

Abstract
The declining effectiveness of current antibiotics due to the emergence of resistant bacterial strains dictates a pressing need for novel classes of antimicrobial therapies, preferably against molecular sites other than those in which resistance mutations have developed. Dihydropteroate synthase (DHPS) catalyzes a crucial step in the bacterial pathway of folic acid synthesis, a pathway that is absent in higher vertebrates. As the target of the sulfonamide class of drugs that were highly effective until resistance mutations arose, DHPS is known to be a valuable bacterial Achilles heel that is being further exploited for antibiotic development. Here, we report the discovery of the first known allosteric inhibitor of DHPS. NMR and crystallographic studies reveal that it engages a previously unknown binding site at the dimer interface. Kinetic data show that this inhibitor does not prevent substrate binding but rather exerts its effect at a later step in the catalytic cycle. Molecular dynamics simulations and quasi-harmonic analyses suggest that the effect of inhibitor binding is transmitted from the dimer interface to the active-site loops that are known to assume an obligatory ordered substructure during catalysis. Together with the kinetics results, these structural and dynamics data suggest an inhibitory mechanism in which binding at the dimer interface impacts loop movements that are required for product release. Our results potentially provide a novel target site for the development of new antibiotics.
The enzyme dihydropteroate synthase (DHPS) is encoded by the folP gene and catalyzes the formation of 7,8-dihydropteroate from p-aminobenzoic acid (pABA) and 6-hydroxymethyl-7,8-dihydropterin-pyrophosphate (DHPP). This reaction is a key step in the folate biosynthetic pathway of bacteria and primitive eukaryotes, and the absence of the folate pathway in higher eukaryotes makes it an attractive focus for antimicrobial drug discovery.1 DHPS is the target for the highly effective sulfa drugs that have been used for over 70 years.2 However, the emergence of sulfa drug resistance has seriously compromised their utility3,4 and has spawned a number of efforts to develop new classes of DHPS inhibitors.5
For a number of years, we have been pursuing novel inhibitors of DHPS that target the conserved and structured pterin-binding pocket with the goal of producing broad-spectrum drugs that are less prone to generating resistance than the flexible pABA binding site to which sulfonamide drugs bind. Although we have identified a number of inhibitors, one challenge has been that the pterin binding pocket is highly selective for the pterin substrate and pterin analogues, and our traditional screening and medicinal chemistry approaches have tended to yield pterin-like molecules with limited solubility.6 In addition, we recently demonstrated that DHPS performs catalysis via a complicated SN1 mechanism that involves the transient folding of two flexible loops.7 The intermediate-state structure that we characterized revealed that the pterin pocket is designed to generate and then stabilize a cationic pterin intermediate species and also revealed a transient pyrophosphate-binding pocket.
In light of these findings, we recently initiated a fragment-based approach to identify new inhibitory scaffolds of DHPS. Fragment based lead discovery (FBLD) is particularly useful for identifying novel chemotypes for difficult targets such as DHPS where traditional high-throughput methods have been challenging.8,9 In the current study, we screened a fragment library against Bacillus anthracis DHPS (BaDHPS) with the goal of identifying novel scaffolds that bind within the pterin pocket. However, a significant subset of the binding molecules was found to bind elsewhere on the protein, and we identified this novel ligand binding site at the dimer interface using X-ray crystallography. A derivative of one of the interfacial binding molecules displayed significant inhibitory activity, and kinetic studies, NMR analyses, and molecular dynamics (MD) calculations suggest that ‘breathing motions’ within the DHPS dimer and communication between the dimer interface and the active-site loops create this allosteric inhibition.
Results and Discussion
Initial Identification of an Allosteric Inhibitor
Using the Maybridge Ro3 library of 1,100 compounds, we performed a fragment screen of BaDHPS using the WaterLOGSY (water-ligand observed via gradient spectroscopy) NMR protocol.10 A total of 74 fragments were revealed as potential binders. Using a previously characterized high affinity pterin pocket-binding inhibitor (2-(7-amino-1-methyl-4,5-dioxo-1,4,5,6-tetrahydropyrimido[4,5-c]pyridazin-3-yl)propanoic acid),11 as well as the substrate pABA, we then used competitive-WaterLOGSY (c-WaterLOGSY)12 to determine which of these binders access active site pockets. c-WaterLOGSY revealed that 10 fragments clearly bind to regions of DHPS other than the active site (compounds 1–10, Figure 1). The fragments that do appear to access the active site are currently being studied and will be reported elsewhere. Compounds 1–10 all share the common structural feature of a trifluoro group attached to a phenyl group, which suggests that they access the same site on DHPS.
Figure 1.

2D structures of the fragment hits that were identified to bind at the dimer interface of DHPS.
Activity and Kinetic Assays
Using previously described inhibition assays,7,11 these 10 compounds were tested for inhibitory activity against BaDHPS and the DHPS enzymes from Yersinia pestis (YpDHPS) and Staphylococcus aureus (SaDHPS). Compound 1 was found to effectively inhibit all three DHPS enzymes at 250 μM (BaDHPS, 86%; YpDHPS, 61%; SaDHPS, 46%), whereas binders 2–10 displayed only modest inhibitory activities. Compound 1 in the Maybridge fragment library was reported to be 4-(trifluoromethyl) benzylamine, but when it was chemically and structurally characterized, the sample was found to contain a mixture of 1 and the ‘dimeric’ Schiff base N-(4-(trifluoromethyl)benzylidene)-1-(4-(trifluoromethyl)benzylamine (compound 11, Figure 1), with the latter in excess. Consequently, compound 1 was repurchased (Sigma Aldrich) and verified as 97% pure, and this sample showed no inhibition of any of the three DHPS enzymes at 250 μM. To verify that this was not simply due to a lack of binding, we used surface plasmon resonance (SPR) and WaterLOGSY titration to measure the Kd value of the pure compound. Both methods verified that 1 binds with very similar Kd values: 187 ± 2 μM using SPR and 168 ± 35 μM using WaterLOGSY (Figure 2A–C). In contrast, when 11 was synthesized in-house, it showed 100% inhibition of all three enzymes at 250 μM, and the IC50 values for BaDHPS, YpDHPS, and SaDHPS were measured as 50, 31, and 17 μM, respectively (Figure 3). Measuring the Kd value of 11 using SPR proved to be technically challenging due to solubility issues, but it was possible to measure a value of 130 ± 1 μM using isothermal titration calorimetry (ITC) (Figure 2D). This Kd value was measured in the absence of substrates, but ITC experiments in the presence of substrates revealed that 11 enables pABA to bind DHPS with a 2-fold increase in binding affinity (Figure 2E and F).
Figure 2.

Binding affinities of compounds 1 and 11 to BaDHPS. (A) SPR sensorgram and (B) binding isotherm for the BaDHPS:1 complex. The fragment was injected as a 3-fold dilution series starting at 200 μM, and the data were fit to a 1:1 interaction model. (C) WaterLOGSY titration of compound 1: solid triangle, compound 1; solid diamonds, compound 1 in the presence of BaDHPS; solid circle, difference. (D, E, F) ITC binding experiments of (D) compound 11 titrated into a solution of BaDHPS, (E) pABA titrated into BaDHPS in the presence of PtPP, and (F) pABA titrated into BaDHPS in the presence of PtPP and compound 11.
Figure 3.
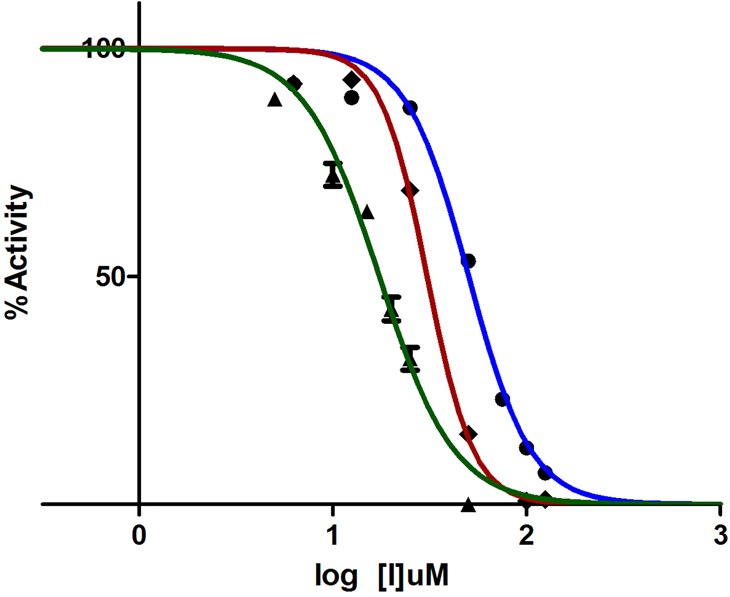
IC50 determination of compound 11 versus BaDHPS (blue), YpDHPS (red), and SaDHPS (green). The enzyme activities were measured in the presence of 0–125 μM compound 11.
To further understand the way in which 11 exerts its inhibitory effect, two Michaelis–Menten kinetic experiments were conducted on all three enzymes in which the rates of DHPS catalysis were measured at increasing concentrations of 11. In the first experiment, the concentration of DHPP was varied and the pABA concentration was held constant, and this was reversed in the second experiment. The results were difficult to interpret for the first experiment, and this was not surprising because we have demonstrated that the DHPS catalytic mechanism involves an ordered SN1 process in which DHPP is the compulsory lead substrate without which the pABA-binding pocket fails to form.7 However, the second experiment revealed that both the Km and Vmax values decrease with increasing concentrations of 11 (Table 1 and Supplementary Figure S1). In the presence of 11 and with DHPP present in the pterin-binding pocket, pABA binds DHPS with increased affinity (Km decreases with increasing concentrations of 11). The actual Km and Vmax values vary somewhat between the three orthologs, but the trend is clear and consistent. One interpretation of this result, consistent with the enzyme mechanism, is that the binding of 11 to an allosteric site of DHPS promotes the ordering of the two active site loops. This would enhance the binding of pABA to the active site, thereby decreasing Km, but would also slow the release of product, thereby decreasing Vmax.
Table 1. Kinetic Analyses of DHPS in the Presence of Compound 11a.
| concn
of compd 11 (μM) |
||||||
|---|---|---|---|---|---|---|
| 0 |
50 |
100 |
||||
| Vmaxobs (nM/min) | Kmobs (μM) | Vmaxobs (nM/min) | Kmobs (μM) | Vmaxobs (nM/min) | Kmobs (μM) | |
| BaDHPS | 125.8 | 1.2 | 106.7 | 1.0 | 70.81 | 0.74 |
| YpDHPS | 177.3 | 2.1 | 131.3 | 1.8 | 40.95 | 1.2 |
| SaDHPS | 583.6 | 7.7 | 211.2 | 2.7 | 31.19 | 0.38 |
In these experiments, the concentration of the pterin substrate (DHPP) was held constant (20 μM), and the concentration of pABA was increased from 0 to 10 μM.
Crystallographic Analyses
To determine whether compounds 1–11 engage the same site on DHPS as suggested by their similar structures, selected compounds were soaked into BaDHPS crystals and the complex structures were determined. Previous studies have shown that BaDHPS crystals are amenable to the soaking protocol for crystallographic analysis,6,7,13 and we were able to obtain high quality crystal structures of BaDHPS in complex with 4, 5, 6, and 11 (Figure 4). Data collection and refinement statistics are shown in Supplementary Tables S1 and S2. In the BaDHPS P6222 crystal lattice, there are two monomers in the asymmetric unit that create two dimers around 2-fold symmetry axes, and clear electron density was visible for all four compounds at these dimer interfaces. The compound density sits exactly on the crystallographic 2-fold symmetry axis and formally corresponds to a pair of 2-fold related molecules, each with half occupancy. Electron density on crystallographic symmetry axes can be misleading, but we are confident that this density corresponds to the bound molecule for three reasons. First, the shapes of the densities closely match those of the bound compounds with the common trifluorophenyl group of each compound buried at the center of the interface in the same location making hydrophobic contacts with Leu235/235′, Met264/264′, and the methylene groups of Glu236/236′ and Glu260/260′ (Figure 4 and Supplementary Figure S2). Second, identical crystals soaked in small molecules that bind within the pterin pocket have previously never displayed this electron density. A specific example of a published crystal structure6 was carefully re-evaluated to confirm this (Supplementary Figure S2). Finally, the electron density is present on both of the 2-fold axes in the unit cell that create the two independent DHPS dimers, and their shapes are very similar.
Figure 4.
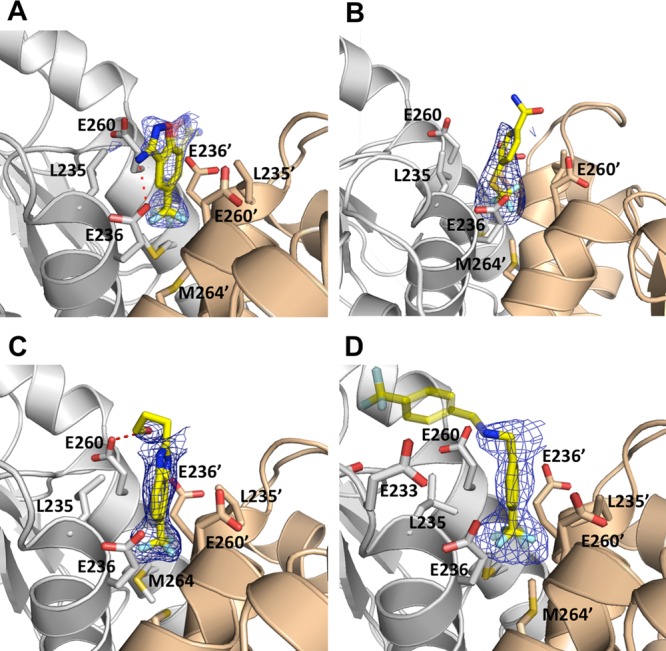
Crystal structures of BaDHPS in complex with fragment molecules bound at the dimer interface. (A) Compound 4. (B) Compound 5. (C) Compound 6. (D) Compound 11; the distal half of the compound is shown in transparency to reflect that its position is derived from weak electron density and docking studies (see text). The two DHPS monomers are shown in gray and wheat, and key residues that interact with the fragments are shown as sticks. The Fo – Fc simulated annealing omit electron density maps are contoured at 2σ.
Although the electron density for 11 clearly shows that the proximal half of the molecule binds at the dimer interface, the very weak electron density for the distal half precluded a reliable interpretation in terms of a molecular model. Liquid chromatography–mass spectrometric analysis showed that 11 is stable in conditions that recapitulate the crystallization conditions (pH and time), which excludes the possibility that the weak electron density for the distal half reflects compound cleavage. To generate a potential docked conformation of the entire molecule, we used the docking program ‘Glide’ from the Schrödinger software suite.14 In all 10 top-scoring poses, the proximal half of 11 is buried within the dimer interface as observed in the electron density, while the distal half is bent toward loop7 on one of the two monomers in a manner consistent with the weak electron density. It is noteworthy that in the MD simulations described below, the distal part is mobile as suggested by the weak electron density and moves between bent conformations that engage one or the other monomer.
The bent conformation is very attractive because loop7 interacts with loop1 in the intermediate-state structure,7 and this suggests that 11 functions as an inhibitor by physically linking the interface with the active site loops. This model is consistent with three observations. First, the kinetic data described above indicate that 11 stabilizes the pABA-binding pocket that is created by the ordering of loop1 and loop2. Second, compounds 1–10 would be expected to be poor inhibitors of DHPS as observed because they lack the distal half and merely occupy the dimer interface without engaging loop7. Third, loop1 and loop2 are trapped in nonfunctional conformations in the BaDHPS crystal lattice and are unable to optimally interact with and stabilize loop7. This, in turn, would compromise the binding of the distal half of 11 to loop7 and explain its weak electron density. To explore this model further, we used NMR and computational approaches.
NMR Analyses
To study the effects of 11 on loop conformation and dynamics, we investigated whether BaDHPS would be suitable for NMR analyses. We first performed a 2D 1H–15N HSQC-TROSY NMR experiment with perdeuterated 15N-labeled BaDHPS, and approximately 40% of the residues were observable in the spectrum (Figure 5A). We therefore prepared 2H,13C,15N-labeled BaDHPS and performed 3D HNCA and HN(CO)CA experiments with the goal of assigning these observable resonances. Not counting 16 assigned peaks within the N-terminal His tag, 72 resonances in the 1H–15N HSQC-TROSY spectrum of the protein were assigned (Supplementary Figure S3 and Supplementary Table S3). These include the residues within loops1, 2, 4, 5, and 6, the N-terminal two-thirds of helix α1, strand β2, the C-terminal half of helix α8, and the C-terminus. These assignments encompass the active site and dimer interface and showed that NMR would be an ideal tool to monitor loop conformational changes and to investigate whether the active site and the interface are indeed dynamically linked.
Figure 5.

NMR analyses of BaDHPS and its complexes. (A) 1H–15N–TROSY-HSQC spectrum of 300 μM apo BaDHPS. (B) 1H–15N–TROSY-HSQC spectrum of 300 μM BaDHPS in the presence of PtPP and pABA. (C) Residues that shift in the presence of substrates PtPP and pABA mapped onto the intermediate-state crystal structure of the closely similar YpDHPS.7 The residues are shown as spheres and color-coded according to position on the structure; loop1, orange; loop2, cyan; dimer interface, red; other locations, green. The active site is shown only for one monomer for clarity and is indicated by pABA and DHPP represented by sticks (yellow carbon atoms) at the N-terminus of helix α-loop7 (yellow). Note the close association between loop1 and the tip of loop7 (yellow) in this structure. (D) 1H–15N–TROSY-HSQC spectrum of 300 μM BaDHPS in the presence of PtPP, pABA, and compound 11. Compared to panel B, resonances in panel D are generally more intense, and three of the most prominent additional resonances are circled.
We first confirmed that the active site loops become more ordered in the presence of substrate as previously observed by crystallography.7 To prevent substrate turnover in these experiments, we used the inactive DHPP analogue 6-hydroxymethyl-pterine-pyrophosphate (PtPP) that we and others have shown to bind within the DHPS pterin pocket.13,15 Weak peak perturbations were observed by the addition of 0.4 mM PtPP, but more profound perturbations were observed when 0.4 mM pABA was subsequently added (Figures 5B). In general, the spectrum of the BaDHPS/PtPP/pABA ternary complex is more dispersed, and many more resonances appear compared to the 1H–15N HSQC-TROSY of the free enzyme. This is consistent with the formation of a folded loop1–loop2 substructure within the ternary complex within which pABA binds, as observed in the intermediate-state crystal structure of DHPS.7 Importantly, the addition of PtPP and pABA also elicits major perturbations in signals of residues at the dimer interface including the C-terminus (Figure 5C). This result supports our proposal that the dimer interface and the loop1/loop2 pABA binding site are dynamically linked.
Upon addition of 11, further perturbations of resonances from loop1, loop2, the dimer interface, and the C-terminus are observed (Figure 5D). Specifically, more well-dispersed resonances appear stemming from ordered regions of the protein, and many of the existing resonances increase in intensity. These changes strongly suggest that the addition of 11 further stabilizes the substrate-bound substructure formed by loop1 and loop2. These NMR analyses have demonstrated a communication between the dimer interface and the active site such that molecules bound at one site perturb the other. The observation that many resonances in the free BaDHPS were unobservable suggested that BaDHPS undergoes motions on the intermediate chemical shift time scale (i.e., μs–ms). Motions on this time scale have been shown to be important in catalysis and allostery.16−20 The increased intensity of some resonances and the appearance of new resonances upon addition of 11 may indicate that loop and interface motions are slowed from the invisible intermediate time scale to the slow-exchange time scale, freezing out catalysis-supporting motions and inhibiting the enzyme. To gain more insight into the dynamics of the system, we performed MD simulations.
Computational Analyses
MD simulations were performed on four systems: the BaDHPS enzyme alone (E), the enzyme with 11 bound (IE), the enzyme with the substrates bound (ES), and the enzyme with both inhibitor and substrates bound (IES). The basic starting structure for the enzyme and for the inhibitor were from the X-ray structure of the compound 11 complex reported here. The active site loop1 and loop2, the substrates, and the catalytically essential Mg2+ ion are not present in this structure, and these were modeled from the structure of YpDHPS in complex with the SN1 intermedates, DHP+, pABA, and PPi.7 This is consistent with the scenario in which inhibition occurs at a later step of catalysis, as suggested by the kinetic and NMR data. The resulting trajectories were analyzed by the quasi-harmonic approach21,22 that identifies sets of correlated motions grouped together as “quasi-harmonic modes” analogous to harmonic vibrational modes.
In the E simulation, the largest fluctuations occur in loop1 and loop2 at the active site, and this is largely captured in the highest-amplitude quasi-harmonic mode (Figure 6), although it also persists in the next several modes (Supplementary Figure S4A). An animation of this mode reveals that these loop motions are coupled to an overall breathing motion in which the monomers rock toward and away from each other, pivoting about the symmetric dimer interface (Supplementary Movie S1). In the presence of the inhibitor (IE simulation), this intermonomer motion is strongly damped, and the mode changes to one in which active-site movements are correlated to those in the inhibitor binding site (Supplementary Movie S2), particularly the loop7 residues 231–235 (Figure 6B). These residues are indeed adjacent to the distal half of 11 in our interpretation of the corresponding crystal structure (Figure 4D). Inspection of the IES model shows that contacts between these loop7 residues, including the highly conserved Arg234, and residues Phe33 and Ser34 of loop1 may participate in transmitting these motions (Figure 7).
Figure 6.

Molecular dynamics simulations of the BaDHPS dimer and its complexes. Shown are (A) the root-mean-square fluctuations (RMSF) of the α-carbons at each residue position during the simulations and (B) the contribution of the highest-amplitude quasi-harmonic mode to those fluctuations. The analysis was performed for all the α-carbons of the homodimer, but only one monomer is shown here. Blue line, enzyme alone simulation (E); red line, enzyme plus 11 (IE); green line, enzyme plus substrates (ES); magenta line, enzyme plus substrates plus 11 (IES). Loop1 is highlighted in orange, loop2 in cyan, and loop7 (which includes the small helix α-Loop7) in yellow.
Figure 7.
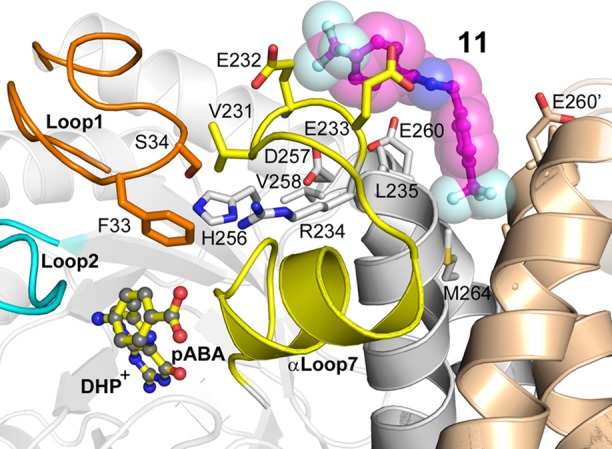
A model showing how the distal half of compound 11 (magenta carbons, stick and surface representations) is proposed to exert a long-range inhibitory effect on the active site of BaDHPS via its interaction with the ordered loop1–loop7 substructure (orange-yellow) that is known to be required for catalysis.7 In this model, the structures of loop1 and loop2 (cyan) at the active site and the bound substrates DHP+ and pABA (shown as sticks) are based on the known structure of the YpDHPS intermediate-state catalytic complex.7
Analysis of the ES simulations shows that substrate binding significantly decreases the motion of loop1 but leaves loop2 mobile, while the additional binding of inhibitor (IES) significantly reduces the loop2 motion (Figure 6A). Animations of the largest quasi-harmonic modes from the ES and IES simulations (Supplementary Movies S3 and S4) show that overall rocking or twisting motions, already reduced by substrate binding, are further reduced by inhibitor binding. These largest modes capture most of the overall RMS fluctuation (Figure 6A vs B). A more detailed examination of these modes shows that a motion of residues 256–258 in the ES simulation is strongly damped upon inhibitor binding (Figure 6B, green vs magenta). This region includes His256 that coordinates the leaving PPi and is important for the SN1 catalytic mechanism. These results suggest that inhibition occurs at a step after both substrates have bound, such as a chemical step or subsequent product release, consistent with the kinetic and NMR data.
The MD simulations support the notion that natural long-range coupled motions within the dimer mediate communication between the dimer interface and the monomer active sites. Specifically, the simulations have shown that an intermonomer rocking motion is reduced upon inhibitor binding and also that correlations are increased between the flexible loops in the active site and the region that spans the N-terminal part of helix α7 and the preceding loop7 residues in the inhibitor-binding site. These analyses suggest a mechanism of allosteric inhibition in which inhibitor binding both rigidifies the protein and alters the pattern of dynamic correlations between dimer-interface and active-site movement. This appears to inhibit a step subsequent to the binding of the second substrate, and the kinetic data suggest that this step is product release.
The apparent connection between DHPS catalysis and the dimer interface prompted us to look more closely at our previous YpDHPS crystal structures of the dimer in the apo conformation,7 the product-analogue-bound conformation, and the intermediate-state conformation in which DHPP and pABA had bound, PPi had been cleaved but not released, and the pterin-pABA bond had not been formed. This comparison revealed that the monomers rotate around the axis of helix α7 in transitioning between the apo and intermediate structures, with the product-bound structure midway between these extremes (Supplementary Figure S5). Examination of loop1 and the loop7 region immediately preceding helix α7 in these structures also shows that the latter moves closer to loop1 on going from the apo state to the intermediate state and moves back in the product-bound state (Supplementary Figure S6). Finally, in the product analogue-bound structure, loop1 rotates out of the active site to form an “open” conformation that allows the product to be released. Together with the MD and NMR results, these observations suggest that when 11 interacts with loop7, it leads to stronger loop1–loop7 interactions that restrict the movement of loop1 out of the active site, thereby inhibiting product release. A similar phenomenon has been observed in ribonuclease A.23
Concluding Remarks
Allosteric sites on enzymes provide novel opportunities for drug discovery, and a major advantage of exploiting these sites is the ability to avoid mechanistic aspects of the active site that hamper the discovery and development of small molecule inhibitors.24 Allosteric sites on DHPS potentially offer a new approach to therapeutically inhibit this valuable antimicrobial target that has a particularly complex and mobile active site. Our studies have revealed that the DHPS dimer interface offers an excellent opportunity for drug discovery. Although DHPS has never been shown to be an allosteric enzyme or to have allosteric effectors, it appears that the natural motions of the dimer are linked to the active site and can be manipulated with small molecules to affect catalytic activity. This is actually supported by and explains mutations that are found in several sulfonamide resistant strains of S. aureus in which lysine and glutamic acid residues are inserted into the distal end of helix α8 at the dimer interface.15 Compound 11 inhibits all three of the DHPS enzymes that we have studied, and this suggests that future antimicrobials that target the interface are likely to be broad-spectrum. Compound 11 currently lacks antimicrobial activity and has limited aqueous solubility, and the metabolic stability of the scaffold is a potential concern. However, using the structural and molecular dynamics information presented here, structure-guided synthetic efforts are currently underway to increase the affinity, physicochemical properties and potency of derivative small molecules. The bipartite nature of the binding site, with a proximal deep aryl pocket at the dimer interface adjacent to multiple potential H-bond donors/acceptors and a distal site comprising exposed residues within loop7, implies that each can be independently optimized. An important initial step in this process is ‘fragment hopping’ to identify new and potentially tighter binding scaffolds for each of these sites.8,9 This is currently underway for the proximal pocket where crystallography can readily be used to monitor progress. For the distal site where structural analysis has proven to be more challenging, we are using the scaffold present in compound 11 as a proximal ‘anchor’ and synthesizing a library of derivative compounds to screen for more potent distal scaffolds that can be subsequently characterized by crystallography. The potential of allosteric inhibitors as antimicrobial agents is well validated clinically; for example, anti-HIV drugs Evavirenz, Nevirapine, and Delaviridine all target an allosteric site within the viral reverse transcriptase.24 This bodes well for the development of allosteric modulators of DHPS, particularly those that can be combined with other antifolates to minimize the risk of resistance development.
Methods
Protein Production
BaDHPS, YpDHPS, and SaDHPS were overexpressed and purified as previously described.7,1315N,2H-Labeled BaDHPS and 13C,15N,2H-labeled BaDHPS were overexpressed according to established procedures25 and were purified using the same procedures as unlabeled BaDHPS.
NMR Spectroscopy: WaterLOGSY Screen
The Maybridge Ro3 fragment library (1100 compounds at the time of purchase) was dissolved in DMSO-d6 into 50 mM stock solutions. The fragments were screened in pools of 5 at a concentration of 150 μM each, and the 216 samples were prepared in 20 mM phosphate buffered saline (PBS), pH 7.0, 10% D2O, 0.01% Triton-X 100. All spectra were recorded with a Varian Inova 600 MHz NMR spectrometer equipped with a triple resonance inverse probe at 298 K. The pulse sequence used was the homonuclear one-dimensional WaterLOGSY with a water flip-back pulse.10 Data collection parameters for the WaterLOGSY experiments were 200 scans, mixing time = 1.25 s, relaxation time = 2.6 s, and spectral windows of 13 ppm. All data were processed using TopSpin (Bruker Biospin). Three spectra were recorded for each sample, a 1D 1H reference, a 1D WaterLOGSY in the absence of protein, and a 1D WaterLOGSY in the presence of 10 μM BaDHPS. Samples for c-WaterLOGSY contained 20 μM concentration of the high affinity pterin-binding inhibitor reported previously (2-(7-amino-1-methyl-4,5-dioxo-1,4,5,6-tetrahydropyrimido[4,5-c]pyridazin-3-yl)propanoic acid)11 or 20 μM pABA in the presence of 100 μM pyrophosphate, 10 μM DHPS, and 300 μM concentration of the fragment to be screened. WaterLOGSY titration experiments were performed in the presence of 10 μM DHPS and decreasing concentrations of compound 1 (2, 1, 0.5, 0.25, 0.125, 0.0625, 0 mM).
NMR Spectroscopy: 2D and 3D Experiments
See Supporting Methods.
X-ray Crystallography
BaDHPS crystals were grown as described previously.13 The four complex cocrystal structures were obtained by soaking the compounds into BaDHPS crystals overnight at ∼10 mM in mother liquor. Soaked crystals were briefly immersed in cryoprotectant (a mixture of 50% mineral oil and 50% Paratone-N) before flash freezing in liquid nitrogen. Diffraction data were collected at the SER-CAT beamlines 22-ID and 22-BM at the Advanced Photon Source and processed using HKL2000.26 Structures were refined using REFMAC27 and PHENIX28 and optimized with COOT.29
Enzyme Inhibition Assays and Enzyme Kinetics
The enzyme activities of BaDHPS, YpDHPS, and SaDHPS were all determined by a linked assay in which the released pyrophosphate was converted to inorganic phosphate by pyrophosphatase. The inorganic phosphate was detected using the PiColorlock Gold Kit (Innova Biosciences) with a NanoDrop 2000C spectrophotometer (Thermo Scientific). The 100-μL reaction mixtures contained 5 μM pABA, 5 μM DHPP, 10 mM MgCl2, 5% DMSO, 50 mM HEPES pH 7.6, 0.01 U yeast inorganic pyrophosphatase (Fermentas Life Sciences), and 5 nM DHPS. Inhibitor compounds were dissolved in DMSO, and inhibition was tested at 250 μM. Inorganic phosphate was measured after 20 min of incubation at 37 °C. To determine the half maximal inhibitory concentration (IC50) values, DHPS activities were measured in the presence of various concentrations of the compounds. The mechanism of inhibition by compound 11 was studied with all three DHPS enzymes. At three different concentrations of the inhibitor (0, 50, 100 μM), the initial catalytic rates were determined at 0, 0.5, 1, 2, 4, 6, 8, 10 μM pABA with a fixed concentration of 20 μM DHPP. Data were analyzed using GraphPad Prism software.
Computational Methods
Starting structures for the molecular dynamics simulations were based on the crystallographic structure of the complex with compound 11 reported here, with additional modeling of loops and substrates based on our previously reported structures,7 as described in Supplemental Methods. Simulations were done in explicit solvent with periodic boundary conditions and the Amber ff99SB force field.30 For each of the four complexes, E, IE, ES and IES, 16 ns production simulations were carried out. For E and IE, longer simulations were also run (150 and 40 ns, respectively), but these showed patterns of fluctuation and correlation very similar to those reported for the 16 ns runs. Simulation details, as well as the methods of quasi-harmonic mode analysis, are provided in Supporting Methods.
Surface Plasmon Resonance (SPR) Experiments
See Supporting Methods.
Isothermal Titration Calorimetry (ITC)
BaDHPS was dialyzed against 50 mM HEPES, pH 7.6, 5 mM MgCl2. Experiments were performed using an ITC200 (Microcal) calorimeter at 298 K. All titrations were measured in 40 mM HEPES, 4 mM MgCl2, 5% DMSO (10% in the titration of 11) at 298 K. Titration of 2.7 μL of 1 mM compound 11 into a solution of 10 μM BaDHPS was performed over 14 injections. pABA titrations in the presence or absence of 11 were performed over 19 injections of 2 μL each. Cell and syringe concentrations were 20 μL BaDHPS, 0.1 mM PtPP (with and without 0.5 mM 11) and 0.25 mM pABA, 0.1 mM PtPP (with and without 0.5 mM 11, respectively).
Chemical Synthesisof Compound 11, (E)-N-(4-(Trifluoromethyl)benzylidene)-1-(4-(trifluoromethyl)phenyl) Methanamine
To 206 mg (1.18 mmol) of (4-(trifluoromethyl)phenyl) methanamine in 1 mL of dichloromethane was added 283 mg (2.35 mmol) of magnesium sulfate, followed by 205 mg (1.78 mmol) of 4-(trifluoromethyl)benzaldehyde. The mixture was stirred under nitrogen for 5 h, filtered through Celite with washing of the insolubles with additional dichloromethane. The organic layer was concentrated in vacuo to afford 222 mg of clean product. No further purification was required. 1H NMR (400 MHz, chloroform-d) δ 8.39 (s, 1H), 7.88–7.78 (m, 2H), 7.62 (d, J = 8.1 Hz, 2H), 7.54 (d, J = 8.1 Hz, 2H), 7.40 (d, J = 8.6 Hz, 2H), 4.83 (s, 2H). Purity (SFC-MS UV-PDA (200–400 nm)) >95%.
Acknowledgments
This work was supported by National Institutes of Health Grant AI070721 (S.W.W. and R.E.L.), Cancer Center (CORE) Support Grant CA21765, and the American Lebanese Syrian Associated Charities (ALSAC). We thank R. Kriwacki for advice, T. Mittag for critical reading of the manuscript, C. Grace for NMR technical assistance, C. Jeffries for LC-MS analysis, and Z. Li, J. Bollinger, B. Waddell. and the St. Jude Molecular Interactions Core for technical assistance. X-ray diffraction data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID and 22-BM beamlines at the Advanced Photon Source, Argonne National Laboratory, and we thank SER-CAT staff for their assistance. Supporting SER-CAT institutions may be found at www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
Supporting Information Available
Methods for 2D and 3D NMR Spectroscopy experiments, computational simulations and quasi-harmonic mode analysis, and Surface Plasmon Resonance (SPR) experiments. X-ray crystallography data collection and refinement statistics, BaDHPS NMR assignments, Michaelis–Menten kinetics, dominant quasi-harmonic modes from simulations E, IE, ES and IES, global and local motions within YpDHPS upon catalysis, and animation movies of the most dominant quasi-harmonic mode from simulations E, IE, ES and IES. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
Coordinates for compound 4 bound BaDHPS: 4NHV; coordinates for compound 5 bound BaDHPS: 4NIL; coordinates for compound 6 bound BaDHPS: 4NIR; coordinates for compound 11 bound BaDHPS: 4NL1.
Author Present Address
# Infection - iMED, AstraZeneca, Waltham, MA 02452.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Matherly L. H. (2001) Molecular and cellular biology of the human reduced folate carrier. Prog. Nucleic Acid Res. Mol. Biol. 67, 131–162. [DOI] [PubMed] [Google Scholar]
- Domagk G. (1935) Ein beitrag zur chemotherapie der bakteriellen infektionen. Dtsch. Med. Wochenschr. 7, 250–253. [Google Scholar]
- Sköld O. (2000) Sulfonamide resistance: mechanisms and trends. Drug Resist Updat 3, 155–160. [DOI] [PubMed] [Google Scholar]
- Sköld O. (2001) Resistance to trimethoprim and sulfonamides. Vet. Res. 32, 261–273. [DOI] [PubMed] [Google Scholar]
- Hammoudeh D. I.; Zhao Y.; White S. W.; Lee R. E. (2013) Replacing sulfa drugs with novel DHPS inhibitors. Future Med. Chem. 5, 1331–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Hammoudeh D.; Yun M. K.; Qi J.; White S. W.; Lee R. E. (2012) Structure-based design of novel pyrimido[4,5-c]pyridazine derivatives as dihydropteroate synthase inhibitors with increased affinity. ChemMedChem 7, 861–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun M.-K.; Wu Y.; Li Z.; Zhao Y.; Waddell M. B.; Ferreira A. M.; Lee R. E.; Bashford D.; White S. W. (2012) Catalysis and sulfa drug resistance in dihydropteroate synthase. Science 335, 1110–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlanson D. A.; McDowell R. S.; O’Brien T. (2004) Fragment-based drug discovery. J. Med. Chem. 47, 3463–3482. [DOI] [PubMed] [Google Scholar]
- Scott D. E.; Coyne A. G.; Hudson S. A.; Abell C. (2012) Fragment-based approaches in drug discovery and chemical biology. Biochemistry 51, 4990–5003. [DOI] [PubMed] [Google Scholar]
- Dalvit C.; Fogliatto G.; Stewart A.; Veronesi M.; Stockman B. (2001) WaterLOGSY as a method for primary NMR screening: practical aspects and range of applicability. J. Biomol. NMR 21, 349–359. [DOI] [PubMed] [Google Scholar]
- Hevener K. E.; Yun M. K.; Qi J.; Kerr I. D.; Babaoglu K.; Hurdle J. G.; Balakrishna K.; White S. W.; Lee R. E. (2010) Structural studies of pterin-based inhibitors of dihydropteroate synthase. J. Med. Chem. 53, 166–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalvit C.; Fasolini M.; Flocco M.; Knapp S.; Pevarello P.; Veronesi M. (2002) NMR-based screening with competition water-ligand observed via gradient spectroscopy experiments: Detection of high-affinity ligands. J. Med. Chem. 45, 2610–2614. [DOI] [PubMed] [Google Scholar]
- Babaoglu K.; Qi J.; Lee R. E.; White S. W. (2004) Crystal structure of 7,8-dihydropteroate synthase from Bacillus anthracis: mechanism and novel inhibitor design. Structure 12, 1705–1717. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- Hampele I. C.; D’Arcy A.; Dale G. E.; Kostrewa D.; Nielsen J.; Oefner C.; Page M. G.; Schonfeld H. J.; Stuber D.; Then R. L. (1997) Structure and function of the dihydropteroate synthase from Staphylococcus aureus. J. Mol. Biol. 268, 21–30. [DOI] [PubMed] [Google Scholar]
- Eisenmesser E. Z.; Millet O.; Labeikovsky W.; Korzhnev D. M.; Wolf-Watz M.; Bosco D. A.; Skalicky J. J.; Kay L. E.; Kern D. (2005) Intrinsic dynamics of an enzyme underlies catalysis. Nature 438, 117–121. [DOI] [PubMed] [Google Scholar]
- Henzler-Wildman K. A.; Thai V.; Lei M.; Ott M.; Wolf-Watz M.; Fenn T.; Pozharski E.; Wilson M. A.; Petsko G. A.; Karplus M.; Hubner C. G.; Kern D. (2007) Intrinsic motions along an enzymatic reaction trajectory. Nature 450, 838–844. [DOI] [PubMed] [Google Scholar]
- Henzler-Wildman K. A.; Lei M.; Thai V.; Kerns S. J.; Karplus M.; Kern D. (2007) A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature 450, 913–916. [DOI] [PubMed] [Google Scholar]
- Velyvis A.; Yang Y. R.; Schachman H. K.; Kay L. E. (2007) A solution NMR study showing that active site ligands and nucleotides directly perturb the allosteric equilibrium in aspartate transcarbamoylase. Proc. Natl. Acad. Sci. U.S.A. 104, 8815–8820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhabha G.; Lee J.; Ekiert D. C.; Gam J.; Wilson I. A.; Dyson H. J.; Benkovic S. J.; Wright P. E. (2011) A dynamic knockout reveals that conformational fluctuations influence the chemical step of enzyme catalysis. Science 332, 234–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichiye T.; Karplus M. (1991) Collective motions in proteins - a covariance analysis of atomic fluctuations in molecular-dynamics and normal mode simulations. Proteins: Struct., Funct., Genet. 11, 205–217. [DOI] [PubMed] [Google Scholar]
- Fragiadakis D.; Casalini R.; Bogoslovov R. B.; Robertson C. G.; Roland C. M. (2011) Dynamic heterogeneity and density scaling in 1,4-polyisoprene. Macromolecules 44, 1149–1155. [Google Scholar]
- Doucet N.; Watt E. D.; Loria J. P. (2009) The flexibility of a distant loop modulates active site motion and product release in ribonuclease A. Biochemistry 48, 7160–7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J. A.; Wells J. A. (2004) Searching for new allosteric sites in enzymes. Curr. Opin. Struct. Biol. 14, 706–715. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Filippov I.; Richter C.; Luo R.; Kriwacki R. W. (2005) Solution NMR studies of an intrinsically unstructured protein within a dilute, 75 kDa eukaryotic protein assembly; probing the practical limits for efficiently assigning polypeptide backbone resonances. ChemBioChem 6, 2242–2246. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D: Biol. Crystallogr. 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D: Biol. Crystallogr. 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D: Biol. Crystallogr. 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Hornak V.; Abel R.; Okur A.; Strockbine B.; Roitberg A.; Simmerling C. (2006) Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.