

Abstract
Autophagy is a lysosomal bulk degradation pathway for cytoplasmic cargo, such as long-lived proteins, lipids, and organelles. Induced upon nutrient starvation, autophagic degradation is accomplished by the concerted actions of autophagy-related (ATG) proteins. Here we demonstrate that two ATGs, human Atg2A and Atg14L, colocalize at cytoplasmic lipid droplets (LDs) and are functionally involved in controlling the number and size of LDs in human tumor cell lines. We show that Atg2A is targeted to cytoplasmic ADRP-positive LDs that migrate bidirectionally along microtubules. The LD localization of Atg2A was found to be independent of the autophagic status. Further, Atg2A colocalized with Atg14L under nutrient-rich conditions when autophagy was not induced. Upon nutrient starvation and dependent on phosphatidylinositol 3-phosphate [PtdIns(3)P] generation, both Atg2A and Atg14L were also specifically targeted to endoplasmic reticulum-associated early autophagosomal membranes, marked by the PtdIns(3)P effectors double-FYVE containing protein 1 (DFCP1) and WD-repeat protein interacting with phosphoinositides 1 (WIPI-1), both of which function at the onset of autophagy. These data provide evidence for additional roles of Atg2A and Atg14L in the formation of early autophagosomal membranes and also in lipid metabolism.
Keywords: autophagy, autophagosome, Atg2A, Atg14L, double-FYVE containing protein 1, WIPI-1
Cellular autophagy is an evolutionarily highly conserved degradation pathway for both stochastic and selective degradation of cytoplasmic cargo within the lysosomal compartment. As such, this process of self-eating secures cellular homeostasis and survival, while malfunction contributes to the initiation and development of many age-related human diseases including diabetes, tumorigenesis, and neurodegeneration (1–4). Autophagy is constitutively active on a low basal level resulting in constant cytoplasmic turnover and the specific elimination of macromolecule aggregates and damaged organelles. Following a great variety of cellular insults such as nutrient starvation, autophagy is induced above basal level and produces monomers and energy for subsequent recycling processes.
Autophagy is hallmarked by the formation of double-membrane vesicles called autophagosomes that are generated from initial preautophagosomal membrane precursors or phagophores. Elongation of the phagophore and subsequent vesicle closure complete the formation of autophagosomes that sequester the cytoplasmic cargo and communicate with the lysosomal compartment to acquire acidic hydrolases for cargo breakdown (5–7).
The interconnection between autophagy and energy metabolism, including lipid metabolism, is of intense interest, especially in the context of dysfunctional autophagy in metabolic disorders (8, 9). Lipophagy, the degradation of lipid droplets (LDs) through autophagy, was recently discovered (10, 11). LDs are intracellular lipid storage reservoirs and major contributors to lipid homeostasis (12, 13). Lipophagy facilitates the release of fatty acids resulting from LD degradation and also prevents excessive accumulation of cellular lipids (8, 14).
Lipids, in particular phosphoinositides, play an important role in the regulation of autophagy (15–17). The generation of phosphatidylinositol 3-phosphate [PtdIns(3)P] is evidenced to represent an essential and conserved initiation step of autophagy (18–22). In detail, phagophore formation is preceded by localized PtdIns(3)P production upon activation of the lipid kinase phosphatidylinositol 3-kinase class III (PtdIns3KC3) in complex with Beclin 1, autophagy-related protein 14L (Atg14L), and vacuolar protein sorting (Vps) 15 (20, 23–28). Thereby, the PtdIns3KC3 complex is targeted via Atg14L to the endoplasmic reticulum (ER) (29) where PtdIns(3)P effectors permit subsequent membrane rearrangements that lead to phagophore and autophagosome formation. Different PtdIns(3)P binding proteins involved in early steps of phagophore formation include the FYVE-domain proteins Alfy, involved in selective autophagy of protein aggregates (30, 31), and DFCP1, which decorates ER-associated omegasome structures formed prior to the phagophore (32). Human WD-repeat proteins interacting with phosphoinositides (WIPIs), 7-bladed β-propeller proteins (33, 34), include the PtdIns(3)P effectors WIPI-1 and WIPI-2, both of which are functionally essential for autophagy (35). WIPI-1 was found to specifically localize to both ER and plasma membrane (PM) when autophagy is induced by nutrient starvation; WIPI-2 also localizes to the PM upon starvation, in addition to membranes situated close to the Golgi cisternae (36). Both WIPI-1 and WIPI-2 subsequently become membrane proteins of the forming phagophore (34, 37–39) and of the inner and outer membrane of autophagosomes, as found by freeze-fracture immuno-electron microscopy (EM) (36).
Yeast Atg2 was identified through functional screening and shown to play an essential role in both autophagy and the cytoplasm-to-vacuole pathway (40, 41). Subsequently, it was demonstrated that yeast Atg2 is targeted via Atg18, the ancestral human WIPI-1/-2 protein, to the phagophore (42, 43). Recently, it was shown that yeast Atg2 can be targeted to the phagophore also in the absence of Atg18, but dependent on PtdIns(3)P (44). Human Atg2 proteins are essential for autophagosome formation (45), suggesting that WIPI proteins and Atg2 share overlapping functions at the phagophore.
Although the membrane origin of autophagosomes is still unclear, landmark experiments showed that phagophore formation initiates from discrete ER regions referred to as cradle (46, 47) where several ATGs colocalize (48). Interestingly, analogous ER regions have also been postulated to facilitate lipid transfer between LDs and the ER (12). In this context, it was suggested that LD biogenesis is connected to phagophore formation, likely involving unknown adaptor proteins that facilitate such functional interactions (8).
Here we provide evidence that both human Atg2A and Atg14L colocalize at LDs under conditions when autophagy is not induced above constitutive basal level. Upon nutrient starvation and dependent on PtdIns(3)P production, both Atg14L and Atg2A also localize to early autophagosomal membranes, decorated with the PtdIns(3)P effectors DFCP1 and WIPI-1. Interestingly, our high-content imaging analysis further showed that the numbers of cellular LDs increase upon nutrient starvation. In this context, we discuss the function of ATGs in both LD and autophagosome biogenesis.
MATERIALS AND METHODS
DNA constructs
The human Atg2A cDNA (gi:83404909, IRAKp961E12232Q2, ImaGenes) was amplified by PCR with the oligonucleotides 5′gagagagaattcttcacgatggctgtggccatg’3 and 5′gagaggatcctcagtcttgggcactgtccgt’3 and subcloned (BamHI/EcoRI) into pEGFP.C1 (Clontech) to generate green fluorescent protein (GFP)-Atg2A. To obtain myc-Atg2A, the human Atg2A was isolated from GFP-Atg2A (BamHI/EcoRI), and ends were blunted and subcloned (EcoRV) into pCMV-Tag3b (Stratagene). To generate Atg2A-myc, the Atg2A cDNA was amplified by PCR using pEGFP.C1-Atg2A as a template along with the oligonucleotides 5′gagagaggatccaccatgtcacgatggctgtggccatg’3 and 5′ ctctctgtcgacgtcttgggcactgtccgagcg’3 and subcloned (BamHI/SalI) into pCMV-Tag1 (Stratagene). Human Atg14L cDNA (gi:80475011, IRCMp5012D055D, ImaGenes) was amplified by PCR with the oligonucleotides 5′gagagaattctgcgtctcccagtgggaagggagcc’3 and 5′gagagtcgacttaacggtgtccagtgtaagctttaaacc’3 and subcloned (EcoRI/SalI) into i) pEGFP.C1 (Clontech) to generate GFP-Atg14L or ii) pCMV-Tag3A (Stratagene) to generate myc-Atg14L. Construct integrities were verified by PCR, restriction digest, and automated DNA sequence analysis. GFP-WIPI-1 (34) and myc-DFCP1 (39) were described earlier. Roger Tsien, University of California, San Diego, provided mCherry-tubulin.
Cell culture and DNA transfection
U2OS, HeLa, and G361 cells were obtained from ATCC and cultured in DMEM (Cat. No. 31966, Life Technologies), supplemented with 10% FCS (Cat. No. A15-101, PAA), 100 U/ml penicillin/100 µg/ml streptomycin (Cat. No. 15140, Life Technologies), and plasmocin (Cat. No. ant-mpp, InvivoGen) at 37°C, 5% CO2. For DNA transfections, cells were seeded on cover slips, and according to the manufacturer’s protocol, transfections were conducted with Lipofectamine 2000 (Cat. No. 11668, Life Technologies) or Promofectin (Cat. No. PK-CT-2000-100, PromoCell). U2OS cells stably expressing GFP-Atg2A were selected by using 0.6 mg/ml G418 (Cat. No. 11811, Life Technologies).
siRNA transfection
ATG2A siRNA (sc-96345), ATG-14 siRNA (sc-92229), and control siRNA-A (sc-37007) were purchased from Santa Cruz. HeLa or G361 cells were transfected with 25 nM siRNA using Lipofectamine RNAimax (11668, Invitrogen) according to the manufacturer’s reverse transfection protocol. siRNA was diluted in 20 µl Opti-MEM, and 0.2 µl of the transfection reagent was added. The transfection mixture was incubated for 20 min and combined with 100 µl DMEM/10% FCS containing 1× 104 HeLa cells or 3× 104 G361 cells in 96-well plates. Forty-eight hours after transfection, cells were subjected to starvation treatments and high-content LD analysis.
Autophagy assays
Autophagy was induced by using nutrient-free (NF) medium (Earl’s balanced salt solution, Cat. No. E2888, Sigma-Aldrich) or by administration of 330 nM rapamycin (RM; Cat. No. R0395, Sigma-Aldrich) for 3 h (37°C, 5% CO2). Autophagy was inhibited by administration of 233 nM wortmannin (WM; Cat. No. W1628, Sigma-Aldrich) for 3 h (37°C, 5% CO2). GFP-WIPI-1 puncta-formation analysis was conducted as previously described (37), and likewise applied for GFP-Atg14L. Oleic acid (OA; 400 μM, Cat. No. O3008, Sigma-Aldrich) was used to pretreat the cells for 24 h prior to autophagy-modulating treatments.
Antibodies and fluorescent dyes
The following primary antibodies were used in this study: anti-tubulin (B-5-1-2) (Cat. No. T5168, Sigma-Aldrich), anti-myc (9E10) (Cat. No. sc-40 or sc-789, Santa Cruz Biotechnology), anti-GFP antibody (Cat. No. 11814460001, Roche), anti-adipocyte differentiation-related protein (ADRP) (Cat. No. 610102, ProGen), anti-LC3 (Cat. No. 0231-100/LC3-5F10, Nano Tools), and anti-GAPDH (Cat. No. ACR001P, Acris). The following secondary antibodies were used: anti-rabbit IgG Alexa 488 (Cat. No. A11008, Life Technologies), anti-mouse (Cat. No. A11003, Life Technologies) or anti-rabbit (Cat. No. A11010, Life Technologies) IgG Alexa 546, anti-rabbit IgG Alexa 633 (Cat. No. A21070, Life Technologies), and anti-mouse (Cat. No. NA931V, GE Healthcare) or anti-rabbit (Cat. No. NA934V, GE Healthcare) IgG-HRP conjugated antibodies. The following reagents were purchased from Life Technologies: HCS LipidTOX Green neutral lipid stain (Cat. No. H34475), HCS LipidTOX Red neutral lipid stain (Cat. No. H34476), and TO-PRO-3 (Cat. No. T3605).
Confocal laser scanning microscopy
Immunostaining and confocal laser scanning microscopy (LSM) were previously described (34). For the visualization of LDs cells were incubated with HCS LipidTOX Green or HCS LipidTOX Red neutral lipid stain (1:1,000) for 30 min at room temperature. For quantitative colocalization analysis, image projections from confocal LSM sections (in distances of 0.5 µm) were acquired with identical laser intensities and detector gains. Subsequently, images were background subtracted, and ADRP LD signals were thresholded and analyzed for individual cells using the ImageJ colocalization threshold plug-in. Alternatively, images were analyzed by using Image Pro Plus software (Media Cybernetics). Using Volocity 3.1 (Improvision), individual confocal LSM sections (in distances of 0.2 µm) were applied for 3D reconstruction and fly-through movie presentations.
Automated high-throughput fluorescence image acquisition and analysis
G361, HeLa, or U2OS cells were cultured in 96-well plates subjected to starvation treatments, fixed with 3.7% paraformaldehyde in PBS for 15 min, stained with 4,6-diamidino-2-phenylindole (DAPI; 5 µg/ml in PBS) (Cat. No. 4099, Applichem) for 15 min, incubated with HCS LipidTOX Green (1:1,000 in PBS) for 30 min at room temperature, and subjected to automated image acquisition using an In Cell Analyzer 1000 high content platform (GE Healthcare) as previously described (39, 49). Twenty DAPI and GFP image fields (each containing about 20 cells) were automatically acquired per well (Nikon 40× Planfluor objective) and subjected to automated image analysis using the In Cell Analyzer 1000 Workstation 3.4 software. The number of LDs per cell was determined by using different parameters for nuclei, cell, and inclusions. The cells were recognized by the nuclei (DAPI channel) and GFP channel. The characteristic cell area was set to 800 µm2 (G361) or 1,500 µm2 (HeLa), with a sensitivity of 25. For the detection of inclusions within the recognized cells, only inclusions with a size between 0.5 µm and 5 µm were counted [sensitivity 25 (G361) or 40 (HeLa)]. Additionally, HeLa cells with an inclusion intensity of <600 were excluded from the analysis.
Live-cell video microscopy
Live-cell imaging was conducted as described earlier (50), and media supplemented with 11.4 mM ascorbic acid to reduce phototoxicity. Movies with five images per second were generated. ImageJ software with the MTrackJ plug-in was used for still image representation and to calculate migration distances of selected structures.
EM
For freeze-fracture immune-EM, unfixed stably transfected GFP-Atg2A U2OS cells were scraped from the culture vessels, centrifuged to remove excess medium, and recentrifuged briefly (<30 s) in 30% glycerol. Cells were mounted in 30% glycerol on gold-nickel alloy carriers and immediately rapidly frozen in Freon 22 cooled with liquid nitrogen. The samples were fractured in a BA310 freeze-fracture unit (Balzers AG) at –100°C. Replicas of the fractured cells were immediately made by electron beam evaporation of platinum-carbon and carbon at angles of 38° and 90° and to thicknesses of 2 and 20 nm, respectively. The replicas were incubated overnight in 5% SDS to remove cellular material except for those molecules adhering directly to the replicas (51). They were then washed in distilled water and incubated briefly in 5% BSA before immunolabeling. Freeze-fracture replicas of the cells were immunogold labeled with primary rabbit polyclonal antibodies raised against the entire sequence of GFP (ab290, Abcam). The secondary antibodies used were goat anti-rabbit antibodies coupled to 18 nm colloidal gold (conjugates from Jackson ImmunoResearch) (52). Control specimens were prepared without the primary antibodies. Examination of the immunogold-labeled freeze-fracture replicas was carried out using a Philips 410 transmission electron microscope. Observations on freeze-fracture immunogold replicas were based on examination of >200 cells from three separate experiments.
For standard EM, subconfluent G361 cells were treated with 500 µM OA for 24 h and fixed in 4% paraformaldehyde (sc-281692, Santa Cruz) and further in 2% glutaraldehyde and 0.5% osmium tetroxide in 0.1 M PBS. Subsequently, fixed cells were dehydrated with ethanol, and embedded in Epon as previously described (53). Thin sections were (ultramicrotome) contrasted with uranyl acetate and lead citrate and examined using an EM410 electron microscope (Philipis) and documented digitally (Ditabis).
Quantitative real-time RT-PCR
Total RNA was extracted from 1× 105 to 1× 106 cells (G361 or HeLa) using InnuPrep RNA Mini Kit (Cat. No. 845-KS-2040250, Analytik Jena), and 0.2 μg of total RNA was reverse transcribed using pd(N)6 random primers (Roche Diagnostics AG, Rotkreuz, Switzerland). Gene expression assays for ATG2A, ATG2B, and ATG14L used in a 96-well format on the StepOnePlus Sequence detection system were Hs00390076_m1, Hs00216083_m1, and Hs00208732_m1, respectively (Life Technologies, Zug, Switzerland). Hydroxymethylbilane synthase (HMBS) primers and probes have been described previously (54), and data analysis was performed as described (55).
Western blotting
Cells were rinsed with PBS (37°C) and lysed with preheated (100°C) 100 mM Tris (pH 6.8), 2.5 mM EDTA (pH 8), 25% glycerol, 4% SDS, 100 mM DTT, 5% β-mercaptoethanol, and 0.05% bromophenol blue, followed by chromatin shearing using a 23G needle. Total protein extracts were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes (Cat. No. IPVH00010, Millipore). Standard ECL detection was performed with Immobilon Western Chemiluminescent HRP Substrate (Cat. No. WBKLS0100, Millipore).
Sucrose density centrifugation
LD purification was performed as described previously (56). Up to 1.5× 107 OA-treated (500 μM, 24 h) cells were lysed in 3 ml disruption buffer [25 mM Tris (pH 7.4), 100 mM KCl, 1 mM EDTA, 5 mM EGTA, 10 µg/ml leupeptin, 0.7 μg/ml pepstatin A, 0.1 mM PMSF (Cat. No. 04 693 132 001, Roche Complete)] by dounce homogenization. Cell lysates were centrifuged (1,000 g) for 10 min at 4°C to pellet cell nuclei and debris. The supernatant was mixed with an equal volume of disruption buffer supplemented with 1.08 M sucrose, transferred to 14 ml centrifugation tubes (Cat. No. 331374, Beckman), and overlayed with 2 ml 0.27 M sucrose [25 mM Tris (pH 7.4), 1 mM EDTA, 1 mM EGTA, 0.27 M sucrose], 2 ml 0.135 M sucrose solution, and 2 ml top solution [25 mM Tris (pH 7.4), 1 mM EDTA, 1 mM EGTA]. Samples were centrifuged at 150,500 g (Beckman Coulter OPTIMA XL-100K ultracentrifuge, Beckman SW-40Ti rotor) for 2 h at 4°C and fractionated into lipid-floating, intermediate, and cytosolic fractions.
Bioinformatics analyses
Cluster analysis: Human Atg2A protein (gi:239047271) was used for a Basic Local Alignment Search Tool search against the NCBI nonredundant protein database ‘nr’ (version November 2012). All sequences producing High Scoring Segment-Pairs (HSPs) up to E-values of 10 were extracted as full-length sequences (813 sequences) and analyzed in Cluster Analysis of Sequences (CLANS) (57). The sequence similarity groups identified in the CLANS map were used as a basis to identify all sequences of the Atg2 protein family and select the animal as well as suitable outgroup sequences from which to infer a phylogeny.
Phylogenetic inference: All sequences from the animal Atg2 sequence similarity groups, as well as the selected outgroup sequences, were combined into one file and aligned using Multiple Sequence Comparison by Log-Expectation (MUSCLE). Fragment or truncated sequences present in the alignment were manually identified and removed prior to phylogenetic inference. The phylogeny was inferred based on the neighbor-joining approach (1,000 bootstrap replicates) using the ASATURA software, the JTT substitution matrix, and no mutational saturation cutoff.
RESULTS
Human Atg2A is expressed in vesicular structures that migrate bidirectionally along microtubules
We generated plasmids for transient and stable expression of N-terminal tagged GFP- or myc-Atg2A and C-terminal tagged Atg2A-myc in human tumor cell lines (U2OS, G361, and HeLa). In addition, human U2OS cells derived from G418-selected clones that stably express GFP-Atg2A protein at a low level (GFP-Atg2A/U2OS hereafter), were also generated for our studies. By Western blotting of protein extracts from stable GFP-Atg2A/U2OS cells, we detected GFP-Atg2A at ∼240 kDa, as expected from the calculated molecular mass for human Atg2A (213 kDa) and GFP (27 kDa) (Fig. 1A). In support, myc-tagged Atg2A was detected at ∼220 kDa (data not shown).
Fig. 1.

Vesicular localization of human Atg2A at the cytoplasmic face of LDs. A: Cell extracts from U2OS cells (1) and a generated monoclonal U2OS cell line stably expressing GFP-Atg2A (2) were separated by SDS-PAGE and subjected to Western blotting using anti-GFP (upper panel) and anti-GAPDH (lower panel) antibodies. B: Representative confocal LSM of stable GFP-Atg2A/U2OS cells. The cell boundary is marked by a dotted line (left image), and the vesicular localization of GFP-Atg2A in the cytoplasm (boxed area) is highlighted by magnification (right image). Scale bar, 20 µm. C: Live cell microscopy of stable GFP-Atg2A/U2OS cells transiently expressing mCherry-tubulin. Cells were treated with NF medium supplemented with 11.4 mM vitamin C to reduce photobleaching and phototoxicity. Displayed are representative still images of a magnified cell area from an imaged cell (supplementary Fig. IC; supplementary Video IV). The migration distance (in µm) of an individual GFP-Atg2A vesicle (arrows in upper panel images; scale bar, 5 µm) was tracked within the investigated time frame using Image J/MTrackJ plug-in and plotted against time (in seconds) (lower panel). D: Localization of Atg2A at LDs. Magnified imaging (upper panel; scale bar, 5 µm) of myc-Atg2A (red) at the surface of LDs (green) visualized by intensity profiling (lower panel) using confocal LSM. The 3D visualization is also available (supplementary Video V). Further supporting material is available (supplementary Fig. I; supplementary Videos I–III).
By confocal LSM of transiently expressed GFP-Atg2A the vesicular localization of Atg2A in the cytoplasm was apparent (Fig. 1B). This result was verified by live-cell microscopy of GFP-Atg2/U2OS cells (supplementary Fig. IA; supplementary Videos I–III) and by confocal LSM of myc-Atg2A (supplementary Fig. IB) or Atg2A-myc (data not shown). Because yeast Atg2 was identified as an essential protein of the autophagic machinery, we conducted treatments that modulate the autophagic activity throughout this study, along with appropriate controls. In general, we used NF medium or RM to induce autophagy above basal level, or WM to inhibit the formation of autophagosomes by inhibiting PtdIns(3)P generation. Independent of the cellular autophagic status Atg2A displayed a vesicular localization pattern (supplementary Fig. I; supplementary Videos I–III). We quantified the area of vesicular Atg2A structures per cell to assess if the autophagic status influences the abundance of vesicular Atg2A structures (supplementary Figs. I and IVA) and found that autophagy induction resulted in a consistent increase in vesicular Atg2A compared with control medium (CM) or autophagy inhibition (supplementary Figs. I and IVA). As vesicular GFP-Atg2A structures displayed high mobility (supplementary Videos I–III), indicative for microtubule-assisted movement, we expressed mCherry-tubulin in GFP-Atg2A/U2OS cells followed by live-cell microscopy (Fig. 1C, upper image panels; supplementary Fig. IC and supplementary Video IV). Indeed, GFP-Atg2A vesicles moved distances of ∼3–5 µm bidirectionally on microtubule tracks (Fig. 1C, lower panel).
Human Atg2A localizes to cytoplasmic LDs
In order to define the identity of vesicular Atg2A structures, we conducted colocalization studies with a variety of cellular markers (e.g., lysosomes, mitochondria, or FYVE-positive endosomes; data not shown) and found that Atg2A specifically localizes to LDs. In detail, by differential interference contrast microscopy we found that LDs colocalized with myc-Atg2A vesicles (supplementary Fig. ID). This result was further confirmed by visualizing transiently expressed myc-Atg2A in U2OS cells treated with LipidTOX Green, which labels neutral lipids (supplementary Fig. IE). We verified that LipidTOX Green specifically detects LDs by stimulation of LD formation using OA (supplementary Fig. IIA) and EM analysis of OA-loaded cells (supplementary Fig. IIB). Clearly, myc-Atg2A was expressed at the surface of LDs as fluorescence intensity profiling showed maximal myc-Atg2A signal intensity (Fig. 1D, in red) at the cytoplasmic face of LDs (Fig. 1D, in green). Localization of myc-Atg2A at the cytoplasmic face of LDs labeled with LipidTOX Green is further highlighted by 3D reconstruction and fly-through movie presentations using confocal LSM sections (supplementary Video V). By freeze-fracture immuno-EM of the GFP-Atg2A/U2OS cell line with anti-GFP antibodies, we were unable to detect GFP-Atg2A at LDs (supplementary Fig. IIC), demonstrating that Atg2A does not become a membrane protein of the LD lipid monolayer but associates with LDs as a peripheral membrane protein.
Further, fluorescence intensity profiling using confocal LSM identified a profound colocalization of endogenous adipophilin (ADRP), a bona fide LD marker (58), with vesicular GFP-Atg2A (supplementary Fig. IIIA). This result is analyzed in more detail subsequently.
ADRP-positive LDs are decorated with GFP-Atg2A
Upon OA treatment, the amount of ADRP protein increases as well as the number and size of ADRP-positive LDs (59). We conducted quantitative confocal LSM visualizing GFP-Atg2A and endogenous ADRP. We used GFP-Atg2A-transfected G361 cells and treated the cells with or without OA for 24 h. Subsequently, the cells were treated for 3 h with nutrient-rich CM, NF medium, or WM. No differences in colocalization patterns of GFP-Atg2A and endogenous ADRP regarding autophagy-modulating conditions were observed (data not shown), and nutrient starvation results (NF, OA + NF) are presented (Figs. 2 and 3; supplementary Fig. III). Clearly, GFP-Atg2A prominently colocalized with endogenous ADRP at LDs, observed as typical ring-like structures (Fig. 2). By comparing confocal LSM images, the amount of ADRP-positive LDs decorated with GFP-Atg2A seemed to increase upon OA treatment (OA + NF) (Fig. 2A, B). As expected, OA-mediated stimulation of LD formation increased the number of ADRP-positive LDs (OA + NF) when compared with conditions without OA (NF), as the fluorescence area of endogenous ADRP significantly increased from 1.7% (NF) to 8.4% (OA + NF) in relation to the total cell area (Fig. 3A, right panels). The fluorescence GFP-Atg2A area also significantly increased upon OA treatment from 2.3% (NF) to 8.8% (OA + NF) (Fig. 3A, left panels). As GFP-Atg2A colocalized with ADRP in both the absence and presence of OA (OA + NF) (Fig. 2; supplementary Fig. III), we quantified the colocalization of GFP-Atg2A and ADRP. To measure the overall association of GFP-Atg2A and endogenous ADRP, we measured the Pearson’s correlation coefficient (PCC) for individual cells (n = 34 cells per treatment; PCC values + 1 = 100% colocalization) using the ImageJ colocalization threshold tool. In the absence of OA and under conditions that induce autophagy (NF), the PCC value was 0.33 and significantly increased to 0.49 when autophagy was induced upon the stimulation of LD formation by OA for 24 h (OA + NF) (Fig. 3B). The Mander’s overlap coefficient (MOC) for GFP-Atg2A (0.036) overlapping with ADRP also significantly increased when LD formation was stimulated (0.119) (Fig. 3C). Such increase in overlap was not observed for ADRP overlapping GFP-Atg2A (Fig. 3D) and reached an MOC of 0.92 before and 0.94 after OA treatment. This result argues that i) ADRP-positive LDs are prominently decorated with GFP-Atg2A and that ii) GFP-Atg2A localizes not exclusively to the LD compartment. Consistent with both this result and expectation, we also detected GFP-Atg2A in the autophagic compartment as detailed subsequently.
Fig. 2.

Atg2A localizes to ADRP-positive LDs independent of treatments that modulate autophagy. GFP-Atg2A/U2OS cells were incubated with or without OA for 24 h, followed by treatments with NF medium for 3 h. Subsequently, the cells were immunostained with anti-ADRP/IgG-conjugated Alexa 546 to detect endogenous ADRP and with TO-PRO-3 to mark cell nuclei, followed by confocal LSM (n = 3). A: Representative images from NF-treated cells. B: Representative images from OA + NF-treated cells. The cell boundaries are marked by a dotted line in the upper panels, and the typical ring-like ADRP-positive LD appearance is highlighted by further magnification in the lower panels. Scale bar, 20 µm. Supporting material is available (supplementary Figs. II and IIIA).
Fig. 3.

OA treatment increased Atg2A targeting to ADRP-positive LDs. G361 cells transiently expressing GFP-Atg2A were incubated with or without OA for 24 h, followed by treatments with NF medium for 3 h. Cells were immunostained with anti-ADRP/IgG Alexa 546 antibodies analyzed by confocal LSM (n = 3). A: Image projections of confocal sections were quantified with regard to fluorescence areas by using Image Pro Plus, providing the percentage of the total inclusion area (ADRP or GFP-Atg2A vesicles) in relation to the total cell area. NF, n = 21 cells; OA + NF, n = 18 cells (from 3 independent experiments). B: Using the ImageJ colocalization threshold tool, the PCC for individual cells (n = 34 cells per treatment) was measured. The MOC for GFP-Atg2A overlapping with ADRP (C) and ADRP overlapping with GFP-Atg2A (D) was also measured using the ImageJ colocalization threshold. Mean ± SEM. P values: n.s. (not significant) P ≥ 0.05, *** P ≤ 0.001. Supporting material is available (supplementary Fig. IIIB).
Independent of the autophagic status, Atg2A colocalized with Atg14L at LDs
Next we conducted colocalization studies for Atg2A with ATGs functioning during the initiation of autophagosome formation of Atg14L, WIPI-1, and DFCP1. (Figs. 4 and Figs. 6). First, we investigated the localization of myc-Atg2A with regard to GFP-Atg14L, a phagophore marker that is partially also present in vesicular structures apart from phagophores (26). We expressed GFP-Atg14L in U2OS cells (Fig. 4A) followed by treatments with nutrient-rich CM, NF medium, or WM and quantified the number of GFP-Atg14L puncta-positive cells (Fig. 4B, left panels). In parallel, we also analyzed transiently expressed GFP-WIPI-1 (Fig. 4A). As expected, nutrient starvation using NF medium resulted in a significant increase of GFP-WIPI-1 puncta-positive cells, and WM-mediated inhibition of PtdIns(3)P generation (WM) resulted in a significant decrease (Fig. 4B, right panels). This result was also achieved by quantifying GFP-Atg14L puncta-positive cells. However, more control cells already displayed a prominent amount of GFP-Atg14L puncta (an average of 40% puncta-positive cells), and WM administration did not completely abolish the presence of GFP-Atg14L puncta. Nutrient starvation, however, significantly increased the number of GFP-Atg14L puncta-positive cells to an average of 63% (Fig. 4B, left panels).
Fig. 4.

Atg2A colocalizes with Atg14L at LDs independent of autophagy. A: Protein extracts from U2OS cells transiently expressing GFP-Atg14L or GFP-WIPI-1 prepared at different hours posttransfection were subjected to Western blotting using anti-GFP or anti-tubulin antibodies (n = 3). B: From 400 cells per treatment (n = 4) GFP-Atg14L or GFP-WIPI-1 puncta formation was determined (36 h posttransfection) and expressed as the percentage of puncta-positive cells. Mean ± SD; P value: *** P ≤ 0.001. C: U2OS cells transiently coexpressing GFP-Atg14L (green) and myc-Atg2A (red) for 24 h were treated with nutrient-rich CM, NF medium, or WM for 3 h, followed by indirect immunofluorescence analysis by confocal LSM using anti-myc/IgG-conjugated Alexa 546 antibodies [CM, NF (n = 4), WM (n = 3)]. The cell boundaries are marked by a dotted line (left panels), and boxed areas highlighted by magnification (right panels). D: U2OS cells expressing GFP-Atg14L for 24 h were incubated with LipidTOX Red and analyzed by confocal LSM (n = 4) followed by intensity profiling. GFP-Atg14L localization at LDs (1) and cytoplasmic puncta (2) is indicated. The 3D visualization is also available (supplementary Video VI). Scale bars, 20 µm. Further supporting material is also available (supplementary Fig. IV).
Fig. 6.

Upon starvation, Atg2A and Atg14L colocalize at DFCP1- and WIPI-1-positive early autophagosomal membranes. A: U2OS cells transiently expressing GFP-Atg14L (green), myc-Atg2A (red), and HA-WIPI-1 (blue) were starved in NF medium for 3 h and immunostained with anti-myc/IgG-conjugated Alexa 546 and anti-WIPI-1/IgG-conjugated Alexa 633 antibodies. Images (left panels) were acquired by confocal LSM followed by intensity profiling (right panel). Colocalization of GFP-Atg14L, myc-Atg2A, and HA-WIPI-1 (1, 2, white arrow) and additional GFP-Atg14L/myc-Atg2A LDs (red arrow) are indicated. B: In parallel to A, cells were treated with WM for 3 h. GFP-Atg14L/myc-Atg2A-positive LDs (1, 2) are indicated, whereas HA-WIPI-1 (blue) is distributed throughout the cytoplasm. C: U2OS cells transiently transfected with GFP-Atg2A (green), myc-DFCP-1 (red), and HA-WIPI-1 (blue) were starved in NF medium for 3 h, immunostained with anti-myc/IgG-conjugated Alexa 546 and anti-WIPI-1/IgG-conjugated Alexa 633 antibodies, and analyzed by confocal LSM (n = 2). The intensity profile is displayed (right panel), and colocalization of GFP-Atg2A, myc-DFCP1, and HA-WIPI-1 is indicated (1, 2, white arrow). GFP-Atg2A LDs are also indicated (red arrow). Presented are magnified areas from corresponding full cell images (supplementary Fig. V).
On this basis, we transiently coexpressed GFP-Atg14L and myc-Atg2A in U2OS cells and treated the cells with nutrient-rich CM, NF medium, or WM. We found that both ATGs colocalized at vesicular structures that were present irrespective of the autophagic status, indicating that these structures represent the colocalization of myc-Atg2A and GFP-Atg14L at LDs (Fig. 4C). In support, fluorescence intensity profiling of GFP-Atg14L-expressing cells incubated with LipidTOX Red demonstrated that in part GFP-Atg14L localizes at the cytoplasmic face of LDs (Fig. 4D), and this result was further highlighted by 3D reconstruction and fly-through movie presentation (supplementary Video VI). Of note, although vesicular GFP-Atg14L (Fig. 4B) and myc-Atg2A structures (supplementary Fig. IVA) increased upon autophagy induction, colocalization of GFP-Atg14L and myc-Atg2A was not significantly changed (68% to 77% of GFP-Atg14L colocalized with myc-Atg2A, whereas 52% to 63% of myc-Atg2A colocalized with GFP-Atg14L) (supplementary Fig. IVB, C). This indicates that Atg2A and Atg14L localize in part to the autophagic compartment upon autophagy induction and in addition to the LD compartment irrespective of the autophagic activity.
Detection of GFP-Atg2A and GFP-Atg14L in the ADRP-positive LD fraction separated by density gradient centrifugation
We conducted cell fractionation upon density centrifugation prepared from GFP-Atg2A/U2OS cells transiently transfected with myc-Atg14L. As expected, endogenous ADRP was exclusively present in the LD fraction. GFP-Atg2A was also detected in this fraction of floating ADRP-positive LDs; however, GFP-Atg2A was more prominently detected in cytoplasmic fractions. To a minor extent, we also detected myc-Atg14L, while the GAPDH control was restricted to cytoplasmic fractions (Fig. 5).
Fig. 5.
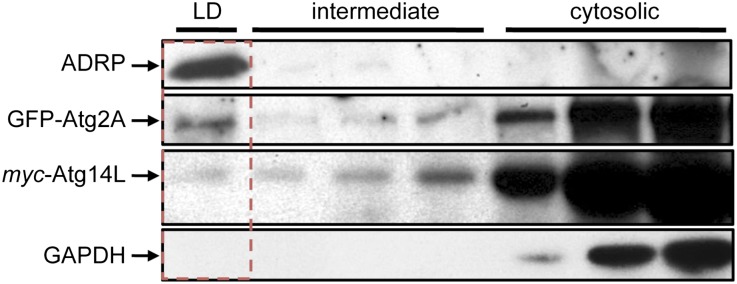
Density gradient centrifugation reveals the presence of ADRP, GFP-Atg2A, and myc-Atg14L in the floating LD fraction. Stable GFP-Atg2A/U2OS cells were transiently transfected with myc-Atg14L and incubated with OA for 24 h. Cell lysates were fractionated upon sucrose density centrifugation (n = 2). The floating LD fraction (boxed with a dotted red line) and intermediate and first cytoplasmic fractions were analyzed by Western blotting using anti-ADRP, anti-GFP, anti-myc, and anti-GAPDH antibodies.
PtdIns(3)P-dependent recruitment of Atg2A to WIPI-1-positive phagophores upon nutrient starvation
Next, we analyzed the localization of Atg2A and Atg14L to the autophagic compartment. We visualized myc-Atg2A, GFP-Atg14L, and HA-WIPI-1 by confocal LSM using U2OS cells (Fig. 6A, B; supplementary Fig. VA, B). Upon autophagy induction by NF medium treatments of 3 h, myc-Atg2A, GFP-Atg14L, and HA-WIPI-1 colocalized at cytoplasmic puncta (white arrow, puncta 1 and 2), easily distinguishable from LDs that were positive for myc-Atg2A and GFP-Atg14L, but negative for HA-WIPI-1 (red arrow) (Fig. 6A, left image panels). Fluorescence intensity profiling underlined this finding (Fig. 6A, right panel). WM administration abolished the triple colocalization of myc-Atg2A, GFP-Atg14L, and HA-WIPI-1, while colocalization of myc-Atg2A and GFP-Atg14L at LDs was maintained (Fig. 6B). We conducted parallel experiments by using GFP-Atg2A, HA-WIPI-1, and myc-DFCP1 (marking prephagophore omegasome structures) and found that, apart from the localization of GFP-Atg2A at LDs (red arrow), GFP-Atg2A, myc-DFCP1, and HA-WIPI-1 colocalized at cytoplasmic puncta (white arrow, puncta 1 and 2) upon the induction of autophagy (Fig. 6C; supplementary Fig. VC) which was also abrogated by WM administration (data not shown). Similarly, upon nutrient starvation myc-Atg2A colocalized with endogenous WIPI-1 in cytoplasmic puncta but not with myc-Atg2A at LDs (Fig. 7A). WM-mediated autophagy inhibition abrogated colocalization of endogenous WIPI-1 and myc-Atg2A, whereas LD localization of Atg2A was maintained (Fig. 7B).
Fig. 7.

Myc-tagged Atg2A is recruited to endogenous WIPI-1 upon the induction of autophagy. G361 cells transiently expressing myc-Atg2A were treated with NF medium (A) or WM (B) for 3 h; fixed; immunostained with anti-myc, anti-WIPI-1, and anti-mouse Alexa 546 antibodies; and analyzed by confocal microscopy. Representative images of three independent experiments are shown. Scale bar, 10 µm
Nutrient starvation leads to an increase of LDs in the human tumor cell lines G361 and U2OS
The previously discussed results demonstrated that Atg2A and Atg14L colocalize at LDs irrespective of the autophagic status of the cell, but that a subpopulation of both ATGs is targeted to early autophagosomal membranes enriched in PtdIns(3)P, and positive for DFCP1 and WIPI-1, upon the induction of autophagy. As we detailed our analysis using conditions when autophagy was induced by nutrient starvation using NF medium, we investigated the effect of NF on the number of LDs by conducting automated, high-throughput fluorescence image acquisition and analysis (49) of cells stained with LipidTOX Greens in 96-well plates (Fig. 8). In HeLa, G361, and U2OS cells treated with NF medium, the number of LDs per cell was found to increase significantly after 3 h (U2OS and G361) and after 24 h (all cell lines) (Fig. 8A). To functionally address the role of Atg2A and Atg14L in the biogenesis of LDs, we performed siRNA-mediated depletion of Atg2A or Atg14L in HeLa (supplementary Fig. VIA) and in G361 (supplementary Fig. VIB) cells. We assessed the number of LDs upon treatment with CM or NF medium using high-content image analysis. Atg2A depletion in HeLa cells followed by nutrient starvation using NF medium resulted in a significant increase of LD numbers in individual cells (Fig. 8B, left panels). Atg14L depletion significantly increased the number of LDs in i) HeLa cells in CM as well as upon nutrient starvation using NF medium (Fig. 8B, left panels) and ii) G361 cells in CM (Fig. 8B, right panels). In G361 cells, the size of LDs significantly increased upon Atg14L depletion and treatments using CM as well as NF medium (supplementary Fig. VID). From this, we conclude that Atg2A and Atg14L have dual functions, acting in the regulation of autophagosome formation as well as contributing to LD metabolism (Fig. 8C).
Fig. 8.

Starvation treatment increases LD formation. HeLa, G361, and U2OS cells were treated with CM or NF medium for 3 h, fixed, and stained with DAPI (blue) and LipidTOX Green (green). Automated image acquisition and analysis was conducted using a high-content imaging platform (In Cell Analyzer 1000). A: Up to 9,305 cells were quantified (n = 3–5) to assess the number of LDs per individual cell (left panel). Representative images for G361 cells are presented (right panel). Scale bar, 20 µm. B: HeLa and G361 cells were transiently transfected with scrambled control siRNA (siControl), siRNA targeting Atg2A (siAtg2A), or Atg14L (siAtg14L), and downregulation of Atg2A and Atg14L mRNA was verified by quantitative PCR (supplementary Fig. VIA, B). Cells transfected with siRNA were incubated with CM or NF medium for 3 h and fixed, and LD abundance was quantified by high-content analysis (n = 3–4). Mean ± SD. P values: n.s. (not significant) P ≥ 0.05, * P < 0.05, ** P ≤ 0.01, *** P ≤ 0.001. Supporting material is available (supplementary Fig. VIC, D). C: A model for the roles of human Atg2A and Atg14L in LD biogenesis and autophagy.
DISCUSSION
In Saccharomyces cerevisiae, PtdIns(3)P-dependent recruitment of Atg2 to autophagosome formation sites has been demonstrated (43, 44). Similarly, PtdIns(3)P-dependent targeting of human Atg2A to WIPI-1 positive phagophores upon starvation-induced autophagy was also found in this study, and WM treatment abrogated this specific recruitment of Atg2A. In addition we demonstrate that Atg2A is targeted to the surface of ADRP-positive LDs that move bidirectionally on microtubules, independent of autophagic status. Further, we identified Atg14L as a novel LD-associated protein, colocalizing there with Atg2A irrespective of autophagic status. Evidence has been provided that Atg14L resides at the ER and regulates localized PtdIns(3)P generation, which is then bound by the WIPIs. Apart from being the site of phagophore formation, LDs are also proposed to form at the ER (60), and thus sites of autophagosomal membrane biogenesis and LD formation seem to be functionally connected. LDs have further been detected in very close association with the forming phagophore (47).
So far, three ATGs have been found to be targeted to LDs, LC3 (61), Atg2A (45) (this study), and Atg14L (this study), indicating that these ATGs either display dual functions in autophagy and LD biogenesis or that they functionally connect both pathways. Therefore, by high-content analysis, we quantified the number of LDs in human HeLa, G361, and U2OS tumor cells upon short-term nutrient starvation and detected a prominent increase in the number and size of LDs per individual cell, resembling LD formation. This indicates that autophagy induction and LD formation can simultaneously occur in starved cells. Hence starvation-induced autophagy might provide energy to boost lipid storage, preparing the cell to compensate for prolonged energy shortage at a later stage. This phenomenon might even be evolutionarily conserved as Caenorhabditis elegans accumulates LDs upon dauer formation (62, 63). Starvation-induced nonspecific degradation of proteins and organelles can provide acetyl-CoA and also fatty acids and cholesterol required for neutral lipid synthesis. Also, starvation is known to trigger autophagosomal degradation of glycogen stores (64), which could liberate additional energy for lipid synthesis. Furthermore, our starvation treatments include lipoprotein depletion, which could decrease ER cholesterol levels resulting in sterol regulatory element binding protein (SREBP)-mediated stimulation of lipid synthesis (65). However, autophagy was also found to degrade LDs, thereby contributing to the regulation of lipid metabolism in liver cells (10, 61) and adipocyte differentiation (66), suggesting that the function of autophagosomal proteins at LDs and in adipogenesis is complex.
Whereas Atg2A and Atg14L are targeted to LDs independent of the autophagic status, both proteins localize to DFCP1 and WIPI-1-positive omegasome and phagophore membranes, distinct to LD structures, upon autophagy stimulation. In addition, we could demonstrate that Atg2A and Atg14L depletion resulted in increased LD abundance upon treatment with CM (Atg14L depletion) or NF medium (Atg2A depletion and Atg14L depletion). Therefore, our study provides evidence that Atg2A as well as Atg14L fulfill at least two functions in human cells: one exerted as a peripheral membrane protein at the surface of ADRP-positive LDs and the other involving the regulation of autophagy, dependent on PtdIns(3)P generation (Fig. 8C). It is possible that LD-localized Atg2A and Atg14L impair excessive lipid storage inside LDs and ensure efficient energy use (e.g., generated by autophagy induction). Another function of Atg2A and Atg14L at the LD surface might involve modulation of the fatty acid and cholesterol esterification/hydrolysis cycle, contributing to the regulation of lipid storage. Our results are in line with recent work on mammalian Atg2, shown to play an essential role in autophagosome formation and to also regulate LD size and distribution (45). However, it is also possible that ATGs, such as Atg2A and Atg14L, fulfill distinct roles at LDs and autophagosomal membranes, but that they also mediate a functional interconnection during simultaneous formation of LDs and autophagsomes, maybe by mediating intermembrane lipid traffic.
Because Atg2 shares protein sequence homologies with Vps13 (supplementary Fig. VII), a protein shown to promote endosome/trans-Golgi network (TGN) membrane protein cycling (67), it might be plausible that Vps13 and Atg2 proteins generally function in membrane formation and morphology, and that distinct members provide specificity for particular vesicle subsets. In this context, it has been proposed that the yeast Atg18/Atg2 complex is required to generate negative curvature at the forming autophagosome or fulfills an essential function at the elongating tips of the phagophore (43). Likewise it was recently found that Vps13 regulates membrane bending and promotes the expansion of the prospore membrane in yeast (68). From this, one would assume that malfunctions of either of the human Atg2/Vps13 proteins should be correlated to distinct human pathologies with defects in intracellular membrane biogenesis. Indeed, hereditary mutations of human Vps13 orthologs cause the diseases chorea acanthocytosis and Cohen syndrome (69, 70). As Atg2 functions in LD and autophagosome biogenesis, it can be assumed that functional or genetic alterations of human Atg2, as yet unidentified, might contribute to particular human pathologies correlated with modulated autophagic activity (1).
Supplementary Material
Acknowledgments
The authors thank Roger Tsien for the mCherry-tubulin encoding plasmid. The authors kindly acknowledge Andrei Lupas for discussions on the bioinformatics analysis.
Footnotes
Abbreviations:
- ADRP
- adipocyte differentiation-related protein
- ATG
- autophagy related
- CM
- control medium
- DAPI
- 4,6-diamidino-2-phenylindole
- DFCP1
- double-FYVE containing protein 1
- EM
- electron microscopy
- ER
- endoplasmic reticulum
- FYVE
- Fab1 YOTB Vac1p EEA1 domain
- GFP
- green fluorescent protein
- LD
- lipid droplet
- LSM
- laser scanning microscopy
- MOC
- Mander’s overlap coefficient
- NF
- nutrient-free
- OA
- oleic acid
- PCC
- Pearson’s correlation coefficient
- PtdIns(3)P
- phosphatidylinositol 3-phosphate
- VPS
- vacuolar protein sorting
- WIPI
- WD-repeat protein interacting with phosphoinositides
- WM
- wortmannin
This work was supported by grants from the German Research Foundation (DFG, SFB 773), the Federal Ministry for Education and Science (BMBF, BioProfile), and the Landesstiftung and the Forschungsschwerpunktprogramm Baden Wuerttemberg, Germany (T.P-C.). The Landesgraduiertenfoerderung Baden Wuerttemberg, Germany, supported S. G. Pfisterer with a predoctoral fellowship.
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of seven figures and six videos.
REFERENCES
- 1.Mizushima N., Levine B., Cuervo A. M., Klionsky D. J. 2008. Autophagy fights disease through cellular self-digestion. Nature. 451: 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levine B., Kroemer G. 2008. Autophagy in the pathogenesis of disease. Cell. 132: 27–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beau I., Mehrpour M., Codogno P. 2011. Autophagosomes and human diseases. Int. J. Biochem. Cell Biol. 43: 460–464 [DOI] [PubMed] [Google Scholar]
- 4.Moscat J., Diaz-Meco M. T. 2011. Feedback on fat: p62-mTORC1-autophagy connections. Cell. 147: 724–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mizushima N. 2007. Autophagy: process and function. Genes Dev. 21: 2861–2873 [DOI] [PubMed] [Google Scholar]
- 6.Jahreiss L., Menzies F. M., Rubinsztein D. C. 2008. The itinerary of autophagosomes: from peripheral formation to kiss-and-run fusion with lysosomes. Traffic. 9: 574–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Z., Klionsky D. J. 2010. Eaten alive: a history of macroautophagy. Nat. Cell Biol. 12: 814–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh R., Cuervo A. M. 2011. Autophagy in the cellular energetic balance. Cell Metab. 13: 495–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mathew R., White E. 2011. Autophagy in tumorigenesis and energy metabolism: friend by day, foe by night. Curr. Opin. Genet. Dev. 21: 113–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh R., Kaushik S., Wang Y., Xiang Y., Novak I., Komatsu M., Tanaka K., Cuervo A. M., Czaja M. J. 2009. Autophagy regulates lipid metabolism. Nature. 458: 1131–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koga H., Kaushik S., Cuervo A. M. 2010. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 24: 3052–3065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin S., Parton R. G. 2006. Lipid droplets: a unified view of a dynamic organelle. Nat. Rev. Mol. Cell Biol. 7: 373–378 [DOI] [PubMed] [Google Scholar]
- 13.Fujimoto T., Ohsaki Y., Cheng J., Suzuki M., Shinohara Y. 2008. Lipid droplets: a classic organelle with new outfits. Histochem. Cell Biol. 130: 263–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Czaja M. J. 2010. Autophagy in health and disease. 2. Regulation of lipid metabolism and storage by autophagy: pathophysiological implications. Am. J. Physiol. Cell Physiol. 298: C973–C978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simonsen A., Wurmser A. E., Emr S. D., Stenmark H. 2001. The role of phosphoinositides in membrane transport. Curr. Opin. Cell Biol. 13: 485–492 [DOI] [PubMed] [Google Scholar]
- 16.Noda T., Matsunaga K., Taguchi-Atarashi N., Yoshimori T. 2010. Regulation of membrane biogenesis in autophagy via PI3P dynamics. Semin. Cell Dev. Biol. 21: 671–676 [DOI] [PubMed] [Google Scholar]
- 17.Obara K., Ohsumi Y. 2011. PtdIns 3-kinase orchestrates autophagosome formation in yeast. J. Lipids. 2011: 498768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seglen P. O., Gordon P. B. 1982. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA. 79: 1889–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blommaart E. F., Krause U., Schellens J. P., Vreeling-Sindelarova H., Meijer A. J. 1997. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur. J. Biochem. 243: 240–246 [DOI] [PubMed] [Google Scholar]
- 20.Petiot A., Ogier-Denis E., Blommaart E. F., Meijer A. J., Codogno P. 2000. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. 275: 992–998 [DOI] [PubMed] [Google Scholar]
- 21.Kihara A., Noda T., Ishihara N., Ohsumi Y. 2001. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J. Cell Biol. 152: 519–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Obara K., Ohsumi Y. 2008. Dynamics and function of PtdIns(3)P in autophagy. Autophagy. 4: 952–954 [DOI] [PubMed] [Google Scholar]
- 23.Codogno P., Meijer A. J. 2005. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. 12 (Suppl. 2): 1509–1518 [DOI] [PubMed] [Google Scholar]
- 24.Abeliovich H., Dunn W. A., Jr, Kim J., Klionsky D. J. 2000. Dissection of autophagosome biogenesis into distinct nucleation and expansion steps. J. Cell Biol. 151: 1025–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Itakura E., Kishi C., Inoue K., Mizushima N. 2008. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14L and UVRAG. Mol. Biol. Cell. 19: 5360–5372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsunaga K., Saitoh T., Tabata K., Omori H., Satoh T., Kurotori N., Maejima I., Shirahama-Noda K., Ichimura T., Isobe T., et al. 2009. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 11: 385–396 [DOI] [PubMed] [Google Scholar]
- 27.Zhong Y., Wang Q. J., Li X., Yan Y., Backer J. M., Chait B. T., Heintz N., Yue Z. 2009. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 11: 468–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pattingre S., Tassa A., Qu X., Garuti R., Liang X. H., Mizushima N., Packer M., Schneider M. D., Levine B. 2005. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 122: 927–939 [DOI] [PubMed] [Google Scholar]
- 29.Matsunaga K., Morita E., Saitoh T., Akira S., Ktistakis N. T., Izumi T., Noda T., Yoshimori T. 2010. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J. Cell Biol. 190: 511–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Filimonenko M., Isakson P., Finley K. D., Anderson M., Jeong H., Melia T. J., Bartlett B. J., Myers K. M., Birkeland H. C., Lamark T., et al. 2010. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol. Cell. 38: 265–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simonsen A., Birkeland H. C., Gillooly D. J., Mizushima N., Kuma A., Yoshimori T., Slagsvold T., Brech A., Stenmark H. 2004. Alfy, a novel FYVE-domain-containing protein associated with protein granules and autophagic membranes. J. Cell Sci. 117: 4239–4251 [DOI] [PubMed] [Google Scholar]
- 32.Axe E. L., Walker S. A., Manifava M., Chandra P., Roderick H. L., Habermann A., Griffiths G., Ktistakis N. T. 2008. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 182: 685–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jeffries T. R., Dove S. K., Michell R. H., Parker P. J. 2004. PtdIns-specific MPR pathway association of a novel WD40 repeat protein, WIPI49. Mol. Biol. Cell. 15: 2652–2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Proikas-Cezanne T., Waddell S., Gaugel A., Frickey T., Lupas A., Nordheim A. 2004. WIPI-1alpha (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene. 23: 9314–9325 [DOI] [PubMed] [Google Scholar]
- 35.Codogno P., Mehrpour M., Proikas-Cezanne T. 2012. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat. Rev. Mol. Cell Biol. 13: 7–12 [DOI] [PubMed] [Google Scholar]
- 36.Proikas-Cezanne T., Robenek H. 2011. Freeze-fracture replica immunolabelling reveals human WIPI-1 and WIPI-2 as membrane proteins of autophagosomes. J. Cell. Mol. Med. 15: 2007–2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Proikas-Cezanne T., Ruckerbauer S., Stierhof Y. D., Berg C., Nordheim A. 2007. Human WIPI-1 puncta-formation: a novel assay to assess mammalian autophagy. FEBS Lett. 581: 3396–3404 [DOI] [PubMed] [Google Scholar]
- 38.Polson H. E., de Lartigue J., Rigden D. J., Reedijk M., Urbé S., Clague M. J., Tooze S. A. 2010. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy. 6: 506–522 [DOI] [PubMed] [Google Scholar]
- 39.Mauthe M., Jacob A., Freiberger S., Hentschel K., Stierhof Y. D., Codogno P., Proikas-Cezanne T. 2011. Resveratrol-mediated autophagy requires WIPI-1 regulated LC3 lipidation in the absence of induced phagophore formation. Autophagy. 7: 1448–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang C. W., Kim J., Huang W. P., Abeliovich H., Stromhaug P. E., Dunn W. A., Jr, Klionsky D. J. 2001. Apg2 is a novel protein required for the cytoplasm to vacuole targeting, autophagy, and pexophagy pathways. J. Biol. Chem. 276: 30442–30451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shintani T., Suzuki K., Kamada Y., Noda T., Ohsumi Y. 2001. Apg2p functions in autophagosome formation on the perivacuolar structure. J. Biol. Chem. 276: 30452–30460 [DOI] [PubMed] [Google Scholar]
- 42.Reggiori F., Tucker K. A., Stromhaug P. E., Klionsky D. J. 2004. The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev. Cell. 6: 79–90 [DOI] [PubMed] [Google Scholar]
- 43.Obara K., Sekito T., Niimi K., Ohsumi Y. 2008. The ATG18–ATG2 complex is recruited to autophagic membranes via PtdIns(3)P and exerts an essential function. J. Biol. Chem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kobayashi T., Suzuki K., Ohsumi Y. 2012. Autophagosome formation can be achieved in the absence of Atg18 by expressing engineered PAS-targeted Atg2. FEBS Lett. 586: 2473–2478 [DOI] [PubMed] [Google Scholar]
- 45.Velikkakath A. K., Nishimura T., Oita E., Ishihara N., Mizushima N. 2012. Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol. Biol. Cell. 23: 896–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hayashi-Nishino M., Fujita N., Noda T., Yamaguchi A., Yoshimori T., Yamamoto A. 2009. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 11: 1433–1437 [DOI] [PubMed] [Google Scholar]
- 47.Ylä-Anttila P., Vihinen H., Jokitalo E., Eskelinen E. L. 2009. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy. 5: 1180–1185 [DOI] [PubMed] [Google Scholar]
- 48.Tooze S. A., Yoshimori T. 2010. The origin of the autophagosomal membrane. Nat. Cell Biol. 12: 831–835 [DOI] [PubMed] [Google Scholar]
- 49.Pfisterer S. G., Mauthe M., Codogno P., Proikas-Cezanne T. 2011. Ca2+/calmodulin-dependent kinase (CaMK) signaling via CaMKI and AMP-activated protein kinase contributes to the regulation of WIPI-1 at the onset of autophagy. Mol. Pharmacol. 80: 1066–1075 [DOI] [PubMed] [Google Scholar]
- 50.Proikas-Cezanne T., Pfisterer S. G. 2009. Assessing mammalian autophagy by WIPI-1/Atg18 puncta formation. Methods Enzymol. 452: 247–260 [DOI] [PubMed] [Google Scholar]
- 51.Robenek H., Severs N. J. 2008. Recent advances in freeze-fracture electron microscopy: the replica immunolabeling technique. Biol. Proced. Online. 10: 9–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robenek H., Buers I., Hofnagel O., Lorkowski S., Severs N. J. 2009. GFP-tagged proteins visualized by freeze-fracture immuno-electron microscopy: a new tool in cellular and molecular medicine. J. Cell. Mol. Med. 13: 1381–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robenek H., Robenek M. J., Buers I., Lorkowski S., Hofnagel O., Troyer D., Severs N. J. 2005. Lipid droplets gain PAT family proteins by interaction with specialized plasma membrane domains. J. Biol. Chem. 280: 26330–26338 [DOI] [PubMed] [Google Scholar]
- 54.Britschgi C., Rizzi M., Grob T. J., Tschan M. P., Hugli B., Reddy V. A., Andres A. C., Torbett B. E., Tobler A., Fey M. F. 2006. Identification of the p53 family-responsive element in the promoter region of the tumor suppressor gene hypermethylated in cancer 1. Oncogene. 25: 2030–2039 [DOI] [PubMed] [Google Scholar]
- 55.Tschan M. P., Shan D., Laedrach J., Eyholzer M., Leibundgut E. O., Baerlocher G. M., Tobler A., Stroka D., Fey M. F. 2010. NDRG1/2 expression is inhibited in primary acute myeloid leukemia. Leuk. Res. 34: 393–398 [DOI] [PubMed] [Google Scholar]
- 56.Yu W., Cassara J., Weller P. F. 2000. Phosphatidylinositide 3-kinase localizes to cytoplasmic lipid bodies in human polymorphonuclear leukocytes and other myeloid-derived cells. Blood. 95: 1078–1085 [PubMed] [Google Scholar]
- 57.Frickey T., Lupas A. 2004. CLANS: a Java application for visualizing protein families based on pairwise similarity. Bioinformatics. 20: 3702–3704 [DOI] [PubMed] [Google Scholar]
- 58.Brasaemle D. L., Barber T., Wolins N. E., Serrero G., Blanchette-Mackie E. J., Londos C. 1997. Adipose differentiation-related protein is an ubiquitously expressed lipid storage droplet-associated protein. J. Lipid Res. 38: 2249–2263 [PubMed] [Google Scholar]
- 59.Xu G., Sztalryd C., Lu X., Tansey J. T., Gan J., Dorward H., Kimmel A. R., Londos C. 2005. Post-translational regulation of adipose differentiation-related protein by the ubiquitin/proteasome pathway. J. Biol. Chem. 280: 42841–42847 [DOI] [PubMed] [Google Scholar]
- 60.Farese R. V., Jr, Walther T. C. 2009. Lipid droplets finally get a little R-E-S-P-E-C-T. Cell. 139: 855–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shibata M., Yoshimura K., Furuya N., Koike M., Ueno T., Komatsu M., Arai H., Tanaka K., Kominami E., Uchiyama Y. 2009. The MAP1–LC3 conjugation system is involved in lipid droplet formation. Biochem. Biophys. Res. Commun. 382: 419–423 [DOI] [PubMed] [Google Scholar]
- 62.Hellerer T., Axang C., Brackmann C., Hillertz P., Pilon M., Enejder A. 2007. Monitoring of lipid storage in Caenorhabditis elegans using coherent anti-Stokes Raman scattering (CARS) microscopy. Proc. Natl. Acad. Sci. USA. 104: 14658–14663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Riddle D. L., Albert P. S. 1997. Genetic and environmental regulation of dauer larva development. In C. elegans II. 2nd edition. D. L. Riddle, T. Blumenthal, B. J. Meyer, and J. R. Priess, editors. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. Chapter 26. [PubMed] [Google Scholar]
- 64.Kotoulas O. B., Kalamidas S. A., Kondomerkos D. J. 2004. Glycogen autophagy. Microsc. Res. Tech. 64: 10–20 [DOI] [PubMed] [Google Scholar]
- 65.Goldstein J. L., DeBose-Boyd R. A., Brown M. S. 2006. Protein sensors for membrane sterols. Cell. 124: 35–46 [DOI] [PubMed] [Google Scholar]
- 66.Singh R., Xiang Y., Wang Y., Baikati K., Cuervo A. M., Luu Y. K., Tang Y., Pessin J. E., Schwartz G. J., Czaja M. J. 2009. Autophagy regulates adipose mass and differentiation in mice. J. Clin. Invest. 119: 3329–3339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brickner J. H., Fuller R. S. 1997. SOI1 encodes a novel, conserved protein that promotes TGN-endosomal cycling of Kex2p and other membrane proteins by modulating the function of two TGN localization signals. J. Cell Biol. 139: 23–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Park J. S., Neiman A. M. 2012. VPS13 regulates membrane morphogenesis during sporulation in Saccharomyces cerevisiae. J. Cell Sci. 125: 3004–3011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rampoldi L., Dobson-Stone C., Rubio J. P., Danek A., Chalmers R. M., Wood N. W., Verellen C., Ferrer X., Malandrini A., Fabrizi G. M., et al. 2001. A conserved sorting-associated protein is mutant in chorea-acanthocytosis. Nat. Genet. 28: 119–120 [DOI] [PubMed] [Google Scholar]
- 70.Kolehmainen J., Black G. C., Saarinen A., Chandler K., Clayton-Smith J., Traskelin A. L., Perveen R., Kivitie-Kallio S., Norio R., Warburg M., et al. 2003. Cohen syndrome is caused by mutations in a novel gene, COH1, encoding a transmembrane protein with a presumed role in vesicle-mediated sorting and intracellular protein transport. Am. J. Hum. Genet. 72: 1359–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.