

Abstract
Ischemic stroke is caused by critical reductions in blood flow to brain or spinal cord. Microglia are the resident immune cells of the central nervous system, and they respond to stroke by assuming an activated phenotype that releases cytotoxic cytokines, reactive oxygen species, proteases, and other factors. This acute, innate immune response may be teleologically adapted to limit infection, but in stroke this response can exacerbate injury by further damaging or killing nearby neurons and other cell types, and by recruiting infiltration of circulating cytotoxic immune cells. The microglial response requires hours to days to fully develop, and this time interval presents a clinically accessible time window for initiating therapy. Because of redundancy in cytotoxic microglial responses, the most effective therapeutic approach may be to target the global gene expression changes involved in microglial activation. Several classes of drugs can do this, including histone deacetylase inhibitors, minocycline and other PARP inhibitors, corticosteroids, and inhibitors of TNFα and scavenger receptor signaling. Here we review the pre-clinical studies in which these drugs have been used to suppress microglial activation after stroke. We also review recent advances in the understanding of sex differences in the CNS inflammatory response, as these differences are likely to influence the efficacy of drugs targeting post-stroke brain inflammation.
Keywords: Brain, corticosteroid, female, HDAC, inflammation, ischemia, minocycline, PARP
INTRODUCTION
Ischemic stroke is caused by critical reductions in blood flow to one or more arteries of the brain or spinal cord, and is a leading cause of morbidity and mortality worldwide. The only clinically-validated treatment for stroke now available is acute thrombolysis, and the utility of this approach is constrained by the need to initiate treatment within 3–6 hours of symptoms and the risk of causing cerebral hemorrhage. These factors have limited the number of stroke patients receiving this treatment to less than 5%, even in areas where this treatment modality is readily available. It is therefore important to develop new approaches that can be used for a much larger fraction of stroke patients.
Ischemic brain injury results from a cascade of events initiated by energy depletion and culminating in cell death. Contributing factors in this cascade include glutamate excitotoxicity, oxidative stress, and inflammation. Interventions targeting glutamate excitotoxicity and oxidative stress (or their sequelae) have been shown to reduce acute ischemic cell death in animal models of stroke [1], but these approaches are generally ineffective if not initiated very soon after ischemia onset. Inflammation, however, requires many hours to days to fully develop, and is thus a much more practical target for treating a large fraction of stroke patients [2].
Microglial cells are the resident immune cells in the central nervous system [3]. These cells continuously scan their environment with highly motile processes and are thought to be the immediate sensors of brain pathology [4]. After ischemia, microglia undergo phenotypic transformation to an “activated” phenotype [5]. Acutely activated microglia produce factors such as reactive oxygen species, cytokines, and proteases that may kill neighboring cells and disrupt the blood–brain barrier [6–9]. This initial innate immune response occurs with almost any disruption in brain homeostasis, and it is thought to be teleologically adapted as a first line defense against infection. In stroke, however, this initial response may be maladaptive.
Signals released by microglia also trigger infiltration of circulating immune cells to the injured area. The effects of these circulating immune cells and of the systemic immune response in general on stroke outcome are complex. Infiltrating neutrophils and T-lymphocytes can aggravate acute injury [10–12], circulating B-lymphocytes may exert a countering, anti-inflammatory effect [13], and all of these cell types can in turn affect microglial activation [14]. Moreover, infiltrating macrophages in brain can be difficult to distinguish from activated microglia. These and other effects of the systemic immune response on stroke are beyond the scope of this review; however, it should be recognized that because of these interactions, drugs affecting post-stroke microglial activation may act in part though effects on the systemic immune system.
Prior comprehensive reviews have examined the many different signaling pathways that trigger microglial activation, their resulting genomic responses, and the several effector mechanisms by which microglia can have cytotoxic effects on surrounding cells [5, 9, 15, 16]. Drugs targeting any of these processes could in principle have salutary effects, but the multiplicity of signaling pathways and effector mechanisms renders it unlikely that targeting any single one would be optimally effective. An alternative approach is to target the coordinated gene expression changes that underlie microglial activation. Here we review the preclinical (animal model) data supporting efficacy of drugs targeting this aspect of microglial activation after stroke. A summary of representative studies is provided as (Table 1). In evaluating this literature we have distinguished between the different stroke models employed because they produce cell death by somewhat different mechanisms, and they have varying fidelity to the clinical condition of stroke.
Table 1.
Drug Treatment Effects on Post-IschemIc Inflammation
| Target | Drug | Dose 1 (mg/kg) | Route | Time to First dose | Treatment Duration | Stroke Model2 | Species | Inflammatory Markers | Other Outcomes | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| PARP | PJ34 | 15 | i.p. | 8 hours | 3 days | GI/R | Rat | CD11b(+) cells | Cell death | [32] |
| 15 | i.p. | 2 days | 7 days | GI/R | Rat | CD11b(+) cells | Cell death, behavior | [33] | ||
| PARP and others | Minocycline | 45 | i.p. | 2 hours | second dose at 4 hours | FI/R | Rat-pup | I L-1β, CINC-1, IL-18, MCP-1; ED1(+) cells | Cell death | [128] |
| 50 then 25 | i.p. | 4 days | 4 weeks | FI/R | Rat | ED1(+) cells | Neurogenesis, behavior | [57] | ||
| 45 | i.p. | 0.5 hours | 2 days | FI/R | Rat | CD11b(+) and MPO(+) cells; 5- LOX | Cell death, behavior | [129] | ||
| 20 then 10; 45 then 23 | i.p. | 2 hours | 6 days | HI/R | Rat-pup | Iba1(+) cells | O1(+), O4(+), MBP(+) cells | [130] | ||
| 10 | i.p. | 1 day | 13 days | FI/R | Mouse | Iba1(+) cells, HMGB1 | Survival, cell death, behavior | [56] | ||
| 10 | i.p. | 5 days | 9 days | FI/R | Mouse | Iba1(+) cells, plasma HMGB1 | Survival, behavior | [131] | ||
| 45 then 22.5 | i.p. | 2 hours | 5 days | HI/R | Rat pup | P2X4R, and Iba1 | [43] | |||
| 45 then 22.5 | i.p. | 2 hours | 5 days | HI/R | Rat pup | Iba-1(+) cells, TNFα, IL- 1β | 5-HT and SERT expression | [44] | ||
| HDAC | SB | 300 | s.c. | 0 hours | second dose at 12 hours | FI | Rat | OX42(+) cells, iNOS, COX2 | Behavior | [72] |
| VPA | 300 | s.c. | 0 hours | second dose at 12 hours | FI | Rat | OX42(+), ED1(+), iNOS(+) cells | Behavior | [72] | |
| 300 | i.p. | 0 hours | 6 days | GI/R | Rat | OX42(+); IL-1β, TNFα expression | Cell death, behavior | [132] | ||
| CD36 | SS31 | 2 | i.p. | 0 hours | 2 days | FI/R | Mouse | CD36 expression | Cell death | [83] |
| TNFα | 3,6′-DT | 56 | i.p. | 1 or 3 hours | 3 days | FI/R | Mouse | Iba-1(+) and MPO(+) cells; TNFα, IL- 1β and iNOS expression | Cell death | [87] |
The table excludes studies in which drugs were given prior to ischemia.
Abbreviations: i.p., intraperitoneal injection; s.c., subcutaneous injection; PARP, poly(ADP-ribose) polymerase; HDAC, histone deacetylase; SS31, D-Arg-Dmt-Lys-Phe-NH2; MPO, myeloperoxidase; TNFα, tumor necrosis factor alpha; IL-18, interleukin 18; MCP-1, monocyte chemoattractant protein 1; VPA, valproic acid; SB, sodium butyrate; ICE, IL-1β-converting enzyme; COX, cyclooxygenase-2; PGE2, prostaglandin E2; IL-1β, interleukin 1beta; CINC-1, cytokine-induced chemoattractant protein 1; 3,6′-DT, 3,6′-Dithiothalidomide; 5-LOX, 5-lipoxygenase; iNOS, inducible nitric oxide synthase.; 5-HT, serotonin; SERT, serotonin transporter.
In some studies the initial dose was higher than subsequent doses.
Stroke models are GI/R, global ischemia/reperfusion; FI/R, focal ischemia/reperfusion; HI/R, hypoxia-ischemia reperfusion; FI, focal ischemia, without reperfusion. See text for details.
PRECLINICAL STROKE MODELS
Variations of four stroke models are commonly employed. In “focal ischemia”, the middle cerebral artery is occluded to produce a focal infarct, as occurs in clinical ischemic stroke. The vessel may be permanently occluded, (“permanent ischemia”), or re-opened after an interval of 30–90 minutes (“transient ischemia” or “focal ischemia-reperfusion”). The latter has become popular because it produces a mild injury in which is easier to identify neuroprotective effects; however it is only rarely true in human stroke that circulation is reestablished within such a short time interval. “Hypoxia-ischemia” is a variation on this model, in which focal ischemia-reperfusion is coupled with hypoxia. This is most commonly used in neonatal animals as a model of perinatal birth asphyxia. In “global ischemia”, animals are subjected to a brief (5 – 20 minute) reduction of blood flow to either the entire brain or just the forebrain, typically by a combination of bilateral carotid artery occlusion and hypotension. This is not a model of stroke per se, but more closely replicates the brain injury produced by cardiac arrest or prolonged hypotension. Global ischemia does not cause infarction (cell necrosis), but instead leads to selective neuronal death of certain vulnerable neuronal populations in the hippocampus and cerebral cortex. All four of these stroke models produce robust post-ischemic microglial activation, but the biological drivers and the effects of the inflammatory response may differ among these models.
PHARMACOLOGICAL AGENTS USED TO SUPPRESS POST-STROKE MICROGLIAL ACTIVATION
a. Corticosteroids and Non-steroidal Anti-inflammatory Drugs
Acute immunosuppression is typically accomplished in non-CNS tissues by the use of corticosteroids or nonsteroidal anti-inflammatory agents. Corticosteroids have been shown to inhibit microglial activation in several neurological disease models including spinal cord injury, traumatic brain injury, and acute stress [17–19]. It is likely that corticosteroids would similarly suppress microglial activation after stroke, but surprisingly only a single published study has addressed this question, and in that study corticosteroids were administered prior to ischemia [20]. The lack of research in this area may reflect the fact that corticosteroids have well-recognized systemic effects that may have impair stroke recovery, such as hyperglycemia, muscle wasting, osteoporosis, and increased vulnerability to infections. It has also been reported that corticosteroids have a direct proapoptotic effect on certain neuronal populations [21], and in some settings they can paradoxically exacerbate inflammation [22, 23]. In (uncontrolled) clinical trials, corticosteroid treatment did not affect either the mortality or aggregated functional outcomes of patients with acute ischemic stroke [24].
Whether nonsteroidal anti-inflammatory drugs can block post-ischemic microglial activation also remains uncertain. However, the drug indomethacin was shown to increase the survival of progenitor cells and allow a higher fraction to differentiate into oligodendrocytes and neurons, with a reduction of inflammatory cells after focal cerebral ischemia [25]. It was not established in that study whether these effects are attributable to reduced microglial activation, or to direct effects of the drug on the progenitor cells.
b. PARP Inhibitors
Poly (ADP-ribose) polymerases (PARPs) catalyze the transfer of ADP-ribose units from NAD+ to target proteins including histones and transcriptional factors. The resulting poly(ADP-ribosyl)ation of these substrate proteins regulates chromatin structure, DNA metabolism, and gene expression [26, 27]. In particular, PARP regulates the activity of NF-αB and other transcription factors, which in turn regulate many aspects of the inflammatory response [28, 29]. PARP inhibitors are small molecules which compete with NAD+ at the enzyme catalytic site. Many PARP inhibitors have good CNS penetration, and some of them have entered clinical trials for cancer and other diseases [30].
Cell culture studies have shown that PARP inhibitors block NF-αB transcriptional activity and block the upregulation in microglial iNOS expression, MMP9 release, morphological changes, and neurotoxicity that is otherwise induced by lipopolysaccharide and other pro-inflammatory mediators [28, 31]. Several studies have examined the effects of PARP inhibitors after stroke and have found that they suppress microglial activation and improve neuronal survival [32–35]. However, the mechanistic interpretation of these results is complicated by the fact that PARP inhibitors also have acute neuroprotective effects that are mediated by a mechanism unrelated to their anti-inflammatory effects [36], such that reduced inflammation seen with these agents could be secondary to reduced cell death rather than vice-versa. However, these neuroprotective effects, like those of almost all neuroprotective agents, require administration within 3 to 6 hours after onset of ischemia, and drugs given after this time interval can be assumed to act by mechanisms other than acute neuroprotection. Studies performed in this manner, with PARP inhibitor therapy initiated 8–48 hours after ischemia and continued for several days, showed reduced microglial activation and improved histological and behavioral outcomes assessed up to 8 weeks after ischemia [32, 33, 37].
c. Minocycline
Like the PARP inhibitors, minocycline and related tetracycline derivatives such as doxycycline have both anti-inflammatory and neuroprotective effects in cell culture preparations [38–41]. They similarly have both antiinflammatory and neuroprotective effects in animal models of stroke [8, 38, 42–47]. The beneficial effects of minocycline on stroke outcome have been attributed to a bewildering variety of biochemical processes, including upregulation of mitochondrial bcl-2 expression [48], reduced mitochondrial calcium uptake, calcium-induced mitochondrial swelling, calcium-induced cytochrome-C release and mitochondrial permeability transition [49–51], direct scavenging of reactive oxygen species [52], and inhibition of mitogen activated protein kinases [53, 54]. However, the biochemical mechanisms by which minocycline affects these processes have not been established, and many may be secondary effects. Indeed, the fact that minocycline, like PARP inhibitors, has both neuroprotective and anti-inflammatory effects may stem from the fact minocycline is itself a potent PARP inhibitor [55]. Minocycline, like other competitive PARP inhibitors, contains an aromatic ring-linked carboxamide group or carbamoyl group analogous to the NAD+ substrate of PARP (Fig. 1).
Fig. (1). Structure / activity relationships of tetracycline PARP-1 inhibitors.
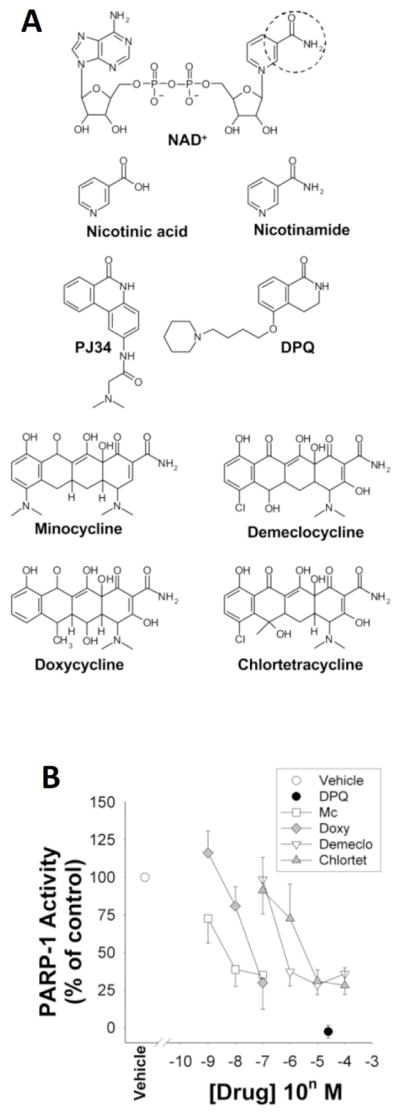
A. An aromatic ring-linked carboxamide group (circled) or carbamoyl group built in a polyaromatic heterocyclic skeleton is shared by the natural PARP-1 substrate NAD+, the competitive PARP-1 inhibitors nicotinamide, DPQ, and PJ34, and the tetracycline derivatives. Nicotinic acid is not a PARP-1 inhibitor and lacks this amide group. B. Activity of isolated, recombinant PARP-1 in the presence of these agents. Studies were performed in the presence of 210 αM NAD+ substrate, as described [55].
Abbreviations: PARP-1, poly(ADP-ribose) polymerase-1; Mc, minocycline; Doxy, doxycycline; Demeclo, demeclocycline; Chlortet, chlortetracyceline; DPQ, 3, 4-dihydro-5- [4-(1-piperidinyl)butoxy]-1(2H)-isoquinolinone; PJ34, N-(6-oxo-5,6-dihydrophenanthridin-2-yl)-N, N-dimethylacetamide hydrochloride. Figure modified from [55].
Only a few studies have initiated minocycline therapy at time points greater than 6 hours after ischemia, i.e. at times that limit the likelihood that observed anti-inflammatory effects are secondary to neuroprotective effects. Treatment initiated 1 day after focal ischemia and continued for 13 days resulted in reduced microglial activation, improved scores on behavioral tests, and improved survival rate [56]. Similarly, minocycline treatment begun 4 days after transient focal ischemia and continued for 4 weeks improved motor and cognitive function, in association with reduced microglia activation and enhanced neurogenesis [57].
Minocycline crosses the blood-brain-barrier and is clinically well-tolerated at anti-inflammatory doses [58]. In a clinical trial, ischemic stroke patients given 200 mg of minocycline for 5 days, beginning 6 – 24 hours after onset of stroke symptoms, showed better functional outcomes relative to placebo-treated patients [59].
d. HDAC Inhibitors
Gene expression is regulated in part by the enzymatic addition and removal of acetyl groups at specific lysine residues by histone acetyltransferase (HAT) and histone deacetylases HDACs [60]. Emerging evidence suggests that HDAC activity may have a master regulatory role in diverse biological activities, including inflammation [61–66]. HDAC inhibition suppresses the inflammatory response in part by inhibiting transcription factor binding at promoter sites of pro-inflammatory genes such as iNOS and COX [67]. HDAC inhibition has been shown to reduce neuronal injury, alleviate post-stroke inflammation, and improve functional outcome in multiple models of focal ischemia [68–71]. A direct effect on inflammation is supported by studies showing that post-insult treatment with the HDAC inhibitors valproic acid and sodium butyrate suppressed microglial activation, reduced the number of microglia, and inhibited other inflammatory markers in the ischemic brain [72]. HDAC inhibitors can also enhance memory and synaptic plasticity in the CNS, suggesting that in addition to salvaging tissue, HDAC inhibition might also be a useful strategy for promoting functional recovery [73–75]. However, the enthusiasm for pan-HDAC inhibition in treating neurological conditions is tempered by their toxicity toward many CNS cell types [68, 76, 77]. Accordingly, an aim of ongoing research is to selectively target specific HDAC isoforms.
e. Inhibitors of Scavenger Receptor and Toll-Like Receptors (TLRs)
After ischemia, danger-associated molecular pattern molecules such as high mobility group box 1 (HMGB1) are released from dead cells and activate pattern recognition receptors, including toll-like receptors (TLRs) and the scavenger receptor CD36, which are key molecular sensors for the innate immune response in brain [78–81]. TLR-mediated intracellular signaling pathways converge to activate NF-κB and c-Jun N-terminal kinases (JNKs), which induce transcription of many genes important in the inflammatory response. The cooperative signaling of TLR2 heterodimers (TLR2/1) and CD36 is a critical factor in the inflammatory response and tissue damage evoked by cerebral ischemia [80]. Microglial expression of CD36 protein is increased in the ischemic brain, and CD36-null mice show attenuated microglial activation, reduced NF-κB activation [81], and reduced infarct size [82]. Drugs that block CD36 signaling are not yet available, but a peptide that blocks CD36 upreguation was found to attenuate ischemic brain injury [83]. Drugs targeting TLRs are also not yet available, but studies using TLR2−/− mice suggest this could be an effective approach for suppressing post-stroke inflammation. TLR2−/− mice have reduced microglial activation and proliferation after stroke, and decreased brain levels of monocyte chemotactic protein-1 [78].
f. Tumor Necrosis Factor-alpha (TNF-α) Receptor Antagonists
TNF-α is a potent stimulator of microglial activation [84], and effective TNFα antagonists are now clinically available. TNF-α exerts its effects by binding to two cell-surface receptors, the p55 and p75 TNF receptors (TNFR), commonly referred to as TNFR1 and TNFR2, which are expressed on both neurons and microglia. The expression of both TNF-α and TNF-α receptors is increased in the ischemic injury zone within hours after stroke onset [85]. Several different strategies for inhibiting TNF-α signaling by pharmacologic agents, neutralizing antibodies or soluble receptors have been reported to reduce infarct volume in preclinical models [86–91]. Some of these studies also reported reduced microglial activation, but it is not possible to ascertain whether the reduced microglial activation reduced neuronal death, or vice versa. It should be noted, however, that TNF-α antagonists can also have deleterious effects on stroke outcome. Hippocampal neurogenesis evaluated two weeks after the ischemic onset after ischemic injury was abolished by anti-TNF-α antibodies administered between day 8 and 14 after stroke [92]. These affects may be attributable to pro-survival effects of TNFR2 activation. A therapeutic strategy that selectively targets TNFR1-mediated signaling, which promotes apoptosis and inflammation, while retaining TNFR2-mediated pro-surviving signaling may lessen adverse effects of anti-TNFα therapies [93]. Antibody to TNF-α or based on TNF-α receptors are now clinically available and in use for autoimmune disorders, but have not yet been used in controlled trials for stroke.
SEXUAL DIMORPHISM IN STROKE AND POSTSTROKE INFLAMMATION
Females exhibit smaller infarcts and better outcomes after experimental brain ischemia than males [94–96]. Sex differences can result from differences in sex steroid hormone levels, sex steroid-induced developmental differences, or hormone - independent differences specified by XX versus XY chromosomal gene dosage [97]. The steroid hormones 17β-estradiol and progesterone influence brain development, and the number and morphology of microglia and the expression of several cytokines, chemokines and their receptors vary with sex and age [98]. Sex steroid receptors are expressed on neurons, astrocytes, and oligodendrocytes in addition to microglia [99–102]. Results of cell culture studies indicate that both estrogens [99] and XX genotype [103] reduce death in cultured neurons exposed to ischemia-like conditions, indicating effects independent of inflammation. However, treatment with 17β-estradiol and progesterone can suppress microglia-mediated neurotoxic effects in culture [101, 104–107], suggesting that an anti-inflammatory effect may also contribute to the neuroprotective effects observed in vivo.
Evidence also suggests that sexual dimorphism exists in the post-stroke inflammatory response with respect to microglial activation, peripheral immune cell infiltration, and expression of pro-inflammatory factors such as cyclooxygenase-2, NADPH oxidase, and vascular cell adhesion molecule-1 [108–111]. In a rat focal ischemia-reperfusion model, 17β-estradiol and progesterone reduced infarct size and improved behavioral function in males and ovariectomized females to a similar extent [112]. These effects were associated with attenuated microglial activation and down-regulated pro-inflammatory cytokines and chemokines (IL-6, CCL2 and CCL5) [112]. In a rat permanent focal ischemia model, progesterone inhibited the expression of Iba1 and COX-2 after stroke in vivo, suggesting that progesterone can exert neuroprotective action by inhibiting the activation of microglia and the over expression of COX-2 after stroke [104].
SEXUAL DIMORPHISM IN RESPONSE TO ANTIINFLAMMATORY TREATMENTS
Given the known differences in male and female inflammatory responses, it might be expected that males and females would respond differently to therapeutic interventions targeting these inflammatory responses. The few studies done in this area point to large differences. The protective effect of PARP-1 inhibitors (including minocycline) is strikingly dependent on sex, with males preferentially protected compared with females [113–116]. Unpublished work from our lab suggests this dichotomy extends also to the effects of these agents on the post-stroke inflammatory response [117]. The reason for these differences has not been established, but one study suggests that estrogen may anchor PARP to ER-α and to the DNA and prevent its recognition of DNA strand breaks and following PARP activation [118]. It remains possible however that sex differences in brain inflammation and in PARP effects on the brain inflammatory response result from developmental rather than hormonal mechanisms [115].
CONCLUSIONS AND CAVEATS
Microglia undergo activation in response to brain ischemia. Cell culture studies show that activated microglia can kill neighboring neurons, and the in vivo studies reviewed here strongly suggest that this occurs in vivo as well. The relatively long time interval (many hours) between ischemia onset and a fully developed microglial activation state makes targeting of this microglial response clinically feasible, and several pharmacologic agents are now available that can effectively block microglial activation at a global, gene-expression level. Together these observations lend credence to the idea that targeting microglial activation after stroke may provide an effective way to limit brain injury caused by stroke.
However, caveats should be noted with respect to extrapolation of these findings to the clinical realm. First, almost all of the preclinical studies in this area have been done with male animals. As noted above, the brain inflammatory response may differ in fundamental ways between males and females, and the effect of anti-inflammatory interventions may likewise differ between males and females. To add to this complexity, the degree to which these differences may persist in post-menopausal females, which is the group most prone to stroke, is unknown.
A second caveat is that microglial “activation” is not a univalent state; the morphological and gene expression changes associated with microglial activation vary enormously with the nature, strength, and duration of the stimulus [119], and activated microglia are very difficult to distinguish from infiltrating peripheral macrophages. Evidence also suggests that brain microglial populations are heterogeneous, and may respond differently to similar stimuli [120]. Activated microglia may be classified as M1 or M2 phenotypes on the basis of surface markers and other differences [121], although hybrid and other phenotypes also occur. The M1 phenotype is characterized by the expression of high levels of pro-inflammatory cytokines and aggravation of inflammatory responses, while M2 macrophages have antiinflammatory functions and promote tissue remodeling [122]. Markers for both phenotypes increase during the first few days after stroke, but their rates of later decline may vary [123]. M2 and possibly other microglial phenotypes can also support neuronal survival [123] and recruit endogenous neural stem cells to the lesion site [124], effects that may be impaired by non-specific anti-inflammatory agents. Microglia similarly play a crucial role in brain recovery after injury through their effects on debris clearance, angiogenesis, and neurite outgrowth [125–127]. For these reasons, the efficacy of anti-inflammatory treatment after stroke may be critically influenced by the timing and duration of this treatment approach.
Acknowledgments
This work was supported by the Chinese Research Scholarship Council (Y.C.) the U.S. National Institutes of Health (grant # NS041421, R.A.S.), and the U.S. Department of Veterans Affairs.
Footnotes
Send Orders for Reprints to reprints@benthamscience.net
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflicts of interest.
References
- 1.Ginsberg MD. Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology. 2008;55(3):363–389. doi: 10.1016/j.neuropharm.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22(9):391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 3.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19(8):312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 4.Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91(2):461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 5.Stoll G, Jander S, Schroeter M. Inflammation and glial responses in ischemic brain lesions. Prog Neurobiol. 1998;56(2):149–171. doi: 10.1016/s0301-0082(98)00034-3. [DOI] [PubMed] [Google Scholar]
- 6.del Zoppo GJ, Frankowski H, Gu YH, Osada T, Kanazawa M, Milner R, Wang X, Hosomi N, Mabuchi T, Koziol JA. Microglial cell activation is a source of metalloproteinase generation during hemorrhagic transformation. J Cereb Blood Flow Metab. 2012;32(5):919–932. doi: 10.1038/jcbfm.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8(1):57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 8.Yenari MA, Xu L, Tang XN, Qiao Y, Giffard RG. Microglia potentiate damage to blood-brain barrier constituents: improvement by minocycline in vivo and in vitro. Stroke. 2006;37(4):1087–1093. doi: 10.1161/01.STR.0000206281.77178.ac. [DOI] [PubMed] [Google Scholar]
- 9.Yenari MA, Kauppinen TM, Swanson RA. Microglial activation in stroke: therapeutic targets. Neurotherapeutics. 2010;7(4):378–391. doi: 10.1016/j.nurt.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Subramanian S, Zhang B, Kosaka Y, Burrows GG, Grafe MR, Vandenbark AA, Hurn PD, Offner H. Recombinant T cell receptor ligand treats experimental stroke. Stroke. 2009;40(7):2539–2545. doi: 10.1161/STROKEAHA.108.543991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gelderblom M, Weymar A, Bernreuther C, Velden J, Arunachalam P, Steinbach K, Orthey E, Arumugam TV, Leypoldt F, Simova O, Thom V, Friese MA, Prinz I, Holscher C, Glatzel M, Korn T, Gerloff C, Tolosa E, Magnus T. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood. 2012;120(18):3793–3802. doi: 10.1182/blood-2012-02-412726. [DOI] [PubMed] [Google Scholar]
- 12.McColl BW, Rothwell NJ, Allan SM. Systemic inflammatory stimulus potentiates the acute phase and CXC chemokine responses to experimental stroke and exacerbates brain damage via interleukin- 1- and neutrophil-dependent mechanisms. J Neurosci. 2007;27(16):4403–4412. doi: 10.1523/JNEUROSCI.5376-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chamorro A, Meisel A, Planas AM, Urra X, van de Beek D, Veltkamp R. The immunology of acute stroke. Nat Rev Neurol. 2012;8(7):401–410. doi: 10.1038/nrneurol.2012.98. [DOI] [PubMed] [Google Scholar]
- 14.Ren X, Akiyoshi K, Dziennis S, Vandenbark AA, Herson PS, Hurn PD, Offner H. Regulatory B cells limit CNS inflammation and neurologic deficits in murine experimental stroke. J Neurosci. 2011;31(23):8556–8563. doi: 10.1523/JNEUROSCI.1623-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17(7):796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140(6):918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cerqueira SR, Oliveira JM, Silva NA, Leite-Almeida H, Ribeiro-Samy S, Almeida A, Mano JF, Sousa N, Salgado AJ, Reis RL. Microglia Response and In Vivo Therapeutic Potential of Methylprednisolone-Loaded Dendrimer Nanoparticles in Spinal Cord Injury. Small. 2012 [Google Scholar]
- 18.Zhang Z, Artelt M, Burnet M, Schluesener HJ. Dexamethasone attenuates early expression of three molecules associated with microglia/macrophages activation following rat traumatic brain injury. Acta Neuropathol. 2007;113(6):675–682. doi: 10.1007/s00401-007-0195-8. [DOI] [PubMed] [Google Scholar]
- 19.Sugama S, Takenouchi T, Fujita M, Kitani H, Conti B, Hashimoto M. Corticosteroids limit microglial activation occurring during acute stress. Neuroscience. 2012;232C:13–20. doi: 10.1016/j.neuroscience.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 20.McRae A, Bona E, Hagberg H. Microglia-astrocyte interactions after cortisone treatment in a neonatal hypoxia-ischemia model. Brain Res Dev Brain Res. 1996;94(1):44–51. doi: 10.1016/0165-3806(96)00043-0. [DOI] [PubMed] [Google Scholar]
- 21.Sapolsky RM. Stress, Glucocorticoids, and Damage to the Nervous System: The Current State of Confusion. Stress. 1996;1(1):1–19. doi: 10.3109/10253899609001092. [DOI] [PubMed] [Google Scholar]
- 22.Sorrells SF, Caso JR, Munhoz CD, Sapolsky RM. The stressed CNS: when glucocorticoids aggravate inflammation. Neuron. 2009;64(1):33–39. doi: 10.1016/j.neuron.2009.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sorrells SF, Sapolsky RM. An inflammatory review of glucocorticoid actions in the CNS. Brain Behav Immun. 2007;21(3):259–272. doi: 10.1016/j.bbi.2006.11.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sandercock PA, Soane T. Corticosteroids for acute ischaemic stroke. Cochrane Database Syst Rev. 2011;(9):CD000064. doi: 10.1002/14651858.CD000064.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoehn BD, Palmer TD, Steinberg GK. Neurogenesis in rats after focal cerebral ischemia is enhanced by indomethacin. Stroke. 2005;36(12):2718–2724. doi: 10.1161/01.STR.0000190020.30282.cc. [DOI] [PubMed] [Google Scholar]
- 26.Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell. 2010;39(1):8–24. doi: 10.1016/j.molcel.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ji Y, Tulin AV. The roles of PARP1 in gene control and cell differentiation. Curr Opin Genet Dev. 2010;20(5):512–518. doi: 10.1016/j.gde.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chiarugi A, Moskowitz MA. Poly(ADP-ribose) polymerase-1 activity promotes NF-kappaB-driven transcription and microglial activation: implication for neurodegenerative disorders. J Neurochem. 2003;85(2):306–317. doi: 10.1046/j.1471-4159.2003.01684.x. [DOI] [PubMed] [Google Scholar]
- 29.Ha HC, Hester LD, Snyder SH. Poly(ADP-ribose) polymerase- 1 dependence of stress-induced transcription factors and associated gene expression in glia. Proc Natl Acad Sci U S A. 2002;99(5):3270–3275. doi: 10.1073/pnas.052712399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kummar S, Ji J, Morgan R, Lenz HJ, Puhalla SL, Belani CP, Gandara DR, Allen D, Kiesel B, Beumer JH, Newman EM, Rubinstein L, Chen A, Zhang Y, Wang L, Kinders RJ, Parchment RE, Tomaszewski JE, Doroshow JH. A phase I study of veliparib in combination with metronomic cyclophosphamide in adults with refractory solid tumors and lymphomas. Clin Cancer Res. 2012;18(6):1726–1734. doi: 10.1158/1078-0432.CCR-11-2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kauppinen TM, Swanson RA. Poly(ADP-ribose) polymerase-1 promotes microglial activation, proliferation, and matrix metalloproteinase- 9-mediated neuron death. J Immunol. 2005;174(4):2288–2296. doi: 10.4049/jimmunol.174.4.2288. [DOI] [PubMed] [Google Scholar]
- 32.Hamby AM, Suh SW, Kauppinen TM, Swanson RA. Use of a poly(ADP-ribose) polymerase inhibitor to suppress inflammation and neuronal death after cerebral ischemia-reperfusion. Stroke. 2007;38(2 Suppl):632–636. doi: 10.1161/01.STR.0000250742.61241.79. [DOI] [PubMed] [Google Scholar]
- 33.Kauppinen TM, Suh SW, Berman AE, Hamby AM, Swanson RA. Inhibition of poly(ADP-ribose) polymerase suppresses inflammation and promotes recovery after ischemic injury. J Cereb Blood Flow Metab. 2009;29(4):820–829. doi: 10.1038/jcbfm.2009.9. [DOI] [PubMed] [Google Scholar]
- 34.Endres M, Wang ZQ, Namura S, Waeber C, Moskowitz MA. Ischemic brain injury is mediated by the activation of poly(ADP-ribose)polymerase. J Cereb Blood Flow Metab. 1997;17(11):1143–1151. doi: 10.1097/00004647-199711000-00002. [DOI] [PubMed] [Google Scholar]
- 35.Takahashi K, Greenberg JH, Jackson P, Maclin K, Zhang J. Neuroprotective effects of inhibiting poly(ADP-ribose) synthetase on focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 1997;17(11):1137–1142. doi: 10.1097/00004647-199711000-00001. [DOI] [PubMed] [Google Scholar]
- 36.Koh DW, Dawson TM, Dawson VL. Mediation of cell death by poly(ADP-ribose) polymerase-1. Pharmacol Res. 2005;52(1):5–14. doi: 10.1016/j.phrs.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 37.Won SJ, Suh SW, Swanson RA. The effect of post-treatment with the PARP inhibitor, INO-1001 on glial activation and neuronal death after transient focal cerebral ischemia. Soc Neuroscience Abstr. 2011:460.10. [Google Scholar]
- 38.Yrjanheikki J, Tikka T, Keinanen R, Goldsteins G, Chan PH, Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci U S A. 1999;96(23):13496–13500. doi: 10.1073/pnas.96.23.13496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tikka TM, Koistinaho JE. Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. J Immunol. 2001;166(12):7527–7533. doi: 10.4049/jimmunol.166.12.7527. [DOI] [PubMed] [Google Scholar]
- 40.Suk K. Minocycline suppresses hypoxic activation of rodent microglia in culture. Neurosci Lett. 2004;366(2):167–171. doi: 10.1016/j.neulet.2004.05.038. [DOI] [PubMed] [Google Scholar]
- 41.Kobayashi K, Imagama S, Ohgomori T, Hirano K, Uchimura K, Sakamoto K, Hirakawa A, Takeuchi H, Suzumura A, Ishiguro N, Kadomatsu K. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. 2013;4:e525. doi: 10.1038/cddis.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci U S A. 1998;95(26):15769–15774. doi: 10.1073/pnas.95.26.15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wixey JA, Reinebrant HE, Carty ML, Buller KM. Delayed P2X4R expression after hypoxia-ischemia is associated with microglia in the immature rat brain. J Neuroimmunol. 2009;212(1–2):35–43. doi: 10.1016/j.jneuroim.2009.04.016. [DOI] [PubMed] [Google Scholar]
- 44.Wixey JA, Reinebrant HE, Spencer SJ, Buller KM. Efficacy of post-insult minocycline administration to alter long-term hypoxia-ischemia-induced damage to the serotonergic system in the immature rat brain. Neuroscience. 2011;182:184–192. doi: 10.1016/j.neuroscience.2011.03.033. [DOI] [PubMed] [Google Scholar]
- 45.Arvin KL, Han BH, Du Y, Lin SZ, Paul SM, Holtzman DM. Minocycline markedly protects the neonatal brain against hypoxic-ischemic injury. Ann Neurol. 2002;52(1):54–61. doi: 10.1002/ana.10242. [DOI] [PubMed] [Google Scholar]
- 46.Xu L, Fagan SC, Waller JL, Edwards D, Borlongan CV, Zheng J, Hill WD, Feuerstein G, Hess DC. Low dose intravenous minocycline is neuroprotective after middle cerebral artery occlusion-reperfusion in rats. BMC Neurol. 2004;4:7. doi: 10.1186/1471-2377-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang SX, Lertvorachon J, Hou ST, Konishi Y, Webster J, Mealing G, Brunette E, Tauskela J, Preston E. Chlortetracycline and demeclocycline inhibit calpains and protect mouse neurons against glutamate toxicity and cerebral ischemia. J Biol Chem. 2005;280(40):33811–33818. doi: 10.1074/jbc.M503113200. [DOI] [PubMed] [Google Scholar]
- 48.Wang J, Wei Q, Wang CY, Hill WD, Hess DC, Dong Z. Minocycline up-regulates Bcl-2 and protects against cell death in mitochondria. J Biol Chem. 2004;279(19):19948–19954. doi: 10.1074/jbc.M313629200. [DOI] [PubMed] [Google Scholar]
- 49.Fernandez-Gomez FJ, Galindo MF, Gomez-Lazaro M, Gonzalez-Garcia C, Cena V, Aguirre N, Jordan J. Involvement of mitochondrial potential and calcium buffering capacity in minocycline cytoprotective actions. Neuroscience. 2005;133(4):959–967. doi: 10.1016/j.neuroscience.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 50.Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, Sarang S, Liu AS, Hartley DM, Wu DC, Gullans S, Ferrante RJ, Przedborski S, Kristal BS, Friedlander RM. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417(6884):74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]
- 51.Wang X, Zhu S, Drozda M, Zhang W, Stavrovskaya IG, Cattaneo E, Ferrante RJ, Kristal BS, Friedlander RM. Minocycline inhibits caspase-independent and -dependent mitochondrial cell death pathways in models of Huntington’s disease. Proc Natl Acad Sci U S A. 2003;100(18):10483–10487. doi: 10.1073/pnas.1832501100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kraus RL, Pasieczny R, Lariosa-Willingham K, Turner MS, Jiang A, Trauger JW. Antioxidant properties of minocycline: neuroprotection in an oxidative stress assay and direct radicalscavenging activity. J Neurochem. 2005;94(3):819–827. doi: 10.1111/j.1471-4159.2005.03219.x. [DOI] [PubMed] [Google Scholar]
- 53.Nikodemova M, Duncan ID, Watters JJ. Minocycline exerts inhibitory effects on multiple mitogen-activated protein kinases and IkappaBalpha degradation in a stimulus-specific manner in microglia. J Neurochem. 2006;96(2):314–323. doi: 10.1111/j.1471-4159.2005.03520.x. [DOI] [PubMed] [Google Scholar]
- 54.Pi R, Li W, Lee NT, Chan HH, Pu Y, Chan LN, Sucher NJ, Chang DC, Li M, Han Y. Minocycline prevents glutamate- induced apoptosis of cerebellar granule neurons by differential regulation of p38 and Akt pathways. J Neurochem. 2004;91(5):1219–1230. doi: 10.1111/j.1471-4159.2004.02796.x. [DOI] [PubMed] [Google Scholar]
- 55.Alano CC, Kauppinen TM, Valls AV, Swanson RA. Minocycline inhibits poly(ADP-ribose) polymerase-1 at nanomolar concentrations. Proc Natl Acad Sci U S A. 2006;103(25):9685–9690. doi: 10.1073/pnas.0600554103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hayakawa K, Mishima K, Nozako M, Hazekawa M, Mishima S, Fujioka M, Orito K, Egashira N, Iwasaki K, Fujiwara M. Delayed treatment with minocycline ameliorates neurologic impairment through activated microglia expressing a high-mobility group box1-inhibiting mechanism. Stroke. 2008;39(3):951–958. doi: 10.1161/STROKEAHA.107.495820. [DOI] [PubMed] [Google Scholar]
- 57.Liu Z, Fan Y, Won SJ, Neumann M, Hu D, Zhou L, Weinstein PR, Liu J. Chronic treatment with minocycline preserves adult new neurons and reduces functional impairment after focal cerebral ischemia. Stroke. 2007;38(1):146–152. doi: 10.1161/01.STR.0000251791.64910.cd. [DOI] [PubMed] [Google Scholar]
- 58.Fagan SC, Waller JL, Nichols FT, Edwards DJ, Pettigrew LC, Clark WM, Hall CE, Switzer JA, Ergul A, Hess DC. Minocycline to improve neurologic outcome in stroke (MINOS): a dose-finding study. Stroke. 2010;41(10):2283–2287. doi: 10.1161/STROKEAHA.110.582601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lampl Y, Boaz M, Gilad R, Lorberboym M, Dabby R, Rapoport A, Anca-Hershkowitz M, Sadeh M. Minocycline treatment in acute stroke: an open-label, evaluator-blinded study. Neurology. 2007;69(14):1404–1410. doi: 10.1212/01.wnl.0000277487.04281.db. [DOI] [PubMed] [Google Scholar]
- 60.Thiagalingam S, Cheng KH, Lee HJ, Mineva N, Thiagalingam A, Ponte JF. Histone deacetylases: unique players in shaping the epigenetic histone code. Ann N Y Acad Sci. 2003;983:84–100. doi: 10.1111/j.1749-6632.2003.tb05964.x. [DOI] [PubMed] [Google Scholar]
- 61.Matsuoka H, Fujimura T, Hayashi M, Matsuda K, Ishii Y, Aramori I, Mutoh S. Disruption of HDAC4/N-CoR complex by histone deacetylase inhibitors leads to inhibition of IL-2 gene expression. Biochem Pharmacol. 2007;74(3):465–476. doi: 10.1016/j.bcp.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 62.Georgopoulos K. From immunity to tolerance through HDAC. Nat Immunol. 2009;10(1):13–14. doi: 10.1038/ni0109-13. [DOI] [PubMed] [Google Scholar]
- 63.Grausenburger R, Bilic I, Boucheron N, Zupkovitz G, El-Housseiny L, Tschismarov R, Zhang Y, Rembold M, Gaisberger M, Hartl A, Epstein MM, Matthias P, Seiser C, Ellmeier W. Conditional deletion of histone deacetylase 1 in T cells leads to enhanced airway inflammation and increased Th2 cytokine production. J Immunol. 2010;185(6):3489–3497. doi: 10.4049/jimmunol.0903610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lundh M, Christensen DP, Rasmussen DN, Mascagni P, Dinarello CA, Billestrup N, Grunnet LG, Mandrup-Poulsen T. Lysine deacetylases are produced in pancreatic beta cells and are differentially regulated by proinflammatory cytokines. Diabetologia. 2010;53(12):2569–2578. doi: 10.1007/s00125-010-1892-8. [DOI] [PubMed] [Google Scholar]
- 65.Marumo T, Hishikawa K, Yoshikawa M, Hirahashi J, Kawachi S, Fujita T. Histone deacetylase modulates the proinflammatory and -fibrotic changes in tubulointerstitial injury. Am J Physiol Renal Physiol. 2010;298(1):F133–141. doi: 10.1152/ajprenal.00400.2009. [DOI] [PubMed] [Google Scholar]
- 66.Ito K, Hanazawa T, Tomita K, Barnes PJ, Adcock IM. Oxidative stress reduces histone deacetylase 2 activity and enhances IL-8 gene expression: role of tyrosine nitration. Biochem Bioph Res Co. 2004;315(1):240–245. doi: 10.1016/j.bbrc.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 67.Faraco G, Pittelli M, Cavone L, Fossati S, Porcu M, Mascagni P, Fossati G, Moroni F, Chiarugi A. Histone deacetylase (HDAC) inhibitors reduce the glial inflammatory response in vitro and in vivo. Neurobiol Dis. 2009;36(2):269–279. doi: 10.1016/j.nbd.2009.07.019. [DOI] [PubMed] [Google Scholar]
- 68.Chuang DM, Leng Y, Marinova Z, Kim HJ, Chiu CT. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009;32(11):591–601. doi: 10.1016/j.tins.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gibson CL, Murphy SP. Benefits of histone deacetylase inhibitors for acute brain injury: a systematic review of animal studies. J Neurochem. 2010;115(4):806–813. doi: 10.1111/j.1471-4159.2010.06993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Faraco G, Pancani T, Formentini L, Mascagni P, Fossati G, Leoni F, Moroni F, Chiarugi A. Pharmacological inhibition of histone deacetylases by suberoylanilide hydroxamic acid specifically alters gene expression and reduces ischemic injury in the mouse brain. Mol Pharmacol. 2006;70(6):1876–1884. doi: 10.1124/mol.106.027912. [DOI] [PubMed] [Google Scholar]
- 71.Ren M, Leng Y, Jeong M, Leeds PR, Chuang DM. Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: potential roles of histone deacetylase inhibition and heat shock protein induction. J Neurochem. 2004;89(6):1358–1367. doi: 10.1111/j.1471-4159.2004.02406.x. [DOI] [PubMed] [Google Scholar]
- 72.Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J Pharmacol Exp Ther. 2007;321(3):892–901. doi: 10.1124/jpet.107.120188. [DOI] [PubMed] [Google Scholar]
- 73.Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T, Wood MA. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci. 2007;27(23):6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459(7243):55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim MS, Akhtar MW, Adachi M, Mahgoub M, Bassel-Duby R, Kavalali ET, Olson EN, Monteggia LM. An essential role for histone deacetylase 4 in synaptic plasticity and memory formation. J Neurosci. 2012;32(32):10879–10886. doi: 10.1523/JNEUROSCI.2089-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bardai FH, D’Mello SR. Selective toxicity by HDAC3 in neurons: regulation by Akt and GSK3beta. J Neurosci. 2011;31(5):1746–1751. doi: 10.1523/JNEUROSCI.5704-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov. 2008;7(10):854–868. doi: 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- 78.Lehnardt S, Lehmann S, Kaul D, Tschimmel K, Hoffmann O, Cho S, Krueger C, Nitsch R, Meisel A, Weber JR. Toll-like receptor 2 mediates CNS injury in focal cerebral ischemia. J Neuroimmunol. 2007;190(1–2):28–33. doi: 10.1016/j.jneuroim.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 79.Brea D, Blanco M, Ramos-Cabrer P, Moldes O, Arias S, Perez-Mato M, Leira R, Sobrino T, Castillo J. Toll-like receptors 2 and 4 in ischemic stroke: outcome and therapeutic values. J Cereb Blood Flow Metab. 2011;31(6):1424–1431. doi: 10.1038/jcbfm.2010.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Abe T, Shimamura M, Jackman K, Kurinami H, Anrather J, Zhou P, Iadecola C. Key role of CD36 in Toll-like receptor 2 signaling in cerebral ischemia. Stroke. 2010;41(5):898–904. doi: 10.1161/STROKEAHA.109.572552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kunz A, Abe T, Hochrainer K, Shimamura M, Anrather J, Racchumi G, Zhou P, Iadecola C. Nuclear factor-kappaB activation and postischemic inflammation are suppressed in CD36-null mice after middle cerebral artery occlusion. J Neurosci. 2008;28(7):1649–1658. doi: 10.1523/JNEUROSCI.5205-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cho S, Park EM, Febbraio M, Anrather J, Park L, Racchumi G, Silverstein RL, Iadecola C. The class B scavenger receptor CD36 mediates free radical production and tissue injury in cerebral ischemia. J Neurosci. 2005;25(10):2504–2512. doi: 10.1523/JNEUROSCI.0035-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cho S, Szeto HH, Kim E, Kim H, Tolhurst AT, Pinto JT. A novel cell-permeable antioxidant peptide, SS31, attenuates ischemic brain injury by down-regulating CD36. J Biol Chem. 2007;282(7):4634–4642. doi: 10.1074/jbc.M609388200. [DOI] [PubMed] [Google Scholar]
- 84.Gregersen R, Lambertsen K, Finsen B. Microglia and macrophages are the major source of tumor necrosis factor in permanent middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab. 2000;20(1):53–65. doi: 10.1097/00004647-200001000-00009. [DOI] [PubMed] [Google Scholar]
- 85.Botchkina GI, Meistrell ME, 3rd, Botchkina IL, Tracey KJ. Expression of TNF and TNF receptors (p55 and p75) in the rat brain after focal cerebral ischemia. Mol Med. 1997;3(11):765–781. [PMC free article] [PubMed] [Google Scholar]
- 86.Shohami E, Ginis I, Hallenbeck JM. Dual role of tumor necrosis factor alpha in brain injury. Cytokine Growth Factor Rev. 1999;10(2):119–130. doi: 10.1016/s1359-6101(99)00008-8. [DOI] [PubMed] [Google Scholar]
- 87.Yoon JS, Lee JH, Tweedie D, Mughal MR, Chigurupati S, Greig NH, Mattson MP. 3,6′-dithiothalidomide improves experimental stroke outcome by suppressing neuroinflammation. J Neurosci Res. 2013;91(5):671–680. doi: 10.1002/jnr.23190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hosomi N, Ban CR, Naya T, Takahashi T, Guo P, Song XY, Kohno M. Tumor necrosis factor-alpha neutralization reduced cerebral edema through inhibition of matrix metalloproteinase production after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2005;25(8):959–967. doi: 10.1038/sj.jcbfm.9600086. [DOI] [PubMed] [Google Scholar]
- 89.Lavine SD, Hofman FM, Zlokovic BV. Circulating antibody against tumor necrosis factor-alpha protects rat brain from reperfusion injury. J Cereb Blood Flow Metab. 1998;18(1):52–58. doi: 10.1097/00004647-199801000-00005. [DOI] [PubMed] [Google Scholar]
- 90.Barone FC, Arvin B, White RF, Miller A, Webb CL, Willette RN, Lysko PG, Feuerstein GZ. Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke. 1997;28(6):1233–1244. doi: 10.1161/01.str.28.6.1233. [DOI] [PubMed] [Google Scholar]
- 91.Sumbria RK, Boado RJ, Pardridge WM. Brain protection from stroke with intravenous TNFalpha decoy receptor-Trojan horse fusion protein. J Cereb Blood Flow Metab. 2012;32(10):1933–1938. doi: 10.1038/jcbfm.2012.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Heldmann U, Thored P, Claasen JH, Arvidsson A, Kokaia Z, Lindvall O. TNF-alpha antibody infusion impairs survival of stroke-generated neuroblasts in adult rat brain. Exp Neurol. 2005;196(1):204–208. doi: 10.1016/j.expneurol.2005.07.024. [DOI] [PubMed] [Google Scholar]
- 93.McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation. 2008;5:45. doi: 10.1186/1742-2094-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hurn PD, Macrae IM. Estrogen as a neuroprotectant in stroke. J Cereb Blood Flow Metab. 2000;20(4):631–652. doi: 10.1097/00004647-200004000-00001. [DOI] [PubMed] [Google Scholar]
- 95.Yuan M, Siegel C, Zeng Z, Li J, Liu F, McCullough LD. Sex differences in the response to activation of the poly (ADP-ribose) polymerase pathway after experimental stroke. Exp Neurol. 2009;217(1):210–218. doi: 10.1016/j.expneurol.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Roof RL, Hall ED. Gender differences in acute CNS trauma and stroke: neuroprotective effects of estrogen and progesterone. J Neurotrauma. 2000;17(5):367–388. doi: 10.1089/neu.2000.17.367. [DOI] [PubMed] [Google Scholar]
- 97.Phoenix CH, Goy RW, Gerall AA, Young WC. Organizing action of prenatally administered testosterone propionate on the tissues mediating mating behavior in the female guinea pig. Endocrinology. 1959;65:369–382. doi: 10.1210/endo-65-3-369. [DOI] [PubMed] [Google Scholar]
- 98.Schwarz JM, Sholar PW, Bilbo SD. Sex differences in microglial colonization of the developing rat brain. J Neurochem. 2012;120(6):948–963. doi: 10.1111/j.1471-4159.2011.07630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Elzer JG, Muhammad S, Wintermantel TM, Regnier-Vigouroux A, Ludwig J, Schutz G, Schwaninger M. Neuronal estrogen receptor-alpha mediates neuroprotection by 17beta-estradiol. J Cereb Blood Flow Metab. 2010;30(5):935–942. doi: 10.1038/jcbfm.2009.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Arevalo MA, Santos-Galindo M, Acaz-Fonseca E, Azcoitia I, Garcia-Segura LM. Gonadal hormones and the control of reactive gliosis. Horm Behav. 2013;63(2):216–221. doi: 10.1016/j.yhbeh.2012.02.021. [DOI] [PubMed] [Google Scholar]
- 101.Bruce-Keller AJ, Keeling JL, Keller JN, Huang FF, Camondola S, Mattson MP. Antiinflammatory effects of estrogen on microglial activation. Endocrinology. 2000;141(10):3646–3656. doi: 10.1210/endo.141.10.7693. [DOI] [PubMed] [Google Scholar]
- 102.Mann SA, Versmold B, Marx R, Stahlhofen S, Dietzel ID, Heumann R, Berger R. Corticosteroids reverse cytokine-induced block of survival and differentiation of oligodendrocyte progenitor cells from rats. J Neuroinflammation. 2008;5:39. doi: 10.1186/1742-2094-5-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Du L, Bayir H, Lai Y, Zhang X, Kochanek PM, Watkins SC, Graham SH, Clark RS. Innate gender-based proclivity in response to cytotoxicity and programmed cell death pathway. J Biol Chem. 2004;279(37):38563–38570. doi: 10.1074/jbc.M405461200. [DOI] [PubMed] [Google Scholar]
- 104.Jiang C, Cui K, Wang J, He Y. Microglia and cyclooxygenase- 2: possible therapeutic targets of progesterone for stroke. Int Immunopharmacol. 2011;11(11):1925–1931. doi: 10.1016/j.intimp.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 105.Muller E, Kerschbaum HH. Progesterone and its metabolites 5- dihydroprogesterone and 5–3-tetrahydroprogesterone decrease LPS-induced NO release in the murine microglial cell line, BV-2. Neuro Endocrinol Lett. 2006;27(5):675–678. [PubMed] [Google Scholar]
- 106.Drew PD, Chavis JA. Female sex steroids: effects upon microglial cell activation. J Neuroimmunol. 2000;111(1–2):77–85. doi: 10.1016/s0165-5728(00)00386-6. [DOI] [PubMed] [Google Scholar]
- 107.Baker AE, Brautigam VM, Watters JJ. Estrogen modulates microglial inflammatory mediator production via interactions with estrogen receptor beta. Endocrinology. 2004;145(11):5021–5032. doi: 10.1210/en.2004-0619. [DOI] [PubMed] [Google Scholar]
- 108.Brait VH, Jackman KA, Walduck AK, Selemidis S, Diep H, Mast AE, Guida E, Broughton BR, Drummond GR, Sobey CG. Mechanisms contributing to cerebral infarct size after stroke: gender, reperfusion, T lymphocytes, and Nox2-derived superoxide. J Cereb Blood Flow Metab. 2010;30(7):1306–1317. doi: 10.1038/jcbfm.2010.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Johann S, Beyer C. Neuroprotection by gonadal steroid hormones in acute brain damage requires cooperation with astroglia and microglia. J Steroid Biochem Mol Biol. 2013;137(1):39–45. doi: 10.1016/j.jsbmb.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 110.Wood H. Neuroimmunology: estrogen receptor ligands suppress inflammatory responses in astrocytes and microglia. Nat Rev Neurol. 2011;7(7):355. doi: 10.1038/nrneurol.2011.87. [DOI] [PubMed] [Google Scholar]
- 111.Crain JM, Watters JJ. Estrogen and P2 Purinergic Receptor Systems in Microglia: Therapeutic Targets for Neuroprotection. Open Drug Discov J. 2010;2:148–167. doi: 10.2174/1877381801002010148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dang J, Mitkari B, Kipp M, Beyer C. Gonadal steroids prevent cell damage and stimulate behavioral recovery after transient middle cerebral artery occlusion in male and female rats. Brain Behav Immun. 2011;25(4):715–726. doi: 10.1016/j.bbi.2011.01.013. [DOI] [PubMed] [Google Scholar]
- 113.McCullough LD, Zeng Z, Blizzard KK, Debchoudhury I, Hurn PD. Ischemic nitric oxide and poly (ADP-ribose) polymerase- 1 in cerebral ischemia: male toxicity, female protection. J Cereb Blood Flow Metab. 2005;25(4):502–512. doi: 10.1038/sj.jcbfm.9600059. [DOI] [PubMed] [Google Scholar]
- 114.Li J, McCullough LD. Sex differences in minocycline-induced neuroprotection after experimental stroke. J Cereb Blood Flow Metab. 2009;29(4):670–674. doi: 10.1038/jcbfm.2009.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hagberg H, Wilson MA, Matsushita H, Zhu C, Lange M, Gustavsson M, Poitras MF, Dawson TM, Dawson VL, Northington F, Johnston MV. PARP-1 gene disruption in mice preferentially protects males from perinatal brain injury. J Neurochem. 2004;90(5):1068–1075. doi: 10.1111/j.1471-4159.2004.02547.x. [DOI] [PubMed] [Google Scholar]
- 116.Liu F, Lang J, Li J, Benashski SE, Siegel M, Xu Y, McCullough LD. Sex differences in the response to poly(ADP-ribose) polymerase-1 deletion and caspase inhibition after stroke. Stroke. 2011;42(4):1090–1096. doi: 10.1161/STROKEAHA.110.594861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Won SJ, Suh SW, Kauppinen TM, Yoo BH, Swanson RA. PARP-1 inhibition blocks ischemia-induced microglial activation in male but not female rats. Soc Neuroscience Abstr. 2010:152.8. [Google Scholar]
- 118.Mabley JG, Horvath EM, Murthy KG, Zsengeller Z, Vaslin A, Benko R, Kollai M, Szabo C. Gender differences in the endotoxin- induced inflammatory and vascular responses: potential role of poly(ADP-ribose) polymerase activation. J Pharmacol Exp Ther. 2005;315(2):812–820. doi: 10.1124/jpet.105.090480. [DOI] [PubMed] [Google Scholar]
- 119.Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- 120.Carson MJ, Bilousova TV, Puntambekar SS, Melchior B, Doose JM, Ethell IM. A rose by any other name? The potential consequences of microglial heterogeneity during CNS health and disease. Neurotherapeutics. 2007;4(4):571–579. doi: 10.1016/j.nurt.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Michelucci A, Heurtaux T, Grandbarbe L, Morga E, Heuschling P. Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: Effects of oligomeric and fibrillar amyloid-beta. J Neuroimmunol. 2009;210(1–2):3–12. doi: 10.1016/j.jneuroim.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 122.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, Gao Y, Chen J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43(11):3063–3070. doi: 10.1161/STROKEAHA.112.659656. [DOI] [PubMed] [Google Scholar]
- 124.Belmadani A, Tran PB, Ren D, Miller RJ. Chemokines regulate the migration of neural progenitors to sites of neuroinflammation. J Neurosci. 2006;26(12):3182–3191. doi: 10.1523/JNEUROSCI.0156-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Eyo UB, Dailey ME. Microglia: Key Elements in Neural Development, Plasticity, and Pathology. J Neuroimmune Pharmacol. 2013;8(3):494–509. doi: 10.1007/s11481-013-9434-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Madinier A, Bertrand N, Mossiat C, Prigent-Tessier A, Beley A, Marie C, Garnier P. Microglial involvement in neuroplastic changes following focal brain ischemia in rats. PLoS One. 2009;4(12):e8101. doi: 10.1371/journal.pone.0008101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhao BQ, Wang S, Kim HY, Storrie H, Rosen BR, Mooney DJ, Wang X, Lo EH. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat Med. 2006;12(4):441–445. doi: 10.1038/nm1387. [DOI] [PubMed] [Google Scholar]
- 128.Fox C, Dingman A, Derugin N, Wendland MF, Manabat C, Ji S, Ferriero DM, Vexler ZS. Minocycline confers early but transient protection in the immature brain following focal cerebral ischemia-reperfusion. J Cereb Blood Flow Metab. 2005;25(9):1138–1149. doi: 10.1038/sj.jcbfm.9600121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chu LS, Fang SH, Zhou Y, Yu GL, Wang ML, Zhang WP, Wei EQ. Minocycline inhibits 5-lipoxygenase activation and brain inflammation after focal cerebral ischemia in rats. Acta Pharmacol Sin. 2007;28(6):763–772. doi: 10.1111/j.1745-7254.2007.00578.x. [DOI] [PubMed] [Google Scholar]
- 130.Carty ML, Wixey JA, Colditz PB, Buller KM. Post-insult minocycline treatment attenuates hypoxia-ischemia-induced neuroinflammation and white matter injury in the neonatal rat: a comparison of two different dose regimens. Int J Dev Neurosci. 2008;26(5):477–485. doi: 10.1016/j.ijdevneu.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 131.Hayakawa K, Irie K, Sano K, Watanabe T, Higuchi S, Enoki M, Nakano T, Harada K, Ishikane S, Ikeda T, Fujioka M, Orito K, Iwasaki K, Mishima K, Fujiwara M. Therapeutic time window of cannabidiol treatment on delayed ischemic damage via high-mobility group box1-inhibiting mechanism. Biol Pharm Bull. 2009;32(9):1538–1544. doi: 10.1248/bpb.32.1538. [DOI] [PubMed] [Google Scholar]
- 132.Xuan A, Long D, Li J, Ji W, Hong L, Zhang M, Zhang W. Neuroprotective effects of valproic acid following transient global ischemia in rats. Life Sci. 2012;90(11–12):463–468. doi: 10.1016/j.lfs.2012.01.001. [DOI] [PubMed] [Google Scholar]