

Abstract
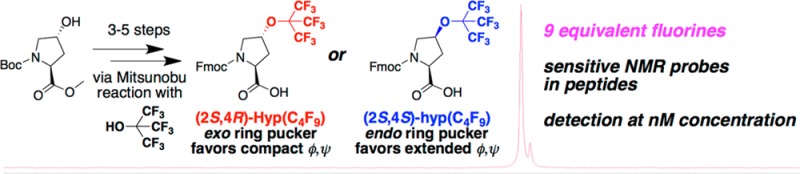
(2S,4R)- and (2S,4S)-perfluoro-tert-butyl 4-hydroxyproline were synthesized (as Fmoc-, Boc-, and free amino acids) in 2–5 steps. The key step of each synthesis was a Mitsunobu reaction with perfluoro-tert-butanol, which incorporated a perfluoro-tert-butyl group, with nine chemically equivalent fluorines. Both amino acids were incorporated in model α-helical and polyproline helix peptides. Each amino acid exhibited distinct conformational preferences, with (2S,4R)-perfluoro-tert-butyl 4-hydroxyproline promoting polyproline helix. Peptides containing these amino acids were sensitively detected by 19F NMR, suggesting their use in probes and medicinal chemistry.
Fluorinated amino acids have unique properties, due to the hydrophobicity and electronegativity of fluorine and the magnetic properties of the sensitive spin-1/2 19F nucleus.1−10 The special nature of fluorine has led to broad interest in the incorporation of fluorinated amino acids in small molecules, peptides, and proteins, for applications in medicinal chemistry, in the design of stabilized proteins, and in NMR and MRI approaches to protein detection and imaging.11−34 Fluorinated amino acids allow the specific, quantitative detection of individual species in complex media, including the observation of protein synthesis and folding in living E. coli cells, due to the low background of fluorine in most environments. The sensitivity of fluorine NMR probes depends on the concentration of the molecule, the coupling patterns of the 19F nuclei to other fluorines and to hydrogens in the molecule, and the number of chemically equivalent fluorines in the molecule. For probes to be employed biologically, it is ideal to be able to work at the lowest concentration of molecule possible due to the typical nanomolar protein concentrations and affinities observed in protein–protein interactions. The most sensitive protein-based probes of biological activity employed to date have utilized aryl trifluoromethyl groups, which exhibit sharp singlets by 19F NMR of intensity 3 (one aryl-CF3 group) or 6 (two symmetrically related, chemically equivalent aryl-CF3 groups).35−37 In contrast, in the most widely used direct analogue of a canonical α-amino acid, hexafluoroleucine (6 fluorines), the two trifluoromethyl groups are diastereotopic, exhibiting separate resonances, and each is coupled to the methine hydrogen, resulting in reduced peak intensity due to each trifluoromethyl group existing as a doublet (four total peaks observed).18,38−41
We previously described the synthesis of tetrapeptides containing the novel amino acids (2S,4R)- and (2S,4S)-perfluoro-tert-butyl 4-hydroxyproline (Figure 1).42 Peptides with these amino acids were prepared by the method of proline editing, in which a hydroxyproline amino acid within a fully synthesized peptide is site-specifically modified via reaction on solid phase to incorporate novel functionalities.42,43 The perfluoro-tert-butyl group of these amino acids, in contrast to most other fluorinated amino acids, exhibits fluorines that are singlets by 19F NMR, here with nine chemically equivalent fluorines that are uncoupled to any other nuclei.37,42,44,45 The unique magnetic nature of the perfluoro-tert-butyl group, combined with the large chemical shift dispersion of 19F nuclei and the general absence of fluorine in typical biological environments, suggests the possibility of highly sensitive detection of molecules containing this functional group in diverse media.
Figure 1.
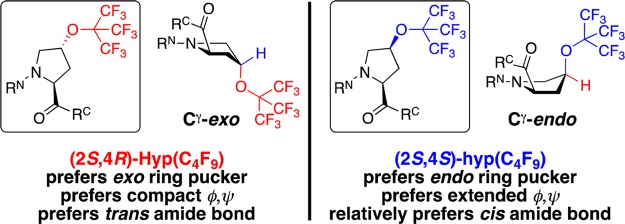
(2S,4R)- and (2S,4S)-perfluoro-tert-butyl 4-hydroxyprolines and their expected conformational preferences, in which the 4-substituents are in a pseudoaxial position on the pyrrolidine ring due to stereoelectronic effects of the 4-substitution, based on data in Ac-TYXN-NH2 peptides (X = 4-substituted proline).42 Hyp = 4R-substituted (trans relative stereochemistry) hydroxyproline, indicated by use of capitalized three-letter code and red color; hyp = 4S-substituted (cis relative stereochemistry) hydroxyproline, indicated by lower-case three-letter code and blue color. Proline exhibits a mixture of exo and endo ring puckers. RN and RC indicate the N-terminal and C-terminal peptide sequences, respectively.
In addition to the magnetic properties of the perfluoro-tert-butyl group, in general, tert-butyl groups are broadly attractive in medicinal chemistry due to functional group symmetry and the consequent reduced conformational entropy penalty upon target binding.46 However, in amino acids and peptides, tert-butyl ethers, the most easily installed tert-butyl groups, are not stable to standard acidic cleavage/deprotection reactions. In contrast, perfluoro-tert-butyl ethers are not subject to carbocation formation and elimination reactions and thus are expected to be a chemical stable functionality that may be incorporated within small molecules, peptides, and proteins.44,45
Perfluoro-tert-butyl hydroxyproline residues in model Ac-TYXN-NH2 peptides, X = either 4R-perfluoro-tert-butyl hydroxyproline or 4S-perfluoro-tert-butyl hydroxyproline, exhibited structural effects of proline substitution that were consistent with previously observed stereoelectronic effects in 4-substituted prolines, in which conformational preferences are dependent on the stereochemistry and the electron-withdrawing nature of the 4-substituent (Figure 1).12,13,42,47 Notably, 4S-perfluoro-tert-butyl hydroxyproline exhibited one of the largest conformational preferences in these model peptides that were designed to promote cis prolyl amide bonds, with similar populations of cis and trans amide bonds (Ktrans/cis = 1.2, compared to Ktrans/cis = 2.7 for the peptide containing proline, ΔΔGtrans/cis = +0.48 kcal mol–1) observed by 1H and 19F NMR, indicating the potential of the perfluoro-tert-butyl group, appropriately installed, to broadly modulate protein structure. In peptides and proteins, the structural effects of 4-substituted prolines are a balance of both the sterics and the electron-withdrawing nature of the proline 4-substituent, which determines the preference for proline ring pucker conformation (exo versus endo), which is coupled to the protein main chain conformation (compact versus extended, respectively) (Figure 1).48
In the previous syntheses of perfluoro-tert-butyl hydroxyproline residues within model tetrapeptides, while the desired peptide product was obtained, the solid-phase Mitsunobu reaction proceeded in relatively modest yield, particularly for the synthesis of the peptide with 4R-perfluoro-tert-butyl hydroxyproline. Of all 123 peptides synthesized via the proline editing method, the synthesis of the peptide with 4R-perfluoro-tert-butyl hydroxyproline proceeded with the lowest yield and required the application of low-load resin to achieve sufficient conversion, presumably due to the substantial steric challenges of the Mitsunobu reaction using a nucleophilic perfluoro-tert-butyl alkoxide. In order to develop general methods to incorporate perfluoro-tert-butyl groups within peptides, we herein describe synthetic methods to prepare both Fmoc- and Boc-protected 4R- and 4S-perfluoro-tert-butyl hydroxyprolines and examine the conformational preferences of these amino acids within different secondary structure contexts where they might be functionally employed.
The commercially available compound Boc-(2S,4R)-4-hydroxyproline methyl ester (1) was used as the starting material for the synthesis of both amino acids. Because of the stereospecific nature of the Mitsunobu reaction,49 with inversion of stereochemistry at the site of substitution, access to the 4R-substituted amino acid requires conversion of the 4R-hydroxyproline to the 4S-hydroxyproline, followed by a second inversion, to generate the 4R-product. Inversion of the hydroxyproline alcohol stereochemistry was accomplished by Mitsunobu reaction with 4-nitrobenzoic acid, followed by nitrobenzoate azidolysis with sodium azide, to generate the protected 4S-hydroxyproline (3), using conditions developed previously (Scheme 1).42,50−52 This alcohol was then subjected to Mitsunobu reaction with perfluoro-tert-butanol (pKa 5),42,44,45,53 to generate the protected 4R-perfluoro-tert-butyl hydroxyproline (4). This product was converted to the Boc-amino acid 5, the free amino acid 6, and the Fmoc-amino acid 7 using standard approaches, generating the protected derivatives for the incorporation of this amino acid in peptides or small molecules by solution or solid-phase peptide synthesis.
Scheme 1. Synthesis of (2S,4R)-Perfluoro-tert-butyl 4-Hydroxyproline (hyp(C4F9)) as Free, Boc-, and Fmoc-Amino Acids.

The 4S-substituted perfluoro-tert-butyl hydroxyproline was analogously synthesized via Mitsunobu reaction of perfluoro-tert-butanol with the same protected 4R-hydroxyproline (1) starting material used previously (Scheme 2). The product of this reaction (8) was converted to the Boc- (9), free (10), and Fmoc-amino acids (11).
Scheme 2. Synthesis of (2S,4S)-Perfluoro-tert-butyl 4-Hydroxyproline (hyp(C4F9)) as Free, Boc-, and Fmoc-Amino Acids.

The Fmoc-4R- and Fmoc-4S-perfluoro-tert-butyl hydroxyproline amino acids were each incorporated within peptides in two different structural contexts, a proline-rich peptide used to identify polyproline helix propensity (Ac-GPPXPPGY-NH2) and a Baldwin-type alanine-rich peptide to identify α-helix propensity (Ac-XKAAAAKAAAAKAAGY-NH2).54−56 These peptide sequences were employed to determine the conformational preferences of these amino acids within the polyproline helix and α-helix secondary structures, both of which are widely employed in molecular recognition. Matching the conformational preferences of the amino acid to the structural context in which the amino acid may be employed (protein design in “chi space”)57 can maximize the effectiveness of the amino acids toward defined applications.
Peptide synthesis with these amino acids proceeded sluggishly using HBTU as a coupling reagent. The desired peptides were obtained when amide coupling of the Fmoc-perfluoro-tert-butyl hydroxyproline was conducted using COMU as a coupling reagent and an extended reaction time.58 For the proline-rich peptides, subsequent amide coupling reactions were conducted using HATU. The relatively lower reactivity in amide bond formation associated with these amino acids was presumably due to the steric hindrance of the perfluoro-tert-butyl group.
In the Ac-GPPXPPGY-NH2 peptide series, the peptide with 4R-perfluoro-tert-butyl hydroxyproline exhibited positive (λmax = 228 nm) and negative (λmin = 205 nm) bands typical for polyproline helix (PPII) (Figure 2). The mean residue ellipticity at 228 nm ([θ]228 = 3770 deg·cm2·dmol–1) indicated that this amino acid had a greater propensity for the polyproline helix than proline ([θ]228 = 2950 deg·cm2·dmol–1).55 In contrast, the peptide with 4S-perfluoro-tert-butyl hydroxyproline exhibited a weaker positive band ([θ]228 = 1700 deg·cm2·dmol–1) and a red-shifted λmax = 230 nm, both of which indicate that this amino acid has a substantially lower polyproline helix propensity than proline. These data are consistent with previous examinations on the stereoelectronic effects of 4-substituted proline residues on polyproline helix stability, where 4R-hydroxyproline and 4R-fluoroproline promote PPII relative to proline, whereas 4S-hydroxyproline and 4S-fluoroproline relatively destabilize PPII compared to proline.19,60 These data indicate that the perfluoro-tert-butyl hydroxyprolines similarly exhibit distinct conformational preferences that allow stereochemical selection to optimize their applications.
Figure 2.
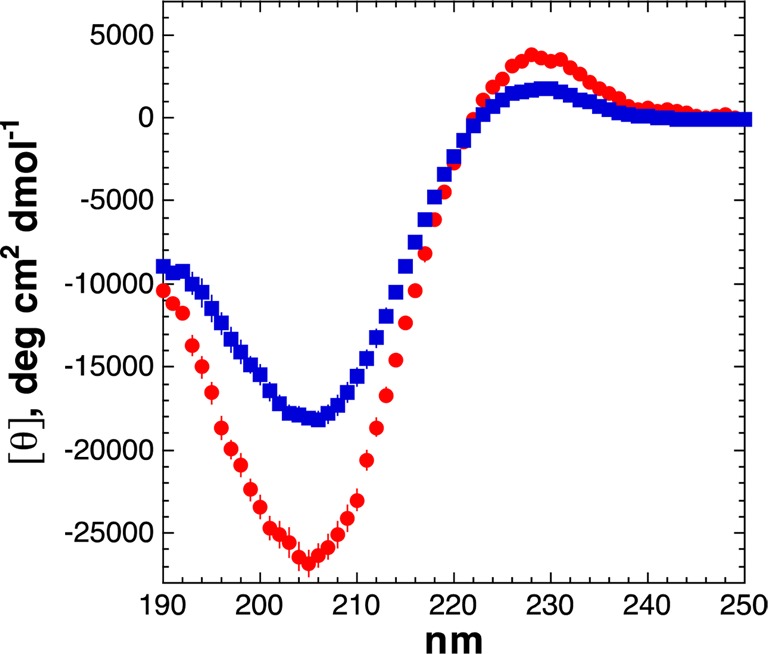
CD spectra of peptides with (2S,4R)-Hyp(C4F9) (red circles) and (2S,4S)-hyp(C4F9) (blue squares) as the guest (X) residue in Ac-GPPXPPGY-NH2 peptides. Polyproline helix (PPII) is indicated by the magnitude of the positive band at ∼228 nm.59 The peptide with X = Pro exhibits an intermediate structure between the perfluoro-tert-butyl hydroxyprolines.55
Proline is compatible with α-helices at their N-terminus, where they can function as start signals for α-helices.56,61,62 In Ac-XKAAAAKAAAAKAAGY-NH2 peptides, both perfluoro-tert-butyl hydroxyprolines exhibited evidence of α-helical structure (Figure 3). However, in the compact conformation of the α-helix, both peptides were less α-helical than the peptide with proline at the first residue (Pro:56 [θ]222 = −12030 deg·cm2·dmol–1, [θ]222/[θ]208 = 0.87, 38% α-helix; 4R-Hyp(C4F9): [θ]222 = −6840 deg·cm2·dmol–1, [θ]222/[θ]208 = 0.67, 24% α-helix; 4S-hyp(C4F9): [θ]222 = −4990 deg·cm2·dmol–1, [θ]222/[θ]208 = 0.64, 19% α-helix). The reduced α-helicity of the 4S-perfluoro-tert-butyl hydroxyproline compared to proline was expected based on the conformational preferences of electron-withdrawing 4S-substituents on proline, which promote a more extended conformation and an endo ring pucker, which is not preferred in α-helices.63 The reduced α-helicity of the 4R-perfluoro-tert-butyl hydroxyproline compared to proline was not expected based on proline conformational analysis. However, these data are consistent with trends across a range of fluorinated amino acids, where highly fluorinated amino acids strongly disfavor α-helix independent of side chain structure.18,64 In addition, in the 4R-perfluoro-tert-butyl hydroxyproline, the perfluoro-tert-butyl group would project toward the subsequent residues of the α-helix, potentially providing a steric basis for reduced α-helicity.
Figure 3.
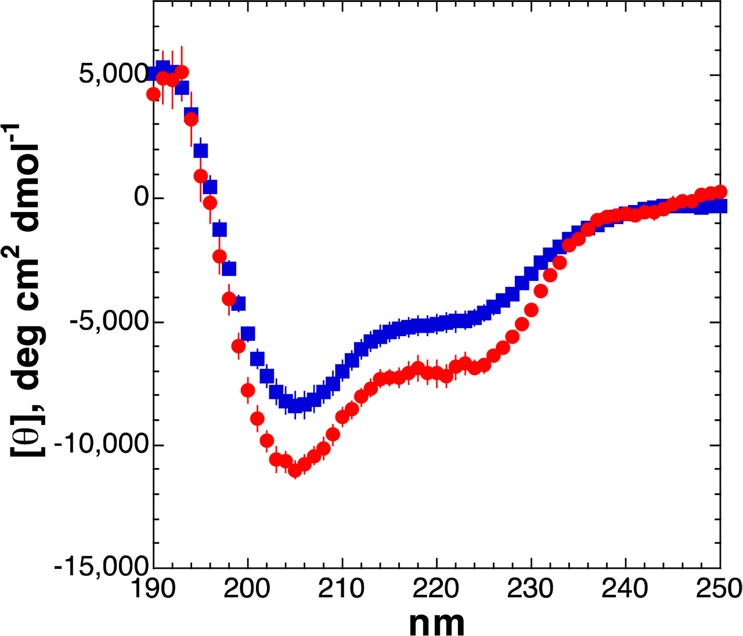
CD spectra of peptides with (2S,4R)-Hyp(C4F9) (red circles) and (2S,4S)-hyp(C4F9) (blue squares) as the guest (X) residue in Ac-XKAAAAKAAAAKAAGY-NH2 model α-helical peptides.56 Population of α-helix is indicated by the magnitude of the negative band at 222 nm or the ratio [θ]222/[θ]208. Data on the peptide with X = Pro are in ref (56).
Perfluoro-tert-butyl amino acids have substantial potential as probes in 19F-based magnetic imaging due to the sensitivity of the perfluoro-tert-butyl group (9 equivalent fluorines that do not couple to any other nuclei) and the large chemical shift dispersion inherent to 19F NMR. All peptides were examined by 19F NMR spectroscopy. In the Ac-GPPXPPGY-NH2 peptides (Figure 4), both peptides exhibited one major resonance and multiple minor resonances. X–Pro amide bonds are inherently prone to cis–trans isomerism, with 10% of Pro–Pro amide bonds cis in the PDB. Thus, these peptides have 32 (25) potential species present in slow exchange, considering combinations of cis versus trans amide bonds at each of the five X–Pro amides. In the 1H NMR spectra of these peptides, evidence of multiple species due to cis–trans isomerism is observable, but due to the complexity of the 1H NMR spectra, the data only clearly indicate the presence of three or four species. In the simpler 19F NMR spectrum, where all peaks are singlets, the relative populations of the major species are confirmed.30,65,66 Herein, however, additional minor species may be identified by 19F NMR that are not observed by 1H NMR. These data indicate that perfluoro-tert-butyl hydroxyprolines may function as sensitive probes of species not observable by standard 1H NMR approaches.
Figure 4.
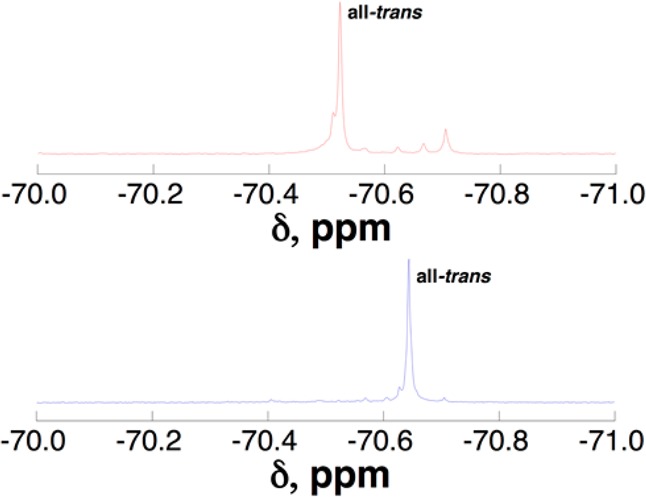
19F NMR spectra of Ac-GPPXPPGY-NH2 peptides with X = (2S,4R)-Hyp(C4F9) (top) and (2S,4S)-hyp(C4F9) (bottom). The NMR spectra indicate the presence of cis–trans isomerism about the X–Pro amide bonds. The major species contains all trans amide bonds.55
19F NMR spectra of the Ac-XKAAAAKAAAAKAAGY-NH2 peptides (Figure 5) indicated one major species (trans Ac–Pro amide bond) and one minor species (cis Ac–Pro amide bond), with a higher population of cis amide bond for the 4S- than the 4R-perfluoro-tert-butyl hydroxyproline, as expected (Figure 1). In order to identify the sensitivity of detection of perfluoro-tert-butyl hydroxyprolines, in consideration of future applications of these amino acids in imaging, the NMR spectrum of the peptide with 4R-perfluoro-tert-butyl hydroxyproline was examined as a function of peptide concentration. The 19F NMR spectrum of this peptide was rapidly obtained (5 min, 128 scans, signal-to-noise = 7.3) at a peptide concentration of 200 nM (Figure S7, Supporting Information). The sensitivity of the perfluoro-tert-butyl group indicated by these experiments suggests broad potential applications of these amino acids in imaging.
Figure 5.
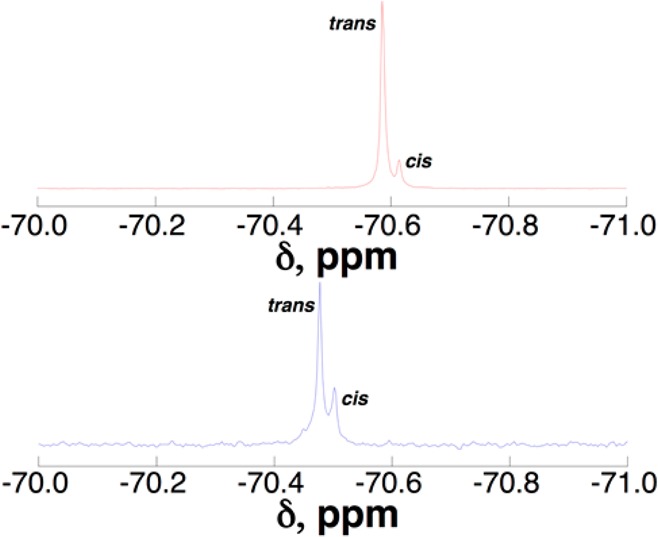
19F NMR spectra of Ac-XKAAAAKAAAAKAAGY-NH2 peptides with (2S,4R)-Hyp(C4F9) (top) and (2S,4S)-hyp(C4F9) (bottom). The peaks indicate the trans (major) versus cis (minor) Ac–Pro amide bonds.
We have described the synthesis of 4R- and 4S-perfluoro-tert-butyl hydroxyproline as free amino acids and as Boc- and Fmoc-amino acids, for incorporation of the amino acids using the major approaches to peptide synthesis. These amino acids have distinct conformational preferences, suggesting alternative applications for each in small molecules, peptides, and proteins. These amino acids represent unique handles for molecular recognition and for use in sensitive 19F NMR and MRI imaging.
Experimental Section
Compounds 2 and 3 were synthesized using minor modifications to methods described.51,52
Boc-(2S,4S)-p-nitrobenzoate-4-hydroxyproline Methyl Ester (2)
Compound 1 (3.90 g, 15.7 mmol), triphenylphosphine (Ph3P) (7.40 g, 28.3 mmol), and p-nitrobenzoic acid (3.10 g, 18.8 mmol) were dissolved in anhydrous THF (157 mL). The reaction was conducted under a nitrogen atmosphere. The solution was cooled to 0 °C. Diisopropylazodicarboxylate (DIAD) (6.35 g, 6.20 mL, 31.4 mmol) was added dropwise over 30 min. The solution was removed from the ice bath, allowed to warm to room temperature, and stirred for an additional 6 h. The solvent was removed under reduced pressure. The crude product was dissolved in ethyl acetate (100 mL) and washed with brine (2 × 200 mL). The solvent was removed, and the crude product was redissolved in CH2Cl2 (75 mL). The crude product was purified via column chromatography (CH2Cl2) to yield compound 2 (4.7 g, 12.0 mmol) as a colorless oil in 76% yield. The NMR data corresponded to the literature values.51,52
Boc-(2S,4S)-4-hydroxyproline Methyl Ester (3)
Compound 2 (2.0 g, 5.0 mmol) was dissolved in acetone (50 mL). Sodium azide (0.49 g, 7.5 mmol) was added, and the solution was heated at reflux for 14 h. The solution was allowed to cool to room temperature, and the solvent was removed under reduced pressure. The crude product was dissolved in ethyl acetate (50 mL) and washed with distilled water (2 × 50 mL). The solvent was removed, and the crude product was redissolved in CH2Cl2 for purification. Compound 3 (0.80 g, 3.3 mmol) was purified via column chromatography (2% methanol in CH2Cl2 v/v) to obtain a colorless oil in 65% yield. The NMR data corresponded to the literature values.51,52
Boc-(2S,4R)-perfluoro-tert-butyl-4-hydroxyproline Methyl Ester (4)
Compound 3 (2.23 g, 9.10 mmol) and Ph3P (2.86 g, 10.9 mmol) were dissolved in toluene (91 mL) under a nitrogen atmosphere. The solution was cooled to 0 °C and stirred on ice for 10 min. DIAD (2.20 g, 2.15 mL, 10.9 mmol) was added dropwise to the solution over 15 min. Perfluoro-tert-butanol (4.30 g, 2.54 mL, 18.2 mmol) and DIPEA (2.35 g, 3.16 mL, 18.2 mmol) were added to the solution, which was then stirred on ice for another 5 min. The solution was removed from the ice bath, warmed to 45 °C in an oil bath, and stirred for 24 h. The solvent was removed under reduced pressure, and the crude product was dissolved in ethyl acetate (50 mL). The crude product was washed with brine (2 × 75 mL) and dried over sodium sulfate. The solvent was removed under reduced pressure, and the crude product was redissolved in CH2Cl2 (50 mL). The crude product was purified via column chromatography (0–7% ethyl acetate in hexanes v/v) to obtain compound 5 (0.790 g, 2.28 mmol) as a colorless oil in 25% yield. 1H NMR (400 MHz, CDCl3) δ 4.91 (s, 1H), 4.49–4.47 (dd, J = 8.6, 6.4 Hz, 0.4H, cis), 4.40–4.36 (dd, J = 7.8, 7.8 Hz, 0.6H, trans), 3.84–3.75 (m, 1H), 3.75 (s, 3H), 3.69–3.66 (d, J = 12.5 Hz, 0.6H, trans), 3.60–3.58 (d, J = 12.3 Hz, 0.4H, cis), 2.47–2.44 (m, 1H), 2.28–2.23 (m, 1H), 1.46 (s, 4H, cis), 1.42 (s, 5H, trans). 13C NMR (150.8 MHz, CDCl3) δ 171.9, 171.7, 153.1, 152.4, 119.1 (q, J = 293 Hz), 79.8, 77.1, 56.5, 56.1, 51.5, 51.4, 51.2, 36.6, 35.6, 27.2. 19F NMR (376.3 MHz, CDCl3) δ −70.47 (trans conformation), −70.53 (cis conformation). HRMS (LIFDI-TOF) m/z: [M]+ calcd for C15H18F9NO5 463.1041, found 463.1051.
Boc-(2S,4R)-perfluoro-tert-butyl-4-hydroxyproline (5)
Compound 4 (0.36 g, 0.78 mmol) and LiOH (0.22 g, 0.93 mmol) were dissolved in a solution of water (4 mL) and 1,4-dioxane (4 mL). The solution was stirred at room temperature for 14 h. The mixture was acidified to pH 2 with dilute HCl and extracted with ethyl acetate (2 × 10 mL). The solvent was removed under reduced pressure and redissolved in CH2Cl2. The crude product was purified via column chromatography (0–2% methanol in CH2Cl2 v/v) to obtain compound 5 (0.22 g, 0.49 mmol) as an off-white solid in 63% yield. 1H (600 MHz, CDCl3) δ 4.92 (s, 1H), 4.53–4.51 (dd, J = 7.7, 7.0 Hz, 0.6H), 4.42–4.39 (dd, J = 7.4, 7.4 Hz, 0.4H), 3.83–3.80 (m, 0.4H), 3.74–3.66 (m, 1.6H), 2.51–2.48 (m, 1H), 2.44–2.40 (m, 0.6H), 2.35–2.32 (m, 0.4H), 1.48 (s, 5H), 1.43 (s, 4H). 13C NMR (150.8 MHz, CDCl3)δ 177.7, 174.7, 155.9, 153.4, 120.1 (q, J = 293 Hz), 82.3, 81.3, 57.4, 52.6, 37.4, 35.7, 28.2. 19F NMR (564.5 MHz, CDCl3) δ −70.49 (trans conformation), −70.55 (cis conformation). HRMS (LIFDI-TOF) m/z: [M + H]+ calcd for C14H17F9NO5 450.0963, found 450.0946.
(2S,4R)-Perfluoro-tert-butyl-4-hydroxyproline Hydrochloride (6)
Compound 4 (1.3 g, 2.2 mmol) was dissolved in a solution of 1,4-dioxane (15 mL) and 4 M HCl (15 mL). The solution was allowed to stir at reflux for 6 h. The solvent was removed under reduced pressure. Compound 6 (0.78 g, 2.2 mmol) was used as a crude reagent in the next step without purification. Alternatively, compound 5 could be subjected to identical conditions to yield compound 6. 1H NMR (600 MHz, MeOD-d4) δ 5.22 (br s, 1H), 4.55–4.51 (dd, J = 9.6, 8.8 Hz, 1H), 3.81–3.79 (dd, J = 13.5, 5.0 Hz, 1H), 3.46–3.44 (d, J = 13.5 Hz, 1H), 2.53–2.52 (m, 1H), 2.51–2.50 (m, 1H). 13C NMR (150.8 MHz, MeOD-d4) δ 168.7, 120.0 (J = 293 Hz), 79.0, 58.1, 51.8, 35.7. 19F NMR (376.3 MHz, MeOD-d4) δ −71.56. HRMS (LIFDI-TOF) m/z: [M]+ calcd for C9H9F9NO3 350.0439, found 350.0420.
Fmoc-(2S,4R)-perfluoro-tert-butyl-4-hydroxyproline (7)
Crude compound 6 (1.01 g, 2.90 mmol) was dissolved in a solution of 1,4-dioxane (15 mL) and H2O (15 mL). Fmoc-OSu (1.17 g, 3.48 mmol) and K2CO3 (0.80 g, 5.80 mmol) were added to the solution, and the resultant mixture was stirred for 14 h at room temperature. The 1,4-dioxane was removed under reduced pressure, and the crude product was acidified with 2 M HCl (10 mL). The crude product was extracted with ethyl acetate (2 × 20 mL). The solvent was removed, and the crude product was redissolved in CH2Cl2. The product was purified via column chromatography (0–4% methanol in CH2Cl2 v/v) to obtain the compound 8 as a white solid (0.83 g, 1.45 mmol) in 50% yield. 1H (600 MHz, CDCl3) δ 7.77–7.76 (d, J = 7.5 Hz, 1.3H, trans), 7.74–7.73 (d, J = 7.6 Hz, 0.7H, cis) 7.55–7.52 (dd, J = 12.4, 7.6 Hz, 2H), 7.42–7.38 (m, 2H), 7.32–7.29 (dd, J = 7.4, 7.3 Hz, 2H), 4.92 (s, 0.7H, trans), 4.89 (s, 0.3H, cis), 4.59–4.57 (dd, J = 7.8, 7.4 Hz, 0.7H, trans), 4.52–4.43 (m, 2H), 4.38–4.37 (dd, J = 7.4, 7.4 Hz, 0.3H, cis), 4.30–4.27 (dd, J = 7.0, 6.6 Hz, 0.7H, trans), 4.17–4.16 (dd, J = 6.6, 6.6 Hz, 0.3H, cis), 3.83–3.73 (m, 1H), 3.71 (s, 1H), 2.49–2.45 (m, 1.7H, mixture of cis and trans), 2.32–2.29 (m, 0.3H, cis). 13C NMR (150.8 MHz, CDCl3) δ 173.1, 156.1, 143.5, 143.4, 141.4, 127.9, 127.2, 124.9, 124.8, 120.12 (q, J = 293 Hz), 120.09, 77.9, 68.6, 67.6, 57.6, 56.7, 53.0, 52.7, 47.2, 47.0, 37.6, 35.8. 19F NMR (564.5 MHz, CDCl3) δ −70.38 (trans conformation), −70.43 (cis conformation). HRMS (CI-TOF) m/z: [M]+ calcd for C24H18F9NO5 571.1041, found 571.1027.
Boc-(2S,4S)-perfluoro-tert-butyl-4-hydroxyproline Methyl Ester (8)
Compound 2 (3.09 g, 12.6 mmol) and Ph3P (3.96 g, 15.1 mmol) were dissolved in toluene (126 mL) under a nitrogen atmosphere. The solution was cooled to 0 °C and stirred on ice for 10 min. DIAD (3.05 g, 2.98 mL, 15.1 mmol) was added dropwise to the solution over 15 min. Perfluoro-tert-butanol (5.95 g, 3.52 mL, 25.2 mmol) and DIPEA (3.18 g, 4.38 mL, 25.2 mmol) were added to the solution, which was stirred on ice for another 5 min. The solution was removed from the ice bath, warmed to 45 °C in an oil bath, and stirred for 24 h. The solvent was removed under reduced pressure, and the crude product was dissolved in ethyl acetate (50 mL). The crude product was washed with brine (2 × 75 mL) and dried over sodium sulfate. The solvent was removed under reduced pressure, and the crude product was redissolved in CH2Cl2 (50 mL). The crude product was purified via column chromatography (0–7% ethyl acetate in hexanes v/v) to obtain compound 5 (2.07 g, 4.47 mmol) as a colorless oil in 36% yield. 1H NMR (400 MHz, CDCl3) δ 4.84 (br s, 1H), 4.52–4.50 (dd, J = 8.3, 3.7 Hz, 0.4H, minor), 4.41–4.38 (dd, J = 9.0, 3.1 Hz, 0.6H, major), 3.84–3.78 (dd, J = 12.5, 5.6 Hz, 0.6H, major), 3.76–3.75 (d, J = 6.5 Hz, 0.4H, minor), 3.72 (s, 3H), 3.68–3.65 (d, J = 13.0 Hz, 0.6H, major), 3.58–3.55 (d, J = 13.0 Hz, 0.4H, minor), 2.53–2.38 (m, 2H), 1.48 (s, 4H, major), 1.43 (s, 5H, minor). 13C NMR (150.8 MHz, CDCl3) δ 172.9, 172.8, 154.2, 154.0, 129.4, 129.3, 124.8, 124.6, 124.5, 120.1 (q, J = 293 Hz), 105.1, 80.9, 80.8, 66.6, 66.2, 57.5, 57.1, 52.3, 37.6, 36.6, 28.3, 28.2. 19F NMR (376.3 MHz, CDCl3) δ −70.42 (minor), −70.44 (major). HRMS (LIFDI-TOF) m/z: [M]+ calcd for C15H18F9NO5 463.1041, found 463.1042.
Boc-(2S,4S)-perfluoro-tert-butyl-4-hydroxyproline (9)
Compound 8 (1.75 g, 3.78 mmol) and LiOH (0.1086 g, 4.53 mmol) were dissolved in a solution of water (20 mL) and 1,4-dioxane (20 mL). The solution was stirred at room temperature for 14 h. The reaction mixture was acidified to pH 2 with dilute HCl and extracted with ethyl acetate (2 × 75 mL). The solvent was removed under reduced pressure, and the crude product was redissolved in CH2Cl2. The crude product was purified via column chromatography (0–2% methanol in CH2Cl2 v/v) to obtain compound 9 (1.05 g, 2.34 mmol) as an off-white solid in 62% yield. 1H (600 MHz, CDCl3) δ 4.86 (s, 1H), 4.51–4.50 (d, J = 6.7 Hz, 0.4H, minor), 4.44–4.42 (d, J = 7.8 Hz, 0.6H, major), 3.82–3.76 (m, 1H), 3.72–3.67 (m, 0.6H, major), 3.56–3.55 (m, 0.4H, minor), 2.52–2.44 (m, 2H), 1.49 (s, 3.6H), 1.44 (s, 5.4H). 13C NMR (150.8 MHz, CDCl3) δ 177.4, 175.5, 154.7, 153.5, 120.1 (q, J = 293 Hz), 80.9, 57.5, 57.1, 52.9, 52.7, 52.1, 37.7, 37.6, 36.1, 28.1. 19F NMR (564.5 MHz, CDCl3) δ −70.38 (major), −70.44 (minor). HRMS (LIFDI-TOF) m/z: [M + H]+ calcd for C14H17F9NO5 450.0963, found 450.0958.
(2S,4S)-Perfluoro-tert-butyl-4-hydroxyproline Hydrochloride (10)
Compound 9 (2.0 g, 4.3 mmol) was dissolved in 1,4-dioxane (15 mL), and 4 M HCl (15 mL) was added. The solution was allowed to stir at reflux for 6 h. The solvent was removed under reduced pressure. Compound 10 (1.5 g, 4.3 mmol) obtained was used as a crude reagent in the next step. Alternatively, compound 8 could be subjected to the same conditions to yield compound 10. 1H NMR (600 MHz, MeOD-d4) δ 5.20 (s, 1H), 4.62–4.60 (dd, J = 9.6, 3.4 Hz, 1H), 3.74–3.71 (dd, J = 13.4, 4.6 Hz, 1H), 3.56–3.53 (d, J = 13.9 Hz, 1H), 2.74–2.68 (ddd, J = 14.6, 10.0, 5.0 Hz, 1H), 2.54–2.51 (d, J = 14.6 Hz, 1H). 13C NMR (150.8 MHz, MeOD-d4) δ 169.3, 120.0 (J = 293 Hz), 78.9, 58.3, 52.1, 36.1. 19F NMR (564.5 MHz, MeOD-d4) δ −71.65. HRMS (LIFDI-TOF) m/z: [M]+ calcd for C9H9F9NO3 350.0439, found 350.0413.
Fmoc-(2S,4S)-perfluoro-tert-butyl-4-hydroxyproline (11)
Crude compound 10 (0.50 g, 1.4 mmol) was dissolved in 1,4-dioxane (7 mL). Fmoc-OSu (0.50 g, 1.4 mmol) and K2CO3 (0.39 g, 2.9 mmol) were added, and the resultant solution was stirred for 14 h at room temperature. The 1,4-dioxane was removed under reduced pressure, and the crude product was acidified with 2 M HCl (10 mL). The crude product was extracted with ethyl acetate (2 × 20 mL). The solvent was removed, and the crude product was redissolved in CH2Cl2. The crude mixture (0.40 g, 0.72 mmol) was purified via column chromatography (0–4% methanol in CH2Cl2 v/v) to obtain compound 11 (0.38 g, 0.70 mmol) as a white solid in 50% yield. 1H (600 MHz, CDCl3) δ 7.77–7.69 (d, J = 7.6 Hz, 2H), 7.60–7.51 (m, 2H), 7.42–7.28 (m, 4H), 4.92 (s, 0.5H), 4.88 (s, 0.5H), 4.61–4.56 (m, 1H), 4.53–4.49 (m, 0.5H), 4.43–4.31 (m, 2H), 4.16–4.13 (dd, J = 6.3, 6.1 Hz, 0.5H), 3.90–3.86 (m, 1H), 3.76–3.63 (m, 1H), 3.74 (s, 1H), 2.57–2.41 (m, 2H). 13C NMR (150.8 MHz, CDCl3) δ 176.3, 175.7, 154.7, 154.3, 143.9, 143.8, 143.60, 143.58, 141.4, 141.3, 127.8, 127.74, 127.70, 127.13, 127.10, 127.0, 125.1, 125.0, 124.8, 121.1, 119.97 (J = 293 Hz), 119.96, 78.2, 67.9, 67.8, 67.0, 57.3, 57.0, 53.4, 52.9, 47.1, 37.7, 36.5, 21.9. 19F NMR (564.5 MHz, CDCl3) δ −70.42, −70.49. HRMS (LIFDI-TOF) m/z: [M]+ calcd for C24H18F9NO5 571.1041, found 571.1038.
Peptide Synthesis and Characterization
Peptides were synthesized on NovaGel PEG-polystyrene graft Rink amide resin (EMD Millipore) by standard solid-phase peptide synthesis. This resin resulted in higher peptide yields than standard polystyrene resin. Amide coupling reactions with Fmoc-perfluoro-tert-butyl 4-hydroxyproline amino acids were conducted using 4 equiv each of amino acid and COMU, with the reaction allowed to proceed for 36 h. Subsequent amide coupling reactions in Ac-GPPXPPGY-NH2 peptides were conducted as double coupling reactions (2 h for the first coupling, 1 h for the second coupling) using HATU as a coupling reagent. All peptides were acetylated on the N-terminus (5% acetic anhydride in pyridine, 3 mL, 3 × 5 min) and contained a C-terminal amide. Peptides were cleaved from the resin and deprotected for 3 h using a 1 mL reaction volume of 90% TFA with 5% H2O and 5% triisopropylsilane. TFA was removed by evaporation, and the peptides were dissolved in 1 mL of 500 mM phosphate buffer (pH 7.2). Crude peptide solutions were filtered using a 0.45 μm syringe filter before injection on the HPLC. Peptides were purified by reverse-phase HPLC (Vydac semipreparative C18, 10 × 250 mm, 5 μm particle size, 300 Å pore) using a 60 min linear gradient of 100% to 40% buffer A (98% H2O, 2% acetonitrile, 0.06% TFA) in buffer B (20% H2O, 80% acetonitrile, 0.05% TFA). Peptide purity was verified by the presence of a single peak on reinjection. Analytical data for peptides: Ac-GPP((2S,4R)-Hyp(C4F9))PPGY-NH2 [tR, 37.8 min; expected mass, 1056.4, observed mass, 1078.4 (M + Na)+]; Ac-GPP((2S,4S)-hyp(C4F9))PPGY-NH2 [tR, 36.8 min; expected mass, 1056.4, observed mass, 1078.4 (M + Na)+]; Ac-((2S,4R)-Hyp(C4F9))KAAAAKAAAAKAAGY-NH2 [tR, 41.9 min; expected mass, 1706.7, observed mass, 853.5 (M)2+]; Ac-((2S,4R)-Hyp(C4F9))KAAAAKAAAAKAAGY-NH2 [tR, 40.8 min; expected mass, 1706.7, observed mass, 853.6 (M)2+].
Circular Dichroism (CD)
Peptides (10–50 μM final concentrations: Ac-((2S,4R)-Hyp(C4F9))KAAAAKAAAAKAAGY-NH2 10 μM; Ac-((2S,4S)-hyp(C4F9))KAAAAKAAAAKAAGY-NH2 50 μM; Ac-GPP((2S,4R)-Hyp(C4F9))PPGY-NH2 20 μM; Ac-GPP((2S,4S)-hyp(C4F9))PPGY-NH2 50 μM) were dissolved in water containing 5 mM phosphate buffer pH 7.4 and 25 mM KF. Data represent the average of at least three independent trials. Error bars indicate standard error. Data were background-corrected but were not smoothed. Experiments in Figure 2 were conducted at 25 °C.55 Experiments in Figure 3 were conducted at 0.5 °C.56 The temperatures for each experiment were chosen to allow direct comparison to the analogous peptides with proline.
NMR Spectroscopy of Peptides
Experiments were conducted in 90% H2O/10% D2O containing 5 mM phosphate buffer pH 4, 25 mM NaCl, and 100 μM TSP as a reference for 1H NMR spectra. Residual trifluoroacetate was used as an internal reference for 19F NMR spectra. 19F NMR experiments were conducted without decoupling using a 1.5 s relaxation delay on a Brüker 600 MHz (545.5 MHz for 19F) NMR spectrometer equipped with a Brüker SMART probe.
Acknowledgments
We thank the NIH (GM93225) and the University of Delaware for funding.
Supporting Information Available
1H, 13C, and 19F NMR spectra for all new compounds; CD spectra for compounds 4, 5, 7, 8, 9, and 11; and full 1H NMR spectra, 19F NMR spectra, and TOCSY spectra of peptides. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Salwiczek M.; Nyakatura E. K.; Gerling U. I. M.; Ye S. J.; Koksch B. Chem. Soc. Rev. 2012, 41, 2135–2171. [DOI] [PubMed] [Google Scholar]
- Marsh E. N. G.; Suzuki Y.. ACS Chem. Biol. 2014, 9, DOI: 10.1021/cb500111u. [DOI] [PubMed] [Google Scholar]
- Qiu X. L.; Qing F. L. Eur. J. Org. Chem. 2011, 3261–3278. [Google Scholar]
- Durr U. H. N.; Grage S. L.; Witter R.; Ulrich A. S. J. Magn. Reson. 2008, 191, 7–15. [DOI] [PubMed] [Google Scholar]
- Grage S. L.; Durr U. H. N.; Afonin S.; Mikhailiuk P. K.; Komarov I. V.; Ulrich A. S. J. Magn. Reson. 2008, 191, 16–23. [DOI] [PubMed] [Google Scholar]
- Muller K.; Faeh C.; Diederich F. Science 2007, 317, 1881–1886. [DOI] [PubMed] [Google Scholar]
- Lee K. H.; Lee H. Y.; Slutsky M. M.; Anderson J. T.; Marsh E. N. G. Biochemistry 2004, 43, 16277–16284. [DOI] [PubMed] [Google Scholar]
- Wang P.; Tang Y.; Tirrell D. A. J. Am. Chem. Soc. 2003, 125, 6900–6906. [DOI] [PubMed] [Google Scholar]
- Dalvit C. Prog. Nucl. Magn. Reson. Spectrosc. 2007, 51, 243–271. [DOI] [PubMed] [Google Scholar]
- Cobb S. L.; Murphy C. D. J. Fluorine Chem. 2009, 130, 132–143. [Google Scholar]
- Marsh E. N. G. Chem. Biol. 2000, 7, R153–R157. [DOI] [PubMed] [Google Scholar]
- Bretscher L. E.; Jenkins C. L.; Taylor K. M.; DeRider M. L.; Raines R. T. J. Am. Chem. Soc. 2001, 123, 777–778. [DOI] [PubMed] [Google Scholar]
- Renner C.; Alefelder S.; Bae J. H.; Budisa N.; Huber R.; Moroder L. Angew. Chem., Int. Ed. 2001, 40, 923–925. [PubMed] [Google Scholar]
- Yoder N. C.; Kumar K. Chem. Soc. Rev. 2002, 31, 335–341. [DOI] [PubMed] [Google Scholar]
- Kim W. Y.; George A.; Evans M.; Conticello V. P. ChemBioChem. 2004, 5, 928–936. [DOI] [PubMed] [Google Scholar]
- Kim W.; McMillan R. A.; Snyder J. P.; Conticello V. P. J. Am. Chem. Soc. 2005, 127, 18121–18132. [DOI] [PubMed] [Google Scholar]
- Kim W.; Hardcastle K. I.; Conticello V. P. Angew. Chem., Int. Ed. 2006, 45, 8141–8145. [DOI] [PubMed] [Google Scholar]
- Chiu H.-P.; Suzuki Y.; Gullickson D.; Ahmad R.; Kokona B.; Fairman R.; Cheng R. P. J. Am. Chem. Soc. 2006, 128, 15556–15557. [DOI] [PubMed] [Google Scholar]
- Horng J. C.; Raines R. T. Protein Sci. 2006, 15, 74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Cabello J.; Barnett B. P.; Bottomley P. A.; Bulte J. W. M. NMR Biomed. 2011, 24, 114–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer L. E.; Sparr C.; Gilmour R. Angew. Chem., Int. Ed. 2011, 50, 11860–11871. [DOI] [PubMed] [Google Scholar]
- Papeo G.; Giordano P.; Brasca M. G.; Buzzo F.; Caronni D.; Ciprandi F.; Mongelli N.; Veronesi M.; Vulpetti A.; Dalvit C. J. Am. Chem. Soc. 2007, 129, 5665–5672. [DOI] [PubMed] [Google Scholar]
- Zheng H.; Comeforo K.; Gao J. J. Am. Chem. Soc. 2009, 131, 18–19. [DOI] [PubMed] [Google Scholar]
- Jackson J. C.; Hammill J. T.; Mehl R. A. J. Am. Chem. Soc. 2007, 129, 1160–1166. [DOI] [PubMed] [Google Scholar]
- Li C.; Wang G.-F.; Wang Y.; Creager-Allen R.; Lutz E. A.; Scronce H.; Slade K. M.; Ruf R. A. S.; Mehl R. A.; Pielak G. J. J. Am. Chem. Soc. 2010, 132, 321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molski M. A.; Goodman J. L.; Craig C. J.; Meng H.; Kumar K.; Schepartz A. J. Am. Chem. Soc. 2010, 132, 3658–3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo M. D.; Rubini M. PLoS One 2011, 6, e19425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka Y.; Sun Y.; Tsukiji S.; Hamachi I. Chem. Sci. 2011, 2, 511–520. [Google Scholar]
- Mizukami S.; Takikawa R.; Sugihara F.; Hori Y.; Tochio H.; Walchli M.; Shirakawa M.; Kikuchi K. J. Am. Chem. Soc. 2008, 130, 794–795. [DOI] [PubMed] [Google Scholar]
- Mykhailiuk P. K.; Afonin S.; Palamarchuk G. V.; Shishkin O. V.; Ulrich A. S.; Komarov I. V. Angew. Chem., Int. Ed. 2008, 47, 5765–5767. [DOI] [PubMed] [Google Scholar]
- Yuvienco C.; More H. T.; Haghpanah J. S.; Tu R. S.; Montclare J. K. Biomacromolecules 2012, 13, 2273–2278. [DOI] [PubMed] [Google Scholar]
- Janjic J. M.; Srinivas M.; Kadayakkara D. K. K.; Ahrens E. T. J. Am. Chem. Soc. 2008, 130, 2832–2841. [DOI] [PubMed] [Google Scholar]
- In vivo detection of a 19F MRI imaging agent containing three symmetrically arranged perfluoro-tert-butyl ethers (relative signal intensity 27):Jiang Z. X.; Liu X.; Jeong E. K.; Yu Y. B. Angew. Chem., Int. Ed. 2009, 48, 4755–4758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synthesis of a dendrimer containing 243 chemically equivalent fluorines (27 symmetrically arranged perfluoro-tert-butyl ethers):Yue X.; Taraban M. B.; Hyland L. L.; Yu Y. B. J. Org. Chem. 2012, 77, 8879–8887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalvit C.; Ardini E.; Flocco M.; Fogliatto G. P.; Mongelli N.; Veronesi M. J. Am. Chem. Soc. 2003, 125, 14620–14625. [DOI] [PubMed] [Google Scholar]
- Dalvit C.; Mongelli N.; Papeo G.; Giordano P.; Veronesi M.; Moskau D.; Kummerle R. J. Am. Chem. Soc. 2005, 127, 13380–13385. [DOI] [PubMed] [Google Scholar]
- Takaoka Y.; Kiminami K.; Mizusawa K.; Matsuo K.; Narazaki M.; Matsuda T.; Hamachi I. J. Am. Chem. Soc. 2011, 133, 11725–11731. [DOI] [PubMed] [Google Scholar]
- Tang Y.; Tirrell D. A. J. Am. Chem. Soc. 2001, 123, 11089–11090. [DOI] [PubMed] [Google Scholar]
- Bilgicer B.; Xing X.; Kumar K. J. Am. Chem. Soc. 2001, 123, 11815–11816. [DOI] [PubMed] [Google Scholar]
- Lee H. Y.; Lee K. H.; Al-Hashimi H. M.; Marsh E. N. G. J. Am. Chem. Soc. 2006, 128, 337–343. [DOI] [PubMed] [Google Scholar]
- Buer B. C.; Meagher J. L.; Stuckey J. A.; Marsh E. N. G. Protein Sci. 2012, 21, 1705–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey A. K.; Naduthambi D.; Thomas K. M.; Zondlo N. J. J. Am. Chem. Soc. 2013, 135, 4333–4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas K. M.; Naduthambi D.; Tririya G.; Zondlo N. J. Org. Lett. 2005, 7, 2397–2400. [DOI] [PubMed] [Google Scholar]
- Jiang Z. X.; Yu Y. B. J. Org. Chem. 2007, 72, 1464–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Incorporation of a perfluoro-tert-butyl group into an α-amino acid in peptides as perfluoro-tert-butyl homoserine:Buer B. C.; Levin B. J.; Marsh E. N. G. J. Pept. Sci. 2013, 19, 308–314. [DOI] [PubMed] [Google Scholar]
- Bisel P.; Al-Momani L.; Muller M. Org. Biomol. Chem. 2008, 6, 2655–2665. [DOI] [PubMed] [Google Scholar]
- DeRider M. L.; Wilkens S. J.; Waddell M. J.; Bretscher L. E.; Weinhold F.; Raines R. T.; Markley J. L. J. Am. Chem. Soc. 2002, 124, 2497–2505. [DOI] [PubMed] [Google Scholar]
- Shoulders M. D.; Satyshur K. A.; Forest K. T.; Raines R. T. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 559–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swamy K. C. K.; Kumar N. N. B.; Balaraman E.; Kumar K. Chem. Rev. 2009, 109, 2551–2651. [DOI] [PubMed] [Google Scholar]
- Martin S. F.; Dodge J. A. Tetrahedron Lett. 1991, 32, 3017–3020. [Google Scholar]
- Gomez-Vidal J. A.; Forrester M. T.; Silverman R. B. Org. Lett. 2001, 3, 2477–2479. [DOI] [PubMed] [Google Scholar]
- Gomez-Vidal J. A.; Silverman R. B. Org. Lett. 2001, 3, 2481–2484. [DOI] [PubMed] [Google Scholar]
- Sebesta D. P.; Orourke S. S.; Pieken W. A. J. Org. Chem. 1996, 61, 361–362. [Google Scholar]
- Doig A. J.; Baldwin R. L. Protein Sci. 1995, 4, 1325–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A. M.; Zondlo N. J. Biochemistry 2012, 51, 5041–5051. [DOI] [PubMed] [Google Scholar]
- Elbaum M. B.; Zondlo N. J. Biochemistry 2014, 53, 2242–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruby V. J.; Li G.; Haskell-Luevano C.; Shenderovich M. Biopolymers 1997, 43, 219–266. [DOI] [PubMed] [Google Scholar]
- El-Faham A.; Funosas R. S.; Prohens R.; Albericio F. Chem.—Eur. J. 2009, 15, 9404–9416. [DOI] [PubMed] [Google Scholar]
- Woody R. W. J. Am. Chem. Soc. 2009, 131, 8234–8245. [DOI] [PubMed] [Google Scholar]
- Chiang Y. C.; Lin Y. J.; Horng J. C. Protein Sci. 2009, 18, 1967–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presta L. G.; Rose G. D. Science 1988, 240, 1632–1641. [DOI] [PubMed] [Google Scholar]
- MacArthur M. W.; Thornton J. M. J. Mol. Biol. 1991, 218, 397–412. [DOI] [PubMed] [Google Scholar]
- Lovell S. C.; Word J. M.; Richardson J. S.; Richardson D. C. Proteins 2000, 40, 389–408. [PubMed] [Google Scholar]
- Chiu H.-P.; Kokona B.; Fairman R.; Cheng R. P. J. Am. Chem. Soc. 2009, 131, 13192–13192. [DOI] [PubMed] [Google Scholar]
- Thomas C. A.; Talatya E. R.; Bann J. G. Chem. Commun. 2009, 3366–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara K.; Nemoto N.; Motooka D.; Nishi Y.; Doi M.; Uchiyama S.; Nakazawa T.; Nishiuchi Y.; Yoshida T.; Ohkubo T.; Kobayashi Y. J. Phys. Chem. B 2012, 116, 6908–6915. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.