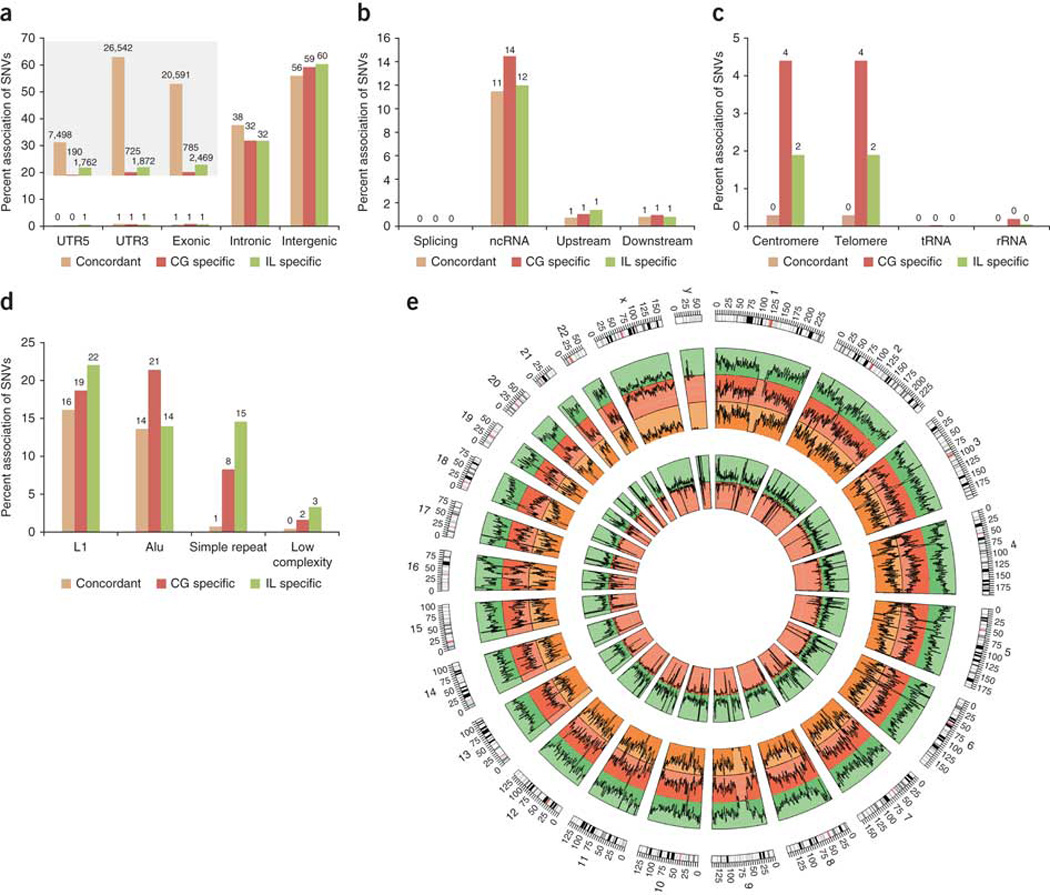
(a) Gene elements: UTR, exonic, intronic and intergenic regions. Inset: number of SNVs associated with UTR5, UTR3 and exonic regions. (b) Gene elements: splicing sites, noncoding RNA and upstream/downstream (<1 kb) regions of genes. (c) Repetitive elements: centromere, telomere, tRNA and rRNA. (d) Repetitive elements: L1, Alu, simple repeat and low-complexity repeat. (e) SNV frequency at different chromosomal locations. Tracks from outer to inner: SNV frequency for Illumina (IL), Complete Genomics (CG), concordant, IL-specific and CG-specific calls. Outermost: chromosome ideogram.
To further ascertain whether the platform-specific SNVs might be located in functionally important regions, we examined whether the variant calls were present in the Varimed database2,17, which contains variants catalogued through genome-wide association studies and other genetic linkage studies. We found that 31 Illumina- and 3 CG-specific SNVs were present in Varimed, from which we were able to estimate associations between diseases and platform-specific SNPs (Supplementary Table 1). One of these, rs2672598, was called in both PBMCs and saliva by the Illumina platform, but not called in either PBMCs or saliva by the CG platform. This SNP is at the 5′ end of HTRA1 and known to increase the risk of age-related macular degeneration by 4.89-fold (P = 3.39 × 10−11)18,19. Another example is the A202T allele in the TERT gene encoding telomerase. This allele has been associated with aplastic anemia20 and was only detected by the Illumina platform. Thus, some platform-specific calls are of high importance.
Association of repetitive regions with variant calling differences
In contrast to coding SNVs, we found that overall the platform-specific SNVs had a substantially stronger association with repetitive elements such as Alu, telomere and simple repeat sequences (Fig. 3c,d). For example, only 0.3% of the concordant SNVs were associated with telomere or centromere sequences, but 4% and 2% of the CG-specific SNVs and Illumina-specific SNVs, respectively, were associated with telomeric or centromeric repeats (Fig. 3c,e). The enrichment of platform-specific SNVs with simple repeats and low-complexity repeats was particularly evident. We found that <1% of the concordant SNVs were associated with simple repeats, but 8% and 15% of the CG-specific SNVs and Illumina-specific SNVs, respectively, were associated with these sequences. Among the platform-specific SNVs, CG had a stronger association with the Alu element and centromere and telomere sequences, whereas Illumina had a stronger association with L1, simple repeat and low-complexity repeat. Overall, these results indicate that many platform-specific SNVs lie in repetitive regions, suggesting that these calls may be due to mapping difficulties and errors.
We also measured GC content and read depth of the SNVs in the gene and repeat regions. The average GC content of the concordant, CG-specific and Illumina-specific SNVs were 0.46, 0.45 and 0.41, respectively. The average read depths were 48, 47 and 44, respectively. Thus, the Illumina-specific SNVs showed a lower GC content and read depth compared to the concordant SNVs. Analysis by gene and repeat regions did not reveal any strong correlation with GC content. However, we found that Illumina-specific SNVs had a strikingly higher read depth in centromeric and telomeric regions, whereas CG had higher read depth in the tRNA and rRNA regions (Supplementary Fig. 2).
Differences in indel calls
We also examined small indel calls from Illumina and CG platforms. Small indels ranged in size from −107 to +36 bp by Illumina and −190 to +48 bp by CG. Illumina calls were made using GATK with the Dindel model21, and CG calls were obtained from their standard pipeline and converted to VCF format22 using the CG conversion tool. A stringent quality score cutoff of 30 was used for each platform. This resulted in a total of 811,903 indel calls with 611,110 for Illumina and 430,258 for CG. We found that only 215,382 (26.5%) indels were detected by both Illumina and CG, whereas 390,060 (48.1%) and 206,461 (25.4%) were Illumina- and CG-specific, respectively (Fig. 4a). Owing to the complexity of indels compared to SNVs, the number of concordant indels was much lower than the number of concordant SNVs. We also observed that the indels detected by both platforms were similar in their size distribution and type (Fig. 4b), though it is noteworthy that the Illumina data showed a slight enrichment of 1-bp insertions, whereas the CG data showed a slight enrichment of 1-bp deletions.
Figure 4. Indel detection and intersection.
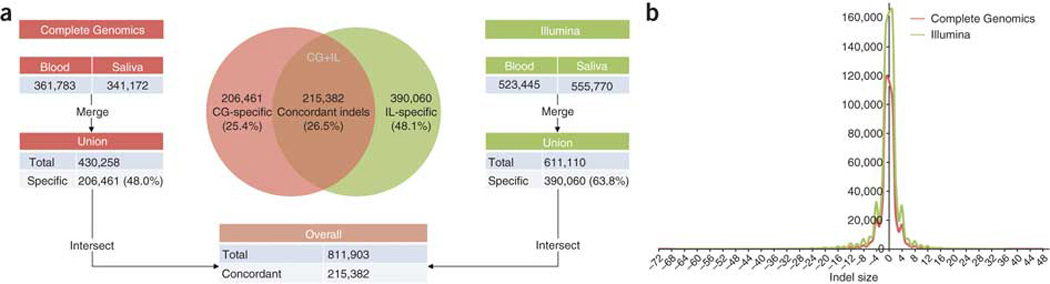
(a) Indels detected from the PBMC and saliva samples in each platform were combined. The unions of indels in each platform were then intersected. Note: 5,668 IL and 8,415 CG indels were removed after 5b-window merging. (b) Indel size distribution. Negative size represents deletion and positive size represents insertion.
Detection accuracy was assessed for concordant and platform-specific indels by comparing them to indels detected by exome sequencing of the same individual23. We validated 2.2% (4,681) of concordant indels but only 1.2% (4,682) of Illumina-specific and 0.3% (561) of CG-specific indels. These lower validation rates for platform-specific indels suggest that they are indeed less robust than those detected by both platforms. Because exome sequencing was performed using the Illumina HiSeq platform, bias toward greater consistency between the Illumina-specific and exome sequencing–specific indels was not unexpected.
We further validated indels by randomly selecting indels for traditional Sanger sequencing. For 24 concordant indels, 15 could be amplified by PCR allowing us to validate 14 of them (93.33%). For 42 platform-specific indels, 19 could be amplified allowing us to validate 10 of 11 Illumina-specific indels and 8 of 8 CG-specific indels. Although the platform-specific indels could be validated at a high rate, the increased frequency of failed PCR amplification for platform-specific versus concordant indels (54.8% versus 37.5%, respectively) suggests that there may have been issues with the sequence context around a larger fraction of the platform-specific calls. We therefore examined whether both the concordant and platform-specific indels overlapped with known repeats. We found that 72% of Illumina-specific and 63% of CG-specific indels overlapped repeats, whereas only 52% of concordant indels overlapped with repeats. Although there is a clear enrichment of platform-specific indels over problematic repeat regions, many bona fide indels were detected by only one platform, as demonstrated by their high validation rate. This suggests that indel detection by both Illumina and CG lacks sensitivity.
DISCUSSION
Overall, we conclude that each genome sequencing approach is generally capable of detecting most SNVs. Based on the transition/transversion ratio and Sanger sequencing, CG appears to be more accurate, but also slightly less sensitive. Illumina, in contrast, covers more bases and makes a higher number of overall calls, but also has more false positives. This may be in part because Illumina has longer reads and is therefore able to map more reads in difficult regions, which leads to both increased sensitivity and decreased specificity. Nonetheless, both methods clearly call variants missed by the other technology. Many of these lie in exons and thus can affect coding potential. In fact, 1,676 genes have platform-specific SNVs in exons; one of the Illumina-specific SNVs lies in a telomerase gene and is likely to affect function. We also found that indel detection is subject to a much larger platform bias, with each platform detecting a large quantity of indels missed by the other platform. It may therefore be beneficial to sequence on both platforms and analyze both data sets together, using evidence from one to bolster discovery in the other.
We demonstrated that the best approach for comprehensive variant detection is to sequence genomes with both platforms if budget permits. We assessed the cost effectiveness of sequencing on both platforms and found that on average it costs about four cents per additional variant (Online Methods). Alternatively, supplementing with exome sequencing can assess the most interpretable part of the genome at higher depth of coverage and accuracy and fill in the gaps in the detection of coding variants23. If genome sequencing is performed on both platforms, platform-specific variants can be validated by Sanger sequencing and array capture experiments or disregarded if they map to difficult regions (that is, simple repeats) or have low quality scores. Using this strategy, variant detection sensitivity and specificity can be maximized, and meaningful variants that may otherwise have been missed can be discovered.
ONLINE METHODS
Sample collection
The subject was enrolled and proper consent was obtained under the institutional reviews board (IRB) protocol IRB-8629 at Stanford University. A whole blood sample was collected and PBMCs were isolated by density gradient centrifugation at 400g for 25 min using the lymphocyte separation media (MP Biomedicals). Twenty milliliters of saliva was also collected from this individual and processed immediately. DNA was isolated from both PBMCs and saliva with the AllPrep DNA/RNA/Protein Mini Kit (QIAGEN).
Whole-genome sequencing
Whole-genome sequencing was done at Illumina and CG. Ten micrograms of genomic DNA isolated from PBMCs and saliva were used for both platforms. 101 bp paired-end reads were generated using the Illumina HiSeq 2000 sequencer, and 35 bp paired-end reads were generated by CG. Illumina sequence reads were mapped to the human reference genome (HG19) with the Burrow Wheel Aligner. Duplicates were removed using the Picard tool (http://www.picard.sf.net).
Sanger DNA sequencing
Sanger DNA PCR and sequencing primers were designed with the Optimus Primer software (http://op.pgx.ca/), and were synthesized at Integrated DNA Technologies. DNA sequencing was performed at ELIM BIOPHARM.
Agilent SureSelect target enrichment and sequencing
A custom Agilent SureSelect target enrichment kit was designed using Agilent eArray. Blood and saliva paired-end Illumina libraries were captured in solution according to the Agilent SureSelect protocol. 101-bp paired-end reads were generated using the Illumina HiSeq 2000 sequencer.
Cumulative coverage assessment
For CG data, coverage statistics were derived from the Coverage and Reference Scores files for each chromosome. Coverage at every base was assessed directly from these files. For Illumina data, coverage was estimated using the BEDTools CoverageBed utility against the human reference genome HG19 with BAM files from which duplicates had been removed.
SNV detection and annotation
For CG, SNVs were derived from the masterVar file. They were extracted and converted to VCF format using the CG masterVar-to-VCF conversion tool available at the CG community website (http://community.completegenomics.com/tools/m/cgtools/219.aspx). For Illumina, SNVs were detected using GATK. The detected SNVs were filtered based on the following filter:
AB > 0.75 ‖QUAL < 50.0 ‖DP > 360 ‖ SB ≥ 0.1‖MQ0 ≥ 4
where
AB = allele balance
QUAL = Phred-scale quality score
DP = depth of coverage
SB = strand bias
MQ0 = number of reads with mapping quality equal to zero
SNVs were combined and compared using custom program scripts. ANNOVAR (http://www.openbioinformatics.org/annovar/) was used to annotate the SNVs with gene and repeat annotations downloaded from the UCSC browser (http://www.genome.ucsc.edu/).
Small indel detection
For CG, small insertions and deletions were derived from the masterVar file. Indels were extracted and converted to VCF format using the CG masterVar-to-VCF conversion tool available at the CG community website. For Illumina, small indels were detected using GATK with the Dindel model for indel detection. Indels from both platforms were filtered based on quality score such that only those with QUAL ≥ 30 remained. Indels were compared using VCFtools (http://www.vcftools.sf.net).
Disease association with SNV
Varimed, a manually curated database (comprising data from 5,478 human genetics papers) of human disease-SNP associations, was used to perform disease association with our SNVs. We queried the subject’s genotypes from the platform-specific SNVs against Varimed, and identified SNVs that were known to increase the subject’s risk of diseases with P < 1 × 10−6. The evidences of their disease associations were evaluated using the number of studies, cohort size, P-value and the odds ratio. For risk genotypes validated in multiple studies, we reported the most significant P-values, the average odds ratio and the total number of distinct subjects in all studies. Subjects reported in different publications were considered as different. The top SNVs associated with diseases were selected with priorities given to SNVs called in both PBMCs and saliva, homozygous risk genotypes and stronger evidence, that is, SNPs validated in multiple studies, with larger cohorts, and more significant P-values.
Cost effectiveness analysis (CEA)
The CEA per additional variant was calculated using the following formula:
Cs/(Nv × Pv)
where
Cs = cost of sequencing on an extra platform
Nv = total additional SNVs detected
Pv = probability of the SNVs being true positives
The cost for sequencing was estimated at $4,000 for each platform. The total additional SNVs were the total platform-specific SNVs. The probability of true-positive SNVs was the Agilent validation rate for the platform-specific calls. The CEA for Illumina was 0.02 and for CG was 0.06.
Supplementary Material
ACKNOWLEDGMENTS
This work is supported by the Stanford Department of Genetics and the US National Institutes of Health.
Footnotes
Accession code. Sequence Read Archive: SRA045736.
Note: Supplementary information is available on the Nature Biotechnology website.
AUTHOR CONTRIBUTIONS
H.Y.K.L. and M.J.C. did the analysis. G.N. and L.H. assisted in the analysis. Rui C. did DNA sequencing. Rong C. did the disease-association study. Rui C. and M.O’H. did the validation experiments. H.Y.K.L., F.E.D., E.A.A., M.B.G., A.J.B., H.P.J. and M.S. coordinated the analysis and revised the manuscript. H.Y.K.L., M.J.C. and M.S. wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/nbt/index.html.
References
- 1.Ajay SS, Parker SC, Ozel Abaan H, Fuentes Fajardo KV, Margulies EH. Accurate and comprehensive sequencing of personal genomes. Genome Research. 2011;21:1498–1505. doi: 10.1101/gr.123638.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashley EA, et al. Clinical assessment incorporating a personal genome. Lancet. 2010;375:1525–1535. doi: 10.1016/S0140-6736(10)60452-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wheeler DA, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 4.McKernan KJ, et al. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res. 2009;19:1527–1541. doi: 10.1101/gr.091868.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roach JC, et al. Analysis of genetic inheritance in a family quartet by wholegenome sequencing. Science. 2010;328:636–639. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pushkarev D, Neff N, Quake S. Single-molecule sequencing of an individual human genome. Nat. Biotechnol. 2009;27:847–852. doi: 10.1038/nbt.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korbel JO, et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Snyder M, Du J, Gerstein M. Personal genome sequencing: current approaches and challenges. Genes Dev. 2010;24:423–431. doi: 10.1101/gad.1864110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rios J, Stein E, Shendure J, Hobbs HH, Cohen JC. Identification by whole-genome resequencing of gene defect responsible for severe hypercholesterolemia. Hum. Mol. Genet. 2010;19:4313–4318. doi: 10.1093/hmg/ddq352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee W, et al. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature. 2010;465:473–477. doi: 10.1038/nature09004. [DOI] [PubMed] [Google Scholar]
- 11.The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 13.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sherry ST, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen R, Davydov EV, Sirota M, Butte AJ. Non-synonymous and synonymous coding SNPs show similar likelihood and effect size of human disease association. PLoS ONE. 2010;5:e13574. doi: 10.1371/journal.pone.0013574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaur I, et al. Variants in the 10q26 gene cluster (LOC387715 and HTRA1) exhibit enhanced risk of age-related macular degeneration along with CFH in Indian patients. Invest. Ophthalmol. Vis. Sci. 2008;49:1771–1776. doi: 10.1167/iovs.07-0560. [DOI] [PubMed] [Google Scholar]
- 19.Tam PO, et al. HTRA1 variants in exudative age-related macular degeneration and interactions with smoking and CFH. Invest. Ophthalmol. Vis. Sci. 2008;49:2357–2365. doi: 10.1167/iovs.07-1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamaguchi H, et al. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N. Engl. J. Med. 2005;352:1413–1424. doi: 10.1056/NEJMoa042980. [DOI] [PubMed] [Google Scholar]
- 21.Albers CA, et al. Dindel: Accurate indel calls from short-read data. Genome Res. 2011;21:961–973. doi: 10.1101/gr.112326.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Danecek P, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clark MJ, et al. Performance comparison of exome DNA sequencing technologies. Nat. Biotechnol. 2011;29:908–914. doi: 10.1038/nbt.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.