

Abstract
Retromer is a multiprotein complex that trafficks cargo out of endosomes. The neuronal retromer traffics the amyloid-precursor protein (APP) away from endosomes, a site where APP is cleaved into pathogenic fragments in Alzheimer’s disease. Here we determined whether pharmacological chaperones can enhance retromer stability and function. First, we relied on the crystal structures of retromer proteins to help identify the ‘weak link’ of the complex and to complete an in silico screen of small molecules predicted to enhance retromer stability. Among the hits, an in vitro assay identified one molecule that stabilized retromer against thermal denaturation. Second, we turned to cultured hippocampal neurons, showing that this small molecule increases the levels of retromer proteins, shifts APP away from the endosome, and decreases the pathogenic processing of APP. These findings show that pharmacological chaperones can enhance the function of a multiprotein complex and may have potential therapeutic implications for neurodegenerative diseases.
The retromer complex acts to sort and traffic cargo from endosomes to the trans-Golgi network or to the cell surface1,2. In yeast, where retromer-mediated transport was originally described, the retromer complex comprises a trimeric core of ‘vacuolar protein sorting’ (VPS) proteins, Vps35–Vps29–Vps26, which binds a dimer of ‘sorting nexin’ (SNX) proteins3. With the subsequent characterization of the retromer complex in mammalian cells, the trimeric core was found to be preserved, but the specific SNXs to which it binds turned out to be more variable. The mammalian Vps35–Vps29–Vps26 core can form a complex with Snx1–Snx2, Snx5–Snx6, Snx3 or, as most recently reported, with Snx27 (refs. 1,2). Additionally, the mammalian retromer complex binds other proteins or protein complexes, such as the Wiskott-Aldrich protein and SCAR homolog (WASH) complex4, which modifies its precise function in trafficking cargo out of endosomes.
Retromer-mediated transport has been implicated in a growing number of neurological diseases, but was first linked to Alzheimer’s disease (AD)5. Deficiencies in Vps35 and Vps26 were identified in the hippocampal formation of AD brains, and, together with SorL1 (also called SorLA) or other Vps10-containing receptors, retromer was proposed to mediate the trafficking and pathogenic processing of APP5. APP is a transmembrane protein that is enzymatically processed via two competitive pathways. The first pathway, implicated in the pathogenesis of AD, is triggered when APP interacts with β-site APP-cleaving enzyme 1 (BACE1), a β-secretase that initiates its pathogenic processing, leading to the accumulation of a number of neurotoxic fragments6: β C-terminal fragment (β-CTF)7, secreted APP β (sAPPβ)8 and amyloid β (Aβ). When β-secretase activity is genetically6,9 or pharmacologically reduced10, APP is diverted to the alternative pathway initiated by α-secretase, increasing the production of the neuroprotective fragment secreted APP α (sAPPα). Interestingly, in contrast to disease-causing APP mutations, a recent mutation in APP was identified that reduces the risk of developing AD9. The mutation was found to act by decreasing BACE1-dependent processing of APP, as shown by a reduction in BACE1-dependent fragments and a concomitant increase in sAPPα9.
APP is a transmembrane protein that is actively sorted and trafficked through membrane compartments of the cell11. After internalization from the cell surface and while it temporarily resides in endosomes, APP is most likely to interact and be cleaved by BACE1 (ref. 11). As hypothesized, retromer-related defects have been shown to reduce trafficking of APP out of endosomes12,13, and several cell culture studies and animal models14–17 have established that deficiencies in Vps26, Vps35 and SorL1 cause an elevation in Aβ and other neurotoxic APP fragments. More specifically, Vps26 was found to interact with a FANSHY sequence in the cytoplasmic domain of SorL1, which in turn binds APP, thereby mediating the trafficking of APP away from endosomes18. Genetic variations in SorL1 (ref. 19) and other retromer-related genes20 are major risk factors for AD, thereby confirming a causal role and suggesting that neurons are particularly vulnerable to retromer dysfunction.
Two observations motivate the current study. First, many studies have shown that knocking down one protein of the Vps35–Vps29–Vps26 trimeric core leads to secondary reductions in other retromer proteins (as shown, for example, in refs. 5,21–23). This suggests that the interaction among the individual proteins are important for complex stability24. Second, studies have shown that increasing retromer levels enhances retromer-mediated trafficking and transport5,25, for example, by decreasing the pathogenic processing of APP5. Accordingly, we hypothesized that increasing the interaction between individual retromer proteins would increase complex stability and enhance retromer-mediated trafficking and transport. Moreover, we hypothesized that pharmacological chaperones could achieve these goals. Pharmacological chaperones are small molecules that bind a protein and, by virtue of stabilizing its three-dimensional structure, protect it from degradation and increase its steady-state concentration in the cell26.
The concept and application of a pharmacological chaperone is not new27, but its use in targeting a multiprotein complex rather than a single protein has not yet been demonstrated. In addition, because the complex is not an enzyme and has no known allosteric sites, identifying chaperone binding sites that would enhance retromer stability presents a challenge. We set to out to identify a pharmacological chaperone that targets an area on retromer proteins that will bolster the entire core structure, so that the trimeric Vps35–Vps29–Vps26 core is less prone to degradation. We relied on the established crystal structures of retromer proteins28 to locate possible binding sites computationally29 and completed a large-scale in silico screen to identify possible small molecules that might act as retromer chaperones. Among a group of candidates, we identified one small molecule that acted as a pharmacological chaperone, stabilizing retromer in vitro by 10 °C and increasing retromer levels in neuronal cultures. We then validated the retromer pharmacological chaperone in an extensive series of studies in hippocampal neuronal culture, showing that the chaperone increased retromer levels and enhanced retromer function, as evidenced by lowering APP in endosomes and shifting it away from BACE1-dependent processing.
RESULTS
Characterizing weak links in retromer complex stability
We established the wild-type (WT) ‘melting’ temperature of each protein in the core retromer complex (Fig. 1a) using differential scanning fluorimetry (DSF). DSF measures the emission of a fluorescent dye, Sypro orange, which fluoresces upon interacting with hydrophobic molecules. In this case, the buried hydrophobic residues inside of the protein are exposed upon denaturation and cause the dye to fluoresce. The denaturation temperatures reported are determined by the temperature that corresponds to the inflection point of the melting curve. At even higher temperatures, the denatured protein molecules aggregate, and fluorescence decreases. This trend is seen in all DSF spectra, and a typical curve will come to a peak and drop back down toward zero. In our hands, this method is efficient and reliable with a precision of better than 1 °C. DSF can be used to assess the binding of potential ligands to proteins because the structural stabilization provided by interaction with a ligand will increase the apparent melting temperature.
Figure 1. Characterization of the weak link in retromer complex stability.
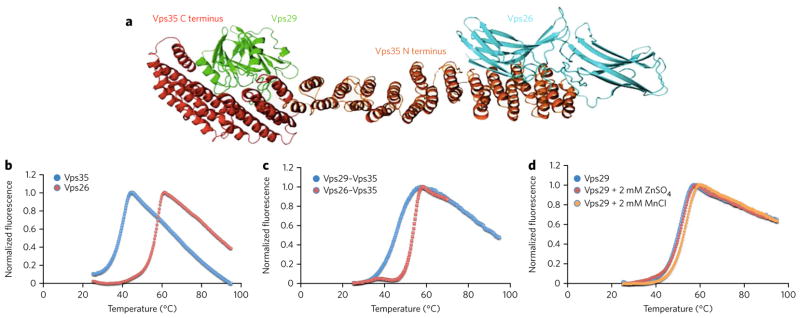
(a) Model of the core retromer complex based on electron microscopy data combined with crystal structures of the component proteins (reprinted with permission from ref. 28. The interaction of Vps35 (red and orange) with Vps29 (green) is known from a structure of their complex22. The structure of the orange portion of Vps35 is hypothesized from sequence similarity to other helical solenoid-like proteins. The interaction between this portion of Vps35 and Vps26 (cyan) is based on modeling alone. (b) Thermal unfolding curve for purified, recombinant Vps35 C-terminal domain (blue curve), showing a denaturation temperature of 38.5 °C. Thermal unfolding curve for purified, recombinant Vps26 showing a denaturation temperature of 56.4 °C (red curve). (c) Thermal unfolding curve for the reconstituted complex of Vps35 and Vps26 (blue curve). The complex denatures at 53.4 °C. Thermal unfolding curve for the reconstituted complex of Vps35 and Vps29 (red curve). The melting temperature is not increased as much as when Vps26 binds Vps35. (d) Thermal unfolding curve for Vps29 in the absence and presence of zinc and manganese.
DSF showed the melting temperature of purified recombinant Vps35 C-terminal domain to be 38.5 °C and showed Vps26 to denature at 56.4 °C (Fig. 1b). When the two proteins are combined in an equimolar ratio, the melting temperature of the complex is 53.4 °C (Fig. 1c). This result indicates that Vps26 binding thermally stabilizes the much larger, elongated and intrinsically less stable Vps35 molecule by about 15 °C. Control measurements incubating Vps35 with proteins such as lysozyme showed no such stabilization (data not shown), indicating that the effect is specific.
Vps29 is known to bind manganese and zinc30. We found that Vps29 denatures at 50 °C. The addition of zinc increases the denaturation temperature to 50.5 °C, whereas the addition of manganese increases the denaturation temperature to 52.6 °C (Fig. 1d). In contrast, the Vps35–Vps29 complex denatures closer to the temperature of Vps35 alone (45.5 °C), consistent with earlier work suggesting that this interface is most likely the weak point in the heterotrimer structure24,31 (Fig. 1c). Supporting this idea, reconstituting the entire Vps26–Vps35–Vps29 core complex from purified recombinant proteins showed that the melting temperature of the heterotrimer was 49 °C (Fig. 2a), which is lower than that of the Vps35–Vps26 complex without Vps29 (Fig. 1c). Thermodynamic studies of the Vps29-Vps35 interaction have shown that it is entropy driven and that large conformational changes occur upon binding24.
Figure 2. Identification of a retromer stabilizing pharmacological chaperone.

(a) Thermal unfolding of the core retromer complex shown in the absence of any compound (blue), retromer in the presence of compound R53 identified from the virtual library (orange) and in the presence of the compound R55 (red) identified from the virtual screen. (b) Chemical structures of the compounds tested for pharmacological activity in vitro as well as in neuronal cell culture. (c) All of the potential binding sites identified computationally by searching for pockets with favorable steric and electrostatic properties were screened with our virtual library. The identified binding sites each gave Glide scores showing favorable binding to multiple compounds from our virtual library. (d) Docked structure of R55 with the Vps35–Vps29 heterodimer. The compound is predicted to bind at the interface between the two proteins, designated site 2 in Figure 2c. R55 is predicted to make interactions with side chains from both Vps35 (orange) and Vps29 (cyan). (e) Structure of site 2 of the Vps29–Vps35 interface shown with the Vps35 (orange) point mutation Q538W. The proposed mutation fills much of the proposed binding site for R55, which is also shown docked to the site. (f) Thermal unfolding curve of the WT retromer complex (blue), the Q538W mutant retromer (red), and the Q538W retromer in the presence of 20 mM R55 (orange). (g) Microscale thermophoresis scan of WT retromer titrated with R55 shows a Kd of ~5 μM.
Identifying target docking sites in the retromer complex
In silico screening (often called ‘docking’) can, in favorable instances, narrow down the number of possible pharmacological chaperones from a large library of small molecules. We chose two computational approaches to address the problem of identifying docking sites with which small-molecule structures could interact. The first is Q-site Finder32, which identifies docking sites (binding pockets) on a protein surface with the highest potential to bind small molecules on the basis of size, favorable van der Waals interaction potential and environment of the pocket. Because the only structure for the full Vps35–Vps26–Vps29 core retromer complex28 is a model derived from electron microscopy, we used the crystal structures of Vps26 and the Vps35–Vps29 complex as the target30,33,34. Results of the pockets identified by Q-site Finder are listed in Supplementary Results, Supplementary Table 1.
The second method used to identify potential ligand docking sites was the computational version of the multiple solvent crystal structures method29,35, where small-molecule probes are used to map the binding surface of the protein. In general, the two methods predicted the same binding pockets, increasing the confidence that the sites used for docking simulations might actually be capable of binding ligands.
Screening for pharmacological chaperones in silico
The above results on the stability of the complex led us to focus our in silico screening on the Vps35–Vps29 structure. All of the docking simulations were carried out using our laboratory library of drug structures, which consists of 46,000 compounds. This library was calculated from a diverse group of ‘lead-like’ compounds, selected to be both optimally diverse and largely commercially available. The library takes into consideration molecular weight, log P, number of rotatable bonds, number of hydrogen bond donors and acceptors and net charge, and it is prefiltered to eliminate compounds that are likely to be cytotoxic as well as promiscuous binders. It was generated to ensure that the similarity cutoff for any two molecules in the set is no more than 70%. An additional criterion for inclusion is a combination of the probability of crossing the blood-brain barrier and known cell penetrability.
Of the ten docking sites found on the Vps35–Vps29 protein complex, six were located at the interface between the two proteins, suggesting that any ligand binding at these sites will most likely interact with residues from both, possibly providing stabilization to the entire complex. Nonetheless, we screened all ten of the sites against the entire library of compound structures. The docking procedure allowed full flexibility in the conformation of the ligand and limited flexibility in the conformations of the amino acid side chains in the binding sites. Only four of the ten predicted docking sites on the Vps35–Vps29 complex generated computed interaction scores with test molecules worth testing in a secondary in vitro screen. Each of these four sites was at the interface of the two proteins.
Validating pharmacological chaperones in vitro
Twenty-four of the top 100 predicted binding compounds were readily available, most of which were predicted to bind at sites 1 or 2, two neighboring sites located at the interface between Vps35 and Vps29 (site 1 is primarily on Vps35 and includes only a single residue from Vps29). These compounds were incubated with purified, reconstituted heterotrimeric retromer complex at various concentrations, and the denaturation temperature of the complex was measured by DSF, as described above. Of the 24 tested, one compound, designated R55 (1; Fig. 2b), improved the thermal stability of the core retromer protein complex by 10 °C at the highest concentration used (Fig. 2a).
R55 was predicted to bind at ‘site 2’, the largest potential ligand-binding site at the interface of Vps29 and Vps35 (Fig. 2c,d and Supplementary Table 1). In the presence of this compound, the entire complex unfolds at 59.5 °C. This ~10-°C increase in stability is comparable to the thermal stabilization obtained for lysosomal enzymes bound to pharmacological chaperones that are currently in clinical trials for the treatment of Gaucher, Fabry and Pompe diseases27. In agreement with the prediction that the compound interacts with residues from both Vps29 and Vps35 (Fig. 2d), it was found to have no effect on the thermal unfolding of the two proteins individually. Control studies with other compounds, such as R53 (2; Fig. 2b), showed no increase in the thermal stability of either the core complex or its components (Fig. 2a), suggesting that the effect of R55 is specific. R55 has the systematic chemical name thiophene-2,5-diylbis(methylene) dicarbamimidothioate dihydrochloride. Binding studies using microscale thermophoresis established its Kd for the retromer core complex to be about 5 μM with a negative thermophoretic trace (Fig. 2).
To confirm the binding site of R55, we looked for mutations that would prevent the compound from binding at the proposed site, site 2. We searched for the best mutation that would hinder binding of R55 to the proposed site sterically. We determined that a Q538W mutation (Fig. 2e) on Vps35 should block site 2 and should prevent R55 from binding. We performed similar DSF thermal denaturation experiments to determine the melting temperature of the Q538W Vps35 retromer mutant. The Q538W mutation causes a slight decrease in thermal stability to the overall retromer complex. We repeated the assay in the presence of up to 20 mM R55 and found that the compound had no effect on the melting temperature of the mutant protein complex (Fig. 2f).
Pharmacological chaperones increase retromer in neurons
To investigate whether enhanced thermal stability of the retromer complex by compound R55 translates into a biological effect in an in vivo system, we turned to cultured primary neurons harvested from the hippocampal formation, a system that has been previously used to investigate retromer trafficking of APP12. First, to establish the compound’s maximum subtoxic dose, we exposed it to neurons derived from P0 WT mice for 48 h. Cell viability studies, using a colorimetric XTT viability assay, showed that doses ranging from 0.5 μM to 50 μM were nontoxic to neurons (data not shown). As 5 μM was the concentration that corresponds to the in vitro Kd of compound R55 to the retromer complex, we used this dose for all of our subsequent neuronal studies unless stated otherwise.
Retromer stabilization in an in vivo system is predicted to increase the levels of individual retromer proteins, and we tested this by exposing neurons to the compound or vehicle (50% DMSO in water) for 48 h. Western blotting analysis showed that, compared to vehicle control, exposure to the compound was associated with a significant increase in the levels of Vps35 (~80%, P < 0.001) and Vps26 (~50%, P < 0.001; Fig. 3a,b). mRNA levels for Vps35 and Vps26 were not affected by R55 (Supplementary Fig. 1a), indicating that the increased protein levels observed for Vps35 and Vps26 proteins is not due to an increase in their respective mRNAs. An inactive compound (R53; Fig. 2b and Supplementary Fig. 1b) that failed to stabilize the retromer complex in vitro altered neither Vps35 nor Vps26 levels in neurons. To visualize the effect of the pharmacological chaperone on retromer levels in neurons, we performed immunocytochemistry studies followed by quantitative confocal microscopy analysis. Exposure of primary hippocampal neurons to R55 for 48 h resulted in a significant ~60% increase in the Vps35 signal (P < 0.05) compared to that in vehicle-treated neurons (Fig. 3c,d). An R55 dose-response curve for neuronal Vps35 levels established a half-maximum effective concentration of ~3.3 μM (Fig. 3e).
Figure 3. The pharmacological chaperone increases retromer levels in neurons.

(a) Left panel, representative western blots of the effect of R55 (5 μM for 48 h) and vehicle on levels of Vps26 and Vps35 in 2-week-old primary cortical neuronal cultures derived from WT mice. (b) Quantitative analysis of the blots. Levels of Vps26 and Vps35 were normalized to total levels of actin (n = 9 per group, P < 0.001). Comparable results were obtained when tubulin was used a control (data not shown). (c) Primary hippocampal neurons were analyzed by confocal microscopy after labeling for MAP2 (blue) and Vps35 (red). Arrows indicate structures showing Vps35 labeling. Scale bars, 10 μm. (d) Bar graph showing a quantitative analysis of Vps35 intensity staining. Levels of Vps35 were normalized to total cell area (n = 14 cells per group). (e) Dose-response curve of the effect of R55 on Vps35 levels performed by western blot analysis (concentration of 0.5 μM, 2.5 μM, 5 μM, 20 μM and 50 μM were used, n = 8 per time point). Results here and in the following figures are shown as mean values ± s.e.m., where *P < 0.05, **P < 0.01 and ***P < 0.005, determined using Student’s t-test.
To further explore the structure-activity relationship, we investigated the effect of compound R33 (Fig. 2b; 3), which has reduced efficacy compared to R55. Our in silico screening data indicated that this analog was also capable of binding the same pocket as R55 but displayed reduced potency in our in vitro measure of retromer stabilization (data not shown). Indeed, exposure of neurons to R33 (5 μM) for 48 h increased Vps35 levels (~22%, P < 0.05), but to an extent less than that observed for R55 (Supplementary Fig. 2). Taken together, these results further strengthen the structure-activity relationship of our compound in the stabilization of the neuronal retromer complex.
A pharmacological chaperone reduces Aβ accumulation
Guided by a previous study that showed that increasing Vps35 levels by genetic manipulation reduces Aβ accumulation5, we set out to test whether R55 would have the same effect. We collected medium from hippocampal neurons exposed to R55 and vehicle and measured endogenous Aβ40 and Aβ42 peptides by ELISA. Compared to vehicle control, the compound significantly reduced the levels of both endogenous Aβ40 (~24%, P < 0.001) and Aβ42 (~28%, P < 0.001; Fig. 4a). When the compound was applied to neurons harboring a pathogenic double mutation in human APP (harvested from the J20 transgenic mouse model of AD36), it significantly reduced both human Aβ40 (~44%, P < 0.01) and Aβ42 (~39%, P < 0.01; Fig. 4b). In this context, the inactive compound R53 did not affect the levels of Aβ accumulation in WT neurons (Supplementary Fig. 3). Furthermore, we observed a dose response of the effect of R55 on Aβ levels (Fig. 4c), where the half-maximum inhibitory concentration (IC50) value for Aβ reduction was found to be ~12 μM.
Figure 4. The pharmacological chaperone decreases Aβ peptide accumulation and reduces the pathogenic pathway of APP.

(a) Effect of R55 (5 μM for 48 h) and vehicle on levels of endogenous mouse Aβ42 and Aβ40 in culture medium from WT primary cortical neurons (n = 12). Amount of Aβ is normalized to total protein content in neurons. (b) Effect of R55 (5 μM for 48 h) and vehicle on levels of human Aβ42 and Aβ40 in culture medium derived from J20 primary cortical neurons (n = 12). (c) Dose-response curve of the effect of R55 on endogenous Aβ production in hippocampal neurons (concentrations of 0.5 μM, 2.5 μM, 5 μM, 20 μM and 50 μM concentrations were used). Mouse and human Aβ samples were analyzed by ELISA. (d) Correlation analysis between R55 dose-dependent levels of neuronal Vps35 and Aβ42 production. Each point in the graph represents an individual measurement from the R55 dose-response curves for retromer and Aβ. (e) Quantification of APP metabolites in medium (sAPPα and sAPPβ) and in cell lysates (FL-APP and βCTF) in primary neurons exposed to R55 (5 μM) for 48 h. FL-APP, sAPPα and βCTF were measured by western blotting, and sAPPβ was measured by ELISA (n = 10 per fragment; Online Methods). *P < 0.05, **P < 0.01 and ***P < 0.005.
Finally, on the basis of the accumulative data obtained in cultured neurons (Figs. 3 and 4), we performed a correlation analysis (Fig. 4d). Results showed that the accumulation of Aβ is inversely correlated to R55 dose-response changes in the levels of retromer (R2 = 0.6, P = 0.009).
A pharmacological chaperone reduces APP processing
The observed reduction in both Aβ40 and Aβ42 is consistent with the interpretation that R55 is acting as hypothesized, that is, by reducing BACE1-dependent processing of APP (Supplementary Fig. 4). To further confirm this interpretation, we collected lysate and medium from hippocampal neurons exposed to R55 and vehicle and respectively measured β-CTF and sAPPβ, the two fragments that are produced by the cleavage of APP by BACE1. Compared to vehicle control, the compound significantly reduced the levels of both endogenous β-CTF (~70%, P < 0.001) and sAPPβ (~25%, P < 0.001; Fig. 4e).
Studies have established that when BACE1 activity is reduced, APP is diverted to the alternative α-secretase pathway6 (Supplementary Fig. 4). Indeed, we find that, compared to vehicle control, R55 exposure caused a significant increase in sAPPα in the medium (~90%, P < 0.05), with the APP fragment indicative of increased α-secretase processing (Fig. 4e). We also observed a relatively modest decrease in full-length APP (Fig. 4e).
A pharmacological chaperone reduces APP in endosomes
It is in endosomes that APP is most likely to be cleaved by BACE1. A previous study documented that retromer deficiency in neurons decreases the trafficking of APP out of endosomes by showing that retromer deficiency increased the intracellular colocalization between APP and the endosomal marker EEA1 (ref. 12). Accordingly, to test the effect that R55 has on APP localization, we performed immunocytochemistry assays with a full-length C-terminal APP antibody in hippocampal neurons, followed by confocal microscopy analysis. Exposure to the compound was associated with a significant decrease in colocalization between APP and the early endosomal marker EEA1 (P < 0.05), suggesting that the compound enhanced the trafficking function of retromer (Fig. 5).
Figure 5. The pharmacological chaperone shifts the endosomal localization of APP and SorL1.

(a) Top, representative confocal microscopy images of WT primary hippocampal neurons immunolabeled for MAP2, APP and EEA1 after a 48-h exposure to vehicle and R55. Insets show a magnification of representative dendritic areas used for the colocalization analysis of APP (red) and EEA1 (green). Arrows indicate structures showing colocalization between APP and EEA1. Bottom, confocal microscopy images of primary hippocampal neurons stained for MAP2, SorL1 and EEA1. Arrows indicate structures showing colocalization between SorL1 and EEA1. (b) Left, quantitative analysis of the colocalization of APP and EEA1 in dendrites (n = 19 cells per group). Right, quantification of colocalization between SorL1 and EEA1 (n = 14 cells per group). *P < 0.05, **P < 0.01.
Because SorL1 is the molecular link between retromer and APP and retromer trafficks APP-SorL1 away from endosomes18, we tested the effect that R55 has on SorL1 localization. Just as for APP, R55 caused a significant decrease in colocalization between SorL1 and the early endosomal marker EEA1 (P < 0.05; Fig. 5).
The effect on APP is retromer dependent
Although the pharmacological chaperone increases retromer levels and reduces Aβ accumulation, we set out to confirm that the effect on Aβ is mediated via retromer. We turned to retromer-deficient hippocampal neurons to address this question. Vps26 knockdown neurons were generated by infecting primary neurons derived from a mouse model containing both alleles of the vps26 gene flanked by a LoxP site (Vps26flox/flox). These Vps26flox/flox primary culture neurons were infected with a lentivirus expressing a Cre recombinase (CRE) or with a virus expressing a catalytically dead version of Cre (Δ) as control (Fig. 6a). The level of Vps26 knockdown with the Cre virus was ~50% compared to neurons infected with the Δ virus. As expected, Vps35 expression was also downregulated (~40%) in our Vps26 knockdown cultures. Notably, R55 partially stabilized Vps35 even in the context of a Vps26 knockdown (Fig. 6b), consistent with its binding to the Vps35–Vps29 interface. Markedly, the levels of Aβ42 (as well as Aβ40, data not shown) are not reduced by R55 in retromer-deficient neurons (R55-CRE condition) when compared to neurons exposed to R55 with the Δ virus (control; Fig. 6c). These results indicate that the Aβ-lowering effect of R55 in neurons is specifically mediated via retromer.
Figure 6. The pharmacological chaperone specifically modulates Aβ production through the retromer pathway.
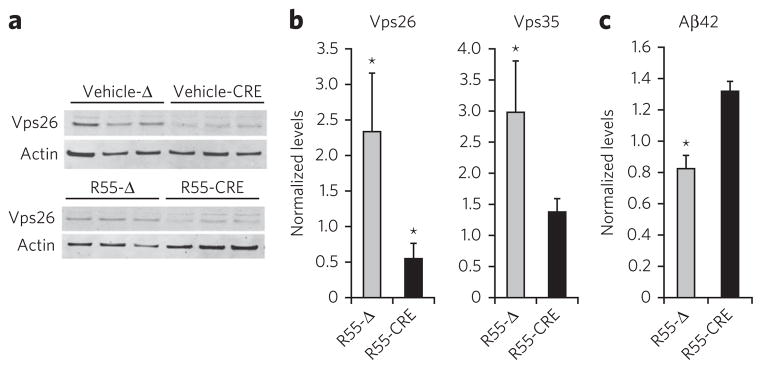
(a) Representative western blots showing a knock-down of Vps26 (vehicle-Cre) in Vps26flox/flox neurons by the use of a lentivirus expressing CRE (three cultures shown for each condition, one per lane), n = 5 per group. (b) Quantitative analysis of the blots for Vps26 and Vps35 normalized to the vehicle-Δ condition. (c) Aβ42 levels in conditioned medium from neurons exposed to either vehicle or R55, and treated with either an active Cre virus (CRE) or the catalytically dead Cre virus (Δ). Aβ samples were analyzed by ELISA (Invitrogen) as suggested by the manufacturer. *P < 0.05.
DISCUSSION
In an effort to test whether small molecules can act as retromer pharmacological chaperones, we first performed a series of in vitro studies to identify and characterize which combination of interacting proteins is most important for regulating the stability of the complex as a whole. Our results agree with a previous observation24,31 showing that the interface between Vps35 and Vps29 is likely to be the weak point in the trimer core of the retromer complex.
Relying on the known three-dimensional structure of the complex between Vps35 and Vps29 (ref. 28), we used complimentary methods to identify docking sites at the interface of these proteins. Once established, we then performed an in silico screen against those sites to identify small-molecule compounds that might act as chaperones in stabilizing the retromer complex. From the top computational ‘hits’, we then completed a series of in vitro studies, isolating the small-molecule compound R55, which does indeed stabilize the reconstituted retromer complex against thermal denaturation. R55 (and its analog R33) is a thiophene thiourea derivative. Little is known about the in vivo stability, pharmacokinetics and biological potency of this family of molecules; they are water soluble and cell penetrant and have been reported to be relatively nontoxic to mice in unpublished data from the National Cancer Institute.
Finally, to test whether retromer stability affects its function, we investigated the effect of this compound in cultured neurons. We specifically focused on hippocampal neurons, because the hippocampal formation is a brain structure enriched in retromer proteins14 and hippocampal cultured neurons have been previously used to investigate retromer trafficking of APP12. Consistent with the predicted effect of retromer complex stabilization at 5-μM concentration, the small molecule was found to increase the levels of retromer proteins in neurons. More notably, the small molecule was found to enhance the function of the retromer, as indicated by decreased APP and SorL1 localization in endosomes. Collectively, these studies suggest that the small molecule acts a retromer pharmacological chaperone.
Because APP is cleaved by BACE1 while it resides in endosomes, we completed an extensive series of biochemical analyses to provide additional evidence that the pharmacological chaperone enhances retromer-dependent trafficking of APP. APP can be cleaved into multiple fragments, and studies have established the precise profile of APP fragments that are indicative of reduced BACE1 cleavage6,9. We show that the retromer pharmacological chaperone recapitulated this profile, characterized by a reduction in BACE1-dependent fragments (β-CTF, sAPPβ, Aβ40 and Aβ42) and a concomitant increase in sAPPα6. Documenting a reduction in BACE1-dependent processing biochemically confirms the microscopic observation that the pharmacological chaperone reduces endosomal APP levels, supporting the interpretation that the pharmacological chaperone enhances retromer’s function.
By showing how and where the individual elements of the complex interact in mediating complex stability, our studies clarify the chemical biology of the retromer complex. Moreover, the results confirm our hypothesis that increasing the interaction between retromer proteins will increase the stability of the whole complex and will enhance retromer’s trafficking function. Finally, we demonstrate for what is to our knowledge the first time that small molecules can act as pharmacological chaperones that can stabilize a multiprotein complex and enhance its function. Although many in vivo properties of the small molecule investigated are unknown, our results show that retromer pharmacological chaperones can, in principle, traffic APP away from a pathogenic and toward a neuroprotective processing pathway, generating a profile of fragments that is genetically shown to be protective against AD9. Retromer pharmacological chaperones are therefore worth exploring in future studies as a potential therapeutic strategy in AD and other diseases4, in particular Parkinson’s disease25, in which retromer has also been implicated.
ONLINE METHODS
In silico screening
The monomeric crystal structure of Vps29–Vps35 (Protein Data Bank entry 2R17) was submitted to the Q-site Finder server. Each of the identified binding sites were docked with our in-house virtual library using the program Glide (Schrödinger LLC). Our library was taken from the “clean leads” library from the ZINC database. We eliminated all of the compounds that were more than 70% similar to decrease the size of the library while maintaining the diversity of compounds. All of the structures were prepared according to Glide guidelines. The sites were screened using the high- throughput virtual screening protocol option.
Protein expression and purification
The VPS35, VPS26, and VPS29 genes were generated using a standard PCR protocol and designed with restriction sites at each end. Primers used for gene amplification for VPS26 were 5′-GGATCCGATGAGTTTTCTTGGAAGG-3′ and 5′-AAGCTTTCACATTTCAGGCTGTTCG-3′. Primers used for gene amplification of VPS29 were 5′-CATATGATGGCTGGGCACAGATTG-3′ and 5′-CCTAGGTTAAGGTTTTTTGTATTCG-3′. Primers used for gene amplification of VPS35 were 5′-GATATCGATGCCTACAACACAGC-3′ and 5′-CCTAGGTTAAAGGATGAGACCTTC-3′. VPS26 was ligated into the first cloning site of PetDuet using BamH1 and Hind3 restriction sites. This allowed for a His6 tag at the N terminus of the protein. VPS29 was ligated into the second cloning site of PetDuet, and was untagged using Nde1 and Avr2 restriction sites. Vps35 was ligated into the second cloning site of PACYCDuet using EcoRV and Avr2. The Q538W retromer mutants were generated using the Quikchange II (Stratagene) site-directed mutagenesis kits and the appropriate primers. Both vectors were cotransformed in BL21-DE3Star Escherichia coli cells. Cultures were grown at 37 °C to OD600 of 0.8, induced with 0.5 mM IPTG and expressed at 18 °C for 16 h. The cells were pelleted by centrifugation and resuspended in buffer containing 50 mM NaH2PO4, pH 8.0, 300 mM NaCl. Cells were lysed using sonication, and cell debris was separated from the sonicated solution using centrifugation at 13,000 r.p.m. for 1 h. The supernatant was purified using a 5-ml His-trap nickel column and washed with a buffer containing 50 mM NaH2PO4, pH 8.0, 300 mM NaCl and 40mM imidazole. The proteins were then eluted using a gradient from 40 mM to 300 mM imidazole in the same 50-mM NaH2PO4, pH 8.0, buffer. Retromer was eluted from the column at 120 mM imidazole. Fractions containing retromer proteins were pooled, concentrated and diluted down to 50 mM NaCl. The proteins were further purified using anion exchange chromatography. Proteins were eluted using a gradient from 50 mM to 500 mM NaCl contained in the same 50 mM NaH2PO4 pH 8.0 buffer. The fractions containing retromer proteins were combined and concentrated down to a volume of <2.0 ml. The concentrated protein sample was further purified using size-exclusion chromatography using 50 mM NaH2PO4 and 300 mM NaCl, pH 8.0. Fractions were analyzed using SDS-PAGE.
DSF
Compounds identified from the in silico screen were obtained from commercial and academic sources. All of the compounds were solubilized to 100 mM using sterile water and DMSO. Experiments were conducted in MicroAmp Fast optical 96-well reaction plates using the Applied Biosystems Step one plus real-time PCR system. Each well contained 50 mM NaH2PO4 300 mM NaCl, pH 8.0, 2 μM purified retromer complex and 20× Sypro orange (Life Technologies) protein gel stain, and compounds were added at varying concentrations. Melting curves were determined by reading fluorescence of the samples in 0.3-°C intervals using a temperature gradient moving from 25 °C to 95 °C.
Neuronal cultures
Mouse hippocampal and cortical cultures were conducted as described in ref. 12. For western blot, ELISA, and qRT-PCR assays, cortical neurons were grown for 14 d. For immunocytochemistry assays, hippocampal neurons were grown for 14 d over an astrocyte feeder layer. For cell viability assays, cortical neurons were grown in 96-well format, at 3 × 104 cells per well. Drug and vehicle were applied at 11 DIV, 5 μM final concentration, for 48 h before analysis. Western blot analysis was performed using ImageJ software (NIH).
APP fragments and Aβ ELISA
Mouse and human Aβ samples were derived from culture medium of WT and APPSwlnd (J20) primary cortical cultures, respectively. Mouse and human Aβ40 and Aβ42 ELISAs were performed according to the manufacturer’s instructions (Life Technologies). Samples from conditioned medium from 14-d-old primary cortical cultures were collected, and AEBSF (0.2 mg/ml) was added to prevent Aβ degradation. Media were spun at 1,000g at 4 °C for 5 min to pellet any cell debris. ELISAs were carried out in duplicates, the averaged values were normalized to total protein as determined by BCA of cell lysates, and the final values were reported in fg/μg and pg/μg for mouse and human Aβ, respectively. FL-APP, β-CTF and sAPPα were measured by western blotting in neuronal lysates and medium, respectively, by using the clone m3.2 antibody from Covance (1:1,000). sAPPβ from medium was measured by ELISA (IBL International).
Immunocytochemistry and confocal microscopy
Primary hippocampal neurons cultured on coverslips were fixed in a final concentration of 2% PFA and 0.5% sucrose (mixed with culture medium) and permeabilized with either 0.1% Triton X-100 (APP) or 0.05% saponin (Vps35 and SorL1) in PBS/serum buffer. Images were captured with a Zeiss LSM 700 META confocal microscope equipped with a 63× Plan-Apochromat objective and HeNE1, HeNe2 and argon lasers. Colocalization analysis of APP and EEA1 was performed using the ‘Colocalization Finder’ plugin (NIH) in ImageJ. The channel intensity was determined on a histogram generated by the plugin, and two regions of interest (ROIs) were quantified using the generated colocalization image. The MAP2 channel was cropped around the soma of each cell, and the resulting images were used as masks on the colocalized image for exclusive quantification in dendrites for one ROI and soma in the other ROI. The total colocalized area was averaged, and the final values are reported in μm2. Colocalization analysis of SorL1 and EEA1 was performed using the ‘JACoB’ plugin (NIH) in ImageJ. The Vps35 staining intensity was determined using the ‘Multi Measure’ option in ImageJ. The intensity of the staining was normalized to the cell area. For staining, we used an anti-C-terminal APP antibody (Calbiochem 171610; 1:1000), an anti-EEA1 antibody (clone n19; Santa Cruz 6415; 1:400), an anti-MAP2 antibody (Abcam 5392; 1:400) and anti-SorL1 antibody (Abcam 16642; 1:500).
Cell viability assay
Neuronal cell viability upon R55 administration was determined per manufacturer’s instructions using the colorimetric XTT Cell viability Kit (Cell Signaling).
qRT-PCR
RNA was isolated from primary cortical neurons using TRIzol (Life Technologies) and purified per manufacturer’s instructions. Reverse transcription was performed using the Maxima First Strand cDNA Synthesis Kit (Fisher), also per manufacturer’s instructions. The PCR reaction was prepared using Solaris qPCR ROX Mix and primers (Fisher), and carried out on a 7900ht Fast Real Time PCR system (Applied Biosystems). The Ct value was read at the peak of the second derivative, and the relative fold change was calculated using the following equation: , where ΔΔCt = ΔCtsample – ΔCtcalibrator, ΔCt = Cttarget – ΔCtreference (R, relative fold change; calibrator, the sample that the others are compared with; reference, internal control such as housekeeping genes). Fold changes were averaged, and the final values are reported as the standardized ratio of treated samples to vehicle.
Cell transfection and lentivirus production
Knockout Vps26–/– or control Vps26flox/flox primary cortical neurons were generated by infecting cells at DIV5, for a period of 10 d, with a lentivirus carrying either the catalytically active CRE or catalytically dead Cre recombinase (Δ, control), respectively. Both Cre recombinases encoded two nuclear localization sequences (NLS) and were fused to eGFP containing a third NLS for enhanced nuclear targeting. Lentiviruses were produced in HEK-293T cells. Briefly, virus-containing medium was collected 72 h after transfection, centrifuged at 2,800g for 5 min, filtered through a 0.45-mm filter (Millipore), supplemented with 10 μg/ml polybrene (Sigma-Aldrich) and applied at a 1:5 ratio to cortical medium. The KO efficiency was further monitored by western blot analysis.
Statistical methods
Student’s t-test was used for all of the other biochemical experiments and immunohistochemical analysis using two-tailed distribution with equal variance (P < 0.05). All of the data are shown as geometric mean with error bars representing ± s.e.m. Significance is indicated as *P < 0.05, **P < 0.01 and ***P < 0.005.
Supplementary Material
Acknowledgments
We thank the US National Institute on Aging/US National Institutes of Health (NIH) grants AG025161 and AG08702, The Alzheimer’s Association, Developmental Therapeutics Program of the National Cancer Institute, Medkoo Biosciences, The Fidelity Biosciences Research Initiative and give special thanks to S. Weninger for advice and encouragement. We also thank The McKnight Endowment for Neuroscience, the Ellison Medical Foundation, and the Gottlieb Family Foundation.
Footnotes
Author contributions
V.J.M. identified the retromer stabilizing site and carried out the in silico screens and designed and performed the in vitro retromer chaperone characterization; D.E.B. designed, coordinated and performed the in vivo retromer chaperone characterization; S.S. assisted with the immunohistochemical staining and genetic recombination in neurons; C.V. assisted with neuronal cultures; M.R.A. assisted with the in vitro thermal studies; V.M.P. assisted with the APP fragment analysis; R.T.S. helped with Aβ measurements; and G.A.P., D.R. and S.A.S. conceived of and supervised the studies and wrote the manuscript.
Competing financial interests
The authors declare no competing financial interests.
Supplementary information and chemical compound information is available in the online version of the paper.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
Contributor Information
Gregory A Petsko, Email: gpetsko@med.cornell.edu.
Dagmar Ringe, Email: ringe@brandeis.edu.
Scott A Small, Email: sas68@columbia.edu.
References
- 1.Seaman MN. The retromer complex—endosomal protein recycling and beyond. J Cell Sci. 2012;125:4693–4702. doi: 10.1242/jcs.103440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cullen PJ, Korswagen HC. Sorting nexins provide diversity for retromer-dependent trafficking events. Nat Cell Biol. 2012;14:29–37. doi: 10.1038/ncb2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seaman MN. Recycle your receptors with retromer. Trends Cell Biol. 2005;15:68–75. doi: 10.1016/j.tcb.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 4.Burd C, Cullen PJ. Retromer: a master conductor of endosome sorting. Cold Spring Harb Perspect Biol. 2014;6:a016774. doi: 10.1101/cshperspect.a016774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Small SA, et al. Model-guided microarray implicates the retromer complex in Alzheimer’s disease. Ann Neurol. 2005;58:909–919. doi: 10.1002/ana.20667. [DOI] [PubMed] [Google Scholar]
- 6.Luo Y, et al. Mice deficient in BACE1, the Alzheimer’s β-secretase, have normal phenotype and abolished β-amyloid generation. Nat Neurosci. 2001;4:231–232. doi: 10.1038/85059. [DOI] [PubMed] [Google Scholar]
- 7.Jiang Y, et al. Alzheimer’s-related endosome dysfunction in Down syndrome is Aβ-independent but requires APP and is reversed by BACE-1 inhibition. Proc Natl Acad Sci USA. 2010;107:1630–1635. doi: 10.1073/pnas.0908953107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nikolaev A, McLaughlin T, O’Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.Jonsson T, et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488:96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- 10.Nishitomi K, et al. BACE1 inhibition reduces endogenous Aβ and alters APP processing in wild-type mice. J Neurochem. 2006;99:1555–1563. doi: 10.1111/j.1471-4159.2006.04178.x. [DOI] [PubMed] [Google Scholar]
- 11.Small SA, Gandy S. Sorting through the cell biology of Alzheimer’s disease: intracellular pathways to pathogenesis. Neuron. 2006;52:15–31. doi: 10.1016/j.neuron.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhalla A, et al. The location and trafficking routes of the neuronal retromer and its role in amyloid precursor protein transport. Neurobiol Dis. 2012;47:126–134. doi: 10.1016/j.nbd.2012.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vieira SI, et al. Retrieval of the Alzheimer’s amyloid precursor protein from the endosome to the TGN is S655 phosphorylation state-dependent and retromer-mediated. Mol Neurodegener. 2010;5:40. doi: 10.1186/1750-1326-5-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muhammad A, et al. Retromer deficiency observed in Alzheimer’s disease causes hippocampal dysfunction, neurodegeneration, and Aβ accumulation. Proc Natl Acad Sci USA. 2008;105:7327–7332. doi: 10.1073/pnas.0802545105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lane RF, et al. Diabetes-associated SorCS1 regulates Alzheimer’s amyloid-β metabolism: evidence for involvement of SorL1 and the retromer complex. J Neurosci. 2010;30:13110–13115. doi: 10.1523/JNEUROSCI.3872-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dodson SE, et al. LR11/SorLA expression is reduced in sporadic Alzheimer disease but not in familial Alzheimer disease. J Neuropathol Exp Neurol. 2006;65:866–872. doi: 10.1097/01.jnen.0000228205.19915.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andersen OM, et al. Neuronal sorting protein–related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci USA. 2005;102:13461–13466. doi: 10.1073/pnas.0503689102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fjorback AW, et al. Retromer binds the FANSHY sorting motif in SorLA to regulate amyloid precursor protein sorting and processing. J Neurosci. 2012;32:1467–1480. doi: 10.1523/JNEUROSCI.2272-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rogaeva E, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39:168–177. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vardarajan BN, et al. Identification of Alzheimer disease-associated variants in genes that regulate retromer function. Neurobiol Aging. 2012;33:2231.e15–2231.e30. doi: 10.1016/j.neurobiolaging.2012.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arighi CN, Hartnell LM, Aguilar RC, Haft CR, Bonifacino JS. Role of the mammalian retromer in sorting of the cation-independent mannose 6-phosphate receptor. J Cell Biol. 2004;165:123–133. doi: 10.1083/jcb.200312055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seaman MN. Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer. J Cell Biol. 2004;165:111–122. doi: 10.1083/jcb.200312034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vergés M, et al. The mammalian retromer regulates transcytosis of the polymeric immunoglobulin receptor. Nat Cell Biol. 2004;6:763–769. doi: 10.1038/ncb1153. [DOI] [PubMed] [Google Scholar]
- 24.Norwood SJ, et al. Assembly and solution structure of the core retromer protein complex. Traffic. 2011;12:56–71. doi: 10.1111/j.1600-0854.2010.01124.x. [DOI] [PubMed] [Google Scholar]
- 25.MacLeod DA, et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron. 2013;77:425–439. doi: 10.1016/j.neuron.2012.11.033. erratum 77, 994 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ringe D, Petsko GA. What are pharmacological chaperones and why are they interesting? J Biol. 2009;8:80. doi: 10.1186/jbiol186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lieberman RL, et al. Structure of acid β-glucosidase with pharmacological chaperone provides insight into Gaucher disease. Nat Chem Biol. 2007;3:101–107. doi: 10.1038/nchembio850. [DOI] [PubMed] [Google Scholar]
- 28.Hierro A, et al. Functional architecture of the retromer cargo-recognition complex. Nature. 2007;449:1063–1067. doi: 10.1038/nature06216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mattos C, Ringe D. Locating and characterizing binding sites on proteins. Nat Biotechnol. 1996;14:595–599. doi: 10.1038/nbt0596-595. [DOI] [PubMed] [Google Scholar]
- 30.Collins BM, Skinner CF, Watson PJ, Seaman MN, Owen DJ. Vps29 has a phosphoesterase fold that acts as a protein interaction scaffold for retromer assembly. Nat Struct Mol Biol. 2005;12:594–602. doi: 10.1038/nsmb954. [DOI] [PubMed] [Google Scholar]
- 31.Swarbrick JD, et al. VPS29 is not an active metallo-phosphatase but is a rigid scaffold required for retromer interaction with accessory proteins. PLoS ONE. 2011;6:e20420. doi: 10.1371/journal.pone.0020420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laurie AT, Jackson RM. Q-SiteFinder: an energy-based method for the prediction of protein-ligand binding sites. Bioinformatics. 2005;21:1908–1916. doi: 10.1093/bioinformatics/bti315. [DOI] [PubMed] [Google Scholar]
- 33.Collins BM, et al. Structure of Vps26B and mapping of its interaction with the retromer protein complex. Traffic. 2008;9:366–379. doi: 10.1111/j.1600-0854.2007.00688.x. [DOI] [PubMed] [Google Scholar]
- 34.Shi H, Rojas R, Bonifacino JS, Hurley JH. The retromer subunit Vps26 has an arrestin fold and binds Vps35 through its C-terminal domain. Nat Struct Mol Biol. 2006;13:540–548. doi: 10.1038/nsmb1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Landon MR, Lancia DR, Jr, Yu J, Thiel SC, Vajda S. Identification of hot spots within druggable binding regions by computational solvent mapping of proteins. J Med Chem. 2007;50:1231–1240. doi: 10.1021/jm061134b. [DOI] [PubMed] [Google Scholar]
- 36.Mucke L, et al. High-level neuronal expression of Aβ 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.