

Abstract
The aim of this study was to determine the size-dependent penetration ability of gold nanoparticles and the potential application of ultrasmall gold nanoparticles for intranucleus delivery and therapy. We synthesized gold nanoparticles with diameters of 2, 6, 10, and 16 nm and compared their intracellular distribution in MCF-7 breast cancer cells. Nanoparticles smaller than 10 nm (2 and 6 nm) could enter the nucleus, whereas larger ones (10 and 16 nm) were found only in the cytoplasm. We then investigated the possibility of using ultrasmall 2 nm nanoparticles as carriers for nuclear delivery of a triplex-forming oligonucleotide (TFO) that binds to the c-myc promoter. Compared to free TFO, the nanoparticle-conjugated TFO was more effective at reducing c-myc RNA and c-myc protein, which resulted in reduced cell viability. Our result demonstrated that the entry of gold nanoparticles into the cell nucleus is critically dependent on the size of the nanoparticles. We developed a strategy for regulating gene expression, by directly delivering TFOs into the nucleus using ultrasmall gold nanoparticles. More importantly, guidelines were provided to choose appropriate nanocarriers for different biomedical purposes.
Keywords: ultrasmall gold nanoparticles, cancer cell nucleus, size-dependent, gene regulation, cancer therapy
Nanoscale materials have been receiving increasing attention in the field of biomedicine. Understanding of the behavior of nanomaterials, especially nanoparticles (NPs), in living system is vitally important for both the design of functionalized nanoparticles and the application of some nanomaterial-based medicines already in use. Due to their superior physiochemical properties, nanomaterials exhibit excellent advantages in biomedical research, such as accumulating preferentially at sites of tumor growth or inflammation, entering cells much more rapidly through different mechanisms compared to small molecules, delivering of various payloads into their targets, and being modified conveniently to perform desirable functions.1−4 For nanoparticles, one of the key parameters is size. The nanoparticle-mediated in vivo and in vitro response is size-dependent. Previous studies showed that nanoparticles with different sizes had distinctive distributions in the organs of living mice,5−8 were taken up into cells by different mechanisms,9,10 and followed a size-specific pathway until they escaped from the cells.11 We have previously demonstrated that smaller nanoparticles (50 nm) can penetrate deeply into tumor tissue more easily and effectively than their larger counterparts (100 nm).12 Among kinds of nanoparticles, gold nanoparticles have received much attention due to their easy fabrication, controllable size and shape, tunable surface functionalization, and good biocompatibility.10,13−16 In the past few years, much effort has been made to develop nanoparticle-based therapeutic approaches. The application of gold nanoparticles is continuously booming, including drug and gene delivery vehicles, diagnostic tools, imaging agent in therapy, and biomarkers in the pharmaceutical field, etc.17−19
Nucleus is the most important organelle in regulating reproduction, growth, metabolism, and death of cells through gene expression. Controlling the nuclear-governed processes has been a major goal for nuclear-targeted therapy. However, most of the nanomaterials enter the cytoplasm exclusively. Information regarding the uptake and penetration of nanoparticles in the nucleus and their application in regulating gene expression is limited. Previously, gold nanostars were used to deliver drugs to the nucleus and induce nuclear phenotype change and apoptosis.20 The use of nuclear-targeting Au NPs was also reported by Mostafa et al. to selectively disturb the division of cancer cells under the observation of cytokinesis arrest.21 Therapeutic or reporter genes attached to magnetic nanoparticles for gene-targeting delivery via high-gradient/high-field magnets demonstrated in vitro transfection of a variety of cell lines.22 However, nuclear targeting was indirectly achieved by conjugating nuclear targeting peptides with nanoparticles or magnetic targeting in these studies.
Gene therapy is a growing field of medicine that introduces genetic materials into the body to treat diseases.23−25 Antisense (AS) gene therapy is a potentially powerful candidate for clinical treatment of various diseases, such as cancer and HIV/AIDS.23,26 Triplex-forming oligonucleotides (TFOs) are known to form triplexes with specific DNA sequences, thereby interfering with gene transcription. However, due to the high activity of DNase in the cytoplasm, most TFOs are degraded before forming a triplex with the target sequence. Thus, the application of TFOs is limited. Previous studies have reported that conjugation with Au NPs can improve the stability of oligonucleotides and prevent their degradation.27−29 However, none of these methods could directly deliver TFOs into the nucleus, and the therapeutic efficiency of TFOs is still maintained at a low level.
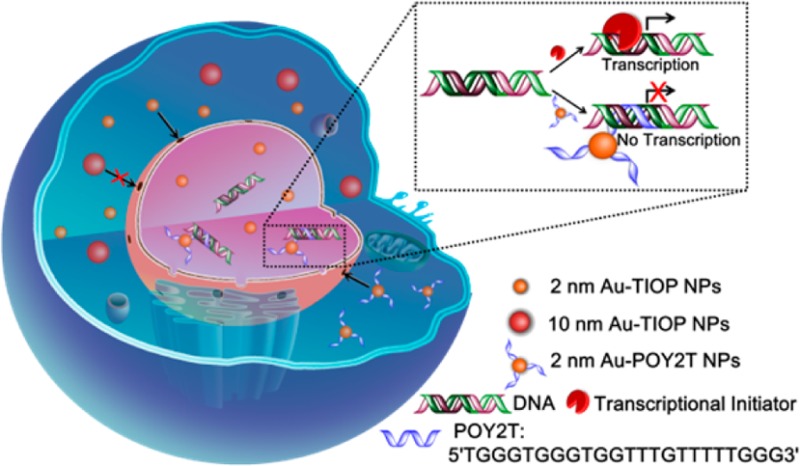
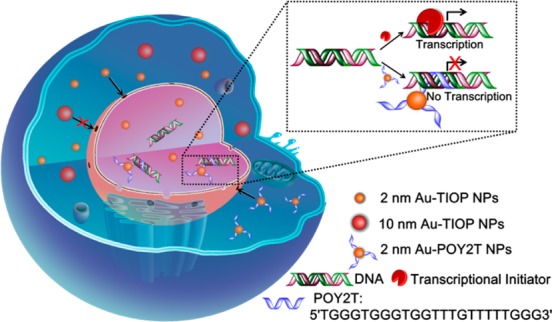
Herein, we first report using tiopronin-covered gold nanoparticles (Au-TIOP NPs) as a typical model to directly target a nucleus based on the nanoparticle’s specific physiochemical properties. Tiopronin is widely used as a thiol drug with good biocompatibility.30 Since the thiol groups of tiopronin can bind to the surface of nanoparticles to prevent coagulation, it is used as a stabilizing agent for metal nanoparticles.31 Additionally, small molecules, peptides, or drugs can be modified to the carboxyl group of tiopronin for different biomedical applications. In this study, we find that only gold nanoparticles smaller than 10 nm (2 and 6 nm) can enter the nucleus. The larger ones (10 and 16 nm) are localized exclusively outside of the nucleus in the cytoplasm (Scheme 1). Importantly, we used the ultrasmall 2 nm Au NPs as a carrier and gene regulator to deliver triplex-forming oligonucleotides into the nucleus directly. Our result shows that the expression of targeted gene was significantly down-regulated by a 2 nm Au NP–TFO complex at a concentration much lower than that of free TFOs.
Scheme 1. Schematic illustration of the distribution and localization behavior of smaller (2 nm) and larger (10 nm) Au-TIOP NPs in MCF-7 cancer cells. The ultrasmall 2 nm Au-TIOP NPs were used as a carrier to enter the nucleus and deliver a TFO (POY2T) to regulate gene expression.
Results and Discussion
Characterization of Au-TIOP NPs with Different Sizes
In order to investigate whether gold nanoparticles with different sizes have distinctive localization behavior and the ability to enter the nucleus in breast cancer cells, tiopronin-coated gold nanoparticles with sizes ranging from 2 to 16 nm were synthesized in this study. Tiopronin is a very well-known pharmaceutical drug used for the treatment of rheumatoid arthritis and cystinuria, and its biocompatibility is quite well-established already.23 As shown in Figure S1 in Supporting Information, 2 and 6 nm Au-TIOP NPs were directly prepared by reduction of sodium borohydride with the protecting agent tiopronin. For the 10 and 16 nm Au-TIOP NPs, citrate-stabilized gold nanoparticles were first prepared, then a surface molecular ligand exchange method was used to add tiopronin.
The morphology of the as-prepared Au-TIOP NPs with different sizes is shown in Figure 1A. TEM images revealed that the gold nanoparticles have uniform spherical morphology and narrow size distribution (Figure S2 in Supporting Information). Compared with the 15 nm BBI gold nanoparticles (Figure S3 in Supporting Information), the sizes of as-prepared nanoparticles were measured and 200 NPs of each size were statistically analyzed and found to be about 2.1 ± 0.6, 5.6 ± 0.8, 10.5 ± 2.1, and 15.8 ± 2.4 nm. In addition, the hydrodynamic diameters of the Au-TIOP NPs were measured by dynamic light scattering (DLS) and are shown in Table 1. It is reasonable that the hydrodynamic diameters were a little larger than the sizes determined by TEM, due to the different measuring principles of each method. For simplicity, the sizes of the Au-TIOP NPs are referred to as 2, 6, 10, and 16 nm in the rest of this paper. The ζ-potentials of each size of Au-TIOP NP were analyzed using a Nano ZS Zetasizer and are shown in Table 1. The ζ-potentials of Au-TIOP NPs were −43.9 mV (2 nm), −41.1 mV (6 nm), −39.1 mV (10 nm), and −35.9 mV (16 nm). All of the Au-TIOP NPs were negatively charged, and there was a minor difference (2–3.2 mV) between each pair of adjacent sizes.
Figure 1.

Characterization of 2, 6, 10, and 16 nm Au-TIOP NPs and Au-FITC NPs with different sizes. (A) TEM images and the corresponding size distribution histograms of the different sizes of Au-TIOP NPs. (B) XRD patterns of as-synthesized Au-TIOP NPs. (C) Gel shift assay of as-synthesized Au-TIOP NPs. (D) Fluorescence spectra of the as-synthesized Au-FITC NPs and free FITC. (E) Photos of as-synthesized Au-FITC NPs and free FITC under a daylight lamp and a UV lamp.
Table 1. Characterization of As-Synthesized Au-TIOP NPs and Au-FITC NPs.
| sample | UV–vis λmax (nm) | hydrodynamic diameter (nm/PDI) | ζ-potential (mV) |
|---|---|---|---|
| 2 nm Au-TIOP NPs | 505 | 2.58/0.39 | –43.9 |
| 2 nm Au-FITC NPs | 456/477 | 4.7/0.46 | –29.2 |
| 2 nm Au-POY2T NPs | 507 | 3.6/0.40 | –30.4 |
| 6 nm Au-TIOP NPs | 518 | 6.11/0.33 | –41.1 |
| 2 nm Au-FITC NPs | 458/480 | 10.02/0.45 | –28.5 |
| 10 nm Au-TIOP NPs | 521 | 11.21/0.20 | –39.1 |
| 10 nm Au-FITC NPs | 458/479 | 15.49/0.27 | –27.4 |
| 16 nm Au-TIOP NPs | 525 | 16.8/0.31 | –35.9 |
| 16 nm Au-FITC NPs | 458/477 | 19.33/0.43 | –26.7 |
X-ray diffraction (XRD) analysis was also used to characterize the four sizes of gold nanoparticles. As shown in Figure 1B, there were five primary diffraction peaks at 38.187, 44.385, 64.576, 77.567, and 81.722, which could be correspondingly indexed to the crystal planes of (111), (200), (220), (311), and (222) of Au. This result was consistent with the XRD patterns of Au with a PDF number of 04-0784 (cubic, a = b = c = 0.2884 nm, d(111) = 0.2355 nm). A previous report showed that the smaller the particle, the wider the corresponding XRD peak.32 This has also been confirmed in this study, as shown in Figure 1B. Due to the preparation of such small nanoparticles and the reduced degree of crystallization, diffraction peaks of the Au NP samples exhibited a degree of broadening. Furthermore, according to the famous Scherrer equation, D = kλ/B cos θ,33 the calculation based on the (111) peak line width at half-maximum intensity from top to bottom roughly showed that the average sizes of the nanoparticles were about 16, 10, 6, and 2 nm. These sizes are also consistent with the results above.
Electrophoresis of colloidal gold nanoparticles was conducted not only to demonstrate the surface charge of the particles but also to determine their relative size.34 As shown in Figure 1C, all of the nanoparticles with good dispersion moved toward the anode side of the electrophoresis tank, meaning that they were all negatively charged. It was also obvious that the 2 nm Au-TIOP NPs traveled the longest distance while the 16 nm Au-TIOP NPs moved the shortest distance within the same time. This can be attributed to the different resistance to the gel, which is related to the particle size. It is reasonable to assume that, since the surface charges were almost the same, the electrophoresis speed or distance traveled by the NPs was determined by their sizes.
Moreover, in order to confirm the dispersion of synthesized Au-TIOP NPs in relevant media, additional stability experiments were conducted. As shown in Figure S4 in Supporting Information, Au-TIOP NPs were well-dispersed in the relevant media in a discrete state, and the absorption peaks of each particle dispersed in different solutions also trended toward identical behaviors (for the ultrasmall size of synthesized nanoparticles, the DMEM culture medium could not be used for this test).
In summary, several methods were used to demonstrate that we successfully prepared spherical-shaped and stable Au-TIOP NPs with diameters of 2, 6, 10, and 16 nm with good PDI. As these nanoparticles differed only in size, they were used for the following studies.
Characterization of Au-FITC NPs with Different Sizes
In order to observe the exact distribution of the nanoparticles in cells through confocal laser scanning microscopy (CLSM), different sizes of Au-TIOP NPs were labeled with FITC. As shown in Figure S5 in Supporting Information, PEG2000 was used as a linker between particles and FITC to prevent fluorescence quenching due to the nanomaterial surface energy transfer effect.35,36 The particle size and morphology of FITC-labeled gold nanoparticles can be visualized by TEM (Figure S6 in Supporting Information). As a result of the attachment of polymers to the surface of the particles, the monodispersity of the gold nanoparticles decreased and localized aggregation occurred. Furthermore, because of the polymeric covering, the size of the nanoparticles increased (Table 1).
UV–vis absorption spectroscopy, a common method to characterize gold nanoparticles, was used to acquire the absorbance spectra of the nanoparticles before and after FITC labeling. As shown in Figure S7 in Supporting Information (a–d, black), the UV–vis absorption peak was red-shifted as the size of the Au-TIOP NPs increased. The measured absorption peaks of the 2, 6, 10, and 16 nm Au-TIOP NPs were 505, 518, 521, and 525 nm, respectively. This result was in agreement with the Mie theory.37 The maximum absorption wavelength of the FITC molecule is between 490 and 495 nm, and its maximum emission wavelength is around 525–530 nm, resulting in a bright yellow-green fluorescence. After fluorescence labeling, as the red curves show (Figure S7 in Supporting Information), all four sizes of Au-TIOP NPs have large absorption peaks between 450 and 480 nm, which are similar to the absorption peak at 492 nm of free FITC. The fluorescence spectra, as shown in Figure 1D, indicated that the FITC-labeled nanoparticles also have different levels of emission and the emission peaks are all consistent with that of free FITC. As shown in Figure 1E, the color of each nanoparticle solution changed after fluorescence labeling. FITC-labeled nanoparticles also emit similar yellow-green fluorescence under ultraviolet light compared with the free FITC solution. This also proved that the prepared gold nanoparticles have fluorescent characteristics that are suitable for use in the CLSM study.
Size is a critically important factor in this study, so more than one method was used to measure the particle size to ensure the credibility of the following experiment. All the data (Figure S8 and Figure S9 in Supporting Information) are summarized in Table 1. As mentioned above, the different UV–vis absorption peaks reflect the different sizes of the gold nanoparticles. Compared with TEM images, DLS characterization gives the true hydrodynamic diameter, which reflects the real dispersed state of gold nanoparticles in solution, especially when they are surface-modified by polymers. For example, the hydrodynamic size of the 6 nm Au-TIOP NPs changed from 6.11 to 10.02 nm after FITC modification, which means that the PEG-FITC polymer added a 3–4 nm thick surface cover to the nanoparticles. The ζ-potential changes were also observed, as shown in Table 1.
Distribution of Au-TIOP NPs with Different Sizes in MCF-7 Cells
Bio-TEM was conducted to observe the distribution of Au-TIOP NPs in MCF-7 cells. After treatment with different sizes of Au-TIOP NPs, cells were harvested and sectioned for bio-TEM analysis. As shown in Figure 2, 2 and 6 nm Au-TIOP NPs were located in both cytoplasm and nucleus. More importantly, they were found to be monodispersed in the cell, not aggregated. In contrast, the larger Au-TIOP NPs (10 and 16 nm) were located in the cytoplasm, and none of them entered the cell nucleus. Additionally, most of them were trapped in vesicles. We reasoned that the size-dependent nuclear distribution of the Au-TIOP NPs is related to the nuclear pore complex (NPC), which is reported to allow cargos smaller than 9 nm to enter the nucleus freely.38 This hypothesis was further proved by our next experiment.
Figure 2.

Bio-TEM images of the localization of 2, 6, 10, and 16 nm Au-TIOP NPs in MCF-7 cells after 24 h treatment.
Bio-TEM results indicate that the smaller 2 and 6 nm Au-TIOP NPs have the ability to enter the nuclei of tumor cells. Thereafter, the quantitative distribution of Au-TIOP NPs in cell nucleus was determined by ICP-MS. Cell nuclei were extracted for ICP-MS analysis after incubation for different times with various concentrations of Au-TIOP NPs. The number of NPs was then calculated according to a previously reported method.16 As shown in Figure 3A(a), when cells were incubated with increasing concentrations (1 to 100 nM in particle number dose) of 2 nm Au-TIOP NPs for 24 h, the number of NPs in cells and nuclei increased and the intranuclear percentage of Au-TIOP NPs in whole cells reached about 40% when the incubation concentration was raised to 100 nM. When the incubation concentration was 100 nM, the amount of 2 nm Au-TIOP NPs in cells and nuclei also increased with increasing incubation time (Figure 3A(b)), which is in accordance with the result in Figure 3A(a). Similar results were obtained for 6 nm Au-TIOP NPs (Figure 3B), but a significant difference was that the number of 6 nm Au-TIOP NPs in the cells and nuclei was about 1/4 of that for cells treated with 2 nm Au-TIOP NPs under the same conditions. This also demonstrated that the smaller 2 nm Au-TIOP NPs can enter into cells and nuclei more easily and quickly than the 6 nm Au-TIOP NPs, which is consistent with the Bio-TEM images.
Figure 3.

Number of 2 and 6 nm Au-TIOP NPs in cells and nuclei. (A) Number of 2 nm Au-TIOP NPs (a) after 24 h treatment at concentrations of 1, 10, and 100 nM; (b) after treatment for 3, 6, 12, and 24 h at a concentration of 100 nM. (B) Number of 6 nm Au-TIOP NPs (a) after 24 h treatment at concentrations of 1, 10, and 100 nM; (b) after treatment for 3, 6, 12, and 24 h at a concentration of 100 nM. All the volumes of Au-TIOP NPs solution added were 1 mL, and the concentration was in particle number dose.
Distribution of Au-FITC NPs with Different Sizes in MCF-7 Cells
The fluorescent molecule FITC was conjugated to Au-TIOP NPs for CLSM. As shown in Figure 4, only 2 nm Au-FITC NP-treated MCF-7 cells exhibited an FITC signal (indicating the location of the nanoparticles) in both cytoplasm and nucleus. Cells treated with the other three larger nanoparticles exhibited an FITC signal in the cytoplasm only. From these results, we concluded that the 2 nm Au-FITC NPs were capable of entering the cell nucleus and 6, 10, and 16 nm Au-FITC NPs were not. This raises the question why the 6 nm nanoparticles were located in the nucleus in the TEM experiment but in the cytoplasm only in the CLSM experiment. This result could be interpreted in terms of the size increase which occurred when the Au-TIOP NPs were conjugated to FITC using PEG as a linker. The hydrodynamic diameter of the 6 nm Au-FITC NPs was actually 10.02 nm after fluorescence labeling, as shown in Table 1. This final size was larger than the 9 nm threshold of the NPC, thus limiting the free entry of the nanoparticles into the nucleus.
Figure 4.

Intracellular localization of Au-FITC NPs with different sizes in MCF-7 cells observed by confocal laser scanning microscopy. MCF-7S cells were cultured in confocal dishes and incubated with Au-FITC NPs (green) of different sizes for 24 h. Cells were then washed with PBS three times, and nuclei were stained by Hoechest 33342 (blue). Cells were fixed by paraformaldehyde and observed by confocal microscopy (exFITC = 488 nm, exHoechest = 405 nm).
Ultrasmall Au-TIOP NPs for Carrying DNA into the Nucleus
Triplex-forming oligonucleotides are a kind of single-stranded DNA which can bind to the major groove of double-stranded DNA and form a triplex.39 The binding between TFOs and double-stranded DNA follows the principle of Hoogsteen base pairing; therefore, a TFO can be designed to bind to a specific double-stranded DNA sequence. TFOs have been used to bind to the transcription initiation site of a target gene and inhibit its transcription, eventually silencing the gene. However, TFOs are quite vulnerable in cells and blood due to high DNase activity. High concentrations of TFOs are needed to achieve gene down-regulation. Many studies reported that DNA stability was increased significantly by conjugating the DNA with gold nanoparticles.29,40,41 The high salt concentration around gold nanoparticles and steric hindrance between DNase and the conjugated DNA strand successfully protected the DNA. Thus, we decided to use our gold nanoparticles as a TFO carrier. As TFOs are only effective in the cell nucleus, we took advantage of the ability of 2 nm Au-TIOP NPs to enter the nucleus and chose 2 nm Au-TIOP NPs as the TFO carrier.
We selected a TFO named POY2T from a previous report.42 POY2T is a 23 nt oligonucleotide (TGGGTGGGTGGTTTGTTTTTGGG) which can bind to the P2 promoter of the c-myc oncogene and down-regulate c-myc expression. Figure S10 in Supporting Information shows that POY2T only inhibits cell proliferation at high concentrations (5 and 10 μM). In order to demonstrate the sequence-specific toxicity of POY2T, another oligonucleotide named POY2M with a scrambled sequence (GGTGTGTTGTTGGTGGTGTGTTG) derived from POY2T was used as a control TFO sequence. It is nontoxic to MCF-7 cells at the same concentration compared with POY2T (Figure S10 in Supporting Information). We concluded that POY2T was toxic to MCF-7 cells by down-regulating c-myc gene expression and inhibiting cell proliferation.43 Therefore, POY2T is an effective TFO for use in our study. 5′ Amino-modified POY2T was linked to Au-TIOP NPs through the carboxyl group of tiopronin (Figure S11 in Supporting Information). The TFO-modified nanoparticles were named as Au-POY2T NPs (Figure 5A and Figure S12 in Supporting Information). MTT assays showed that Au-POY2T NPs inhibit cell viability by about 30% at a low concentration (1 μM), and POY2T was not toxic at this concentration (Figure 5B). Au NPs will interfere with cell functions according to a previous report.44 The potential toxicity of amino modification should also be noticed. NH2-POY2T or Au-TIOP NPs showed no cytotoxicity at the same concentration of 1 μM Au-POY2T according to our result (Figure 5B), which means that the decrease in cell viability after treatment with Au-POY2T NPs was not caused by Au-TIOP NPs or amino modification of POY2T. Moreover, Au-POY2M was set as a control of Au-POY2T to rule out the interference of possible toxic materials introduced during the synthesis. The result shows that Au-POY2M was not more toxic than free POY2M, indicating that the increased toxicity of Au-POY2T NPs was not caused by the conjugating process. It is the outcome of improved stability of POY2T in culture medium and cells after conjugation with Au NPs.
Figure 5.

Cytotoxicity and gene regulation evaluation of Au-POY2T NPs in MCF-7 cells. (A) TEM image of as-synthesized Au-POY2T NPs. (B) Cytotoxicity evaluation of Au-POY2T NPs compared to control, Au NPs, POY2T, and NH2-POY2T (at 1 μM in POY2T). (C) C-myc mRNA level determined by real-time PCR after 24 h treatment of 5 μM POY2T and Au-POY2T. (D) C-myc protein level determined by Western blotting after 24 h treatment of 5 μM POY2T and Au-POY2T.
Real-time PCR and Western blot analyses were conducted to determine c-myc expression after Au-POY2T NP treatment. Real-time PCR results revealed that the c-myc mRNA level was decreased to about 50% in Au-POY2T NP-treated MCF-7 cells, much lower than in the POY2T-treated cells (85%) (Figure 5C and Figure S13 in Supporting Information). Western blotting also confirmed this result. As shown in Figure 5D and Figure S14 in Supporting Information, the c-myc protein level in cells was reduced significantly after treatment with Au-POY2T NPs for 24 h, and POY2T only showed weak gene silencing ability compared to Au-POY2T NPs.
Nucleus, as the center for cell proliferation regulation, plays an important role in tumor metastasis. Killing tumor cells by interfering with their gene expression in the nucleus has long been catching scientist’s eyes. Antisense oligonucleotides are potential candidates for gene therapies because of their easy manipulation. Although the antisense approach is attractive, it suffers from one major disadvantage: the number of mRNA molecules in a cell is huge, which makes it difficult to completely inhibit a specific mRNA. Moreover, blocking translation of mRNA does not prevent mRNA from reproduction because of the feedback mechanisms that lead to increased mRNA transcription in antisense therapy. Triplex-forming oligonucleotides are molecules that bind to specific duplex DNA sequence and can selectively modulate the expression of genes. It is therefore a much more practical approach that directly blocks gene transcription to bring down the mRNA concentration in a more efficient and long-lasting way.45
However, the inefficient delivery of TFOs to the targeted cell greatly challenges TFOs to be used as antigene agents. Oligonucleotides have a much greater molecular mass and are polyanionic compared to small-molecule pharmaceuticals, and these make the cellular uptake of TFOs by passive diffusion inefficient. Therefore, simple addition of TFOs to culture medium could hardly reach the therapeutic concentration. Previously, cell-penetrating peptides have been used to increase the uptake of TFOs. Other ways, such as single-cell microinjection, comixture of oligonucleotides with cationic liposomes, and electroporation, have also been tried.46−50 However, none of these methods could directly deliver TFOs into the nucleus. In our method, we directly delivered TFOs using our 2 nm Au NP carriers, while avoiding the stress caused by microinjection and electroporation, etc.
Moreover, even triplex-forming oligonucleotides are successfully internalized into cells, and they do not effectively down-regulate the targeted gene. It is because unmodified oligonucleotides are prone to be degraded by endogenous DNase enzymes, resulting in little intact TFOs in the media that finally enter the nucleus. Many efforts have been made to improve the stability of triplex-forming oligonucleotides under physiological conditions. Typical approaches include modifying the nucleosides of TFO or incorporation of modified bases. However, while these modifications are carried out to achieve resistance to undesirable DNase degradation, decreased binding to the target sequence or unspecific binding happens sometimes. Gold nanoparticles have a high affinity for oligonucleotides. Our method of using ultrasmall Au-TIOP NPs functionalized with amino-modified TFOs to deliver TFOs stably to cells in the culture media and the binding between gold nanoparticles and the TFOs is very selective and cooperative. Thus, not only enhanced entry of TFOs to the nucleus is achieved but also protection of oligonucleotide is achieved while keeping its specificity and high transcription inhibition efficacy. During the binding of TFO with the DNA double helix in the major groove, Au NPs and other TFOs may cause steric hindrance, but 2 nm Au NPs are even smaller than the width of the major groove (22 Å).51 Oligonucleotide is also considered to be a flexible material, and only a few TFOs are near the hybridization site. Thus, the assumed steric hindrance caused by Au NPs and other TFOs is not likely to stop hybridization of TFO with the target site effectively. Another question that should be discussed is that only one TFO in Au-POY2T takes part in the hybridization, then higher overall doses of TFO are needed theoretically. We think that the improved stability of TFO on Au NPs may be the answer to this question. Mirkin et al. reported that the half-life of DNA on Au NPs was prolonged by 4.3 times compared to that of free DNA.40 Au NPs can protect TFOs effectively on their way from culture medium to cell nuclei, while free TFOs go unprotected. Higher TFO concentration in nuclei could then be achieved by a Au-TFO delivery system at the same treatment concentration. Slower diffusion of larger Au-TFO particles than that of free TFOs according to Stokes’ law is also a problem which may makes Au-TFO work slowly. However, the diffusion speed of Au-TFO and free TFO will not be very different concerning the ultrasmall size of Au NPs. Moreover, stability is the key in TFO-mediated gene regulation, not diffusion speed. We therefore think that slower diffusion speed will not compromise the gene regulation efficiency of Au-TFO.
Conclusion
In this work, gold nanoparticles with different sizes and consistent surface properties were selected as a typical model to explore the interaction between nanoparticles and the nucleus of breast cancer cells. We used various means to systematically study the entry of Au-TIOP NPs into the cell nucleus and concluded that nuclear entry is critically dependent on size. This work presents new evidence about the entry of ultrasmall Au-TIOP NPs into the cell nucleus and provides a new strategy for regulating gene expression by delivering TFOs into the nucleus with ultrasmall gold nanoparticles. Our future work also aims at using these ultrasmall nanoparticles to realize nuclear targeting for biomedical application. More importantly, guidelines were provided to choose nanocarriers for different purposes in the field of biomedicine.
Experimental Section
Chemicals
Sodium borohydride (≥98.0%), gold(III) chloride trihydrate (≥99.9%, HAuCl4·3H2O), trisodium citrate tribasic dihydrate (≥99.0%), N-(2-mercaptopropionyl)glycine (tiopronin), N-hydroxysuccinimide (98%, NHS, C4H5NO3), N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride (EDC, C8H17N3·HCl), and fluorescein isothiocyanate isomer I (≥90%, FITC, C21H11NO5S) were purchased from Sigma-Aldrich (St Louis, USA). l-(+)-Ascorbic acid (99%) was supplied by Acros (USA). Amino-modified poly(ethylene glycol) (PEG-2NH2, MW = 2000, 99%) was supplied by Beijing SeaskyBio Technology Company. Nitric acid and hydrogen peroxide (MOS grade) were provided by Beijing Chemical Reagents Institute (China). Gold stock standard solution (1000 μg/mL) was obtained from the National Analysis Center for Iron and Steel, China. Unless otherwise noted, all chemicals were used as received without further purification, and Milli-Q water (18.2 MΩ·cm, Millipore System Inc.) was used throughout this study.
Cell Culture
The MCF-7 (human breast cancer cell line) cell was maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 4.5 g/L glucose and 10% fetal bovine serum in a water-jacketed CO2 incubator (Forma Series II 3110, Thermo Fisher Scientific Inc., USA) providing a humidified atmosphere containing 5% CO2 at 37 °C.
Cell Viability Assay
MCF-7 cells were seeded at 5 × 103 cells per well in a 96-well plate, preincubated for 24 h, then incubated with POY2T (0.1, 1, 2.5, 5, and 10 μM), POY2M (0.1, 1, 2.5, 5, and 10 μM), Au-POY2T NPs (at 1 μM in POY2T), NH2-POY2T (at 1 μM in POY2T), or Au-TIOP NPs (as a control of Au-POY2T NPs) for 24 h. The medium was then replaced with 100 μL of 0.5 mg/mL MTT, and after 3 h, the MTT solution was replaced with 150 μL of DMSO solution. The absorbance at 570 nm of each well was measured by a microplate reader (Infinite M200, Tecan, Durham, USA). The absorbance at 630 nm was also measured as a reference. Untreated cells in medium were used as control. All standard deviations were calculated from three replicates.
Isolation of Cell Nuclei and Qualitative Determination of Au Content
MCF-7 cells (105) were seeded in 6-well plates 24 h in advance. Afterward, 1 mL of Au-TIOP NPs of 2 and 6 nm at concentrations of 1, 10, and 100 nM (in particle number dose) was added to the wells and incubated for 24 h or incubated for 3, 6, 12, and 24 h at a concentration of 100 nM. Then the cells were washed with PBS, trypsinized, and centrifuged for 3 min at 179g. The treated cells were then resuspended in 2 mL of PBS and divided into two equal groups. One group was dissolved in aqua regia for nitrolysis and then analyzed by ICP-MS for gold element quantification. The other part was handled for cell nucleus extraction using a nuclear extraction kit (SN000, Solarbio, Shanghai, China). In brief, the sample was grinded in lysis buffer for 3 min, centrifuged at 700g for 5 min, resuspended in lysis buffer, precipitated in medium buffer, and stored in store buffer. All procedures were carried out at 4 °C quickly to keep nucleus integrity. The extracted nuclei were also analyzed by ICP-MS after nitrolysis. The percentage of Au NPs located in the cell nucleus was calculated according to the Au content in the isolated nuclei compared to that in the whole cells.
Subcellular Localization of Au-FITC NPs
MCF-7 cells were seeded into 35 mm glass dishes, incubated at 37 °C for 24 h, then cultured with Au-FITC NPs at a gold content concentration of 5 μM at 37 °C for 24 h. Cells were imaged using a TCS SP2 (Leica Microsystems GmbH Wetzlar, Germany) confocal microscope. Hoechest 33342 was used to stain cell nuclei. FITC was excited by a 488 nm laser, while Hoechest 33342 was excited by a 405 nm laser.
Bio-TEM Observation of Au-TIOP NP-Treated MCF-7 Cells
Bio-TEM was carried out to observe the distribution of Au-TIOP NPs in treated MCF-7 cells. Briefly, MCF-7 cells were seeded in 6-well plates and incubated with 1 mL of Au-TIOP NPs at a concentration of 100 nM for 24 h, washed with PBS gently twice, and fixed overnight using 3% glutaraldehyde solution at room temperature. This was followed by secondary fixation with 1% osmium tetraoxide, then dehydration in a gradient ethanol series. Cells were finally embedded in Epon resin after 3 day polymerization at 60 °C. Embedded samples were sectioned (50–60 nm in thickness) and examined under an electron microscope (HT7700, 120 kV, Hitachi, Japan). Uranyl acetate staining was not used in this study to avoid uranyl acetate precipitation interfering with the TEM observation.
C-myc Gene Expression
C-myc gene expression, in both mRNA and protein levels, was quantitatively determined by reverse transcriptase polymerase chain reaction (RT-PCR), real-time PCR, and Western blotting. MCF-7 cells were incubated with Au-POY2T NPs and POY2T at a POY2T concentration of 5 μM for 24 h. For RT-PCR, total mRNA was extracted from MCF-7 cells, and first-strand cDNA was synthesized using a PrimeScript first-strand cDNA synthesis kit (Takara, Shiga, Japan). Real-time PCR analysis of c-myc was conducted for quantitative analysis of the reduction in c-myc transcription. GAPDH was used as a reference gene. Specific forward and reverse primer sequences were as follows: c-myc fwd, 5′-TGAGGAGACACCGCCCAC-3′; c-myc rev, 5′-CAACATCGATTTCTCTCATCTTC-3′; GAPDH fwd, 5′-GACTTCAACAGCAACTCCCAC-3′; GAPDH rev, 5′-TCCACCACCCTGTTGCTGTA-3′. PCR parameters were as follows: one cycle of 2 min at 95 °C, followed by 20 s at 95 °C, 20 s at 55.6 °C, and 40 s at 72 °C for 45 cycles.
For Western blotting, protein lysates from cells treated with 5 μM POY2T and Au-POY2T for 24 h were extracted in TNE lysis buffer (0.5% NP-40, 10 mM Tris, 150 mM NaCl, 1 mM EDTA and protease inhibitors). Total protein (50 mg) was separated by 10% SDS-PAGE gel, transferred to a PVDF membrane, treated with primary rabbit monoclonal c-myc (diluted 1:1000, BS246; Bioworld Technology, Inc., Minnesota, USA), and then immunoblotted with peroxidase-conjugated secondary antibody (1:5000, zsBio, Beijing, China).
Acknowledgments
This work was supported by the Chinese Natural Science Foundation project (81171455), a National Distinguished Young Scholars grant (31225009) from the National Natural Science Foundation of China, the National Key Basic Research Program of China (2009CB930200 and SS2014AA020708), the Chinese Academy of Sciences (CAS) “Hundred Talents Program” (07165111ZX), the CAS Knowledge Innovation Program and the State High-Tech Development Plan (2012AA020804). This work was also supported in part by NIH/NIMHD 8 G12 MD007597, and USAMRMC W81XWH-10-1-0767 grants. The authors also appreciate the support by the “Strategic Priority Research Program” of the Chinese Academy of Sciences (XDA09030301).
Author Contributions
∥ S.H. and S.J. contributed equally to this work.
Supporting Information Available
Synthesis of Au-TIOP NPs and Au-FITC NPs with different sizes, characterization of Au-TIOP NPs and Au-FITC NPs with different sizes, agarose gel electrophoresis, and ICP-MS. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Dreaden E. C.; Alkilany A. M.; Huang X. H.; Murphy C. J.; El-Sayed M. A. The Golden Age: Gold Nanoparticles for Biomedicine. Chem. Soc. Rev. 2012, 41, 2740–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh P.; Han G.; De M.; Kim C. K.; Rotello V. M. Gold Nanoparticles in Delivery Applications. Adv. Drug Delivery Rev. 2008, 60, 1307–1315. [DOI] [PubMed] [Google Scholar]
- Pissuwan D.; Niidome T.; Cortie M. B. The Forthcoming Applications of Gold Nanoparticles in Drug and Gene Delivery Systems. J. Controlled Release 2011, 149, 65–71. [DOI] [PubMed] [Google Scholar]
- Dreaden E. C.; Mackey M. A.; Huang X. H.; Kang B.; El-Sayed M. A. Beating Cancer in Multiple Ways Using Nanogold. Chem. Soc. Rev. 2011, 40, 3391–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K. Y.; Ma H. L.; Liu J.; Huo S. D.; Kumar A.; Wei T.; Zhang X.; Jin S. B.; Gan Y. L.; Wang P. C.; et al. Size-Dependent Localization and Penetration of Ultrasmall Gold Nanoparticles in Cancer Cells, Multicellular Spheroids, and Tumors in Vivo. ACS Nano 2012, 6, 4483–4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jong W. H.; Hagens W. I.; Krystek P.; Burger M. C.; Sips A. J. A. M.; Geertsma R. E. Particle Size-Dependent Organ Distribution of Gold Nanoparticles after Intravenous Administration. Biomaterials 2008, 29, 1912–1919. [DOI] [PubMed] [Google Scholar]
- Pan Y.; Neuss S.; Leifert A.; Fischler M.; Wen F.; Simon U.; Schmid G.; Brandau W.; Jahnen-Dechent W. Size-Dependent Cytotoxicity of Gold Nanoparticles. Small 2007, 3, 1941–1949. [DOI] [PubMed] [Google Scholar]
- Zhang X. D.; Wu D.; Shen X.; Chen J.; Sun Y. M.; Liu P. X.; Liang X. J. Size-Dependent Radiosensitization of PEG-Coated Gold Nanoparticles for Cancer Radiation Therapy. Biomaterials 2012, 33, 6408–6419. [DOI] [PubMed] [Google Scholar]
- Lesniak A.; Salvati A.; Santos-Martinez M. J.; Radomski M. W.; Dawson K. A.; Aberg C. Nanoparticle Adhesion to the Cell Membrane and Its Effect on Nanoparticle Uptake Efficiency. J. Am. Chem. Soc. 2013, 135, 1438–1444. [DOI] [PubMed] [Google Scholar]
- Jiang W.; Kim B. Y. S.; Rutka J. T.; Chan W. C. W. Nanoparticle-Mediated Cellular Response Is Size-Dependent. Nat. Nanotechnol. 2008, 3, 145–150. [DOI] [PubMed] [Google Scholar]
- Zhu M. T.; Li Y. Y.; Shi J.; Feng W. Y.; Nie G. J.; Zhao Y. L. Exosomes as Extrapulmonary Signaling Conveyors for Nanoparticle-Induced Systemic Immune Activation. Small 2012, 8, 404–412. [DOI] [PubMed] [Google Scholar]
- Huo S. D.; Ma H. L.; Huang K. Y.; Liu J.; Wei T.; Jin S. B.; Zhang J. C.; He S. T.; Liang X. J. Superior Penetration and Retention Behavior of 50 nm Gold Nanoparticles in Tumors. Cancer Res. 2013, 73, 319–330. [DOI] [PubMed] [Google Scholar]
- Chauhan V. P.; Popovic Z.; Chen O.; Cui J.; Fukumura D.; Bawendi M. G.; Jain R. K. Fluorescent Nanorods and Nanospheres for Real-Time In Vivo Probing of Nanoparticle Shape-Dependent Tumor Penetration. Angew. Chem., Int. Ed. 2011, 50, 11417–11420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B.; Han G.; Toley B. J.; Kim C.-k.; Rotello V. M.; Forbes N. S. Tuning Payload Delivery in Tumour Cylindroids Using Gold Nanoparticles. Nat. Nanotechnol. 2010, 5, 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrault S. D.; Walkey C.; Jennings T.; Fischer H. C.; Chan W. C. Mediating Tumor Targeting Efficiency of Nanoparticles through Design. Nano Lett. 2009, 9, 1909–1915. [DOI] [PubMed] [Google Scholar]
- Chithrani B. D.; Ghazani A. A.; Chan W. C. W. Determining the Size and Shape Dependence of Gold Nanoparticle Uptake into Mammalian Cells. Nano Lett. 2006, 6, 662–668. [DOI] [PubMed] [Google Scholar]
- Chen J.; Saeki F.; Wiley B. J.; Cang H.; Cobb M. J.; Li Z.-Y.; Au L.; Zhang H.; Kimmey M. B.; Li X.; et al. Gold Nanocages: Bioconjugation and Their Potential Use as Optical Imaging Contrast Agents. Nano Lett. 2005, 5, 473–477. [DOI] [PubMed] [Google Scholar]
- Hirsch L. R.; Stafford R. J.; Bankson J. A.; Sershen S. R.; Rivera B.; Price R. E.; Hazle J. D.; Halas N. J.; West J. L. Nanoshell-Mediated Near-Infrared Thermal Therapy of Tumors under Magnetic Resonance Guidance. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 13549–13554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykman L.; Khlebtsov N. Gold Nanoparticles in Biomedical Applications: Recent Advances and Perspectives. Chem. Soc. Rev. 2012, 41, 2256–2282. [DOI] [PubMed] [Google Scholar]
- Dam D. H. M.; Lee J. H.; Sisco P. N.; Co D. T.; Zhang M.; Wasielewski M. R.; Odom T. W. Direct Observation of Nanoparticle–Cancer Cell Nucleus Interactions. ACS Nano 2012, 6, 3318–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang B.; Mackey M. A.; El-Sayed M. A. Nuclear Targeting of Gold Nanoparticles in Cancer Cells Induces DNA Damage, Causing Cytokinesis Arrest and Apoptosis. J. Am. Chem. Soc. 2010, 132, 1517–1519. [DOI] [PubMed] [Google Scholar]
- Dobson J. Gene Therapy Progress and Prospects: Magnetic Nanoparticle-Based Gene Delivery. Gene Ther. 2006, 13, 283–287. [DOI] [PubMed] [Google Scholar]
- Patil S. D.; Rhodes D. G.; Burgess D. J. DNA-Based Therapeutics and DNA Delivery Systems: A Comprehensive Review. AAPS J. 2005, 7, E61–E77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus M. T.; Sharp P. A. Gene Silencing in Mammals by Small Interfering RNAs. Nat. Rev. Genet. 2002, 3, 737–747. [DOI] [PubMed] [Google Scholar]
- Lebedeva I.; Stein C. A. Antisense Oligonucleotides: Promise and Reality. Annu. Rev. Pharmacol. 2001, 41, 403–419. [DOI] [PubMed] [Google Scholar]
- Jason T. L. H.; Koropatnick J.; Berg R. W. Toxicology of Antisense Therapeutics. Toxicol. Appl. Pharmacol. 2004, 201, 66–83. [DOI] [PubMed] [Google Scholar]
- Rosi N. L.; Giljohann D. A.; Thaxton C. S.; Lytton-Jean A. K. R.; Han M. S.; Mirkin C. A. Oligonucleotide-Modified Gold Nanoparticles for Intracellular Gene Regulation. Science 2006, 312, 1027–1030. [DOI] [PubMed] [Google Scholar]
- Jin R. C.; Wu G. S.; Li Z.; Mirkin C. A.; Schatz G. C. What Controls the Melting Properties of DNA-Linked Gold Nanoparticle Assemblies?. J. Am. Chem. Soc. 2003, 125, 1643–1654. [DOI] [PubMed] [Google Scholar]
- Lytton-Jean A. K. R.; Mirkin C. A. A Thermodynamic Investigation into the Binding Properties of DNA Functionalized Gold Nanoparticle Probes and Molecular Fluorophore Probes. J. Am. Chem. Soc. 2005, 127, 12754–12755. [DOI] [PubMed] [Google Scholar]
- Goldsborough A. S.; Handley M. D.; Dulcey A. E.; Pluchino K. M.; Kannan P.; Brimacombe K. R.; Hall M. D.; Griffiths G.; Gottesman M. M. Collateral Sensitivity of Multidrug-Resistant Cells to the Orphan Drug Tiopronin. J. Med. Chem. 2011, 54, 4987–4997. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Dahl J. A.; Maddux B. L. S.; Hutchison J. E. Toward Greener Nanosynthesis. Chem. Rev. 2007, 107, 2228–2269. [DOI] [PubMed] [Google Scholar]
- Petkov V.; Peng Y.; Williams G.; Huang B. H.; Tomalia D.; Ren Y. Structure of Gold Nanoparticles Suspended in Water Studied by X-ray Diffraction and Computer Simulations. Phys. Rev. B 2005, 72, 195402. [Google Scholar]
- Li Z. P.; Duan X. R.; Liu C. H.; Du B. A. Selective Determination of Cysteine by Resonance Light Scattering Technique Based on Self-Assembly of Gold Nanoparticles. Anal. Biochem. 2006, 351, 18–25. [DOI] [PubMed] [Google Scholar]
- Albanese A.; Chan W. C. W. Effect of Gold Nanoparticle Aggregation on Cell Uptake and Toxicity. ACS Nano 2011, 5, 5478–5489. [DOI] [PubMed] [Google Scholar]
- Ao L. M.; Gao F.; Pan B. F.; He R.; Cui D. X. Fluoroimmunoassay for Antigen Based on Fluorescence Quenching Signal of Gold Nanoparticles. Anal. Chem. 2006, 78, 1104–1106. [DOI] [PubMed] [Google Scholar]
- Loumaigne M.; Praho R.; Nutarelli D.; Werts M. H. V.; Debarre A. Fluorescence Correlation Spectroscopy Reveals Strong Fluorescence Quenching of FITC Adducts on PEGylated Gold Nanoparticles in Water and the Presence of Fluorescent Aggregates of Desorbed Thiolate Ligands. Phys. Chem. Chem. Phys. 2010, 12, 11004–11014. [DOI] [PubMed] [Google Scholar]
- Foss C. A.; Hornyak G. L.; Stockert J. A.; Martin C. R. Template-Synthesized Nanoscopic Gold Particles-Optical-Spectra and the Effects of Particle-Size and Shape. J. Phys. Chem. 1994, 98, 2963–2971. [Google Scholar]
- Paine P. L.; Moore L. C.; Horowitz S. B. Nuclear-Envelope Permeability. Nature 1975, 254, 109–114. [DOI] [PubMed] [Google Scholar]
- Jain A.; Magistri M.; Napoli S.; Carbone G. M.; Catapano C. V. Mechanisms of Triplex DNA-Mediated Inhibition of Transcription Initiation in Cells. Biochimie 2010, 92, 317–320. [DOI] [PubMed] [Google Scholar]
- Seferos D. S.; Prigodich A. E.; Giljohann D. A.; Patel P. C.; Mirkin C. A. Polyvalent DNA Nanoparticle Conjugates Stabilize Nucleic Acids. Nano Lett. 2009, 9, 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han G.; Martin C. T.; Rotello V. M. Stability of Gold Nanoparticle-Bound DNA toward Biological, Physical, and Chemical Agents. Chem. Biol. Drug Des. 2006, 67, 78–82. [DOI] [PubMed] [Google Scholar]
- McGuffie E. M.; Pacheco D.; Carbone G. M. R.; Catapano C. V. Antigene and Antiproliferative Effects of a c-Myc-Targeting Phosphorothioate Triple Helix-Forming Oligonucleotide in Human Leukemia Cells. Cancer Res. 2000, 60, 3790–3799. [PubMed] [Google Scholar]
- Dang C. V. c-Myc Target Genes Involved in Cell Growth, Apoptosis, and Metabolism. Mol. Cell. Biol. 1999, 19, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leifert A.; Pan Y.; Kinkeldey A.; Schiefer F.; Setzler J.; Scheel O.; Lichtenbeld H.; Schmid G.; Wenzel W.; Jahnen-Dechent W.; et al. Differential hERG Ion Channel Activity of Ultrasmall Gold Nanoparticles. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 8004–8009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guntaka R. V.; Varma B. R.; Weber K. T. Triplex-Forming Oligonucleotides as Modulators of Gene Expression. Int. J. Biochem. Cell Biol. 2003, 35, 22–31. [DOI] [PubMed] [Google Scholar]
- Rogers F. A.; Manoharan M.; Rabinovitch P.; Ward D. C.; Glazer P. M. Peptide Conjugates for Chromosomal Gene Targeting by Triplex-Forming Oligonucleotides. Nucleic Acids Res. 2004, 32, 6595–6604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris J. D.; Lemoine N. R. Strategies for Targeted Gene Therapy. Trends Genet. 1996, 12, 400–405. [DOI] [PubMed] [Google Scholar]
- Stein C. A.; Cheng Y. C. Antisense Oligonucleotides as Therapeutic Agents: Is the Bullet Really Magical. Science 1993, 261, 1004–1012. [DOI] [PubMed] [Google Scholar]
- Felgner J. H.; Kumar R.; Sridhar C. N.; Wheeler C. J.; Tsai Y. J.; Border R.; Ramsey P.; Martin M.; Felgner P. L. Enhanced Gene Delivery and Mechanism Studies with a Novel Series of Cationic Lipid Formulations. J. Biol. Chem. 1994, 269, 2550–2561. [PubMed] [Google Scholar]
- Lewis J. G.; Lin K. Y.; Kothavale A.; Flanagan W. M.; Matteucci M. D.; DePrince R. B.; Mook R. A.; Hendren R. W.; Wagner R. W. A Serum-Resistant Cytofectin for Cellular Delivery of Antisense Oligodeoxynucleotides and Plasmid DNA. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 3176–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing R.; Drew H.; Takano T.; Broka C.; Tanaka S.; Itakura K.; Dickerson R. E. Crystal-Structure Analysis of a Complete Turn of B-DNA. Nature 1980, 287, 755–758. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.