

Abstract
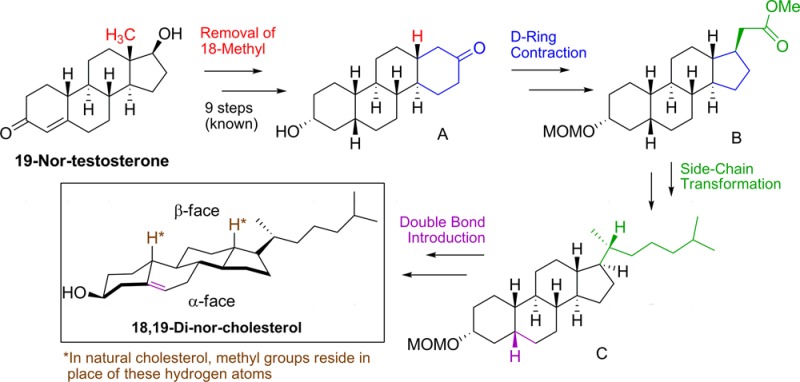
Herein, we report the first synthesis of a demethylated form of cholesterol (18,19-di-nor-cholesterol), in which the C18 and C19 methyl groups of the β-face were eliminated. Recent molecular simulations modeling 18,19-di-nor-cholesterol have suggested that cholesterol’s opposing rough β-face and smooth α-face play necessary roles in cholesterol’s membrane condensing abilities and, additionally, that specific facial preferences are displayed as cholesterol interacts with different neighboring lipids and transmembrane proteins. Inspired by these poorly characterized biochemical interactions, an extensive 18-step synthesis was completed as part of a collaborative effort, wherein synthesizing a “smoothened” cholesterol analogue would provide a direct way to experimentally measure the significance of the β-face methyl groups. Starting from known perhydrochrysenone A, the synthesis of 18,19-di-nor-cholesterol was accomplished with an excellent overall yield of 3.5%. The use of the highly stereoselective Dieckmann condensation and the employment of Evans’ chiral auxiliary were both key to ensuring the success of this synthesis.
Introduction
Cholesterol is a ubiquitous molecule that plays a vital role in maintaining the overall integrity of eukaryotic cells by giving rise to the requisite structure, fluidity, and organization of the membrane.1 It is synthesized endogenously through an extensive multistep biosynthetic pathway,2 with the first sterol in the pathway being lanosterol. Interestingly, of all the sterol intermediates between lanosterol and cholesterol, none possess cholesterol’s ability to order and condense lipid membranes;3−5 however, the details regarding cholesterol’s structural interactions within the membrane are poorly understood. Thus, this remains an area of active investigation.6−9
One of the more intriguing aspects of this planar tetracyclic molecule pertains to its two structurally distinct faces: its β-face contains two protruding methyl groups that are conspicuously absent on its α-face (Figure 1a). There has been much speculation that this facial asymmetry may be essential for cholesterol’s unique membrane condensing abilities as well as being essential to the way in which it interacts with and stabilizes neighboring lipids and transmembrane proteins. To test this theory, models have been used to simulate the properties and interactions of a demethylated form of cholesterol (18,19-di-nor-cholesterol, which lacks the β-face methyl groups).6,9−11 However, direct experimental evidence has not been obtained, as the molecule has never been previously synthesized. Herein, we describe the first synthesis of a “smoothened” 18,19-di-nor-cholesterol, which we have completed as part of a collaborative effort,12 in hopes of providing a way to experimentally measure its effects on lipid membranes and other important transmembrane interactions.
Figure 1.
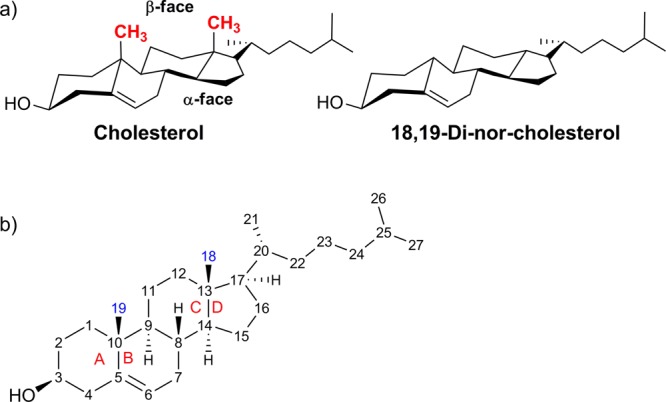
(a) Structures of cholesterol and 18,19-di-nor-cholesterol; the β-face methyl groups of cholesterol (shown in red) are absent in 18,19-di-nor-cholesterol. (b) Cholesterol nomenclature and numbering system. Blue numbers are methyl groups absent in 18,19-di-nor-cholesterol; red letters label each ring. Wedged bonds are referred to as having a β-configuration, and dashed bonds, an α-configuration.
Discussion and Results
Retrosynthetic Analysis
Utilizing a nine-step sequence developed in our laboratory,13 commercially available 19-nor-testosterone was first transformed into perhydrochrysenone derivative A (Scheme 1). In this transformation, the unwanted C18 methyl group was removed while still preserving the trans-C,D ring fusion. Additionally, the cis-A,B ring fusion of compound A allows for the later introduction of the Δ5 double bond. The α-stereochemistry of the hydroxyl group at C3 in compound A is not important, as the 3β-hydroxy group will be established during the Δ5 double bond introduction (see Figure 1b for numbering).
Scheme 1.

As depicted in our retrosynthesis (Scheme 1), starting from A, we can essentially fragment our synthesis into three main parts: D-ring contraction, side-chain installation, and introduction of the Δ5 double bond. Installation of the double bond was chosen as the final step in our synthesis of 18,19-di-nor-cholesterol because of its high reactivity toward a variety of reaction conditions and because it could be obtained from 18,19-di-nor-epicoprostanol (C) via enone formation and double bond deconjugation.14 It was then envisaged that synthon C could be obtained through the addition of an isohexyl side chain to a steroid precursor containing a CH2COOR group at C17. Synthon B fits this description and could be accessed from A by an oxidative D-ring-opening (to yield a 1,4-diester), a Dieckmann condensation to reclose the ring,15 the removal of the 16-ketone group, and a one-carbon homologation reaction at C17 to give synthon B.
The success of this entire approach, however, is contingent upon whether ring closure via the Dieckmann condensation (preceding the C17 homologation reaction) would yield the desired 17β-carbomethoxy group as the major isomeric product (Scheme 2, compound 4). Although four isomers are possible, extrapolating results from previous studies on the synthesis of A-nor-steroids15−18 and examining the steric interactions at play in our di-nor system led us to expect that this transformation would, in fact, yield our desired product (see Supporting Information Scheme S1). This was therefore seen as our most critical, and yet most elegant, synthetic step, as this reaction would simultaneously contract the D-ring and selectively install a C17β functional group while preserving the trans-C,D ring fusion.
Scheme 2.

Construction of Synthon B (Scheme 2)
To begin, we first protected perhydrochrysenone derivative A as the acetate (2) before carrying out a chromium(VI)-mediated, oxidative D-ring-opening and esterification to give 1,4-diester (3). The key step in our synthetic approach was next. Employing Dieckmann condensation conditions,16 compound 3 was heated at 40 °C for 45 min in basic MeOH to yield a single β-ketoester product. A crystal structure confirmed the stereochemistry to be that of the expected 17β-methyl ester (4) (see the Supporting Information).
After removing the 16-keto group via formation of thioketal 5 followed by Raney nickel dethioketalization to yield compound 6, we focused on lengthening the side chain of compound 6 by one carbon. Having little success with the Arndt Eistert homologation reaction,19,20 we chose to proceed via the introduction of a carbonitrile group. We first protected compound 6 as a methoxymethyl ether (7) and then reduced the ester moiety using LiAlH4 to afford primary alcohol 8. A carbonitrile was then introduced via a two-part procedure, wherein a methanesulfonyl ester was formed and then stirred at 65 °C for 16 h with NaCN in DMF to give carbonitrile 9, which was subsequently hydrolyzed to carboxylic acid 10 (Scheme 2).
Construction of Synthon C (Scheme 3)
Scheme 3.

For steroids with a C18 methyl group, the introduction of a side-chain via an α-substitution at C20 is known to favor the desired (R)-diastereomer;21 however, this selectivity is attributed to steric interferences caused by the C18 methyl group,22 which is absent in our 18,19-di-nor series. After confirming that alkylations at C20 are not stereoselective for 18-nor-steroids, we used Evans’ chiral auxiliary [(R)-4-benzyl-2-oxazolidinone] to ensure (R)-stereochemistry at C21. Introduction of this group went smoothly, affording compound 11. However, in a variety of subsequent alkylation attempts,23 we were unable to introduce the entire isohexyl group in satisfactory yield, presumably because of the added bulk of the chiral auxiliary. Therefore, we instead installed the side chain in two pieces, first utilizing the highly reactive allyl iodide, to obtain compound 12 as the desired (R)-diastereomer in 86% yield.
Next, we needed to remove the chiral auxiliary in order to generate the C21 methyl group (currently positioned as the carbonyl carbon). Although one-step reduction procedures failed to remove the chiral auxiliary,24 it was found to be easily removed by first hydrolyzing the benzyloxazolidinone and then reducing the resultant carboxylic acid with LiAlH4 to give primary alcohol 13. At this point, with all critical stereocenters intact, a crystal structure was obtained (see the Supporting Information). Finally, deoxygenation was completed via a simple two-part protocol by first forming the methylsulfonate ester and then displacing this group using LiAlH4 to reveal the C21 methyl group and give compound 14.
Transformation of the allyl group into the isohexyl side chain was the last step in completing the construction of synthon C. Although the side chain was not introduced in one piece via our initial plan, transformation of the allyl group into an isohexyl chain was straightforward and higher yielding overall than the initial isohexyl introduction attempts. Accordingly, the terminal olefin was oxidatively cleaved using O3/Me2S to give the crude aldehyde intermediate, which was immediately treated with isobutyl(triphenyl)phosphonium bromide under Wittig conditions to give the isohexenyl side chain of compound 15 in 85% yield as a single double bond isomer whose stereochemistry we did not determine. A subsequent hydrogenation proceeded near quantitatively, affording 18,19-di-nor-epicoprostanol (16) and completing the synthesis of synthon C (Scheme 3).
Construction of 18,19-Di-nor-cholesterol (1) (Scheme 4)
Scheme 4.

Lastly, we needed to introduce the Δ5 double bond, which is well-known in literature, through formation of a Δ4-3-ketone followed by deconjugation.14,25 Therefore, we first synthesized the 3-keto-precursor (17), which was easily obtained by first removing the methoxymethyl protecting group of compound 16 with anhydrous HCl and then oxidizing the resulting crude alcohol using Jones’ reagent to give ketone 17. However, subsequent attempts to introduce the Δ4-3-ketone proved to be difficult in the absence of the C19 methyl group.
After having little success with a variety of steroid protocols,26 we ultimately adapted a general procedure for the formation of α,β-unsaturated ketones, wherein bromination of a silyl enol ether first yields an α-bromoketone that undergoes a dehydrobromination. After optimizing conditions, compound 17 was treated with trimethylsilyl triflate (TMSOTf) and Et3N to form the TMS-enol ether in situ. Upon cooling the reaction to −78 °C and adding NaHCO3 and N-bromosuccinimide (NBS), the intermediate bromoketone (obtained as a mixture of bromides; see Supporting Information Scheme S2) was heated in the presence of Li2CO3 and LiBr in DMF at 120 °C for 16 h to generate the desired Δ4-3-ketone (18) in a moderate 32% yield, with an unwanted Δ1-3-ketone byproduct obtained in 55% yield (reflecting the ratio of the bromide intermediates).
Although the desired Δ4-3-ketone was the minor product, of the reaction protocols attempted this protocol gave the highest product yield and the least amount of byproducts.26 It was also far more economical than the other attempted procedures, in that we were consistently able to recover and reuse starting material 17 via hydrogenation of the unwanted Δ1-3-ketone. Thus, after hydrogenation, using 10% Pd/C, starting material 17 could be recovered in 54% yield. By running this reaction sequence three consecutive times, we were able to obtain our desired 18,19-di-nor-cholest-4-ene-3-one (18) in a respectable 60% yield, with 16% of our total starting material (17) recovered.
Deconjugation was the final step in our synthesis, which proved to be relatively straightforward using typical literature protocols.14 Compound 18 was dissolved in Ac2O and TMSI to form the dienol acetate, which was then reduced with NaBH4 in EtOH. This simultaneously deconjugated the enone and set our final stereocenter (3β-OH),27 affording our target 18,19-di-nor-cholesterol (1) in 60% yield (3.6% overall from perhydrochrysenone A) (Scheme 4).
Conclusions
The synthesis of the smoothened 18,19-di-nor-cholesterol was achieved starting from commercially available 19-nor-testosterone. The novel use of the Dieckmann condensation was key to accessing our target molecule, allowing us to contract the D-ring to a 5-membered ring and to simultaneously introduce a modifiable functional group and set the stereochemistry at C17 while maintaining the trans-C,D ring fusion. Furthermore, the completion of this molecule can now allow for the direct comparison of cholesterol and demethylated 18,19-di-nor-cholesterol in a variety of biological and biophysical studies, some of which are currently underway.12
Experimental Section
Chromatography was performed using flash chromatography grade silica gel (32–63 μm; Scientific Adsorbents). DCM was distilled over CaH prior to application. Tetrahydrofuran was distilled over Na/benzophenone just prior to application. NMR spectra were recorded in CDCl3 at 400 MHz (1H), 100 MHz (13C), or 400 MHz (2D COSY). Chemical shifts (δ) are reported downfield from internal Me4Si (δ: 0.00). High resolution FAB-MS determinations were made with a matrix m-nitrobenzyl alcohol, using NaI as necessary, utilizing a double-focusing analyzer composed of a magnetic sector followed by a electrostatic sector. IR spectra were recorded as films on a NaCl plate. All other chemicals were used as purchased without further purification unless noted.
(2R,4aS,4bR,6aR,10aR,10bS,12aR)-8-Oxooctadecahydrochrysen-2-yl Acetate (2)
To a stirred solution of A(13) (5.28 g, 19.1 mmol) at 0 °C under N2 in anhydrous pyridine (20 mL) was added acetic anhydride (3.6 mL, 38 mmol) dropwise followed by a catalytic amount of 4-dimethylaminopyridine (116 mg, 0.95 mmol). The reaction mixture was allowed to warm to 25 °C and was stirred for 16 h. The pyridine was then evaporated in vacuo, and the crude residue was then redissolved in DCM (100 mL) and washed with 1 M HCl (3 × 25 mL) followed by H2O (2 × 25 mL). The organic layer was then dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (EtOAc/hexanes, gradient elution) to yield compound 2 as colorless crystals in 96% yield (5.83 g, 18.3 mmol). Compound 2: mp 169–171 °C (EtOAc/hexanes); [α]D20 −6.7 (c = 0.22, CHCl3); IR: 2931, 2905, 2858, 1724, 1243, 1032 cm–1; 1H NMR (CDCl3) δ 4.75 (m, 1H, CHOAc), 2.25–2.43 (m, 4H), 2.04 (s, 3H, OAc), 2.12–1.10 (m, 20H), 0.75–0.92 (m, 2H); 13C NMR (CDCl3) δ 212.1, 170.8, 74.4, 48.8, 47.3, 46.3, 43.4, 41.5, 40.3, 37.4, 35.2, 34.2, 32.3, 31.4, 30.5, 29.5, 26.0, 25.7, 25.3, 21.7; HR-FAB MS [M + Na]+ calcd for C20H30O3Na+: 341.2093, found 341.2099.
(1R,2R,4aR,4bS,7R,8aR,10aS)-7-Hydroxy-tetradecahydro-1,2-phenanthrenediacetic Acid Dimethyl Ester (3)
Compound 3 was prepared according to a literature procedure.28 A stock solution of CrO3/H2O/AcOH (5.0 g/7.10 mL/8.35 mL; w/v/v) was first prepared. Then, in a two-necked flask fitted with a dropping funnel and a reflux condenser, compound 2 (3.05 g, 9.58 mmol) was dissolved into a solution of AcOH/H2O/MeOH 600:20:1 (30 mL) at 60 °C. The stock CrO3/H2O/AcOH solution (13.5 mL) was then added via dropping funnel to compound 2, taking care not to heat the reaction mixture above 65 °C (∼1 drop/5 s). The reaction flask was allowed to stir for 2 h at 60 °C, at which time starting material disappearance was observed by TLC. At this point, heated H2O was added (30 mL, 60 °C), the stirring was stopped, and the reaction was gradually cooled to room temperature. The reaction was allowed to stand for 16 h, during which time the desired compound precipitated. Cold H2O (10 mL) was then added, and the precipitate was then filtered off and washed several times with cold H2O and dried in vacuo. After 3 h of drying, the compound was redissolved in MeOH (100 mL) and cooled to 0 °C under N2. Acetyl chloride (10 mL) was slowly added dropwise to the flask, and after 10 min, the reaction was brought to rt. After 16 h, the reaction was cooled to 0 °C and neutralized with saturated aqueous NaHCO3. The reaction was diluted with H2O (100 mL) and extracted with DCM (4 × 50 mL). The organic layers were combined, dried over Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (EtOAc/hexanes, gradient elution) to yield compound 3 as a white amorphous solid in 44% yield (1.49 g, 4.21 mmol). Compound 3: mp 52–54 °C (amorphous solid, EtOAc/hexanes); [α]D20 +13.4 (c = 0.19, CHCl3); IR: 3401, 2928, 2863, 1737, 1436, 1270, 1161 cm–1; 1H NMR (CDCl3) δ 3.64 (br s, 7H, CHOH, 2 × OMe), 2.55–0.72 (m, 24H); 13C NMR (CDCl3) δ 173.6 (×2), 71.6, 51.5, 51.5, 47.0, 43.7, 40.1, 39.3, 38.7, 37.3, 36.1, 35.1, 35.0, 32.4, 31.5, 29.7, 28.9, 25.5, 25.5; HR-FAB MS [M + Na]+ calcd for C20H32O5Na+: 375.2147, found 375.2150.
(3α,5β,17β)-3-Hydroxy-16-oxo-gonane-17-carboxylic Acid Methyl Ester (4)
Similar to the literature,16 in a flame-dried flask under N2, Na(s) (0.60 g, 26 mmol) was dissolved portionwise into anhydrous MeOH (15 mL). The excess MeOH was then evaporated off, leaving only a small amount (∼2.5 mL) to prevent the generated NaOMe from precipitating. Anhydrous THF (50 mL) was added to the flask followed by the dropwise addition of a solution of compound 3 (2.63 g, 7.46 mmol) in anhydrous THF (100 mL). The reaction was refluxed at 100 °C for 45 min and then promptly cooled to 0 °C and neutralized with 1 M HCl. The reaction mixture was then diluted with H2O (150 mL) and extracted with EtOAc (4 × 50 mL). The organic extracts were then combined, dried over Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (EtOAc/hexanes, gradient elution) to give compound 4 as colorless crystals in 70% yield (1.65 g, 5.22 mmol). Compound 4: mp 163–164 °C (EtOAc/hexanes); [α]D20 −86.8 (c = 0.19, CHCl3); IR: 3399, 2924, 2861, 1756, 1727, 1440, 1271, 1051 cm–1; 1H NMR (CDCl3) δ 3.76 (s, 3H, CO2Me), 3.66 (br s, 1H, H-3), 2.91 (d, 1H, 3J = 12.8 Hz), 2.51 (dd, 1H, 2J = 18.0 Hz, 3J = 6.8 Hz), 1.18–2.11 (m, 19H), 0.92–1.11 (m, 2H); 13C NMR (CDCl3) δ 210.4, 169.7, 71.8, 62.3, 52.6, 48.0, 47.0, 46.3, 43.3, 40.1, 37.6, 36.4, 35.5, 31.3, 30.2, 29.9, 29.3, 26.8, 25.9; HR-FAB MS [M + H]+ calcd for C19H29O4+: 321.2066, found 321.2064.
(3α,5β,17β)-16,16-[1,2-Ethanediylbis(thio)]-3-hydroxy-gonane-17-carboxylic Acid Methyl Ester (5)
Following a literature procedure,30 compound 4 (1.58 g, 4.93 mmol) was dissolved into AcOH (12 mL) and ethanedithiol (1.04 mL, 12.3 mmol). To prevent the reaction mixture from solidifying, the flask was cooled in an ice bath no longer than 2 min before adding BF3·HOAc (3.43 mL, 24.7 mmol) dropwise. The reaction was then allowed to stir at 0 °C for 10 min and then for another 30 min at rt. The flask was then cooled to 0 °C and neutralized with saturated aqueous NaHCO3. The reaction mixture was then diluted with H2O (25 mL) and extracted with DCM (4 × 25 mL). The organic extracts were then combined, dried over Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (EtOAc/hexanes, gradient elution) to give compound 5 as colorless crystals in 95% yield (1.86 g, 4.68 mmol). Compound 5: mp 103–105 °C (EtOAc/hexanes); [α]D20 −65.0 (c = 0.14, CHCl3); IR: 3368, 2923, 2861, 1735, 1438, 1210, 1156, 1056, 1033, 731 cm–1; 1H NMR (CDCl3) δ 3.74 (s 3H, OMe), 3.64 (br s 1H, H-3), 3.40–3.45 (m, 1H), 3.30–3.34 (m, 1H), 3.13–3.22 (m, 2H), 2.86 (d, 1H, 3J = 11.3 Hz), 2.51 (dd, 1H, J = 13.3 Hz, J = 5.5 Hz), 1.18–1.97 (m, 18H), 1.02–1.13 (m, 1H), 0.85–0.97 (m, 2H); 13C NMR (CDCl3) δ 172.4, 71.9, 69.7, 65.9, 52.5, 51.7, 49.7, 48.3, 47.6, 41.1, 40.2, 40.1, 37.3, 36.5, 35.6, 31.4, 30.1, 30.0, 29.6, 26.6, 26.1; HR-FAB MS [M + Na]+ calcd for C21H32O3S2Na+: 419.1691, found 419.1698.
(3α,5β,17β)-3-Hydroxy-gonane-17-carboxylic Acid Methyl Ester (6)
Raney Nickel Activation
Raney nickel (W.R. Grace and Co. Raney4200, slurry in H2O, active catalyst) was successively washed and decanted with deionized H2O (3×) and EtOH (×). Once rinsed thoroughly, the catalyst was kept in a solution of EtOH to keep it from drying.
Compound 5 (1.87 g, 4.71 mmol) was dissolved in EtOH (30 mL). A slurry of activated Raney nickel (17g) in EtOH (30 mL) was added portionwise, and the reaction was refluxed at 80 °C for 1.5 h. The reaction was then cooled to rt, and the crude reaction mixture was filtered through a short column of silica gel (eluting several times with EtOAc/EtOH 1:1) to remove the Raney nickel and concentrated in vacuo. The residue was purified by column chromatography on silica gel (EtOAc/hexanes, gradient elution) to give compound 6 as colorless crystals in 95% yield (1.37 g, 4.47 mmol). Compound 6: mp 123–125 °C (EtOAc/hexanes); [α]D20 +43.0 (c = 0.10, CHCl3); IR: 3350, 2924, 2864, 1735, 1596, 1440, 1208, 1156, 1050 cm–1; 1H NMR (CDCl3) δ 3.65 (s 3H, OMe), 3.64 (br s 1H, H-3), 2.25 (m, 1H), 0.95–1.99 (m, 22H), 0.72–0.90 (m, 2H); 13C NMR (CDCl3) δ 177.1, 71.8, 52.0, 51.6, 50.2, 49.2, 48.5, 40.1, 37.5, 36.4, 35.7, 31.5, 30.3, 29.9, 29.8, 28.6, 27.4, 26.6, 26.2; HR-FAB MS [M + H]+ calcd for C19H31O3+: 307.2278, found 307.2273.
(3α,5β,17β)-3-Methoxymethoxy-gonane-17-carboxylic Acid Methyl Ester (7)
Compound 6 (1.34 g, 4.37 mmol) was dissolved in anhydrous DCM (200 mL) and cooled to 0 °C under N2. N,N-Diisopropylethylamine (3.05 mL, 17.5 mmol) was added followed by the dropwise addition of chloromethyl methyl ether (0.66 mL, 8.8 mmol). The reaction was allowed to warm to rt and left to stir for 16 h. Upon completion, the reaction was washed with H2O (50 mL), brine (50 mL), and H2O (50 mL), dried over Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (EtOAc/hexanes, gradient elution) to give compound 7 as colorless crystals in 98% yield (1.50 g, 4.28 mmol). Compound 7: mp 66–69 °C (cold hexanes); [α]D20 +43.8 (c = 0.18, CHCl3); IR: 2929, 2867, 1733, 1440, 1144, 1107, 1041 cm–1; 1H NMR (CDCl3) δ 4.68 (s 2H, OCH2OCH3), 3.67 (s 3H, CO2Me), 3.54 (m, 1H, H-3), 3.37 (s 3H, OCH2OCH3), 2.25 (m, 1H), 0.97–1.99 (m, 22H), 0.77–0.90 (m, 2H); 13C NMR (CDCl3) δ 177.1, 94.7, 77.0, 55.3, 52.0, 51.7, 50.3, 49.3, 48.5, 40.4, 37.5, 35.8, 33.7, 31.7, 30.4, 29.8, 28.7, 27.5, 27.1, 26.6, 26.3; HR-FAB MS [M + H]+ calcd for C21H35O4+: 351.2535, found 351.2536.
(3α,5β,17β)-3-Methoxymethoxy-gonane-17-methanol (8)
Compound 7 (2.89 g, 8.25 mmol) was dissolved in anhydrous THF (300 mL) and then cooled to 0 °C under N2. Over 30 min, LiAlH4 (1 M in THF; 8.25 mL, 16.5 mmol) was added dropwise to the reaction mixture, and upon completion of LiAlH4 addition, the reaction was found to be complete. The reaction was quenched by the careful addition of H2O (0.63 mL) followed by 15% aqueous NaOH (0.63 mL), and the reaction was allowed to stir for 30 min at rt. Another aliquot of H2O (1.90 mL) was added, and within 5 min a white precipitate formed. The precipitate was filtered off, and the filtrate was dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (EtOAc/hexanes, gradient elution) to give compound 8 as colorless crystals in 91% yield (2.42 g, 7.50 mmol). Compound 8: mp 76–78 °C (EtOAc/hexanes); [α]D20 +41.4 (c = 0.14, CHCl3); IR: 3401, 2928, 2865, 1441, 1144, 1107, 1041 cm–1; 1H NMR (CDCl3) δ 4.69 (s 2H, OCH2OCH3), 3.62–3.73 (m 1H), 3.45–3.57 (m, 2H, H-3), 3.37 (s 3H, OCH2OCH3), 0.72–1.99 (m, 25H); 13C NMR (CDCl3) δ 94.7, 77.0, 66.8, 55.3, 52.3, 48.8, 48.7, 47.1, 40.5, 37.8, 35.9, 33.7, 31.7, 30.8, 30.1, 28.3, 27.1, 27.1, 26.6, 26.3; HR-FAB MS [M + Na]+ calcd for C20H34O3Na+: 345.2406, found 345.2404.
(3α,5β,17β)-3-Methoxymethoxy-18,19-di-nor-pregnane-21-nitrile (9)
To a solution of compound 8 (0.90 g, 2.8 mmol) in anhydrous DCM (30 mL) at 0 °C under N2 was added Et3N (1.17 mL, 8.37 mmol). MsCl (0.43 mL, 5.6 mmol) was dissolved in anhydrous DCM (0.6 mL) and added dropwise to the reaction flask. After 30 min at 0 °C, the reaction was quenched with ice and subsequently washed with cold saturated aqueous NaHCO3 (10 mL), cold brine (10 mL), and cold H2O (10 mL), dried over Na2SO4, and concentrated in vacuo at rt. The reaction flask was allowed to dry for 2 to 3 h under vacuum, and the residue was dissolved in anhydrous DMF (60 mL), and the flask was fitted with a reflux condenser. NaCN (0.68 g, 14 mmol) was crushed into a powder and added to the reaction flask, and the mixture was stirred at 65 °C for 16 h. The reaction was then cooled to 25 °C, and ice was added to the reaction mixture, causing the product to precipitate. The product was then filtered, washed several times with H2O, and dried in vacuo. The crude compound was purified by column chromatography on silica gel (EtOAc/hexanes, gradient elution) to give compound 9 as colorless crystals in 85% yield (0.79 g, 2.4 mmol). Compound 9: mp 113–115 °C (EtOAc/hexanes); [α]D20 +46.1 (c = 0.17, CHCl3); IR: 2934, 2909, 2867, 2851, 2244, 1143, 1106, 1040 cm–1; 1H NMR (CDCl3) δ 4.67 (s 2H, OCH2OCH3), 3.47–3.58 (m 1H, H-3), 3.35 (s, 3H, OCH2OCH3), 2.44 (dd, 1H, 2J = 16.8 Hz, 3J = 5.5 Hz), 2.32 (dd, 1H, 2J = 16.3 Hz, 3J = 6.7 Hz), 1.88–1.99 (m, 4H), 0.74–1.86 (m, 21H); 13C NMR (CDCl3) δ 119.3, 94.7, 76.9, 55.3, 51.7, 50.8, 48.5, 40.6, 40.3, 37.6, 35.8, 33.6, 31.6, 29.7, 29.6, 29.5, 27.7, 27.1, 26.4, 26.2, 21.2; HR-FAB MS [M + Na]+ calcd for C21H33NO2Na+: 354.2409, found 354.2403.
(3α,5β,17β)-3-Methoxymethoxy-18,19-di-nor-pregnan-21-oic Acid (10)
In a flask fitted with a reflux condenser, compound 9 (1.98 g, 5.97 mmol) was dissolved in ethylene glycol (150 mL) at 120 °C. To this was slowly added a solution of 50% aqueous NaOH (20 mL), and the reaction was allowed to stir for 16 h at 120 °C. The reaction was then cooled to 25 °C and acidified to a pH 4 with 6 N HCl. The reaction mixture was then extracted several times with DCM (4 × 50 mL). The organic fractions were then combined, dried over Na2SO4, and concentrated in vacuo. The crude compound was purified through a short column of silica gel (MeOH/DCM, 1% isocratic elution) to give compound 10 as colorless crystals in 85% yield (1.78 g, 5.07 mmol). Compound 10: mp 164–166 °C (DCM/hexanes); [α]D20 +35.7 (c = 0.12, CHCl3); IR: 3367, 2914, 2866, 1701, 1141,1109, 1047 cm–1; 1H NMR (CDCl3) δ 4.69 (s 2H, OCH2OCH3), 3.49–3.59 (m 1H, H-3), 3.37 (s, 3H, OCH2OCH3), 2.52 (dd, 1H, 2J = 14.9 Hz, 3J = 4.7 Hz), 2.14 (dd, 1H, 2J = 15.3 Hz, 3J = 9.4 Hz), 1.45–2.03 (m, 13H), 0.93–1.36 (m, 10H), 0.71–0.85 (m, 3H); 13C NMR (CDCl3) δ 179.7, 94.6, 77.1, 55.3, 51.7, 51.6, 48.8, 41.0, 40.5, 39.1, 37.7, 35.9, 33.7, 31.7, 30.0, 30.0, 29.8, 27.9 27.1, 26.5, 26.3; HR-FAB MS [M + Na]+ calcd for C21H34O4Na+: 373.2355, found 373.2349.
(4R)-3-[2-[3α,5β,17β-[3-Methoxymethoxy-gonan-17-yl]-acetyl]]-4-(phenylmethyl)- 2-oxazolidinone (11)
Following literature methodology,31 to a solution of compound 10 (0.36 g, 1.0 mmol) dissolved in anhydrous THF (15 mL) under N2 at 0 °C was added Et3N (0.79 mL, 5.7 mmol). The reaction was cooled to 0 °C, and pivaloyl chloride (0.19 mL, 1.5 mmol) was added dropwise. The reaction was stirred at 0 °C for 2 h. Then, LiCl (0.22 g, 5.1 mmol) and (R)-4-benzyl-2-oxazolidinone (0.27 g, 1.5 mmol) were added, and the reaction was allowed to slowly warm to 25 °C and was then stirred for 16 h. The reaction was then cooled to 0 °C, quenched with saturated aqueous NaHCO3, and further diluted with H2O (30 mL). The reaction was then extracted with EtOAc (4 × 10 mL). The organic extracts were combined, dried over Na2SO4, and concentrated in vacuo. The crude compound was purified by column chromatography on silica gel (EtOAc/hexanes, gradient elution) to give compound 11 as colorless crystals in 96% yield (0.50 g, 0.99 mmol). Compound 11: mp 143–145 °C (cold EtOAc); [α]D20 +10.2 (c = 0.25, CHCl3); IR: 3392, 2928, 2865, 1783, 1699, 1386, 1212, 1106, 1039 cm–1; 1H NMR (CDCl3) δ 7.11–7.29 (m, 5H), 4.62 (s 2H, OCH2OCH3), 4.57–4.64 (m, 1H, NCH), (overlapping doublets, 2H, OCH2CH), 3.43–3.51 (m, 1H, H-3), 3.30 (s, 3H, OCH2OCH3), 3.23 (dd, 1H, 2J = 13.1 Hz, 3J = 3.3 Hz, CHHPh), 3.03 (dd, 1H, 2J = 16.6 Hz, 3J = 4.1 Hz, CHHC(O)N), 2.66–2.79 (overlapping doublets, 2H, CHHC(O)N, CHHPh), 0.65–2.03 (m, 25 H); 13C NMR (CDCl3) δ 173.5, 153.7, 135.5, 129.6 (×2), 129.1 (×2), 127.5, 94.7, 77.0, 66.3, 55.4, 55.4, 51.6, 51.5, 48.8, 40.6, 40.5, 40.4, 38.1, 37.8, 35.9, 33.7, 31.7, 30.4, 30.0, 29.9, 28.0, 27.1, 26.6, 26.3; HR-FAB MS [M + Na]+ calcd for C31H43NO5Na+: 532.3039, found 532.3043.
(4R)-3-[(2R)-[(3α,5β,17β)-3-Methoxymethoxy-gonan-17-yl]-1-oxo-4-penten-1-yl]-4-(phenylmethyl)-2-oxazolidinone (12)
In a flame-dried flask under N2, compound 11 (1.32 g, 2.59 mmol) was dissolved in anhydrous THF (50 mL), and the reaction mixture was cooled to −78 °C. NaHMDS (1 M in THF; 4.32 mL, 4.53 mmol) was added dropwise, and the reaction was allowed to stir for 1 h at −78 °C. Allyl iodide (0.47 mL, 5.2 mmol) was then added dropwise, and the reaction was stirred at −78 °C for another 1 h. After 1 h, the reaction was quenched with saturated aqueous NH4Cl, H2O was added (50 mL), and the reaction was extracted several times with EtOAc (4 × 50 mL). The organic fractions were then combined, dried over Na2SO4, and concentrated in vacuo. The crude compound was purified by column chromatography on silica gel (EtOAc/hexanes, gradient elution) to give compound 12 as a white foamy solid in 86% yield (1.22 g, 2.23 mmol), with recovery of unreacted starting material 11 in 7%. Compound 12: [α]D20 −28.2 (c = 0.13, CHCl3); IR: 3369, 2927, 2865, 1782, 1697, 1640, 1384, 1208, 1105, 1040 cm–1; 1H NMR (CDCl3) δ 7.15–7.29 (m, 5H), 5.70–5.81 (m, 1H, CH=CH2), 4.93–5.03 (overlapping doublets, 2H, CH=CH2), 4.61 (s, 2H, OCH2OCH3), 4.54–4.62 (m, 1H, NCH), 4.01–4.06 (overlapping doublets, 2H, OCH2CH), 3.92–3.96 (m, 1H, CHC=O), 3.41–3.49 (m, 1H, H-3), 3.29 (s, 3H, OCH2OCH3), 3.24 (dd, 1H, 2J = 13.2 Hz, 3J = 3.2 Hz, CHHPh), 2.56 (dd, 1H, 2J = 13.2 Hz, 3J = 2.8 Hz, CHHPh), 2.38–2.48 (m, 1H, CHHCH=CH2), 2.24–2.32 (m, 1H, CHHCH=CH2), 1.38–1.85 (m, 14H), 0.85–1.21 (m, 9H), 0.60–0.78 (m, 2H); 13C NMR (CDCl3) δ 176.1, 163.4, 135.8, 135.7, 129.6 (×2), 129.1 (×2), 127.4, 117.0, 94.6, 76.9, 65.9, 56.0, 55.3, 52.0, 49.6, 48.7, 46.4, 45.2, 40.3, 38.2, 37.6, 35.8, 34.8, 33.7, 31.6, 30.7, 30.1, 27.6, 27.2, 27.0, 26.4, 26.2; HR-FAB MS [M + H]+ calcd for C34H48NO5+: 550.3533, found 550.3539.
(3α,5β,20R)-3-Methoxymethoxy-18,19-di-nor-chol-23-en-21-ol (13)
Following a reported procedure,32 a solution of compound 12 (1.65 g, 3.00 mmol) was dissolved in THF/H2O 5:1 (40 mL) at 0 °C followed by dropwise addition of 30% aqueous H2O2 (3 mL). This was followed by the portionwise addition of LiOH (0.25 g, 6.0 mmol) over 30 min. The solution was allowed to slowly come to rt. After stirring for 16 h, the reaction was acidified to pH 1 with 1 M HCl and was immediately extracted with EtOAc (4 × 20 mL). The organic extracts were combined, dried over Na2SO4, and concentrated in vacuo for 3 h. The crude intermediate carboxylic acid was then redissolved in anhydrous THF (30 mL) at 0 °C under N2. LiAlH4 (2 M in THF; 3.75 mL, 7.50 mmol) was added dropwise, and the reaction was brought to 25 °C and stirred for 16 h. Upon reaction completion, the reaction was cooled to 0 °C and quenched carefully with 1 M HCl. The reaction was then diluted with H2O (30 mL) and extracted with EtOAc (4 × 20 mL). The organic fractions were then combined, dried over Na2SO4, and concentrated in vacuo. The crude compound was purified by column chromatography on silica gel (EtOAc/hexanes, gradient elution) to give compound 13 as colorless crystals in 91% yield (1.02 g, 2.73 mmol). Compound 13: mp 89–91 °C (EtOAc/hexanes); [α]D20 +37.2 (c = 0.13, CHCl3); IR: 3401, 2928, 2866, 1639, 1440, 1144, 1107, 1040 cm–1; 1H NMR (CDCl3) δ 5.75–5.86 (m, 1H, CH=CH2), 4.95–5.04 (overlapping doublets, 2H, CH=CH2), 4.66 (s, 2H, OCH2OCH3), 3.46–3.62 (m 3H, H-3, CH2OH), 3.34 (s, 3H, OCH2OCH3), 2.10–2.19 (m, 1H, CHHCH=CH2), 0.90–2.12 (m, 25H), 0.70–0.85 (m, 2H); 13C NMR (CDCl3) δ 138.5, 115.8, 94.6, 77.0, 65.2, 55.2, 52.0, 48.8, 48.3, 44.9, 42.6, 40.4, 37.8, 35.8, 33.6, 32.7, 31.7, 30.8, 30.1, 28.2, 27.1, 26.5, 26.2, 25.7; HR-FAB MS [M + Na]+ calcd for C24H40O3Na+: 399.2875, found 399.2883.
(3α,5β)-3-Methoxymethoxy-18,19-di-nor-chol-23-ene (14)
To a solution of compound 13 (1.04 g, 2.76 mmol) in anhydrous DCM (80 mL) at 0 °C under N2 was added Et3N (1.54 mL, 11.0 mmol). MsCl (0.43 mL, 5.5 mmol) was dissolved in anhydrous DCM (30 mL), and this solution was added dropwise to the reaction flask. After 30 min at 0 °C, the reaction was quenched with ice and subsequently washed with cold saturated aqueous NaHCO3 (25 mL), cold brine (25 mL), and cold H2O (25 mL), dried over Na2SO4, and condensed in vacuo at rt. The reaction flask was allowed to dry for 2 to 3 h under vacuum, the residue was then dissolved in anhydrous THF (40 mL), and the flask was fitted with a reflux condenser. LiAlH4 (2 M in THF, 13.8 mL, 27.6 mmol) was added dropwise to the reaction flask, and the mixture was stirred at 50 °C for 1 h. The reaction was then cooled to 0 °C, quenched carefully with 1 M HCl, subsequently diluted with H2O (30 mL), and extracted with EtOAc (4 × 20 mL). The organic fractions were then combined, dried over Na2SO4, and concentrated in vacuo. The crude compound was purified by column chromatography on silica gel (EtOAc/hexanes, gradient elution) to give compound 14 as colorless crystals in 94% yield (0.93 g, 2.6 mmol). Compound 14: mp 55–57 °C (cold hexanes); [α]D20 +41.9 (c = 0.16, CHCl3); IR: 2933, 2910, 2861, 1437, 1147, 1105, 1046, 913 cm–1; 1H NMR (CDCl3) δ 5.70–5.85 (m, 1H), 4.95–4.99 (overlapping doublets, 2H), 4.68 (s, 2H, OCH2OCH3), 3.49–3.59 (m, 1H, H-3), 3.37 (s, 3H, OCH2OCH3), 2.11–2.19 (m, 1H), 0.75–1.98 (m, 27H), 0.87 (d, 3H, 3J = 6.7 Hz); 13C NMR (CDCl3) δ 138.7, 115.3, 94.7, 77.1, 55.3, 52.6, 49.5, 48.8, 48.4, 40.5, 38.5, 37.8, 35.9, 35.9, 33.7, 31.8, 31.6, 30.3, 28.3, 27.1, 26.9, 26.5, 26.3, 18.1; HR-FAB MS [M + Na]+ calcd for C24H40O2Na+: 383.2926, found 383.2925.
(3α,5β)-3-Methoxymethoxy-18,19-di-nor-cholest-23-ene (15)
Combining two reported procedures,33,34 ozone gas was bubbled into a solution of compound 14 (0.62 g, 1.7 mmol) and dissolved in DCM (50 mL) at −78 °C until the reaction turned light blue in color (ca. 15–30 min). At this time, the ozone generator was turned off, and oxygen was allowed to bubble into the flask until the blue ozone color disappeared (ca. 5 min). Acetic acid (10 mL) was added to the flask, and subsequently the DCM was evaporated under reduced pressure. DCM (10 mL) was then added back to the flask containing the acetic acid and crude ozonide intermediate. The flask was then cooled to −78 °C under N2, and dimethyl sulfide (25 mL) was added dropwise. Wanting to avoid possible byproduct formation, the reaction was carefully monitored. After stirring at 25 °C for 5 h, the reaction was found to be incomplete. It was then placed in a freezer (5 °C) for 16 h to slow reaction progress. The reaction was then stirred an additional 8 h at rt followed by another 16 h at 5 °C, after which the starting alkene was found to be completely converted into an aldehyde intermediate. The reaction was diluted with DCM (50 mL), washed with H2O, saturated aqueous NaHCO3, and brine, dried over Mg2SO4, concentrated, and dried under high vacuum for 2 h. During this time, in a separate flask, isobutyltriphenylphosphonium bromide (2.39 g, 3.5 mmol) was dissolved into anhydrous THF (50 mL) under N2 and cooled to 0 °C. NaHMDS (1 M in THF; 3.08 mL, 3.08 mmol) was added dropwise to the reaction flask, and the mixture was stirred for 30 min at 0 °C, becoming bright orange in color. Tris[2-(2-methoxyethoxy)ethyl]amine (TDA-1) (68 μL) was then added to the bright orange ylide followed by the dropwise addition of the crude aldehyde dissolved in anhydrous THF (50 mL). The reaction was stirred for 30 min at 0 °C. Upon reaction completion, the remaining ylide was quenched by the addition of a few drops of saturated aqueous NH4Cl. The THF was evaporated under reduced pressure, EtOAc (50 mL) was added, and the mixture was sequentially washed with H2O, saturated aqueous NaHCO3, and brine, dried over Na2SO4, and concentrated in vacuo. The crude compound was purified by column chromatography on silica gel using DCM/hexanes gradient elution to remove excess triphenylphosphine and transitioning to a EtOAc/hexanes gradient elution to give compound 15 as colorless crystals in 85% yield (0.58 g, 1.5 mmol). Compound 15: mp 74–76 °C (cold hexanes); [α]D20 +44.0 (c = 0.32, CHCl3); IR: 2929, 2866, 1456, 1375, 1145, 1106, 1046, 917 cm–1; 1H NMR (CDCl3) δ 5.16–5.26 (m, 2H), 4.70 (s, 2H, OCH2OCH3), 3.50–3.59 (m, 1H, H-3), 3.38 (s, 3H, OCH2OCH3), 2.53–2.62 (m, 1H), 1.08–2.11 (m, 28H), 0.95 (d, 3H, 3J = 1.2 Hz), 0.94 (d, 3H, 3J = 1.6 Hz), 0.87 (d, 3H, 3J = 6.8 Hz); 13C NMR (CDCl3) δ 138.06, 127.0, 94.7, 77.1, 55.3, 52.6, 49.8, 48.9, 48.5, 40.6, 37.9, 36.5, 35.9, 33.7, 31.8, 31.5, 31.4, 30.3, 28.3, 27.2, 26.9, 26.7, 26.6, 26.3, 23.4, 23.3, 18.4; HR-FAB MS [M + Na]+ calcd for C27H46O2Na+: 425.3396, found 425.3389.
(3α,5β)-3-Methoxymethoxy-18,19-di-nor-cholestane (16)
Compound 15 (0.56 g, 1.4 mmol) and 10% Pd/C (200 mg) were added to a borosilicate sealable glass vessel containing EtOAc (50 mL). The reaction vessel was attached to the Parr hydrogenator (shaker-type), and the air was evacuated and replaced with H2 at 60 psi. The reaction was allowed to shake for 3 h, at which time the H2 was evacuated under vacuum for 30 min. The vessel was repressurized and removed from the hydrogenator, and the reaction mixture was filtered through Celite (taking care to wash the Pd/C several times with EtOAc). The eluent was concentrated and recrystallized from cold hexanes to give compound 16 as colorless crystals in 99% yield (0.56 g, 1.4 mmol). Compound 16: mp 64–65 °C (cold hexanes); [α]D20 +40.2 (c = 0.17, CHCl3); IR: 2928, 2867, 1456, 1375, 1145, 1107, 1045, 916 cm–1; 1H NMR (CDCl3) δ 4.70 (s, 2H, OCH2OCH3), 3.49–3.59 (m, 1H, H-3), 3.38 (s, 3H, OCH2OCH3), 0.73–1.99 (m, 33H), 0.86–0.87 (3, d, 9H); 13C NMR (CDCl3) δ 94.7, 77.1, 55.3, 52.6, 50.0, 48.9, 48.1, 40.6, 39.6, 37.9, 36.0, 35.5, 33.8, 33.7, 31.8, 31.6, 30.3, 28.4, 28.2, 27.2, 26.7, 26.6, 26.4, 25.7, 23.0, 22.8, 18.4; HR-FAB MS [M + Na]+ calcd for C27H48O2Na+: 427.3552, found 427.3556.
(5β)-18,19-Di-nor-cholestan-3-one (17)
To a flask containing compound 16 (306 mg, 0.76 mmol) in MeOH (15 mL) was added AcCl (1.5 mL) dropwise over 10 min. After stirring 16 h at rt, the reaction was neutralized with saturated aqueous NaHCO3, further diluted with H2O (30 mL), and extracted with DCM (4 × 15 mL). The organic extracts were combined and evaporated to give the crude alcohol intermediate, which was immediately redissolved in acetone (30 mL). Jones reagent (∼0.5 mL; 30% CrO3·30% H2SO4·40% H2O) was added dropwise until the solution remained a light brown color. At this time, a few drops of isopropyl alcohol were added to quench the remaining Jones reagent, and the reaction solution was filtered through a short silica column and eluted several times with EtOAc to remove chromium byproducts. The eluent was collected and concentrated and then purified by column chromatography on silica gel (EtOAc/hexanes, gradient elution) to give compound 17 as colorless crystals in 97% yield (263 mg, 0.73 mmol). Compound 17: mp 59–62 °C (amorphous solid, EtOAc/hexanes); [α]D20 = +40.9 (c = 0.17, CHCl3); IR: 2951, 2927, 2911, 2861, 1712, 1463 cm–1; 1H NMR (CDCl3) δ 2.60 (t, 1H, J = 14.0 Hz), 0.86–2.32 (m, 32H), 0.86–0.88 (3, d, 9H); 13C NMR (CDCl3) δ 213.2, 52.4, 50.0, 48.4, 48.0, 43.1, 40.2, 39.6, 38.5, 37.8, 36.7, 35.4, 33.7, 31.4, 30.8, 30.4, 28.3, 28.1, 28.0, 26.6, 25.8, 25.7, 23.0, 22.7, 18.4; HR-FAB MS [M + Na]+ calcd for C25H42ONa+: 381.3133, found 381.3132.
18,19-Di-nor-cholest-4-en-3-one (18)
Compound 17 (225 mg, 0.63 mmol) was added to a flask containing anhydrous DCM (4 mL) and Et3N (0.22 mL, 1.6 mmol) and cooled to 0 °C under N2. Trimethylsilyl trifluoromethanesulfonate (TMSOTf) (0.17 mL, 0.94 mmol) was added dropwise, and the reaction was allowed to stir for 30 min at 0 °C. After formation of the silyl enol ether (vide TLC), the reaction was cooled to −78 °C, and following a general literature procedure,35 NaHCO3(s) (77 mg, 1.3 mmol) and NBS (168 mg, 0.94 mmol) were then added sequentially. The reaction was allowed to warm to −40 °C over a period of 30 min. Upon formation of the intermediate bromide, the reaction was quenched by the addition of saturated aqueous Na2S2O3 (1 mL), removed from the dry ice bath, and allowed to stir at 25 °C for 5 min. The reaction was further diluted with DCM (4 mL) and washed with H2O (2 × 5 mL). The organic phase was then dried over Na2SO4, concentrated in vacuo, and dried under vacuum for 30 min. According to standard protocol,36 the crude bromide compound was then redissolved in anhydrous DMF (7.5 mL), LiBr (162 mg, 1.88 mmol) and Li2CO3 (324 mg, 4.39 mmol) were added, and the reaction was heated to 120 °C for 16 h. Upon the formation of two new products (vide TLC), the DMF was removed, and the crude mixture was purified by column chromatography on silica gel (EtOAc/hexanes, gradient elution) to yield the undesired Δ1-3-ketone in 55% (123 mg, 0.34 mmol) yield and the desired Δ4-3-ketone (18) in 32% (71 mg, 0.20 mmol) yield. The undesired Δ1-3-ketone was then hydrogenated using the conditions in the synthesis of compound 16, regenerating reuseable starting material (17, 122 mg, 0.34 mmol).
In repeating this sequence of protocols two more times, the desired enone (18) was obtained as colorless crystals in 60% yield (135 mg, 0.38 mmol), with starting material (17) recovered in 16% yield (36 mg, 0.10 mmol). Compound 18: mp 65–67 °C (cold hexanes); [α]D20 +64.3 (c = 0.10, CHCl3); IR: 2951, 2930, 2862, 1678, 1619, 1466, 1257 cm–1; 1H NMR (CDCl3) δ 5.81 (s, 1H) 2.33–2.47 (m, 2H), 2.18–2.33 (m, 3H), 1.88–2.06 (m, 4H), 0.81–1.78 (m, 21H), 0.84–0.86 (3, d, 9H); 13C NMR (CDCl3) δ 200.1, 167.2, 124.5, 52.1, 49.7, 49.1, 47.8, 47.3, 42.9, 39.5, 36.7, 35.8, 35.5, 33.7, 31.5, 31.1, 31.0, 28.2, 28.1, 26.7, 26.6, 25.6, 23.0, 22.7, 18.3; HR-FAB MS [M + H]+ calcd for C25H41O+: 357.3157, found 357.3151.
18,19-Di-nor-cholesterol (1)
Similar to a reported literature method,9 to a flask containing Ac2O (6 mL) and NaI (140 mg, 0.93 mmol) at 0 °C under N2 was added trimethylsilyl chloride (0.11 mL, 0.89 mmol) dropwise. The reagents were stirred for 30 min at 0 °C, after which compound 18 (57 mg, 0.18 mmol) was added. The reaction was allowed to slowly warm to rt, and stirring was continued another 16 h. Upon formation of the 18,19-di-nor-cholesta-3,5-dien-3-ol acetate intermediate, the reaction was neutralized with a 1:1 mix of Et3N/diethyl ether, water (5 mL) was added, and the reaction was then extracted with hexanes (4 × 2.5 mL), dried over Na2SO4, and concentrated in vacuo. The residue was immediately purified by column chromatography (EtOAc/hexanes gradient elution; adding ∼0.5% Et3N to each gradient) on a silica gel column packed with hexanes/Et3N (49:1). The dienol acetate intermediate was collected, the solvents were removed, and the compound was redissolved in EtOH and cooled to 0 °C. NaBH4 (60 mg) was added portionwise over 15 min, the reaction was allowed to come to rt, and stirring was then continued for another 1 h. The reaction mixture was then quenched with a few drops of saturated aqueous NH4Cl, and the solvents were evaporated. The crude product was redissolved in DCM (20 mL) and washed with brine (2 × 20 mL). The organic layer was dried over Na2SO4 and concentrated in vacuo. Purification by column chromatography on silica gel (EtOAc/hexanes, gradient elution) gave 18,19-di-nor-cholesterol (1) as colorless crystals in 60% yield (39 mg, 0.11 mmol). Compound 1: mp 124–126 °C (DCM/hexanes); [α]D20 +23.4 (c = 0.19, CHCl3); IR: 3373, 2926, 2853, 1449, 1375, 1056 cm–1; 1H NMR (CDCl3) δ 4.60 (dd, 1H, 3J = 5.6 Hz, H-6), 3.51–3.58 (m, 1H, H-3), 2.50 (m, 1H), 0.80–2.12 (m, 29H), 0.86–0.88 (3, d, 9H); 13C NMR (CDCl3) δ 137.3, 121.8, 71.4, 53.0, 49.9, 47.7, 45.2, 45.2, 43.1, 43.0, 39.6, 35.7, 35.6, 33.9, 32.0, 31.3, 31.1, 30.6, 28.3, 28.2, 26.7, 25.7, 23.0, 22.8, 18.4; HR-FAB MS [M + Na]+ calcd for C25H42ONa+: 381.3133, found 381.3136.
Acknowledgments
This work was funded by the National Institutes of Health (HL67773 (D.F.C.) and 5 T32 HL007275 (L.K.M.)) and by a National Science Foundation Shared Instrument Grant (CHE-0420497).
Supporting Information Available
1H and 13C NMR spectra for compounds 1–18; X-ray projection views for compounds 4 and 13; and CIF files for the crystal structures of compounds 4 and 13. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Yeagle P. L. Biochim. Biophys. Acta 1985, 822, 267.and references therein. [DOI] [PubMed] [Google Scholar]
- Vance D. E.; van den Bosch H. Biochim. Biophys. Acta 2000, 1529, 1. [DOI] [PubMed] [Google Scholar]
- Demel R. A.; van Deenen L. L. M.; Pethica B. A. Biochim. Biophys. Acta 1967, 135, 11. [DOI] [PubMed] [Google Scholar]
- Leathes J. B. Lancet 1925, 208, 853. [Google Scholar]
- Phillips M. C. Prog. Surf. Membr. Sci. 1972, 5, 139. [Google Scholar]
- Martinez-Seara H.; Rog T.; Karttunen M.; Vattulainen I.; Reigada R. PLoS One 2010, 5, e11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly T. A.; Wang M.; Regen S. L. Langmuir 2011, 27, 2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khelashvili G.; Harries D. J. Phys. Chem. B 2013, 117, 2411. [DOI] [PubMed] [Google Scholar]
- Fantini J.; Barrantes F. J. Front. Physiol. 2013, 4, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rog T.; Pasenkiewicz-Gierula M.; Vattulainen I.; Karttunen M. Biophys. J. 2007, 92, 3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyry S.; Rog T.; Karttunen M.; Vattulainen I. J. Phys. Chem. B 2008, 112, 2922. [DOI] [PubMed] [Google Scholar]
- Biophysical measurements will be published separately by the S. L. Regen Lab, Department of Chemistry, Lehigh University, Bethlehem, PA.
- Stastna E.; Rath N. P.; Covey D. F. Org. Biomol. Chem. 2011, 9, 4685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covey D. F.; Jiang X. J. Org. Chem. 2002, 67, 4893. [DOI] [PubMed] [Google Scholar]
- Fuchs B.; Loewenthal H. J. E. Tetrahedron 1960, 11, 1. [Google Scholar]
- Kontonassios D.; Sandris C. Steroids 1982, 39, 411. [DOI] [PubMed] [Google Scholar]
- Nace H. R.; Pyle J. L. J. Org. Chem. 1970, 36, 81. [DOI] [PubMed] [Google Scholar]
- Nace H. R.; Smith A. H. J. Org. Chem. 1973, 38, 1941. [DOI] [PubMed] [Google Scholar]
- Arndt F.; Eistert B. Org. React. 1942, 1, 38. [Google Scholar]
- Levin S.; Nani R. R.; Reisman S. E. J. Am. Chem. Soc. 2011, 133, 774. [DOI] [PubMed] [Google Scholar]
- Jiang C.-S.; Huang C.-G.; Feng B.; Li J.; Gong J.-X.; Kurtan T.; Guo Y.-W. Steroids 2010, 75, 1153. [DOI] [PubMed] [Google Scholar]
- Wicha J.; Bal K. J. Chem. Soc., Perkin Trans. 1 1978, 1, 1282. [Google Scholar]
- The various alkylation conditions tried include (a) LDA, 11, DCM, −78 °C, 1 h, then isohexyltriflate, −78 to −20 °C (no reaction, deprotection of the chiral auxiliary occurs above −40 °C); (b) LDA, 11, THF, −78 °C, 1 h, then isohexylbromide, HMPA, −78 to −40 °C, 2 h (no reaction, deprotection of chiral auxiliary above −40 °C); (c) NaHMDS, 11, THF, −78 °C, 1 h, THF, then isohexylbromide, −78 to 40 °C (no reaction).
- Unsuccessful chiral auxiliary removal conditions include (a) LiAlH4, THF, 0 to 25 °C (forms incorrect product); (b) LiBH4, THF, EtOH, rt, 16 h (reaction is sluggish, forms incorrect product, and does not proceed to completion).
- Abul-Hajj Y. J. J. Org. Chem. 1986, 51, 3059.and references therein. [Google Scholar]
- Procedures attempted (with 19-nor analogues) include (a) (i) Br2, HOAc, rt, 15 min, then (ii) Li2CO3, LiBr, DMF, 150 °C, 2 h (yielded several products; desired product 15–20%); (b) (i) pyridinium tribromide, HOAc, 20 °C, 30 min, then (ii) Li2CO3, LiBr, DMF, 150 °C, 2 h (yielded several spots; desired product 30%); (c) IBX, TFA, DMSO/tol, 40 °C, 4 days (reaction is sluggish and does not go to completion; desired product 15%); (d) (i) KHMDS, THF, rt, 30 min, then BEt3N, NBS, −78 °C to rt, 16 h, then (ii) Li2CO3, LiBr, DMF, 150 °C, 2 h (does not go to completion, very inconsistent, product isolation is difficult; desired product 17–50%).
- Yan J.; Bittman R. J. Lipid Res. 1990, 31, 160. [PubMed] [Google Scholar]
- Organic Reactions in Steroid Chemistry; Fried J., Edwards J. A., Eds.; Van Nostrand Reinhold Co.: New York, 1972; Vol. 2. [Google Scholar]
- Canney D. J.; Lu H.-F.; McKeon A. C.; Yoon K.-W.; Xu K.; Holland K. D.; Rothman S. M.; Ferrendelli J. A.; Covey D. F. Bioorg. Med. Chem. 1997, 6, 43. [DOI] [PubMed] [Google Scholar]
- Kawa K.; Hara A.; Ishikawa Y.; Nishiyama S. Molecules 2011, 16, 5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takacs J. M.; Jaber M. R.; Vellekoop A. S. J. Org. Chem. 1998, 63, 2742. [DOI] [PubMed] [Google Scholar]
- Auchus R. J.; Palmer J. O.; Carrell H. L.; Covey D. F. Steroids 1989, 53, 77. [DOI] [PubMed] [Google Scholar]
- Westover E. J.; Covey D. F. Steroids 2003, 68, 159. [DOI] [PubMed] [Google Scholar]
- Harrowven D. C.; Pascoe D. D.; Guy I. L. Angew. Chem., Int. Ed. 2007, 46, 425. [DOI] [PubMed] [Google Scholar]
- Tochtrop G. P.; DeKoster G. T.; Cistola D. P.; Covey D. F. J. Org. Chem. 2002, 67, 6764. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.